

Abstract
The purpose of this study was to investigate the safety profile of SR29142 when administered as a single agent both prior to chemotherapy and during treatment, and to compare the efficacy of SR29142 administered at two dose levels in adult Japanese patients with leukemia or lymphoma. During this open‐label, multicenter, phase II study, patients received SR29142 for 5 days, administered at either 0.15 or 0.20 mg/kg per day. Chemotherapy was started 4–24 h after the first infusion of SR29142. The primary end‐point was overall response rate, defined as the normalization of plasma uric acid to 7.5 mg/dL or less, from 48 h after the first infusion to 24 h after the last infusion of SR29142. SR29142‐related adverse events including hypersensitivity (allergic) reactions were assessed. Overall, 50 patients received SR29142 at either 0.15 mg/kg per day (n = 25) or 0.20 mg/kg per day (n = 25) followed by chemotherapy. The overall response rate was 100.0% (95% confidence interval, 86.3–100.0%) with 0.15 mg/kg and 96.0% (95% confidence interval, 79.6–99.9%) with 0.20 mg/kg. Both dose levels of SR29142 were equally effective at reducing plasma uric acid levels. In six patients, seven drug‐related adverse events of grade 1/2 occurred before chemotherapy. SR29142‐related, hypersensitivity‐associated reactions occurred in three patients, and rash, anorexia, application site pain and pyrexia occurred in one patient each; only five patients (10%) showed anti‐SR29142 antibodies by day 29. In conclusion, SR29142 is effective at reducing plasma uric acid levels with a tolerable safety profile as a single agent both prior to chemotherapy and during treatment. (Trial register: ClinicalTrials.gov, NCT00631579.) (Cancer Sci 2009; 100: 357–362)
Tumor lysis syndrome (TLS) is a metabolic abnormality caused by the rapid killing of tumor cells during chemotherapy and the subsequent release of intracellular metabolites into the circulation.( 1 , 2 ) It is characterized by hyperuricemia, hyperkalemia, hyperphosphatemia, hypocalcemia and renal dysfunction, and despite the availability of several preventative measures, can be a life‐threatening complication. The formation of uric acid crystals in the renal tubule, tissue precipitation of calcium phosphate, renal tumor infiltration, xanthinuria or use of nephrotoxic drugs may cause renal dysfunction. The prevention of TLS is crucial for the effective treatment of hematological malignancies, especially rapidly proliferating neoplasms including acute lymphoblastic leukemia and high‐grade non‐Hodgkin's lymphoma.( 3 ) Despite the introduction of new drugs, such as rituximab and fludarabine, TLS remains one of the most serious complications in the treatment of low‐grade lymphoma and chronic lymphoblastic leukemia. The overall incidence of TLS in hematological malignancies is reported to be approximately 5%.( 4 )
General procedures to prevent TLS are hydration, alkalinization of urine with sodium bicarbonate and allopurinol. Allopurinol inhibits the formation of uric acid, thereby controlling the plasma uric acid level through inhibition of xanthine oxidase, which converts hypoxanthine to xanthine, and xanthine to uric acid.( 5 ) However, allopurinol requires 24–48 h to exert its effect on uric acid synthesis and does not affect pre‐existing uric acid levels. Furthermore, allopurinol causes increases in serum levels of xanthine and hypoxanthine, which may cause xanthine nephropathy; allopurinol also exhibits drug–drug interactions with several purine‐based anticancer drugs.
Rasburicase (SR29142, EC 1.7.3.3, Sanofi‐Aventis, Paris, France) is a recombinant urate oxidase enzyme produced in the yeast Saccharomyces cerevisiae from the cDNA of Aspergillus flavus. SR29142 lowers uric acid levels by converting uric acid to allantoin, which is approximately fivefold more soluble than uric acid and is easily excreted in the urine. Consequently, SR29142 leads (within 4 h) to a rapid decline in uric acid, thereby reducing the risk of TLS. Results of previous clinical trials showed that SR29142 provides early control of hyperuricemia in patients with hyperuricemia prior to dosing and minimizes the risk of hyperuricemia following cytoreductive chemotherapy.( 6 , 7 ) As SR29142 is a recombinant protein, the development of anti‐SR29142 antibodies and hypersensitivity reactions due to this agent are potential major safety issues; however, the relationship between anti‐SR29142 antibodies and hypersensitivity reactions remains unclear. To date, there is little information from long‐term follow‐up studies regarding the development of anti‐SR29142 antibodies.
The aim of this phase II study was to investigate the safety and efficacy of SR29142 as a single agent administered before chemotherapy in Japanese patients with leukemia or lymphoma. In particular, the incidence of SR29142‐related adverse events (AE), including hypersensitivity reactions prior to initiation of chemotherapy, was studied in order to provide an accurate evaluation of the safety of SR29142 monotherapy excluding the effects of chemotherapy.
Materials and Methods
Study design. This was an open‐label, multicenter, phase II study of repeated‐dose SR29142 (0.15 or 0.20 mg/kg for 5 days) as uricolytic therapy for hyperuricemia in adult patients with leukemia or lymphoma.
The primary objective was to evaluate the safety and efficacy of SR29142 in Japanese patients with malignant lymphoma or acute leukemia. Secondary objectives included determination of the pharmacokinetic profile of SR29142 for 10 patients in each dose group, assessment of anti‐SR29142 antibody production and estimation of the optimal dosage of SR29142 for use in Japanese patients. The study was approved by the institutional review boards of all participating institutions. Written informed consent was obtained from all participants before randomization.
A total of 50 Japanese adult patients were recruited and received SR29142 administered at one of two dose levels: 0.15 or 0.20 mg/kg per day. The pharmacokinetics study was conducted in the first 10 patients at each dose level. The safety and efficacy evaluation board reviewed the safety profile of these first 20 patients in order to determine whether the study could be continued.
Patients were classified as being either at high risk or at potential risk of developing TLS‐associated hyperuricemia. High risk was defined as: hyperuricemia of malignancy (plasma uric acid, >7.5 mg/dL); very aggressive lymphoma/leukemia according to the Proposed Clinical Schema for Malignancies of the Lymphoid System( 8 ) based on the Revised European‐American Lymphoma (REAL);( 9 ) acute myelocytic leukemia; chronic myelocytic leukemia in blast crisis; or high‐grade myelodysplastic syndrome with 10% bone marrow blast involvement and undergoing aggressive treatment similar to acute myelocytic leukemia. Potential risk was defined as aggressive lymphoma/leukemia( 8 ) plus one or more of the following criteria: lactate dehydrogenase of twice the upper limit of normal; stage III/IV disease; or stage I/II disease with one lymph node or tumor of more than 5 cm in diameter. In all cases, vital signs were monitored just before administration, then at 10–20 and 30–40 min after initiation of each administration. Standard laboratory tests were performed at baseline, and on days 1, 3, 5, 8, 15, 22, 29 and 36. Creatinine clearance was evaluated on days 1 and 8.
Patient eligibility. To be eligible for the study adult patients (aged 18–74 years) scheduled for chemotherapy for leukemia or lymphoma needed to meet at least one of the following criteria: acute leukemia with a white blood cell count of 20 000/µL or more; stage III/IV malignant lymphoma (not further specified) regardless of plasma uric acid level; stage II malignant lymphoma with bulky disease (defined as a node or nodal mass ≥10 cm, or with maximum mediastinal mass width at least one‐third of the internal transverse diameter of the thorax at the level of T5/6); any leukemia or lymphoma associated with plasma uric acid of 8.0 mg/dL or more and lactate dehydrogenase twice the upper limit of normal. These patients were considered to have potential or a high risk of TLS. A performance status of 3 or less on the Eastern Cooperative Oncology Group scale and an estimated life expectancy of at least 40 days were also required.
Patients were excluded if: they received allopurinol within 72 h prior to the start of SR29142; were scheduled to receive asparaginase; had a known history of significant allergic reactions; had a documented history of asthma or asthmatic bronchitis; were glucose‐6‐phosphate dehydrogenase deficient; or were pregnant or lactating women.
Treatment modalities. SR29142 (0.15 or 0.20 mg/kg) was administered once daily for 5 consecutive days by i.v. infusion over 30 min. The dosing schedule of 0.15 or 0.20 mg/kg was randomly allocated based on the stratification of underlying disease (lymphoma or acute leukemia) and uric acid level (8 mg/dL vs <8 mg/dL). The drug infusion was started at the same time on day 1 through to day 5. Chemotherapy for lymphoma or leukemia was started within 4–24 h after the first dose of SR29142. Between the start of SR29142 administration and the start of chemotherapy, neither prophylactic treatment with antiemetic drugs nor treatment with sodium bicarbonate for alkalinization of the urine was permitted.
Efficacy evaluation. The primary efficacy end‐point was overall response rate (ORR) for SR29142 treatment, defined as the normalization of uric acid levels as determined by assays of plasma uric acid concentration. Treatment was considered to be successful and the patient considered to be a treatment responder if the plasma uric acid level had decreased to 7.5 mg/dL or less 48 h after the start of the first SR29142 infusion and was maintained until 24 h after the start of the final (day 5) SR29142 infusion. Patients who failed to complete 5 days of treatment were classified as non‐responders, even if their uric acid levels were normal.
Secondary end‐points included the rate of plasma uric acid concentration decline over time following the first administration of SR29142, urinary allantoin levels and excretion rate, and renal function (serum creatinine, creatinine clearance, potassium, and phosphorus or calcium levels). Plasma uric acid levels were determined from blood samples collected via polypropylene tubes containing anticoagulant (heparin). To prevent the enzymatic action of SR29142, samples were placed on ice immediately after collection, centrifuged and frozen until measurement. The standard method used at each institution was performed to determine plasma uric acid. Plasma uric acid sampling was performed on: day 1 (just before treatment administration; 4 and 8 h after starting administration); days 2, 3 and 4 (just before treatment administration); day 5 (just before treatment administration; 8 h after starting administration); day 6 (24 h after starting administration on day 5); day 8 (72 h after starting administration on day 5); and day 15. Other hyperuricemia agents, such as allopurinol, were not to be used until after blood sampling for uric acid assay on day 15. The N‐ethyl‐N‐(2‐hydroxy‐3‐sulfopropyl)‐3,5dimethoxy‐4‐fluoranilline and the peroxidase assay methods were used for determination of plasma uric acid concentrations at each institution. Urinary allantoin concentrations were determined by an electrospray ionization liquid chromatography with tandem mass spectrometry method with a limit of quantification of 13.6 mg/mL. Urinary allantoin sampling was performed 24 h before initial drug administration and on days 1–7. SR29142 plasma concentration was determined by a validated immunoenzymometric assay with a limit of quantification of 0.7 ng/mL.
Safety evaluation. Safety assessments were based on clinical observation, standard laboratory tests, vital signs (blood pressure, pulse and body temperature) and the occurrence of AE. The severity of AE was graded according to the National Cancer Institute–Common Toxicity Criteria version 2.0. All AE that occurred during SR29142 monotherapy and during the administration of concomitant chemotherapy were to be recorded on Case Report Forms by the investigator. The relationship (related/not related) of AE to SR29142 was assessed by the individual investigators. Antibody measurement was performed on days 1, 8 and 29 in all patients. Further measurements were conducted in patients who tested positive. Levels of human immunoglobulin (hIg) anti‐SR29142 antibodies from blood samples were determined by enzyme‐linked immunosorbent assay using the following method. All wells of the microplates were coated with SR29142. Standard and circulating hIg were immunofixed to the coated SR29142 standard and were detected with anti‐hIg–peroxidase conjugate. The peroxidase activity was detected with a chromogenic substrate (O‐phenylene‐diamine); and absorption at 492 nm correlated to the amount of anti‐SR29142 antibodies in the plasma.( 10 )
Statistical analysis. All patients who received at least one dose of SR29142 and had at least one post‐baseline efficacy evaluation were included in the efficacy population. All patients who received at least one dose of SR29142 were included in the safety population. Descriptive statistics are provided throughout using an observed‐cases approach. P‐values (two‐tailed Student's t‐test) and 95% confidence intervals (CI) were calculated, with P < 0.05 regarded as significant.
Assuming that the true response rate would be 95%, the probability of observing at least 23 responders among 25 patients treated with each dose of SR29142 would be 87.3%. If 23 responders were observed, the 95% two‐sided confidence lower limit for the response rate would be 74.0%. It could therefore be concluded with 97.5% confidence that the true response rate would be at least 74.0%. In line with clinical findings showing that any anti‐SR29142 antibodies are usually produced within a month of treatment, only patients who had tested positive for the presence of anti‐SR29142 antibodies on day 29 were to be followed up after the study period.
Dose proportionality for area under concentration–time curve from 0–24 h (AUC0–24) and plasma concentration at the end of infusion (Ceoi) were evaluated using the log‐transformed power model. An estimate and 90% CI for the difference in dose group means were computed within the mixed‐model framework and converted to a ratio of adjusted means by the anti‐log transformation. Within‐patient, between‐patient and total variances were estimated for log AUC0–24 by equating observed and expected mean squares within the linear mixed‐effects model described above. The 95% CI for the variances were computed using the simple χ2‐test method for within‐patient variance, the Modified Large Sample procedure for between‐patient variance, and the Graybill–Wang procedure for total‐patient variance. Variance estimates were expressed as standard deviations. All analyzes were carried out by using SAS ver. 8.2 (SAS Institute, Cary, NC, USA).
Results
Patients. Between April 2003 and June 2004, 50 adult Japanese patients with leukemia and/or lymphoma were enrolled in this study from nine centers. Demographic and baseline characteristics are summarized in Table 1. Overall, demographic characteristics were similar between the two dosage groups. A total of five patients (10%) were hyperuricemic at baseline and approximately half (n = 24) had stage IV lymphoma. Thirteen patients (26%) were defined as having a high risk for TLS‐associated hyperuricemia and 37 patients (74%) were defined as having a potential risk.
Table 1.
Baseline characteristics of 50 eligible patients
| SR29142 | Total(n = 50) | ||
|---|---|---|---|
| 0.15 mg/kg(n = 25) | 0.20 mg/kg(n = 25) | ||
| Age (years) | |||
| Median | 51 | 55 | 54 |
| Range | 19–73 | 23–73 | 19–73 |
| Age class, n (%) | |||
| <65 years | 20 (80) | 18 (72) | 38 (76) |
| ≥65 years | 5 (20) | 7 (28) | 12 (24) |
| Sex, n (%) | |||
| Male | 11 (44) | 13 (52) | 24 (48) |
| Female | 14 (56) | 12 (48) | 26 (52) |
| ECOG performance status, n (%) | |||
| 0 | 17 (68) | 17 (68) | 34 (68) |
| 1 | 3 (12) | 4 (16) | 7 (14) |
| 2 | 5 (20) | 2 (8) | 7 (14) |
| 3 | 0 (0) | 2 (8) | 2 (4) |
| Diagnosis, n (%) | |||
| Lymphoma | 21 (84) | 21 (84) | 42 (84) |
| Stage II | 2 (8) | 2 (8) | 4 (8) |
| Stage III | 6 (24) | 8 (32) | 14 (28) |
| Stage IV | 13 (52) | 11 (44) | 24 (48) |
| Acute lymphocytic leukemia | 2 (8) | 0 (0) | 2 (4) |
| Acute myelogenous leukemia | 2 (8) | 4 (16) | 6 (12) |
| Hyperuricemic at baseline (≥8 mg/dL), n (%) | |||
| Yes | 3 (12) | 2 (8) | 5 (10) |
| No | 22 (88) | 23 (92) | 45 (90) |
| Risk category, n (%) | |||
| High | 6 (24) | 7 (28) | 13 (26) |
| Potential | 19 (76) | 18 (72) | 37 (74) |
ECOG, Eastern Cooperative Oncology Group.
Drug administration. Forty‐nine patients (98%) completed 5 days of treatment. One patient who received the 0.20 mg/kg dose did not complete 5 days of treatment due to a severe AE (elevated liver enzymes).
Control of plasma uric acid and excretion of allantoin. Mean plasma uric acid concentrations of both cohorts over time are presented in Figure 1. Uric acid levels declined immediately after the administration of SR29142 and was maintained below 1 mg/dL at the measurement point (4 h after administration) until 120 h. The uric acid concentration was maintained at low levels thereafter (during the concomitant chemotherapy period) and was normalized by day 15 (Fig. 1).
Figure 1.
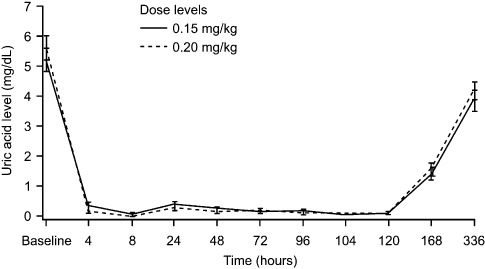
Mean plasma uric acid concentrations by dose over time. Patients with leukemia or lymphoma were randomly allocated (based on stratification by underlying disease and uric acid level) to receive SR29142 administered at either 0.15 or 0.20 mg/kg per day for 5 days, followed by chemotherapy starting from 4 to 24 h after the first infusion of SR29142.
Mean daily urinary allantoin levels in all patients receiving SR29142 at various time points were as follows: pre‐dose, 12.1 mg; day 1, 1280 mg; day 3, 1030 mg; day 5, 897 mg; and day 7368 mg. Compared with background levels, the amounts of allantoin in the urine were increased approximately 100‐fold after SR29142 treatment, with no difference between the two dose levels (data not shown).
Efficacy. The ORR was 100.0% (95% CI, 86.3–100.0%) in the 0.15 mg/kg group and 96.0% (95% CI, 79.6–99.9%) in the 0.20 mg/kg group. The total ORR was 98.0% (95% CI, 89.3–100.0%). One patient in the 0.20 mg/kg treatment group was removed from the study due to a severe AE following the investigator's judgment. Although the uric acid level of this patient decreased to less than 0.1 mg/dL for the 3 days during which they had received SR29142, the final uric acid level was not obtained. Therefore the patient was classified as a non‐responder as defined in the protocol.
Adverse events. Due to the nature and severity of the underlying illness and concomitant chemotherapy, all patients had at least one AE. AE that occurred in 20% or more of patients during the study were similar to those commonly reported for chemotherapy in patients with lymphoma and leukemia (Table 2a). Drug‐related AE judged by the investigators occurred in 23 patients (46%) overall: 10 patients (40%) in the 0.15 mg/kg group and 13 patients (52%) in the 0.20 mg/kg group. The most frequently occurring drug‐related AE were elevated liver enzymes (24%).
Table 2a.
Adverse events occurring in 20% or more of patients
| Adverse event | SR29142 | |||
|---|---|---|---|---|
| 0.15 mg/kg (n = 25) | 0.20 mg/kg (n = 25) | |||
| All gradesn (%) | Grade 3/4n (%) | All gradesn (%) | Grade 3/4n (%) | |
| White blood cells decreased | 24 (96) | 22 (88) | 22 (88) | 21 (84) |
| Neutrophil count decreased | 22 (88) | 22 (88) | 19 (76) | 19 (76) |
| Alopecia | 20 (80) | 0 (0) | 18 (72) | 0 (0) |
| Lymphocyte count decreased | 16 (64) | 15 (60) | 18 (72) | 15 (60) |
| Nausea | 12 (48) | 0 (0) | 15 (60) | 1 (4) |
| Constipation | 10 (40) | 4 (16) | 11 (44) | 3 (12) |
| Aspartate aminotransferase increased | 6 (24) | 0 (0) | 11 (44) | 1 (4) |
| Hemoglobin decreased | 11 (44.0) | 4 (16) | 6 (24) | 0 (0) |
| Platelet count decreased | 8 (32) | 4 (16) | 9 (36) | 3 (12) |
| Alanine aminotransferase increased | 7 (28) | 1 (4) | 8 (32) | 1 (4) |
| Anorexia | 7 (28) | 1 (4) | 8 (32) | 1 (4) |
| Malaise | 7 (28) | 0 (0) | 8 (32) | 0 (0) |
| Vomiting | 6 (24) | 0 (0) | 9 (36) | 0 (0) |
| Diarrhea | 8 (32) | 0 (0) | 4 (16) | 0 (0) |
| Hyperglycemia | 6 (24) | 2 (8) | 5 (20) | 1 (4) |
| Pyrexia | 5 (20) | 0 (0) | 6 (24) | 1 (4) |
| Stomatitis | 3 (12) | 0 (0) | 8 (32) | 1 (4) |
| Blood bilirubin increased | 7 (28) | 0 (0) | 3 (12) | 0 (0) |
| Blood lactate dehydrogenase increased | 5 (20) | 0 (0) | 5 (20) | 1 (4) |
Given the potential risk of anaphylaxis associated with SR29142, it was decided to evaluate the safety of the drug administered as monotherapy during the 5 consecutive days before chemotherapy. Drug‐related AE that occurred before initiation of concomitant chemotherapy are summarized in Table 2(b). Although six patients developed seven drug‐related AE before chemotherapy (SR29142‐related hypersensitivity reactions occurred in three patients during the SR29142‐administration period, and application site pain, pyrexia, anorexia and rash occurred in one patient each), none of these events were categorized as grade 3 or 4 severity and patients recovered immediately.
Table 2b.
SR29142‐related adverse events that occurred before initiation of first chemotherapy
| Adverse events | SR29142 | Total(n = 50)n (%) | |
|---|---|---|---|
| 0.15 mg/kg(n = 25)n (%) | 0.20 mg/kg(n = 25)n (%) | ||
| Application site pain | 0 (0) | 1 (4) | 1 (2) |
| Pyrexia | 1 (4) | 0 (0) | 1 (2) |
| Hypersensitivity | 1 (4) | 2 (8) | 3 (6) |
| Anorexia | 1 (4) | 0 (0) | 1 (2) |
| Rash | 0 (0) | 1 (4) | 1 (2) |
Overall, during the entire study, hypersensitivity reactions, regardless of relationship to study medication, occurred in 35 patients (70%); eight patients (16%) experienced reactions of grade 3 or 4 severity. Hypersensitivity reactions that were likely to be related to SR29142 occurred in 11 patients (22%): five patients in the 0.15 mg/kg group and six patients in the 0.20 mg/kg group. In two patients, both of whom were in the 0.20 mg/kg group, these events were categorized as grade 3 or 4. All hypersensitivity reactions were manageable and resolved with no sequels.
Three patients had serious AE during the study. One patient treated with 0.15 mg/kg developed unstable angina, and in the 0.20 mg/kg group, one patient had sepsis and septic shock, and one patient had elevated liver enzymes (resulting in study discontinuation). Of these events, only the case of increased liver enzymes was considered to be related to SR29142 following assessment by the Efficacy/Safety Evaluation Committee and the investigator. This patient was a 54‐year‐old Japanese woman with stage IV follicular lymphoma. Increased liver enzyme levels (grade 3) were noted before drug administration on day 3 and SR29142 was permanently discontinued. Liver enzyme levels were nearly normalized by day 16. No patients experienced hemolysis or methemoglobinemia and no deaths occurred during the study.
Renal outcome. There were no clinically significant changes from baseline during chemotherapy in the renal parameters: creatinine clearance, potassium, phosphorous, and calcium (data not shown).
Production of anti‐SR29142 antibodies. The time course of anti‐SR29142 antibody production is summarized in Table 3. None of the patients had any anti‐SR29142 antibodies on day 8. Five patients (10%) had the antibodies on day 29 (two in the 0.15 mg/kg group and three in the 0.20 mg/kg group); after 6 months, antibodies were only detected in two patients (both in the 0.20 mg/kg group). After 1 year, one patient had no antibodies; the other had been lost to follow up. Of the 11 patients who experienced SR29142‐related hypersensitivity reactions during the study, one patient tested positive for anti‐SR29142 antibodies. Although hypersensitivity reaction (rash) was observed on day 1, before the start of chemotherapy in this patient, no anti‐SR29142 antibodies had been produced in this period.
Table 3.
Production of SR29142 antibodies
| Sampling date | SR29142 | |||
|---|---|---|---|---|
| 0.15 mg/kg | 0.20 mg/kg | |||
| n | Positive no. (%) | n | Positive no. (%) | |
| Day 1 | 25 | 0 (0) | 25 | 0 (0) |
| Day 8 | 25 | 0 (0) | 24* | 0 (0) |
| Day 29 (± 2 days) | 25 | 2 (8) | 25 | 3 (12) |
| Follow‐up period | ||||
| 3 months (± 2 weeks) | 2 | 0 (0) | 3 | 3 (12) |
| 6 months (± 2 weeks) | 0 | − | 3 | 2 (8) |
| 1 year (± 2 weeks) | 0 | − | 1 | 0 |
One patient was withdrawn from treatment on day 3; this patient was monitored on day 29 but not on day 8.
Pharmacokinetics. The mean SR29142 pharmacokinetics parameters are shown in Table 4. Increased exposure to SR29142, as measured by AUC0–24 and Ceoi, was dose proportional (Fig. 2). For the 1.33‐fold difference in dose between 0.15 and 0.20 mg/kg, the AUC0–24 was 1.28‐fold and 1.31‐fold higher, and Ceoi was 1.11‐fold and 1.27‐fold higher for 0.20 versus 0.15 mg/kg on days 1 and 5, respectively. Steady state was reached between days 2 and 3 and terminal half‐life (t½z) was comparable for both dose groups. The accumulation ratio of AUC0–24 (defined as the ratio of day 5 to 1 AUC0–24) was 1.07 (95% CI, 0.99–1.14), indicating slight accumulation of SR29142.
Table 4.
Pharmacokinetic parameters of SR29142
| SR29142† | ||||||
|---|---|---|---|---|---|---|
| 0.15 mg/kg (n = 11) | 0.20 mg/kg (n = 10) | |||||
| Mean | SD | CV (%) | Mean | SD | CV (%) | |
| Day 1 | ||||||
| AUC0–24 (ng.h/mL) | 45 653 | 7544 | 17 | 59 333 | 15 849 | 27 |
| Ceoi (ng/mL) | 3734 | 1081 | 29 | 4239 | 1556 | 37 |
| Day 5 | ||||||
| AUC0–24 (ng.h/mL) | 48 210 | 9660 | 20 | 65 154 | 22 713 | 35 |
| t½z (h) | 22.5 | 5.8 | 26 | 16.1 | 5.6 | 35 |
| Ceoi (ng/mL) | 3948 | 710 | 18 | 5126 | 1468 | 29 |
| Cmin (ng/mL) | 852 | 269 | 32 | 1033 | 494 | 48 |
SR29142 was administered once daily for 5 consecutive days. AUC0–24, area under the concentration–time curve for 0–24 h; Ceoi, plasma concentration of end of infusion; Cmin, minimum plasma concentration; CV, coefficient of variance; SD, standard deviation; t1/2z, terminal elimination half‐life.
Figure 2.
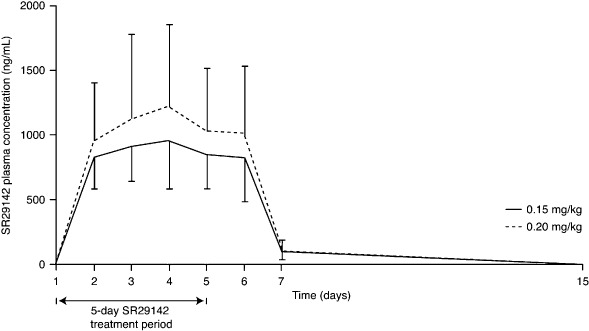
Mean (standard deviation) SR29142 plasma Ceoi concentrations after once‐daily repeated 30‐min i.v. infusions for a total of 5 days. Patients with leukemia or lymphoma were randomly allocated (based on stratification by underlying disease and uric acid level) to receive SR29142 administered at either 0.15 or 0.20 mg/kg per day for 5 days, followed by chemotherapy starting from 4 to 24 h after the first infusion of SR29142.
Discussion
SR29142 is used as a supportive drug in patients with cancer and is administered with concomitant chemotherapeutic agents. This is the first study to evaluate pharmacokinetics and AE related to SR29142 before the initiation of chemotherapy in Japanese patients with hematological malignancies. As expected, the majority of AE can be attributed to the patients’ underlying cancer status and/or concomitant cytotoxic drug administration. In this study, six patients had a total of seven AE, including three hypersensitivity reactions, before the first dose of chemotherapy. These toxicities, however, were of grade 1 or 2 severity and were all manageable. After the first dose of chemotherapy, no unexpected SR29142‐related AE were observed. These results show that single‐agent SR29142 has an associated low‐toxicity profile in adult patients with lymphoma and acute leukemia.
Because SR29142 is a recombinant protein that is exogenous to humans, the production of antibodies to this agent is a potential concern, although the clinical significance of the development of anti‐SR29142 antibodies remains unknown. In this study, 10% of patients developed anti‐SR29142 antibodies in line with previously reported data,( 11 ) and none of the patients had any anti‐SR29142 antibodies on day 8. Importantly, of the 11 patients who experienced hypersensitivity reactions likely to be related to SR29142, only one patient tested positive for SR29142‐related antibodies. In one patient, although hypersensitivity reaction (rash) was observed on day 1, before the start of chemotherapy, it was confirmed that no anti‐SR29142 antibodies were produced during this period.
The concentration of plasma uric acid was controlled rapidly with SR29142 and the ORR was 98%. Additionally, the ability of SR29142 to prevent hyperuricemia was further supported for both doses by the appearance of large amounts of urinary allantoin, the end product of uric acid metabolism by SR29142, and a marker of its activity. The finding that renal function remained stable during the study indicates the ability of SR29142 to indirectly prevent TLS. These results are consistent with those reported in patient populations in European and North American countries.( 11 , 12 , 13 , 14 , 15 , 16 )
The pharmacokinetic findings of the current study support the premise that SR29142 exerts dose‐proportional effects. Furthermore, the accumulation ratio of AUC0–24 showed that there was a slight accumulation of SR29142 during the study. These results are comparable with those reported in European and North American populations.( 17 ) Therefore, no ethnic differences are associated with the pharmacokinetics of SR29142.
Both doses (0.15 and 0.20 mg/kg) of SR29142 were safe and effective under the study conditions. No differences in the toxicity or efficacy profiles were observed between the two dose groups. As this is a phase II study, the optimal dose cannot be defined definitely, but previous findings show that 0.20 mg/kg is well tolerated in adult patients with non‐Hodgkin's lymphoma.( 12 ) Furthermore, recent guidelines for the management of patients with TLS( 18 ) report that rasburicase 0.20 mg/kg is appropriate for seriously ill patients with baseline hyperuricemia or for those who are at high risk of developing TLS, whereas the 0.15 mg/kg dose should be used in patients without baseline hyperuricemia but who have a potential risk of TLS.
In conclusion, SR29142 is highly effective as a supportive drug during chemotherapy to control hyperuricemia, which can induce TLS in adult patients with malignant lymphoma and acute leukemia. SR29142 was well tolerated, with a good safety profile when administered as a single agent prior to the commencement of chemotherapy.
Acknowledgments
We thank the investigators at the following participating institutions as members of the Safety/Efficacy Evaluation Committee: K. Sawada, MD (Akita University School of Medicine); K. Oshimi, MD (Juntendo University School of Medicine); and K. Dan, MD (Nippon Medical University). We also thank Sanofi‐Aventis (Paris, France) for their help. Participating centers are as follows: Aichi Cancer Center (Y. Morishima, M. Ogura), National Hospital Organization, Nagoya Medical Center (M. Hamaguchi); Tokai University School of Medicine (T. Hotta); Hamamatsu University School of Medicine (K. Ohnishi); Tokyo Metropolitan Komagome Hospital (T. Sasaki, H. Sakamaki); Fukuoka University Hospital (K. Tamura); The Jikei University School of Medicine (N. Usui); and Tohoku University Hospital (K. Ishizawa, H. Yokoyama, H. Harigae). This study was sponsored by Sanofi‐Aventis. The authors have no conflicts of interest to declare.
References
- 1. Cairo MS, Bishop M. Tumor lysis syndrome: new therapeutic strategies and classification. Br J Haematol 2004; 127: 3–11. [DOI] [PubMed] [Google Scholar]
- 2. Del Toro G, Moris E, Cairo MS. Tumor lysis syndrome: pathophysiology, definition, and alternative treatment approaches. Clin Adv Hematol Oncol 2005; 3: 54–61. [PubMed] [Google Scholar]
- 3. Cheson BD, Frame JN, Vena D, Quashu N, Sorensen JM. Tumor lysis syndrome: an uncommon complication of fludarabine therapy of chronic lymphocytic leukemia. J Clin Oncol 1998; 16: 2313–20. [DOI] [PubMed] [Google Scholar]
- 4. Annemans L, Moeremans K, Lamotte M et al . Incidence, medical resource utilisation and costs of hyperuricemia and tumour lysis syndrome in patients with acute leukaemia and non‐Hodgkin's lymphoma in four European countries. Leuk Lymphoma 2003; 44: 77–83. [DOI] [PubMed] [Google Scholar]
- 5. Hagemeister F, Huen A. The status of allopurinol in the management of tumor lysis syndrome: a clinical review. Cancer J 2005; 11 (Suppl. 1): S1–S10. [PubMed] [Google Scholar]
- 6. Ronco C, Inguaggiato P, Bordoni V et al . Rasburicase therapy in acute hyperuricemia and renal dysfunction. Contrib Nephrol 2005, 147: 115–23. [DOI] [PubMed] [Google Scholar]
- 7. Oldfield V, Perry CM. Spotlight on rasburicase in anticancer therapy‐induced hyperuricemia. Biodrugs 2006; 20: 197–9. [DOI] [PubMed] [Google Scholar]
- 8. Hiddemann W, Longo DL, Coiffier B et al . Lymphoma classification – the gap between biology and clinical management is closing. Blood 1996; 88: 4085–9. [PubMed] [Google Scholar]
- 9. Harris NL, Jaffe ES, Stein H et al . A revised European‐American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood 1994; 84: 1361–92. [PubMed] [Google Scholar]
- 10. Crowther JR. The ELISA guidebook. In: Walker JM ed. Methods in Molecular Biology, Vol. 149. Totowa, NJ, USA: Humana Press Inc., 2001; 64, 395. [DOI] [PubMed] [Google Scholar]
- 11. Pui CH, Mahmoud HH, Wiley JM et al . Recombinant urate oxidase for the prophylaxis or treatment of hyperuricemia in patients with leukemia or lymphoma. J Clin Oncol 2001; 19: 697–704. [DOI] [PubMed] [Google Scholar]
- 12. Coiffier B, Mounier N, Bologna S et al . Efficacy and safety of rasburicase (recombinant urate oxidase) for the prevention and treatment of hyperuricemia during induction chemotherapy of aggressive non‐Hodgkin's lymphoma: Results of GRAAL1 (Groupe d’Etude des Lymphomes de l’Adulte Trial on Rasburicase Activity in Adult Lymphoma) study. J Clin Oncol 2003; 21: 4402–6. [DOI] [PubMed] [Google Scholar]
- 13. Goldman SC, Holcenberg JS, Finklestein JZ et al . A randomized comparison between rasburicase and allopurinol in children with lymphoma or leukemia at high risk for tumor lysis. Blood 2001; 97: 2998–3003. [DOI] [PubMed] [Google Scholar]
- 14. Jeha S, Kantarjian H, Irwin D et al . Efficacy and safety of rasburicase, a recombinant urate oxidase (Elitek), in the management of malignancy‐associated hyperuricemia in pediatric and adult patients: final result of a multicenter compassionate use trial. Leukemia 2005; 19: 34–8. [DOI] [PubMed] [Google Scholar]
- 15. Pui CH, Jeha S, Irwin D, Camitta B. Recombinant urate oxidase (rasburicase) in the prevention and treatment of malignancy‐associated hyperuricemia in pediatric and adult patients: results of a compassionate‐use trial. Leukemia 2001; 15: 1505–9. [DOI] [PubMed] [Google Scholar]
- 16. Wang LY, Shih LY, Chang H et al . Recombinant urate oxidase (rasburicase) for the prevention and treatment of tumor lysis syndrome in patients with hematologic malignancies. Acta Haematol 2006; 115: 35–8. [DOI] [PubMed] [Google Scholar]
- 17. Pui CH. Rasburicase: a potent uricolytic agent. Expert Opin Pharmacother 2002; 3: 433–42. [DOI] [PubMed] [Google Scholar]
- 18. Coiffier B, Altman A, Pui CH et al . Guidelines for the management of pediatric and adult tumor lysis syndrome: an evidence‐based review. J Clin Oncol 2008; 26: 2767–78. [DOI] [PubMed] [Google Scholar]