

Abstract
Management strategies of chronic phase chronic myelogenous leukemia (CML) have been revolutionized due to the discovery of a selective tyrosine kinase inhibitor, imatinib (Gleevec, STI571), which is substantially improving median survival. However, emergence of imatinib‐resistance has put up a serious problem that requires novel treatment methods. Catechins, polyphenolic compounds in green tea, are gathering much attention due to their potential antitumor effects. So far (–)‐epigallocatechin‐3‐gallate (EGCG), the most abundant component of catechin, has been shown to cause typical apoptosis in several tumor cell lines in most cases through activation of caspases. In this study, we showed that EGCG predominantly caused necrosis‐like cell death via a caspase‐independent mechanism in CML cells, K562 and C2F8, whereas imatinib induced the typical apoptotic cell death. Moreover, this caspase‐independent cell death partially mediated the release of apoptosis‐inducing factor, AIF, and serine protease, HtrA2/Omi, from the mitochondria to cytosol. In addition, EGCG enhanced the imatinib‐induced cell death (P < 0.01) resulting in additive cell death in K562 cells and EGCG alone, effectively reduced the viability of imatinib‐resistant K562 cells (P < 0.01). Catechin is a possible candidate for an antitumor agent that causes cell death in CML cells via a caspase‐independent mechanism. (Cancer Sci 2009; 100: 349–356)
Imatinib (Gleevec, STI571), currently the first choice drug for chronic phase chronic myelogenous leukemia (CML), was developed as a selective tyrosine kinase inhibitor of BCR/ABL fusion protein, contributing to uncontrolled cellular proliferation and anti‐apoptotic mechanism of CML. Imatinib was molecularly designed to bind to the adenosine triphosphate (ATP)‐binding pocket of the BCR/ABL shutting down its downstream signaling, and subsequently facilitates the apoptotic pathway via the release of cytochrome c and caspase activation.( 1 ) However, there has been the continuously arising problem of imatinib‐resistance, mainly due to point mutations within the ATP‐binding site as well as gene amplification of the BCR/ABL gene. Although more potent tyrosine kinase inhibitors such as dasatinib and nilotinib are now undergoing clinical trials to overcome the imatinib‐resistance, emergence of novel mutations counteracting these drugs will always be a concern. Therefore, the search for drugs with novel targets, or those that induce cell death independent of the apoptotic pathway, may be beneficial for the management of CML.
Apoptosis is a cell suicide program essential for development and also used for the chemotherapy‐induced cell death mechanism. Most anticancer agents trigger the mitochondrial apoptotic pathway involving outer mitochondrial membrane permeabilization (MMP).( 2 ) This process is controlled by the pro‐ and anti‐apoptotic members of the Bcl‐2 family and leads to the cytosolic release of mitochondrial intermembrane proteins, including cytochrome c, leading to caspase activation. Other mitochondrial proteins such as apoptosis‐inducing factor (AIF) and endonuclease G (Endo G) are endonucleases that can cause caspase‐independent apoptosis.( 3 ) HtrA2/Omi has a dual role in cell death through inhibition of inhibitor of apoptosis (IAP) proteins leading to apoptosis and in caspase‐independent non‐apoptotic cell death.( 4 )
Polyphenolic compounds in green tea, including (–)‐epigallocatechin‐3‐gallate (EGCG), (–)‐epigallocatechin (EGC), (–)‐epicatechin‐3‐gallate (ECG) and (–)‐epicatechin (EC), have been widely studied since they showed selective cytotoxicity to cancerous cells and not to their counterparts.( 5 ) EGCG is the most abundant and biologically active polyphenol with antioxidant activity in green tea, and the majority of mechanistic studies have focused on this compound.( 6 ) Antitumor activity of EGCG has been reported to result from inhibition of multiple signaling pathways,( 7 ) including cell cycle arrest,( 8 , 9 ) activation of apoptosis via mitochondrial pathway( 10 , 11 , 12 ) and via the death receptor (Fas) pathway,( 13 ) inhibition of telomerase activity( 14 , 15 ) and inhibition of metastasis via binding to LR67.( 16 , 17 , 18 ) Although several mechanisms for EGCG‐induced anticancer activity have been proposed, they generally shared a common final phase of apoptosis.
However, our preliminary data on Wright–Giemsa stained CML cells after EGCG treatment showed atypical cell death morphology where apoptotic morphology was absent. This prompted us to specify the mode of EGCG‐induced cell death in CML cell lines based on biochemical and morphological approaches, not only to clarify its antitumor activity, but also on the hypothesis that if EGCG induced non‐apoptotic cell death, it may be clinically beneficial in overcoming both imatinib‐sensitive and ‐resistant CML cells, often with apoptosis‐resistant characteristics due to constitutively active BCR/ABL suppressing the apoptotic pathway.
In the present work, we used CML cell lines, K562 and C2F8, which were treated with a non‐toxic concentration of EGCG to evaluate the mode of cell death on the criteria of mitochondrial and plasma membrane permeability, caspase activation, mitochondrial death signaling and cell death morphology. We have demonstrated that EGCG predominantly induced necrosis‐like cell death in these CML cell lines without caspase activation and PARP cleavage, the hallmarks of the apoptotic pathway. Also, EGCG effectively caused cell death in imatinib‐resistant K562 cells suggesting that it is a possible candidate to overcome imatinib‐resistant CML cells.
Materials and Methods
Cells. Chronic myelogenous leukemia cell lines, K562 and imatinib‐resistant K562 cells (K562/sti) were generously provided by Dr Yukimasa Shiotsu (Kyowa Hakko Kogyo, Shizuoka, Japan), and C2F8 cells by Dr Tatsuo Furukawa (Nigata University, Nigata, Japan). Cells were cultured in RPMI‐1640 (Sigma, St Louis, MO, USA) supplemented with 10% fetal bovine serum (JRH Biosciences, Lenexa, KS, USA) in the presence of antibiotics (penicillin and streptomycin) (Wako Chemicals, Osaka, Japan) in a humidified incubator of 5% CO2 at 37°C.
Reagents. Imatinib was purchased from Sequoia Research Products (Oxford, UK). A purified preparation of EGCG was generously provided by Dr Yukihiko Hara, Mitsui‐Norin (Shizuoka, Japan). Z‐VAD‐FMK was purchased from Peptide Institute (Osaka, Japan). Propidium iodide (PI), 3,3′‐dihexyloxacarbocyanine iodide (DiOC6[3]), and protease inhibitor cocktail were purchased from Sigma. Z‐VAD‐FMK and imatinib were dissolved in dimethylsulfoxide (DMSO) where the concentration was kept under 0.1% throughout all the experiments to avoid its cytotoxicity. EGCG and DiOC6(3) were dissolved in ethanol followed by appropriate dilution with phosphate‐buffered saline (PBS), and PI was dissolved in water.
Cell viability assay. Cell viability was measured using CellTitre‐Glo Luminescent Cell Viability Assay (Promega, Madison, WI, USA) according to the manufacturer's instructions. Briefly, 1 × 105 cells were incubated with EGCG or imatinib alone or in combination for 48 h before spectrophotometric measurement. Cell survival was expressed as the fraction of control samples.
Cell death and mitochondrial transmembrane potential. Cell membrane permeability was assessed using PI staining and mitochondrial transmembrane potential (ΔΨm) was determined using DiOC6(3) that stains the intact mitochondria. Briefly, 1.0 × 106 cells were incubated with 5 µg/mL PI and 40 nM DiOC6(3) at 37°C for 15 min, and after washing with PBS three times, the fluorescence was measured using FL1 and FL3 channels of FACS Caliber (Becton Dickinson, Franklin Lakes, NJ, USA).
Subcellular fractionation and western blotting. The mitochondria‐free cytosol and mitochondria were isolated according to Yang et al.( 19 ) Briefly, K562 cells were resuspended in ice‐cold isotonic buffer and were passed through a needle. Unlyzed cells and nuclei were pelleted by centrifugation for 10 min at 750 g. The supernatant was then centrifuged at 100 000 g for 15 min. This pellet, representing the mitochondrial fraction, was resuspended in isotonic buffer. The supernatant was again centrifuged at 100 000 g for 1 h to give mitochondria‐free cytosolic fraction.
The cytosolic and mitochondrial protein (30 µg) and cell lysate protein (20 µg) were subjected to 15% sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS‐PAGE) and transferred onto a polyvinylidine difluoride membrane (Hybond‐P) (Amersham Biosciences, Little Chalfont, UK). Antibody binding was detected with a chemiluminescence kit and ECL film (Amersham Biosciences) according to standard protocol. The antibodies presented were: mouse anticytochrome c, mouse anti‐Bcl‐2 and mouse anti‐Bax (Pharmingen, Becton Drive, NJ, USA), mouse anti‐HtrA2/Omi (MBL International, Woburn, MA, USA), rabbit anti‐AIF and mouse anti‐survivin (Santa Cruz Biotechnology, Delaware, NJ, USA), anti‐endo G (Novus Biologicals, Littleton, CO, USA), mouse anti‐caspase‐8, rabbit anti‐caspase 3 and 9, and rabbit anti‐XIAP (Cell Signaling Technology, Danvers, MA, USA), mouse anti‐PARP (WAKO Chemicals, Osaka, Japan) and secondary antibodies conjugated with horseradish peroxidase (DAKO, A/S, Denmark). The blots were stripped and reprobed with rabbit anti‐β‐actin antibody to ensure equality of lane loading (Santa Cruz Biotechnology).
Immunocytochemistry. Before and after EGCG and imatinib treatment alone, cells were collected and washed with PBS. After the fixation in 4% paraformaldehyde, cells were permeabilized in 0.1% triton‐X in PBS for 5 min. These cells were then blocked in 1% bovine serum albumin solution for 30 min. The cells were incubated in rabbit anti‐AIF (Santa Cruz Biotechnology), rabbit anti‐HtrA2/Omi (Alexis Biochemicals, Lausen, Switzerland), mouse anti‐MTCO1 (Abcam, Cambridge, UK) for 1 h at room temperature, and after washing with PBS twice, cells were subsequently incubated with Alexa fluor 594 goat anti‐mouse or Alexa fluor 488 goat anti‐rabbit secondary antibody (Molecular Probes, Eugene, OR, USA) for 1 h at room temperature, and nuclei were stained with FluoroGuard Antifade (Bio‐Rad, Hercules, CA, USA) containing 4′,6‐diamidino‐2‐phenylindole (DAPI) (Sigma). The slides were viewed using a Zeiss LSM 510 laser confocal microscope (Carl Zeiss, Jena, Germany).
Morphological studies of cell death. Detailed cell death morphology was observed using an electron microscope. Control and EGCG‐treated cells were washed with PBS, and fixed in 0.05 M phosphate buffered (pH 7.4) mixture of 2.5% glutaraldehyde, followed by postfixation for 2 h in phosphate buffered 1% osmium tetroxide. The cells were then dehydrated progressively in ethanol/water series followed by propylene oxide and embedded in epoxy resin. Semi‐thin sections were cut with glass knives and stained with toluidine blue to check orientation. Ultrathin sections were stained with uranyl acetate and lead citrate, and examined by a JEM‐1200 EX II electron microscope (JEOL, Tokyo, Japan). Toluidine blue‐stained cells were also used to quantify the percentage of cells showing apoptotic or necrotic cell death morphologies.
Statistical analysis. All data were expressed as the mean ± standard deviation. Differences between samples were analyzed by Student's t‐test. P < 0.01 and P < 0.05 were considered significant.
Results
EGCG induced cell death in CML cells. To verify the effect of EGCG on cell growth, K562 and C2F8 cells were treated with increasing concentrations of EGCG for 48 h, and cell survival was assessed by measuring intracellular ATP content. EGCG reduced the viability of K562 and C2F8 cells dose‐dependently with mean IC50 values of 125 µM and 87.5 µM, respectively, at 48 h; and imatinib with a mean IC50 of 120 nM for both cell lines (Fig. 1A). EGCG (200 µM) reduced the viability of K562 cells to 39 ± 5.5% and of C2F8 cells to 28 ± 4% (Fig. 1A), but had no significant effect on the healthy human peripheral blood mononuclear cells (PBMC) (Supplementary Fig. S1), indicating specific toxicity of EGCG towards leukemic cells.
Figure 1.
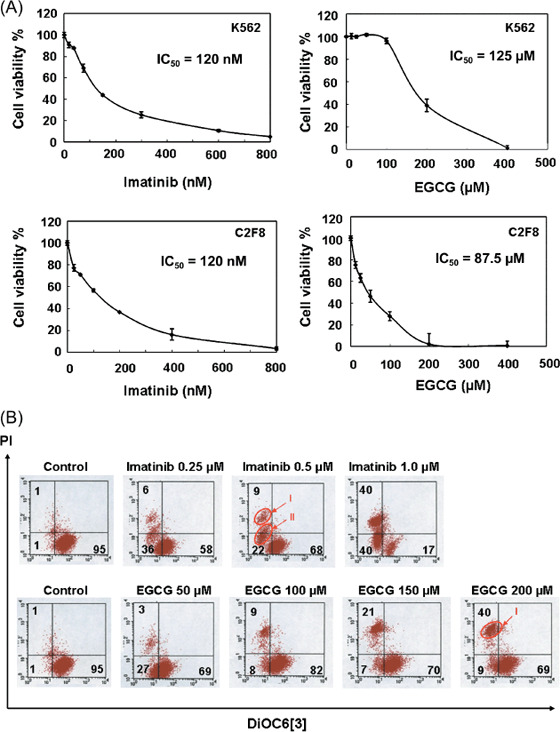
(A) (–)‐Epigallocatechin‐3‐gallate (EGCG) and imatinib both induce cell death in chronic myelogenous leukemia (CML) cells in a dose‐dependent manner. K562 and C2F8 cells were seeded at a density of 5 × 103 cells per well in 96‐well plates, and treated with increasing concentrations of EGCG or imatinib for 48 h, and the number of viable cells were measured based on intracellular adenosine triphosphate (ATP) content. Viable cells are expressed as a percentage of 0.1% ethanol‐treated control cells for each cell line. Values are representatives of three independent experiments. (B) EGCG and imatinib showed distinct cell death patterns. K562 cells were treated with 0.1% ethanol (control), EGCG or imatinib for 48 h, harvested and incubated with 3,3′‐dihexyloxacarbocyanine iodide (DiOC6[3]) and propidium iodide (PI). Mitochondrial transmembrane potential (Δψm) and cell death were determined by dual‐parameter flow cytometry. Results are representative of three independent experiments.
Mitochondrial membrane permeability and cell death were analyzed by flow cytometry (Fig. 1B). Cells were stained with DiOC6(3) and PI, an indicator of mitochondrial and plasma membrane permeability, respectively, for fluorescence‐activated cell sorter (FACS) analysis as previously reported.( 20 ) Untreated K562 cells exhibited a DiOC6(3)high/PI− pattern showing viable cells. K562 cells treated with 200 µM EGCG for 48 h exhibited a DiOC6(3)low/PI+ pattern (population I), showing a dead population with reduced MMP and loss of plasma membrane integrity (Fig. 1B). K562 cells treated with 1 µM imatinib contained a population of DiOC6(3)low/PI− (population II), apoptotic population typically with reduced MMP and still intact plasma membrane integrity, as well as DiOC6(3)low/PI+ pattern (population I) (Fig. 1B). This distinct cell death pattern was also observed for C2F8 cells (Supplementary Fig. S2). EGCG and imatinib induced distinct cell death populations suggesting the distinct modes of cell death.
EGCG‐induced cell death was independent of caspase cascade. To ascertain whether EGCG induced a non‐apoptotic cell death, activation of caspases, key executioners in apoptotic pathway, were examined in EGCG‐induced cell death by western blotting using antibodies that recognize both active and inactive forms of caspases. Although imatinib‐treated K562 and C2F8 cells showed activation of all caspases tested and PARP cleavage at 24 h (Fig. 2A; Supplementary Fig. S3), EGCG‐treated K562 and C2F8 cells showed partial activation of caspases‐9 and caspase‐8, but the most downstream effector caspase‐3 and PARP were clearly not activated with exposure to either 100 µM or 200 µM EGCG at 24 h (Fig. 2A; Supplementary Fig. S3). To confirm this result, the cells were treated with either 200 µM EGCG/1 µM imatinib alone or after pretreatment with Z‐VAD‐FMK, broad caspase inhibitor, for 48 h. Imatinib‐induced apoptosis was prevented by Z‐VAD‐FMK dose‐dependently (P < 0.01) though complete rescue was not achieved (Fig. 2B). However, EGCG‐induced cell death was not prevented at all by any concentrations of Z‐VAD‐FMK (P < 0.01) (Fig. 2B), suggesting that EGCG induced predominantly caspase‐independent cell death.
Figure 2.
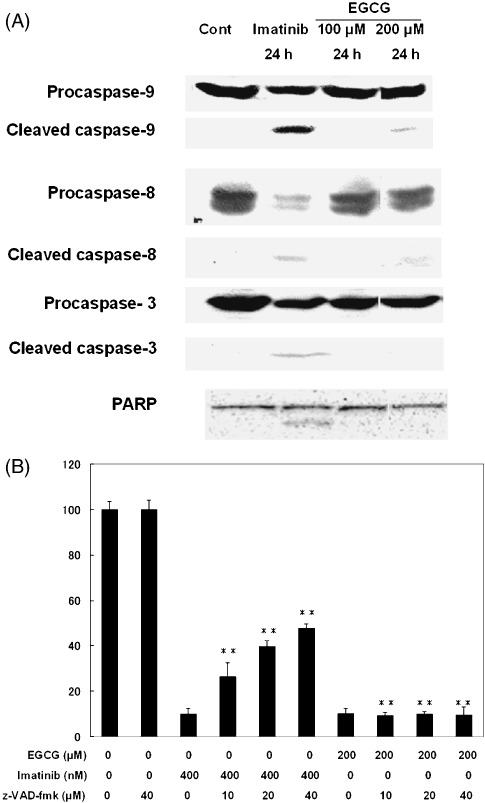
(A) (–)‐Epigallocatechin‐3‐gallate (EGCG)‐induced cell death is caspase‐independent. K562 cells were treated with 0.1% ethanol (control), EGCG or imatinib for 24 h, and subjected to Western blotting. Activation of caspase‐9, caspase‐8 and caspase‐3 are indicated by cleaved fragments of 37, 43 and 15 kDa, respectively. Activation of PARP responsible for caspase‐dependent apoptosis is indicated by cleaved fragment of the 89‐kDa band. (B) Z‐VAD‐FMK fails to prevent EGCG‐induced cell death. K562 cells were pre‐incubated with 0.1% ethanol (control) or increasing doses of Z‐VAD‐FMK for 1 h and subsequently treated with or without 200 µM EGCG or 1 µM imatinib for 48 h. The cell viability was measured based on intracellular adenosine triphosphate (ATP) content. Asterisks indicate significant difference from single imatinib treatment and significant indifference for EGCG‐treated cells (P < 0.01).
EGCG did not alter the expression levels of the Bcl‐2 and IAP family proteins. Because both EGCG‐ and imatinib‐induced cell deaths contained a DiOC6(3)low pattern suggesting the involvement of mitochondrial dysfunction in EGCG‐induced cell death, we examined the upstream regulators of mitochondrial membrane permeability. Mitochondrial integrity is strictly regulated by anti‐ and pro‐apoptotic Bcl‐2 family members such as Bcl‐2 and Bax.( 21 , 22 , 23 ) EGCG treatment did not alter the expression levels of Bcl‐2 and Bax, whereas imatinib treatment resulted in reduction of Bcl‐2 protein expression level at 24 h (Fig. 3A), indicative of facilitation of apoptotic pathway in imatinib‐ but not in EGCG‐induced cell death. Also, IAP proteins, Bcl‐2 family members, are known as negative regulators of apoptosis that inhibit the caspase activity as a safeguard mechanism. XIAP and survivin are two IAP proteins which were downregulated by imatinib treatment at 24 h but their expression levels were not modulated by EGCG treatment (Fig. 3A), suggesting that EGCG did not interfere with the safeguard apoptotic pathway.
Figure 3.
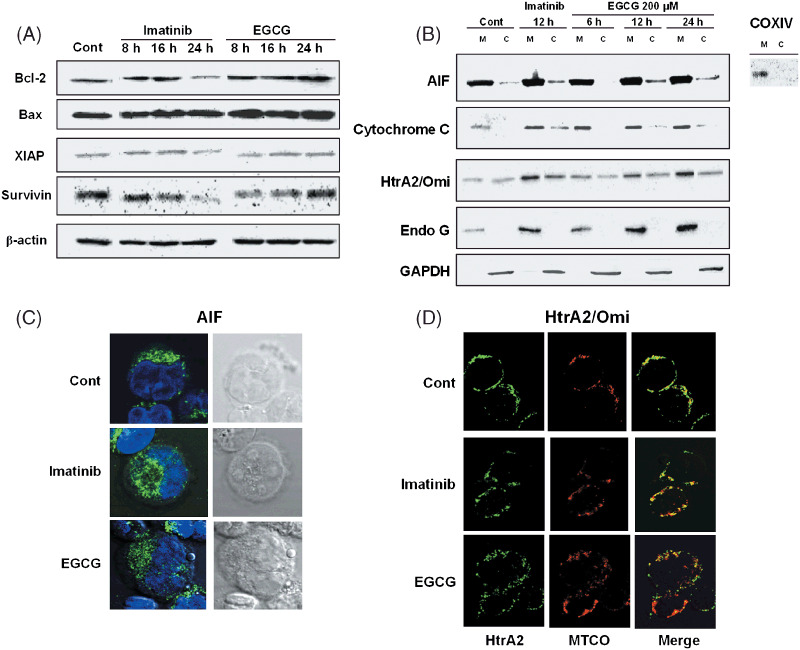
(A) Expression analysis of Bcl‐2 family proteins after (–)‐epigallocatechin‐3‐gallate (EGCG) and imatinib treatment. K562 cells treated with 0.1% ethanol (control), EGCG and imatinib were harvested at indicated times for western blot analysis with indicated antibodies. β‐Actin was used as loading control of cytoplasmic fractions. (B) Release of apoptosis‐inducing factor (AIF) and HtrA2/Omi contribute partly to EGCG‐induced cell death. K562 cells were treated with 0.1% ethanol (control), EGCG and imatinib for the indicated times, and subjected to western blot analysis of mitochondrial proteins following subcellular fractionation. M, mitochondrial fraction; C, cytosolic fraction. COXIV is an antibody against mitochondria‐specific protein used to check the purity of mitochondria fractions. Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) was used as loading control of cytoplasmic fractions. Results are representative of three independent experiments. (C) Nuclear translocation of AIF was visualized after EGCG and imatinib treatment. K562 cells (2 × 105 cells) were treated with 0.1% ethanol, EGCG and imatinib for 24 h, followed by immunofluorescent staining of AIF (green fluorescence) and nuclei staining with 4′,6‐diamidino‐2‐phenylindole (DAPI) (blue fluorescence). (D) Release of HtrA2/Omi from the mitochondria to cytosol was visualized after EGCG and imatinib treatment. K562 cells (2 × 105 cells) were treated with 0.1% ethanol, EGCG and imatinib for 24 h, followed by immunofluorescent staining of HtrA2/Omi (green fluorescence) and mitochondrial staining with MTCO1 antibody (red fluorescence). Endo G, endonuclease G.
Caspase‐independent factors contributed partly to EGCG‐induced cell death. To verify the role of caspase‐independent pro‐apoptotic factors in mitochondria, given the reduction of MMP without caspase‐dependency, we next examined the subcellular expression of the mitochondrial proteins on mitochondrial and cytoplasmic fractions by western blotting. EGCG and imatinib triggered the release of AIF and HtrA2/Omi from mitochondria to cytosol along 6–24 h peaking at around 12 h (Fig. 3B). Cytosolic release of cytochrome c was minimal for EGCG‐treated cells whereas the release was higher in imatinib‐treated cells at 12 h (Fig. 3B). However, EGCG‐triggered cytochrome c release was not sufficient to cause activation of downstream caspase‐3 (Fig. 2A). The endonuclease G release was absent in neither imatinib nor EGCG‐treated K562 cell cytoplasmic fractions. These results suggest that EGCG triggers caspase‐independent cell death, at least in part, via the AIF/HtrA2 pathway.
Subcellular localization of AIF and HtrA2/Omi verified with immunocytochemistry was consistent with the western blot results. Untreated K562 cells showed punctuated AIF expression in the cytoplasmic compartment within the mitochondria while the EGCG‐treated cells at 24 h showed a more diffused expression pattern with a low level of nuclear AIF translocation (Fig. 3C). In HtrA2/Omi and MTCO1 double staining, control cells showed a clear co‐localized expression (yellow) whereas both imatinib and EGCG‐treated cells showed regional disruption of this co‐localization showing the partial release of HtrA2/Omi from the mitochondria to cytosol (green and red) (Fig. 3D).
EGCG predominantly induced necrosis‐like cell death morphology. Because a clearer delineation of the mode of cell death was based on morphological studies,( 24 ) we next examined the morphological differences in EGCG‐treated and imatinib‐treated cell deaths in K562 and C2F8 cells by transmission electron microscopy. Ethanol‐treated control cells exhibited healthy cell morphology with wavy surface, intact nucleus, clear mitochondria with inner cristae and endoplasmic reticulum (ER) (Fig. 4A, 4B). Imatinib‐treated cells exhibited a typical apoptotic feature with smoothing of the cell surface, nuclear fragmentation with compaction of chromatin to the crescents adjacent to the nuclear envelope and rather structurally intact mitochondria (Fig. 4A, 4B). ATP‐depleted cells for 12 h exhibited a typical necrotic cell death with strong cytoplasmic vacuolations, cloudy chromatin but without chromatin condensation, disrupted mitochondria and cristae (Fig. 4A, 4B). EGCG‐treated (200 µM) cells resembled significantly the necrotic morphology without nuclear fragmentation but with significant cytoplasmic vacuolations with severe disruption of cell plasma membrane, mitochondrial membrane, and even detachment of nuclear envelope developing a large vacuole (Fig. 4A, 4B). Both EGCG and ATP depletion caused morphological alterations typical of necrosis such as cytoplasmic vacuolations (65 ± 5% and 67 ± 5%, respectively) with lumpy chromatin with almost no apoptotic bodies (0.6 ± 5%) (Fig. 4C,D), whereas imatinib predominantly caused apoptotic morphology (74 ± 5%). A lower concentration of EGCG (100 µM) also induced early morphological alterations (i.e. appearance of weaker cytoplasmic vacuolations) at 24 h, most likely indicative of early necrotic alterations (Supplementary Fig. S4). Thus, EGCG clearly induced cell death with necrotic characteristics.
Figure 4.
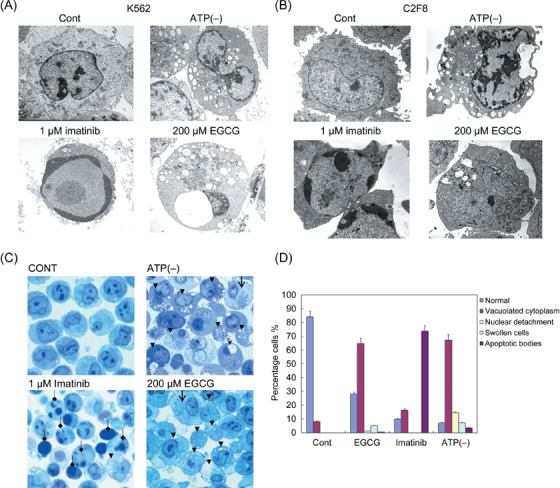
(–)‐Epigallocatechin‐3‐gallate (EGCG) treatment caused necrotic cell death morphology resembling adenosine triphosphate (ATP)‐depleted condition. K562 (A) and C2F8 (B) cells were grown in glucose‐free medium with 5 µg oligomycin for ATP‐depleted condition for 12 h; treated with 1 µM imatinib or 200 µM EGCG for 48 h and compared with the 0.1% ethanol‐treated control K562 and C2F8 cells. (C) Control, EGCG‐treated, imatinib‐treated, and ATP‐depleted K562 cells were stained with toluidine blue for quantification of apoptotic and necrotic cells. Closed arrows indicate cells with vacuolated cells; open arrows indicate swollen cells; arrowheads indicate cells with nuclear envelope detachment; and diamond‐shaped arrows indicate cells with apoptotic bodies. Vacuolated cells, nuclear envelope detachment and swollen cells were categorized as features typical of necrosis. (D) Percentage cells were quantified according to cell death morphologies categorized as above for each 0.1% ethanol‐treated (control), EGCG‐treated, imatinib‐treated, and ATP‐depleted conditions. Results are means ± standard deviation of three independent experiments. Cont, Control.
EGCG enhanced imatinib‐induced cell death. Several reports have suggested the use of tea polyphenols as chemosensitizers that potentiate the cytotoxicity of conventional anticancer drugs.( 25 ) Co‐treatment of low dose of imatinib (50–200 nM) and EGCG induced enhanced cell death in K562 cells (P < 0.01) (Fig. 5A). EGCG alone effectively induced cell death of K562/sti cells in a dose‐dependent manner (Fig. 5B right) with a mean IC50 of 140 µM, slightly higher than that of the parental K562 cells, though the combination treatment was not so effective to imatinib‐resistant K562 cells (K562/sti) (data not shown). These results suggest the possibility of EGCG in combination therapy with imatinib in inducing cell death in CML cells, and its possible effectiveness to imatinib‐resistant CML cells.
Figure 5.
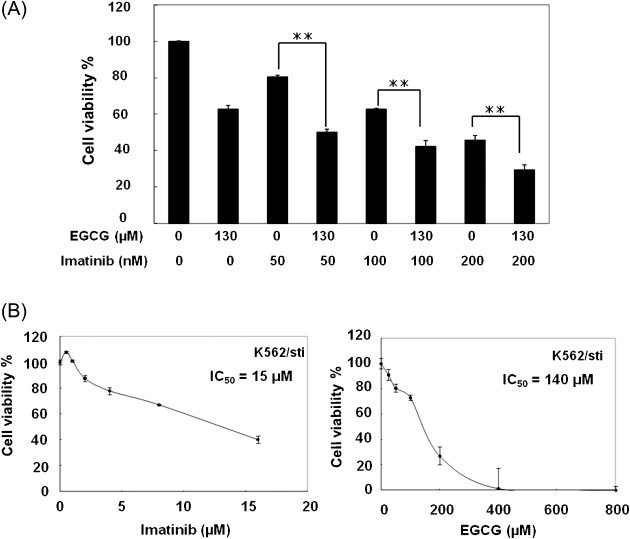
(A) (–)‐Epigallocatechin‐3‐gallate (EGCG) enhanced imatinib‐induced cell death. K562 cells were seeded at a density of 5 × 103 cells per well in 96‐well plates, and cultured with EGCG and imatinib alone/or in combination for 48 h. The number of viable cells were measured based on intracellular ATP content, and expressed as a percentage of 0.1% ethanol‐treated control cells. Results are means ± standard deviation (SD) of three independent experiments. Two asterisks indicate significant difference from the corresponding dose of single imatinib treatment (P < 0.01) and one asterisk indicates significant difference of P < 0.05. (B) EGCG was equally effective to induce cell death in imatinib‐resistant chronic myelogenous leukemia (CML) cells. Imatinib‐resistant K562 (K562/sti) cells were seeded at a density of 5 × 103 cells per well in 96‐well plates, and treated with increasing concentrations of EGCG or imatinib for 48 h, and the number of viable cells were measured based on intracellular ATP content. Viable cells are expressed as a percentage of 0.1% ethanol‐treated control cells for each cell line. Values are representative of three independent experiments.
Discussion
We demonstrated for the first time that EGCG predominantly induced necrosis‐like cell death in CML cell lines via a caspase‐independent mechanism, whereas imatinib induced typical apoptosis in these cell lines with caspase activation as previously reported.( 1 )
Most anticancer agents have been reported to kill tumor cells by inducing apoptosis via mitochondrial apoptotic pathway, or via the death receptors.( 26 ) However, our data clearly showed that EGCG did not modulate the apoptotic pathway, but predominantly induced caspase‐independent cell death in accordance with the necrotic morphology. The CML cell lines used in our study were not defective of apoptotic pathway as shown by the imatinib‐induced apoptosis, indicating that EGCG differentially induced necrosis in these cell lines. Additionally, the specific cytotoxicity of EGCG towards CML cells with minimal cytotoxicity to PBMC suggests that non‐specific drug toxicity was unlikely for this compound. However, a low level of nuclear AIF translocation induced by EGCG treatment suggested the presence of a small population of AIF‐dependent apoptotic cells among the predominant necrotic population. However, according to precise quantification data, apoptotic cell morphology was undetectable after EGCG treatment. Further investigations are necessary to verify whether this AIF activation was only a bystander effect of necrotic cell death. Other previous reports concluded that EGCG induced caspase‐mediated apoptosis in several tumor cells including prostate cancer, ovarian cancer and leukemic cells,( 11 , 27 , 28 ) however, detailed cell death morphology has not been extensively studied that may have overlooked non‐apoptotic cell death.
While apoptotic cell death has become increasingly well defined over the last three decades, notions of necrotic cell death have become progressively more vague.( 29 ) However, recent evidence suggests that necrotic cell death is not only a pathological/accidental cell death but a programmed event.( 30 ) Although molecular mechanism of the necrotic pathway is still poorly understood, a distinct cell death population pattern induced by EGCG has been proved in our study by the criteria of mitochondrial dysfunction and plasma membrane permeability, absence of caspase activation and morphological studies. As speculations for necrotic cell death factors, first, intracellular ATP concentration acted as a switch in the decision between apoptosis and necrosis in Jurkat cells after staurosporine treatment.( 31 ) Because necrosis is thought to occur as a result of bioenergetic catastrophe resulting from ATP depletion to a level incompatible with cell survival, intracellular ATP reduction may be the mediator of EGCG‐induced necrotic cell death. Second, EGCG downregulated the ATPase activity via interaction with the ATP‐binding site of glucose‐regulated protein (GRP78), associated with the multidrug resistance in various cancer cells, and increased sensitivity to etoposide.( 32 ) This raises the possibility of competitive binding between EGCG and ATP to various ATP binding sites including that of cytochrome c/Apaf‐1/caspase‐9 apoptosome rendering the apoptotic pathway, while exerting necrotic cell death.
However, an attempt to delineate the modes of cell death based on mitochondrial cell death signaling such as AIF and HtrA2/Omi has been more challenging. Nuclear translocation of AIF has been observed either in apoptosis or necrosis in staurosporine‐treated Jurkat cells.( 33 ) HtrA2/Omi also has a dual role as caspase activators via inhibition of IAP proteins and as an effector of necrosis‐like programmed cell death exerting its serine protease activity.( 34 ) In our study, cytosolic release of HtrA2/Omi was commonly present both in EGCG‐ or imatinib‐induced cell death showing distinct cell death morphologies. HtrA2/Omi may have acted as a necrosis‐like cell death effector in EGCG‐induced cell death in the absence of caspase activation, probably as caspase activators in imatinib‐induced apoptosis by inhibiting XIAP and survivin. Some commonly known anticancer drugs such as cladribine and paclitaxel triggered both caspase‐dependent and ‐independent pathways in chronic lymphocytic leukemia (CLL) cells.( 35 ) Indeed, different modes of cell death can coexist in the same cell demize( 36 ) pointing out the difficulty in distinguishing different cell death types based on the current knowledge of cell death signaling and requires further investigations on necrotic death signaling.
As to the clinical benefits of EGCG, the efficacy of combination therapy using EGCG with other agents such as curcumin in breast cancer cells, TRAIL in prostate cancer cells, or trastuzumab (monoclonal antibody) in breast cancer cells in vitro and in vivo have been reported.( 37 , 38 , 39 ) In this study, the enhanced cell death effect exerted by the co‐treatment of a low dose of imatinib and EGCG most likely reflects the fact that EGCG and imatinib induce distinct modes of cell death. This suggests that this combination may improve the treatment regimen for CML with reduced imatinib dosage for the treatment. In addition, EGCG effectively induced cell death of imatinib‐resistant K562 cells and did not cause significant normal PBMC death, showing that EGCG may be a possible candidate to treat the imatinib‐resistant CML cells without any significant cytotoxicity to normal cells. This specific cytotoxicity of EGCG towards CML cells is of clinical merit but the concentration of EGCG used in this study is much higher than that of imatinib, which may be unrealistic to achieve in vivo. Also, EGCG is unstable under physiological condition and has poor bioavailability,( 40 ) and it could be a substrate of efflux transporters like multidrug resistance‐associated proteins (MRP) and P‐glycoprotein,( 41 ) indicating its disadvantage as an antitumor agent in the clinical setting. However, i.v. injection of EGCG rather than p.o. administration in rats has been shown to reach such high plasma concentrations of EGCG without any toxic effects,( 42 , 43 ) opening up the possibility of EGCG in future clinical use. Also, a novel chemically modified form of EGCG with added peracetate protections to its reactive hydroxyl groups has shown higher bioavailability, stability and anticancer activity in human breast cancer cells, in vitro and in vivo.( 44 ) With the development of a more stable form of EGCG in progress, it may help to overcome this pharmacokinetic disadvantage in the long‐run.
Moreover, induction of necrosis may also have chemotherapeutic implications because some authors recently suggested the importance of immunogenic cancer cell death within the aims of anticancer chemotherapy,( 45 ) where apoptosis is believed to be poorly immunogenic and necrotic cell death being truly immunogenic.( 46 , 47 , 48 ) Necrotic, but not apoptotic, cancer cells have been reported to enhance the immune response against cancer after being ingested by antigen‐presenting cells (APC).( 49 , 50 ) The mode of cell death is now an important point to consider in chemotherapy against tumor cells.
In conclusion, our data showed that EGCG induced necrotic cell death via a caspase‐independent mechanism in CML cell lines, without influencing any aspects of the apoptotic pathway. Not only has this shed some additional insights into the novel mode of cell death induced by EGCG, it should also be of interest to oncologists and immunologists to further investigate the impact of these distinct modes of cell death in surrounding tissues and consequently in our body.
Supporting information
Fig. S1. Effect of EGCG on PBMC.
Fig. S2. Distinct cell death pattern between EGCG and imatinib treatments.
Fig. S3. Caspases status in EGCG‐induced cell death in C2F8 cells.
Fig. S4. EGCG‐induced early necrotic alternations in C2F8 cells.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Supporting info item
Supporting info item
Supporting info item
Acknowledgments
This work was supported by the Japan Foundation for Promotion of International Medical Research Co‐operation (JF‐PIMRC).
References
- 1. Dan S, Naito M, Tsuruo T. Selective induction of apoptosis in Philadelphia chromosome‐positive chronic myelogenous leukemia cells by an inhibitor of BCR–ABL tyrosine kinase, CGP 57148. Cell Death Differ 1998; 5: 710–5. [DOI] [PubMed] [Google Scholar]
- 2. Decaudin D, Marzo I, Brenner C, Kroemer G. Mitochondria in chemotherapy‐induced apoptosis: a prospective novel target of cancer therapy (review). Int J Oncol 1998; 12: 141–52. [PubMed] [Google Scholar]
- 3. Ravagnan L, Roumier T, Kroemer G. Mitochondria, the killer organelles and their weapons. J Cell Physiol 2002; 192: 131–7. [DOI] [PubMed] [Google Scholar]
- 4. Verhagen AM, Silke J, Ekert PG et al . HtrA2 promotes cell death through its serine protease activity and its ability to antagonize inhibitor of apoptosis proteins. J Biol Chem 2002; 277: 445–54. [DOI] [PubMed] [Google Scholar]
- 5. Ahmad N, Feyes DK, Nieminen AL, Agarwal R, Mukhtar H. Green tea constituent epigallocatechin‐3‐gallate and induction of apoptosis and cell cycle arrest in human carcinoma cells. J Natl Cancer Inst 1997; 89: 1881–6. [DOI] [PubMed] [Google Scholar]
- 6. Bettuzzi S, Brausi M, Rizzi F, Castagnetti G, Peracchia G, Corti A. Chemoprevention of human prostate cancer by oral administration of green tea catechins in volunteers with high‐grade prostate intraepithelial neoplasia: a preliminary report from a one‐year proof‐of‐principle study. Cancer Res 2006; 66: 1234–40. [DOI] [PubMed] [Google Scholar]
- 7. Khan N, Afaq F, Saleem M, Ahmad N, Mukhtar H. Targeting multiple signaling pathways by green tea polyphenol (‐)‐epigallocatechin‐3‐gallate. Cancer Res 2006; 66: 2500–5. [DOI] [PubMed] [Google Scholar]
- 8. Ahmad N, Cheng P, Mukhtar H. Cell cycle dysregulation by green tea polyphenol epigallocatechin‐3‐gallate. Biochem Biophys Res Commun 2000; 275: 328–34. [DOI] [PubMed] [Google Scholar]
- 9. Nakazato T, Ito K, Miyakawa Y et al . Catechin, a green tea component, rapidly induces apoptosis of myeloid leukemic cells via modulation of reactive oxygen species production in vitro and inhibits tumor growth in vivo . Haematologica 2005; 90: 317–25. [PubMed] [Google Scholar]
- 10. Islam S, Islam N, Kermode T et al . Involvement of caspase‐3 in epigallocatechin‐3‐gallate‐mediated apoptosis of human chondrosarcoma cells. Biochem Biophys Res Commun 2000; 270: 793–7. [DOI] [PubMed] [Google Scholar]
- 11. Pan MH, Liang YC, Lin‐Shiau SY, Zhu NQ, Ho CT, Lin JK. Induction of apoptosis by the oolong tea polyphenol theasinensin A through cytochrome c release and activation of caspase‐9 and caspase‐3 in human U937 cells. J Agric Food Chem 2000; 48: 6337–46. [DOI] [PubMed] [Google Scholar]
- 12. Saeki K, Kobayashi N, Inazawa Y et al . Oxidation‐triggered c‐Jun N‐terminal kinase (JNK) and p38 mitogen‐activated protein (MAP) kinase pathways for apoptosis in human leukaemic cells stimulated by epigallocatechin‐3‐gallate (EGCG): a distinct pathway from those of chemically induced and receptor‐mediated apoptosis. Biochem J 2002; 368: 705–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hayakawa S, Saeki K, Sazuka M et al . Apoptosis induction by epigallocatechin gallate involves its binding to Fas. Biochem Biophys Res Commun 2001; 285: 1102–6. [DOI] [PubMed] [Google Scholar]
- 14. Naasani I, Oh‐Hashi F, Oh‐Hara T et al . Blocking telomerase by dietary polyphenols is a major mechanism for limiting the growth of human cancer cells in vitro and in vivo . Cancer Res 2003; 63: 824–30. [PubMed] [Google Scholar]
- 15. Naasani I, Seimiya H, Tsuruo T. Telomerase inhibition, telomere shortening, and senescence of cancer cells by tea catechins. Biochem Biophys Res Commun 1998; 249: 391–6. [DOI] [PubMed] [Google Scholar]
- 16. Shammas MA, Neri P, Koley H et al . Specific killing of multiple myeloma cells by (–)‐epigallocatechin‐3‐gallate extracted from green tea: biologic activity and therapeutic implications. Blood 2006; 108: 2804–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tachibana H, Koga K, Fujimura Y, Yamada K. A receptor for green tea polyphenol EGCG. Nat Struct Mol Biol 2004; 11: 380–1. [DOI] [PubMed] [Google Scholar]
- 18. Umeda D, Yano S, Yamada K, Tachibana H. Green tea polyphenol epigallocatechin‐3‐gallate (EGCG) signaling pathway through 67‐kDa laminin receptor. J Biol Chem 2008; 283: 3050–8. [DOI] [PubMed] [Google Scholar]
- 19. Yang SH, Chien CM, Lu MC, Lin YH, Hu XW, Lin SR. Up‐regulation of Bax and endonuclease G, and down‐modulation of Bcl‐XL involved in cardiotoxin III‐induced apoptosis in K562 cells. Exp Mol Med 2006; 38: 435–44. [DOI] [PubMed] [Google Scholar]
- 20. Hamahata K, Adachi S, Matsubara H et al . Mitochondrial dysfunction is related to necrosis‐like programmed cell death induced by A23187 in CEM cells. Eur J Pharmacol 2005; 516: 187–96. [DOI] [PubMed] [Google Scholar]
- 21. Antonsson B. Mitochondria and the Bcl‐2 family proteins in apoptosis signaling pathways. Mol Cell Biochem 2004; 256–257: 141–55. [DOI] [PubMed] [Google Scholar]
- 22. Festjens N, Van Gurp M, Van Loo G, Saelens X, Vandenabeele P. Bcl‐2 family members as sentinels of cellular integrity and role of mitochondrial intermembrane space proteins in apoptotic cell death. Acta Haematol 2004; 111: 7–27. [DOI] [PubMed] [Google Scholar]
- 23. Van Gurp M, Festjens N, Van Loo G, Saelens X, Vandenabeele P. Mitochondrial intermembrane proteins in cell death. Biochem Biophys Res Commun 2003; 304: 487–97. [DOI] [PubMed] [Google Scholar]
- 24. Leist M, Jaattela M. Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol 2001; 2: 589–98. [DOI] [PubMed] [Google Scholar]
- 25. Garg AK, Buchholz TA, Aggarwal BB. Chemosensitization and radiosensitization of tumors by plant polyphenols. Antioxid Redox Signal 2005; 7: 1630–47. [DOI] [PubMed] [Google Scholar]
- 26. Friesen C, Fulda S, Debatin KM. Cytotoxic drugs and the CD95 pathway. Leukemia 1999; 13: 1854–8. [DOI] [PubMed] [Google Scholar]
- 27. Gupta S, Hussain T, Mukhtar H. Molecular pathway for (–)‐epigallocatechin‐3‐gallate‐induced cell cycle arrest and apoptosis of human prostate carcinoma cells. Arch Biochem Biophys 2003; 410: 177–85. [DOI] [PubMed] [Google Scholar]
- 28. Huh SW, Bae SM, Kim YW et al . Anticancer effects of (–)‐epigallocatechin‐3‐gallate on ovarian carcinoma cell lines. Gynecol Oncol 2004; 94: 760–8. [DOI] [PubMed] [Google Scholar]
- 29. Zong WX, Thompson CB. Necrotic death as a cell fate. Genes Dev 2006; 20: 1–15. [DOI] [PubMed] [Google Scholar]
- 30. Kitanaka C, Kuchino Y. Caspase‐independent programmed cell death with necrotic morphology. Cell Death Differ 1999; 6: 508–15. [DOI] [PubMed] [Google Scholar]
- 31. Leist M, Single B, Castoldi AF, Kuhnle S, Nicotera P. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med 1997; 185: 1481–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ermakova SP, Kang BS, Choi BY et al . (–)‐Epigallocatechin gallate overcomes resistance to etoposide‐induced cell death by targeting the molecular chaperone glucose‐regulated protein 78. Cancer Res 2006; 66: 9260–9. [DOI] [PubMed] [Google Scholar]
- 33. Daugas E, Susin SA, Zamzami N et al . Mitochondrio‐nuclear translocation of AIF in apoptosis and necrosis. FASEB J 2000; 14: 729–39. [PubMed] [Google Scholar]
- 34. Okada M, Adachi S, Imai T et al . A novel mechanism for imatinib mesylate‐induced cell death of BCR‐ABL‐positive human leukemic cells: caspase‐independent, necrosis‐like programmed cell death mediated by serine protease activity. Blood 2004; 103: 2299–307. [DOI] [PubMed] [Google Scholar]
- 35. Marzo I, Perez‐Galan P, Giraldo P, Rubio‐Felix D, Anel A, Naval J. Cladribine induces apoptosis in human leukaemia cells by caspase‐dependent and ‐independent pathways acting on mitochondria. Biochem J 2001; 359: 537–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Blagosklonny MV. Cell death beyond apoptosis. Leukemia 2000; 14: 1502–8. [DOI] [PubMed] [Google Scholar]
- 37. Eddy SF, Kane SE, Sonenshein GE. Trastuzumab‐resistant HER2‐driven breast cancer cells are sensitive to epigallocatechin‐3 gallate. Cancer Res 2007; 67: 9018–23. [DOI] [PubMed] [Google Scholar]
- 38. Siddiqui IA, Malik A, Adhami VM et al . Green tea polyphenol EGCG sensitizes human prostate carcinoma LNCaP cells to TRAIL‐mediated apoptosis and synergistically inhibits biomarkers associated with angiogenesis and metastasis. Oncogene 2008; 27: 2055–63. [DOI] [PubMed] [Google Scholar]
- 39. Somers‐Edgar TJ, Scandlyn MJ, Stuart EC, Le Nedelec MJ, Valentine SP, Rosengren RJ. The combination of epigallocatechin gallate and curcumin suppresses ER alpha‐breast cancer cell growth in vitro and in vivo . Int J Cancer 2008; 122: 1966–71. [DOI] [PubMed] [Google Scholar]
- 40. Chen L, Lee MJ, Li H, Yang CS. Absorption, distribution, and elimination of tea polyphenols in rats. Drug Metab Dispos 1997; 25: 1045–50. [PubMed] [Google Scholar]
- 41. Vaidyanathan JB, Walle T. Cellular uptake and efflux of the tea flavonoid (–)‐epicatechin‐3‐gallate in the human intestinal cell line Caco‐2. J Pharmacol Exp Therap 2003; 307: 745–52. [DOI] [PubMed] [Google Scholar]
- 42. Kohri T, Nanjo F, Susuki M et al . Synthesis of (–)‐[4–3H]Epigallocatechin gallate and its metabolic fate in rats after intravenous administration. J Agric Food Chem 2001; 49: 1042–8. [DOI] [PubMed] [Google Scholar]
- 43. Isbrucker RA, Bausch J, Edwards JA, Wolz E. Safety studies on epigallocatechin gallate (EGCG) preparations. Part 1: Genotoxicity. Food Chem Toxicol 2006; 44: 626–35. [DOI] [PubMed] [Google Scholar]
- 44. Landis‐Piwowar KR, Huo C, Chen D et al . A novel prodrug of the green tea polyphenol (–)‐epigallocatechin‐3‐gallate as a potential anticancer agent. Cancer Res 2007; 67: 4303–10. [DOI] [PubMed] [Google Scholar]
- 45. Obeid M, Tesniere A, Ghiringhelli F et al . Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 2007; 13: 54–61. [DOI] [PubMed] [Google Scholar]
- 46. Bellamy CO, Malcomson RD, Harrison DJ, Wyllie AH. Cell death in health and disease: the biology and regulation of apoptosis. Semin Cancer Biol 1995; 6: 3–16. [DOI] [PubMed] [Google Scholar]
- 47. Igney FH, Krammer PH. Death and anti‐death: tumour resistance to apoptosis. Nat Rev Cancer 2002; 2: 277–88. [DOI] [PubMed] [Google Scholar]
- 48. Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science 1995; 267: 1456–62. [DOI] [PubMed] [Google Scholar]
- 49. Reiter I, Krammer B, Schwamberger G. Cutting edge: differential effect of apoptotic versus necrotic tumor cells on macrophage antitumor activities. J Immunol 1999; 163: 1730–2. [PubMed] [Google Scholar]
- 50. Sauter B, Albert ML, Francisco L, Larsson M, Somersan S, Bhardwaj N. Consequences of cell death: exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J Exp Med 2000; 191: 423–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Effect of EGCG on PBMC.
Fig. S2. Distinct cell death pattern between EGCG and imatinib treatments.
Fig. S3. Caspases status in EGCG‐induced cell death in C2F8 cells.
Fig. S4. EGCG‐induced early necrotic alternations in C2F8 cells.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item