

Abstract
Hepatocellular carcinoma (HCC) is one of the most common cancer‐related causes of death, and is chemoresistant to anticancer drugs. Anti‐angiogenic therapy has been shown to enhance the efficacy of chemotherapy to treat solid tumors. The aim of the present study was to determine whether endostatin, a potent antiangiogenic agent, could enhance the efficacy of doxorubicin to combat HCC. An endostatin expression plasmid was constructed and its expression in vitro and in vivo was detected after gene transfer. Recombinant endostatin inhibited angiogenesis in the chorioallantoic membrane assay, and showed synergistic effects with doxorubicin in inhibiting the in vitro proliferation of endothelial cells, but not that of tumor cells. Both endostatin gene therapy and doxorubicin suppressed the growth of subcutaneous human HepG2 tumors established in BALB/c nude mice, and tumor angiogenesis. Combination therapy with endostatin gene therapy and doxorubicin showed a stronger effect in suppressing tumor growth, and tumor angiogenesis, than the respective monotherapies. Gene transfer of endostatin down‐regulated the expression of both hypoxia‐inducible factor‐1α and vascular endothelial growth factor (VEGF), whereas doxorubicin only down‐regulated VEGF expression. Endostatin and doxorubicin synergized to down‐regulate VEGF expression. Endostatin and doxorubicin combination therapy warrants investigation as a therapeutic strategy to combat HCC. (Cancer Sci 2007; 98: 1381–1387)
Abbreviations:
- Ab
antibody
- ATCC
American Type Culture collection
- BCIP
5‐bromo‐4‐chloro‐3‐indolyl phosphate
- BSA
bovine serum albumin
- CAM
chorioallantoic membrane
- CMV
cytomegalovirus
- DAB
3,3′‐diaminobenzidine tetrahydrochloride
- HCC
hepatocellular carcinoma
- HIF
hypoxia‐inducible factor
- HUVEC
human umbilical vein endothelial cell
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyl tetrazolium bromide
- NBT
nitro blue tetrazolium
- OD
optical density
- PBS
phosphate‐buffered saline
- PVDF
polyvinylidene difluoride
- RPMI
Roswell Park Memorial Institute
- SDS
sodium dodecyl sulfate
- VEGF
vascular endothelial growth factor.
HCC is the third leading global cause of cancer death, with an estimated incidence of over one million new cases per year worldwide.( 1 , 2 ) HCC has a very poor prognosis, with an overall 5‐year survival rate of just 3–5%.( 3 ) Only surgery offers a cure, but tumor resection is feasible for less than 15% of patients, and recurrence rates remain as high as 50% after tumor resection.( 4 ) Chemotherapy offers unsatisfactory response rates. From a recent review by Zhu,( 5 ) none of the currently approved anticancer drugs has shown to be particularly effective against HCC. Response rates are low, and the duration of response is typically short, thus the survival benefit of systemic chemotherapy for HCC remains uncertain. Doxorubicin is perhaps the most widely used agent in the treatment of HCC. However, despite initially encouraging reports from Uganda using single‐agent doxorubicin, subsequent studies have failed to confirm these data.( 5 ) Doxorubicin failed to demonstrate responses in 109 patients with advanced HCC,( 6 ) and only showed a 16% response rate in 475 patients, with a median survival time of 3–4 months.( 7 ) In a recent large phase III study, doxorubicin only showed a 4.0–10.5% response rate in HCC patients, and induced significant hematological and gastrointestinal toxicities.( 8 ) Therefore, new strategies to enhance the efficacy of doxorubicin to treat HCC are needed.
Anti‐angiogenic therapy targeting the tumor blood supply has become a promising approach to combat cancer, given that solid vascular tumors such as HCC must establish an adequate vascular network to acquire the nutrition necessary for growth and metastasis.( 9 ) Endostatin, an Mr 20 000 proteolytic fragment of the C‐terminus of collagen XVIII, is one of the most potent angiogenesis inhibitors isolated from the sera and urine of tumor‐bearing mice.( 10 ) Its antiangiogenic activity appears to be dependent on binding to integrin α5β1,( 11 ) or E‐selectin,( 12 ) or inhibiting metalloproteinases,( 13 ) or its extensive influence on a set of growth‐associated gene networks in endothelial cells.( 14 , 15 ) Endostatin induces the regression of a wide variety of established tumors of mouse, rat and human origin.( 16 ) Endostatin treatment using either the recombinant protein or gene therapy has been shown to suppress HCC tumors in animal models.( 17 , 18 , 19 , 20 ) Furthermore, endostatin stabilized tumor growth after chemotherapy in a NOD/SCID mouse model of human high‐grade non‐Hodgkin lymphoma.( 21 ) It also helped low‐dose carboplatin to prevent metastasis of human testicular germ cell tumor xenografts,( 22 ) enhanced the antitumor efficacy of doxorubicin to prevent liver metastasis in a model of colorectal cancer model,( 23 ) and synergized with adriamycin to suppress tumor progression in an orthotopic murine mammary carcinoma model.( 24 ) Endostatin gene therapy prevented the formation of tumor blood vessels, decreased tumor growth, and synergized with conventional therapy to suppress ovarian and lung cancers.( 25 , 26 ) Here the authors test the hypothesis that endostatin gene therapy enhances the efficacy of doxorubicin to combat HCC.
Materials and Methods
Mice, cell lines and antibodies. Male nude BALB/c mice (H‐2b), 6–8 weeks old, were obtained from the Animal Research Center, Shandong University, China. The human HCC cell line HepG2 was obtained from the ATCC (Rockville, MD, USA). HUVEC were supplied by the Typical Animal Reserve Center of China. The cells were routinely cultured at 37°C in RPMI 1640 medium supplemented with 10% fetal calf serum. The anti‐endostatin (PA1‐601), anti‐HIF‐1α, anti‐VEGF, and anti‐CD31 (MEC13.3) antibodies were purchased from Affinity BioReagents Inc. (Golden, CO, USA), Boster Biological Technology (Wuhan, China), Laboratory Vision Corporation (CA, USA), and Pharmingen (CA, USA), respectively.
Endostatin expression vectors. Complementary DNA encoding mouse endostatin was released from an endostatin plasmid,( 27 ) and subcloned into pcDNA3.1 to construct the endostatin expression vector, End‐pcDNA3.1. It was driven by a CMV immediate‐early promoter, and a secretion signal from the mouse immunoglobulin κ chain was fused to the N‐terminus of endostatin. DNA sequence analysis confirmed that the cDNA sequences were inserted in the proper reading frame, and no mutations had been incorporated.
Western blot analysis to detect protein expression in vitro. The methodology has been described previously.( 28 ) Briefly, COS‐1 or HepG2 cells were grown to 60–70% confluence, and transfected with 4 µg of End‐pcDNA3.1 plasmid using lipofectamine PLUS (Life Technologies, China). Empty pcDNA3.1 vector served as a control. For detection of endostatin expression, the COS‐1 cells and supernatants were collected 48 h later, and the concentrated conditioned media and cell lysates were subjected to western blot analysis as described previously.( 29 ) For detection of HIF‐1α and VEGF expression under hypoxia, CoCl2 was added to HepG2 cells culture medium at the concentration of 200 µM 48 h after End‐pcDNA3.1 transfection. Three hours later, the cells were harvested and cell lysates were subjected to western blot analysis as above.
CAM assay. The chicken embryo CAM assay is widely used as an assay to measure the activity of antiangiogenic agents.( 30 ) Briefly, fertilized eggs from white Leghorn chickens were incubated for 7 days at 37°C and 60% humidity. A square window was opened in the shell and the membrane of the gas chamber was carefully removed to expose the chorioallantoic membrane. Twelve eggs were randomly divided into two groups, and were doped with either 50 µL conditioned media from COS‐1 cell that had been transfected with End‐pcDNA3.1 or pcDNA3.1. The windows were sealed with sterile parafilm and the eggs incubated for 72 h, as above. The CAM were photographed and the number and extent of vessel branch‐points formed by the blood vessels was determined as a measure of angiogenesis.
Proliferation assay. HUVEC (1 × 104) and HepG2 (5 × 103) cells were seeded in 200 µL of RPMI 1640 into 96‐well plates. The medium was replaced 12 h later with 200 µL of fresh RPMI 1640 media containing 10, 20, or 40 µL of conditioned medium from End‐pcDNA3.1‐transfected COS‐1 cells, or doxorubicin at the concentration of 0.001, 0.1, 1 or 10 µg/mL. The cells were cultured for a further 48 h. Cells cultured in 200 µL of RPMI 1640 served as controls. MTT (20 µL) was added to each well followed by a 4‐h incubation, and cells were processed to record the OD value at 570 nm. The proliferation index was calculated according to the formula: experimental OD value/control OD value × 100%. The experiments were repeated thrice.
Animal model and treatments. All surgical procedures and care administered to the animals were in accordance with institutional guidelines. Tumors were established by subcutaneous injection of 1 × 106 HepG2 tumor cells into the mice. Tumors volumes were estimated according to the formula: π/6 × a 2 × b, where a is the short axis, and b the long axis. When tumors reached approximately 100 mm3 at approximately 3 weeks, the mice were randomly assigned to four treatment groups: pcDNA3.1, End‐pcDNA3.1, doxorubicin, and End‐pcDNA3.1 + doxorubicin. To standardize the experiments, mice in each group received both intratumoral and i.p. injections. In the pcDNA3.1 and End‐pcDNA3.1 groups, mice received i.p. injection of 200 µL of PBS, and intratumoral injection of 200 µg of pcDNA3.1 or End‐pcDNA3.1 diluted in 100 µL of FuGENE 6 transfection reagent (Roche, Shanghai, China), respectively. In the doxorubicin group, mice received i.p. injection of 200 µL of doxorubicin (diluted in PBS) at a dose of 12.5 mg/kg and intratumoral injection of 200 µg of pcDNA3.1 diluted in 100 µL of FuGENE 6. In the Endo‐pcDNA3.1 + doxorubicin group, mice received i.p. injection of 200 µL of doxorubicin (diluted in PBS) at a dose of 12.5 mg/kg and intratumoral injection of 200 µg of End‐pcDNA3.1 diluted in 100 µL of FuGENE 6. All experiments included 12 mice per treatment group. FuGENE 6 was shown to be an efficient in vivo transfection reagent in the authors’ previous study.( 31 )
Immunohistochemistry. Tumor cryosections (6 µm) were fixed with acetone, rinsed with PBS, blocked with 3% BSA for 2 h, and incubated overnight with primary antibodies. They were subsequently incubated for 30 min with appropriate secondary antibodies using the Ultra Sensitive TMS‐P kit (Zhongshan Co., Beijing, China), and immunoreactivity developed with Sigma FAST DAB and CoCl2 enhancer tablets (Sigma‐Aldrich, Shanghai, China). Sections were counterstained with hematoxylin, mounted, and examined by microscopy.
Assessment of tumor vascularity. The methodology to determine tumor vascularity has been described previously.( 28 ) Briefly, 6‐µm tumor sections were immunostained with an anti‐CD31 Ab, as described above. Stained vessels were counted in 10 blindly chosen random fields at 400 × magnification, and the mean microvessel density was recorded.
Western blot analysis. The tumor tissues were minced and homogenized in protein lysate buffer. Debris was removed by centrifugation at 10 000 g for 10 min at 4°C. The lysates were resolved on 12% polyacrylamide SDS gels, and electrophoretically transferred to PVDF membranes. The membranes were blocked with 3% BSA overnight, incubated with primary antibodies, and subsequently with alkaline phosphatase‐conjugated secondary antibody. They were developed with BCIP/NBT (Tiangen Biotech Co. Ltd, Beijing, China). Blots were stained with an anti‐tubulin antibody to confirm that each lane contained similar amounts of tumor homogenate.
Statistical analysis. Results were expressed as mean values ± SD, and Student's t‐test was used to evaluate statistical significance. A value of less than 0.05 (P < 0.05) was used for statistical significance.
Results
Expression of recombinant endostatin in vitro. The presence of recombinant endostatin in the cell lysate and conditioned medium of COS‐1 cells transfected with End‐pcDNA3.1 was confirmed using western blot analysis with an anti‐endostatin antibody. The antibody detected an endostatin protein band of 20 kDa in both the cell lysate and conditioned medium of End‐pcDNA3.1 transfectants, whereas no such band was present in the cell lysate or conditioned medium of COS‐1 cells transfected with empty vector pcDNA3.1 (Fig. 1a).
Figure 1.
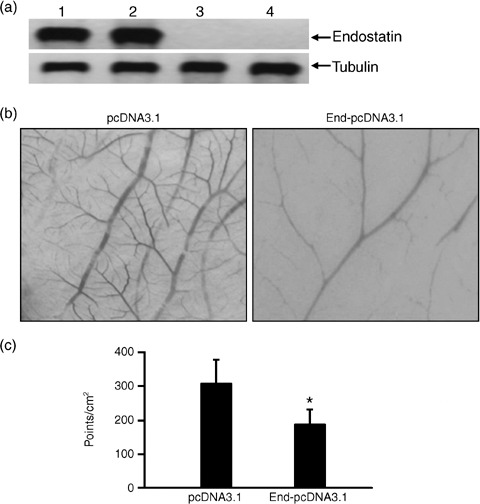
Recombinant endostatin inhibits angiogenesis in the chorioallantoic membrane (CAM) of chicken eggs. (a) Western blot analysis of recombinant endostatin expression in COS‐1 cells transfected with the End‐pcDNA3.1 plasmid. Cell lysates (lanes 1 and 3) and concentrated conditioned media (lanes 2 and 4) of COS‐1 cells transfected with End‐pcDNA3.1 (lanes 1 and 2) and empty pcDNA3.1 plasmid (lanes 3 and 4) were western blotted with anti‐endostatin (upper panel) and anti‐tubulin (lower panel) antibodies. (b) Illustrated are representative photographs of CAM treated with conditioned media from cultures of COS‐1 cells transfected with pcDNA3.1 and End‐pcDNA3.1 plasmids. (c) The numbers of branch‐points of the blood vessels were counted. A significant difference between the End‐pcDNA3.1 and pcDNA3.1 groups is denoted by ‘*’.
Recombinant endostatin inhibits angiogenesis in the CAM. As shown in Fig. 1b, the conditioned media of COS‐1 cells transfected with the End‐pcDNA3.1 significantly inhibited angiogenesis in the CAM, as evidenced by the reduction in vessel branch‐points compared to treatment with the empty vector (P < 0.01; Fig. 1c).
Recombinant endostatin synergizes with doxorubicin to inhibit the proliferation of HUVEC but not HepG2 cells. First, the authors could demonstrate that doxorubicin inhibited proliferation of both HUVEC (Fig. 2a) and HepG2 cells (Fig. 2b) in a dose‐dependent manner. Doxorubicin at the concentration of 0.5 µg/mL was selected for the following experiments, as this concentration suboptimally inhibited the proliferation of HepG2 and HUVEC cells by 30% and 22%, respectively. As shown in Fig. 2c, the conditioned medium of End‐pcDNA3.1 transfectants significantly (P < 0.01) inhibited HUVEC proliferation in a dose‐dependent manner, compared with that of pcDNA3.1 (control). Incubation of the cells with the combination of doxorubicin and the endostatin‐containing conditioned medium reduced the proliferation index of HUVEC compared with doxorubicin alone or the conditioned medium alone (both P < 0.05). However, the conditioned medium of End‐pcDNA3.1 transfectants did not shown any inhibitory effects or synergism with doxorubicin on the proliferation of HepG2 cells (Fig. 2d).
Figure 2.
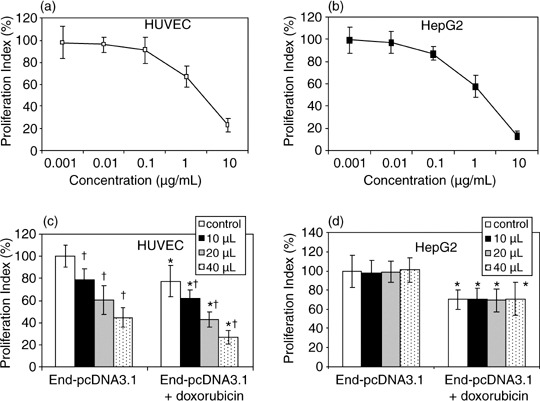
Proliferation of human umbilical vein endothelial cells (HUVEC) and HepG2 cells in vitro. (a,b) HUVEC and HepG2 cells were incubated in RPMI medium containing different concentrations of doxorubicin. (c,d) HUVEC and HepG2 cells were incubated in the presence or absence of doxorubicin in RPMI medium containing different volumes of conditioned media from COS‐1 cells transfected with End‐pcDNA3.1 plasmid. Conditioned media from COS‐1 cells transfected with empty vector pcDNA3.1 served as control. The proliferation of HUVEC cells was assessed, and the proliferation index was calculated. A significant difference between End‐pcDNA3.1 and End‐pcDNA3.1 + doxorubicin treatment groups is denoted by ‘*’, and between pcDNA3.1 and End‐pcDNA3.1 treatment groups by ‘†’.
Endostatin gene transfer results in intense in situ transgene expression. HepG2 tumors injected with End‐pcDNA3.1 plasmid were sectioned 2 days following gene transfer. Representative photographs revealed intense expression of endostatin throughout tumors treated with End‐pcDNA3.1, whereas control sections from pcDNA3.1‐treated tumors were only slightly stained by the anti‐endostatin Ab due to weak expression of endogenous endostatin (Fig. 3a vs b). An endostatin protein of 20 kDa was detected using western blot analysis of homogenates of transfected tumors 2 days after gene injection, but was absent from homogenates of control tumors. The intense expression of endostatin was also detected 7 days after gene injection, but the band became weak 14 days after gene injection. (Fig. 3c).
Figure 3.
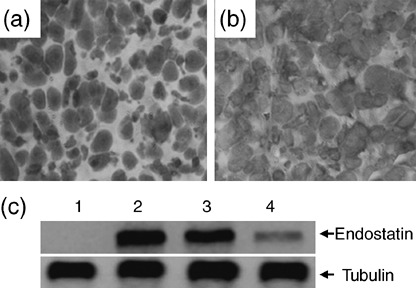
Intense expression of endostatin in situ after intratumoral gene transfer. Illustrated are representative tumor sections prepared 2 days following intratumoral gene transfer of (a) pcDNA3.1 and (b) End‐pcDNA3.1 plasmids. The sections were stained with an anti‐endostatin antibody. (c) Homogenates of tumors 2 (lane 2), 7 (lane 3) or 14 (lane 4) days following intratumoral injection of the End‐pcDNA3.1, or pcDNA3.1 (lane 1) plasmids were western blotted with either anti‐endostatin (upper panel) or ‐tubulin (lower panel) antibodies.
Endostatin gene therapy synergizes with doxorubicin to suppress hepatomas. Tumors were established by subcutaneous injection of HepG2 tumor cells into the mice. Three weeks later, when the tumors reached approximately 100 mm3, the mice were randomly assigned to four treatment groups: pcDNA3.1, End‐pcDNA3.1, doxorubicin, and Endo‐pcDNA3.1 + doxorubicin. As shown in Fig. 4, the tumors treated with empty vector pcDNA3.1 grew remarkably fast, reaching 2235 ± 268 mm3 in volume 6 weeks after implantation, which is not significantly different from the untreated tumors (2360 ± 330 mm3, P > 0.05). In contrast, in the doxorubicin group, the tumors had reached only 953 ± 250 mm3 in volume 6 weeks after implantation, which is significantly smaller than the control tumors (P < 0.01). Endostatin gene therapy also resulted in a significant reduction in tumor volumes (1545 ± 180 mm3), compared with control tumors (P < 0.05). A combination of Endo‐pcDNA3.1 and doxorubicin further suppressed tumor growth such that tumors reached only 426 ± 87 mm3 in size, which is highly significantly smaller than the control tumors (P < 0.001), and significantly smaller than the tumors treated with endostatin and doxorubicin monotherapies (both P < 0.05; Fig. 4).
Figure 4.
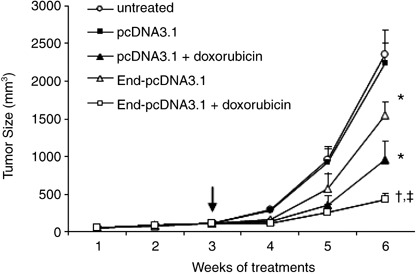
Endostatin gene therapy synergizes with doxorubicin to suppress tumor growth. HepG2 hepatomas were established. When the tumors reached approximately 100 mm3 (indicated by a vertical arrow), they received pcDNA3.1, pcDNA3.1 + doxorubicin, End‐pcDNA3.1, or End‐pcDNA3.1 + doxorubicin treatments. Untreated tumors served as controls. The sizes (mm3) of tumors were monitored and recorded. A significant difference in tumor volumes from control is denoted by ‘*’, and a highly significant difference by ‘†’. ‘‡’ Indicates significant difference from End‐pcDNA3.1 or doxorubicin treatments.
Endostatin gene therapy synergizes with doxorubicin to inhibit tumor angiogenesis. Five mice were killed 2 weeks after treatment from the above four groups of mice, and tumors were sectioned and stained with an anti‐CD31 Ab to visualize microvessels. There were fewer microvessels in tumors treated with endostatin gene therapy (Fig. 5b) or doxorubicin (Fig. 5c), compared with pcDNA3.1‐treated tumors (Fig. 5a); and there were even fewer microvessels in tumors treated using the combination therapy with endostatin and doxorubicin (Fig. 5d), compared with tumors treated using the monotherapies. There was no significant difference in microvessel densities in pcDNA3.1‐treated and untreated tumors (data not shown). Tumor microvessels in sections were counted in blindly chosen random fields to record microvessel density. Gene transfer of endostatin resulted in a significant 40% (P < 0.01) reduction in microvessel density compared with control, and doxorubicin also reduced microvessel density by 24% compared with control (P < 0.05). Furthermore, the combination therapy with End‐pcDNA3.1 and doxorubicin highly significantly reduced the microvessel density by 60%, compared with mock treatment (P < 0.001). The microvessel density in the combination treatment group was also significantly less than that seen in the End‐pcDNA3.1 and doxorubicin treatment groups (both P < 0.05), demonstrating synergism between endostatin and doxorubicin in inhibiting tumor angiogenesis (Fig. 5e).
Figure 5.
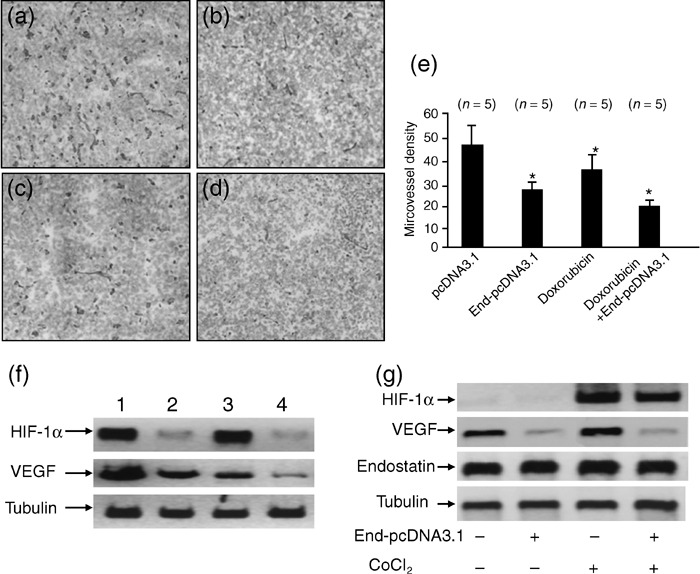
Endostatin gene transfer synergizes with doxorubicin to inhibit tumor angiogenesis. Illustrated are representative tumor sections prepared 2 weeks after treatment from mice receiving (a) pcDNA3.1 (control), (b) End‐pcDNA3.1, (c) doxorubicin, or (d) doxorubicin + End‐pcDNA3.1 treatment. (e) Tumor microvessels in sections were stained with the anti‐CD31 antibody and counted in blindly chosen random fields to record microvessel density. A significant difference in microvessel density between tumors treated with doxorubicin, or End‐pcDNA3.1 versus control is denoted by ‘*’, and a highly significant difference between tumors treated with combinational therapy with doxorubicin + End‐pcDNA3.1 and control by ‘**’. (f) Homogenates of tumors from mice treated with pcDNA3.1 (lane 1), End‐pcDNA3.1 (lane 2), doxorubicin (lane 3), or doxorubicin + End‐pcDNA3.1 (lane 4) were western blotted with anti‐ hypoxia‐inducible factor (HIF)‐1α (upper panel), ‐vascular endothelial growth factor (VEGF; middle panel) and ‐tubulin (lower panel) antibodies. (g) Western blot analysis of HIF‐1α, VEGF and endostatin expression in HepG2 cells in vitro. HepG2 cells were transfected with End‐pcDNA3.1, followed by CoCl2 treatment. Cell lysates were western blotted with anti‐HIF‐1α, VEGF, endostatin and tubulin antibodies.
The authors next investigated whether the above treatments effected tumoral expression of HIF‐1α and VEGF, which are key angiogenic factors. As shown in Fig. 5f, gene transfer of endostatin down‐regulated HIF‐1α expression, whereas doxorubicin was without effect. The combination therapy with endostatin and doxorubicin also down‐regulated HIF‐1α expression. Both endostatin and doxorubicin down‐regulated the expression of VEGF, and together showed a synergism or at least an additive effect in inhibiting the expression of this angiogenic factor (Fig. 5f).
It has been shown that endostatin inhibited the expression of HIF‐1α in cultured endothelial cells.( 15 ) To investigate whether endostatin could also down‐regulate expression of HIF‐1α and VEGF in tumor cells, the HepG2 cells were transfected with End‐pcDNA3.1, followed by treatment with CoCl2, which has been used extensively to study the hypoxic signaling pathway because of its hypoxia‐mimicking effect under normoxic conditions. Throughout the present study, 200 µM CoCl2 was used as a hypoxia‐mimetic reagent for 3 h. Over 95% of the cells excluded trypan blue, indicating that cell viability was not compromised during CoCl2 treatment. The cells transfected with empty pcDNA3.1 and in the absence of CoCl2 treatment served as controls. As shown in Fig. 5g, intense expression of endostatin protein was detected in HepG2 cells after gene transfection. Endostatin gene transfection decreased the expression of both HIF‐1α and VEGF, which has been elevated by CoCl2 exposure, in HepG2 cells. Interestingly, the expression of VEGF was also down‐regulated by endostatin in HepG2 cells without CoCl2 treatment, indicating endostatin might regulate VEGF expression independent of the HIF‐1 pathway (Fig. 5g).
Discussion
The present study has demonstrated that concomitant treatment of mice bearing subcutaneous human HCC tumors with endostatin gene therapy and doxorubicin results in increased inhibition of tumor growth. Endostatin exerts its antitumor effect by down‐regulating expression of HIF‐1α and VEGF, resulting in inhibition of tumor angiogenesis. In accordance, endostatin down‐regulated HIF‐1α by 74% and VEGF by 67% in human microvascular endothelium, as detected using cDNA microarray analysis.( 15 ) Doxorubicin has direct effects on tumor cells as well as endothelium. Thus, in the present study endostatin and doxorubicin synergistically inhibited endothelial cell proliferation, but not tumor cell proliferation in vitro. Whether the interaction between endostatin and chemotherapeutic drugs is synergistic or additive is debatable. Velde et al.( 23 ) reported that combined treatment with recombinant endostatin protein and doxorubicin resulted in additive antitumor effects in a liver metastases model, whereas Plum et al.( 24 ) reported a synergistic activity of this combination in an orthotopically implanted mammary tumor model. Irrespective of the mechanism, the enhanced therapeutic efficacy and benefit obtained by combining endostatin with chemotherapy is without question. Consideration needs to be given to the fact that combining doxorubicin with other cytotoxics could lead to non‐overlapping or overlapping toxicities, which may hinder clinical implementation. In this respect, antiangiogenic therapy is advantageous as it is relatively less toxic compared with cytotoxic drugs. The present results, and those reported previously,( 21 , 22 , 23 , 24 ) support the incorporation of endostatin antiangiogenic therapy into conventional chemotherapy as a means to improve clinical utility. Some authors have speculated that antiangiogenic therapy targeting the tumor vasculature could hinder other blood‐borne therapeutic agents from reaching tumor cells.( 32 , 33 ) However, a combination of the antiangiogenic agent, tumor necrosis factor‐α, and the cytotoxic agent, melphalan, was shown to generate a higher intratumoral concentration of melphalan than melphalan alone.( 34 ) As suggested by Jain,( 35 ) antiangiogenic therapy may ‘normalize’ the tumor vasculature, thereby enhancing the effect of chemotherapy rather than being inhibitory. However, antiangiogenic therapy itself is probably best performed in combination with other forms of therapy, considering the wide range of angiogenic factors produced by tumor cells and the biological heterogeneity of tumor‐induced blood vessels.( 36 ) In the present study, doxorubicin augmented the antiangiogenic effects of endostatin by inhibiting the proliferation of endothelial cells, and down‐regulating VEGF expression. It can't be discounted that doxorubicin's antiangiogenic effect may have simply been due to the killing of tumor cells that produce VEGF.
Use of cytotoxic drugs at a low dose (metronomic) to specifically target the tumor endothelium can potentiate the antiangiogenic effect of chemotherapy. A number of chemotherapeutic drugs have been shown to inhibit the tumor angiogenesis of several different types of cancers when administered at low doses.( 37 ) The combination of metronomic chemotherapy and antiangiogenic therapy enhances anti‐tumor activity by targeting multiple facets of tumor endothelial cells.( 38 ) Metronomic therapy, however, is not currently accepted for cytotoxic drugs in the clinic, as continuous administration at low doses might induce acquired drug resistance. In the present study, a clinically relevant dose of doxorubicin was used to target both the tumor cells and the tumor endothelium. The enhanced anti‐tumor activity displayed by the combination of endostatin and doxorubicin may be exerted at the level of the tumor‐associated endothelium and not the malignant cells. This conclusion is based on the observation that recombinant endostatin did not inhibit the proliferation of the tumor cells in vitro, and the inhibition of tumor cell proliferation by doxorubicin was not affected by the addition of endostatin. The results indicate that endostatin neither had a direct effect on tumor cells nor modulated the anti‐tumor cytotoxic activity of doxorubicin, similar to the results of a previous study that investigated an interaction between endostatin and adriamycin.( 24 ) Combining endostatin with chemotherapy might be advantageous, as endostatin could be employed to reduce the dose of chemotherapeutic agents to spare the patient the side‐effects of cytotoxic drugs, without impairing anti‐tumor efficacy. Further investigation of this possibility is warranted.
Despite many successful experiments with antiangiogenic therapies in suppressing tumors in animal models of cancer, disappointing reports have emerged recently from their clinical application.( 39 , 40 ) Recent studies have revealed potential tumor resistance mechanisms to antiangiogenic therapy, indicating that single‐agent therapeutic strategies are not desirable.( 36 ) The obvious example is in a rat tumor model of Yoshida sarcoma, where intravenous administration of TNP‐470, a classic angiogenesis inhibitor, suppressed the growth of primary tumors, but increased the growth of metastatic foci in distant lymph nodes.( 41 ) The lesson from this study is that angiogenesis inhibitors need to be administered long‐term at relatively high levels. Although a majority of preclinical and clinical antiangiogenic therapies to date have been conducted with purified antiangiogenic factors, gene therapy appears to be more powerful than other forms of antiangiogenic therapy. Gene therapy has the potential to produce the therapeutic agents at high concentration in a local area for a sustained period, thereby avoiding the problems encountered with long‐term administration of recombinant proteins.
Acknowledgments
This work was supported in part by grants from the National Natural Scientific Foundation of China (30471681, 30571808), and the Scientific and Technological Bureau of Heilongjiang Province, China (QC06C075, WH05C02). Fengjun Liu and Gang Tan contributed equally to this work.
References
- 1. Befeler AS, Di Bisceglie AM. Hepatocellular carcinoma: diagnosis and treatment. Gastroenterology 2002; 122: 1609–19. [DOI] [PubMed] [Google Scholar]
- 2. Schafer DF, Sorrell MF. Hepatocellular carcinoma. Lancet 1999; 353: 1253–7. [DOI] [PubMed] [Google Scholar]
- 3. Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin 2005; 55: 74–108. [DOI] [PubMed] [Google Scholar]
- 4. El‐Assal ON, Yamanoi A, Soda Y, Yamaguchi M, Yu L, Nagasue N. Proposal of invasiveness score to predict recurrence and survival after curative hepatic resection for hepatocellular carcinoma. Surgery 1997; 122: 571–7. [DOI] [PubMed] [Google Scholar]
- 5. Zhu AX. Systemic therapy of advanced hepatocellular carcinoma: how hopeful should we be? Oncologist 2006; 11: 790–800. [DOI] [PubMed] [Google Scholar]
- 6. Sciarrino E, Simonetti RG, Le Moli S et al . Adriamycin treatment for hepatocellular carcinoma. Experience with 109 patients. Cancer 1985; 56: 2751–5. [DOI] [PubMed] [Google Scholar]
- 7. Nerenstone SR, Ihde DC, Friedman MA. Clinical trials in primary hepatocellular carcinoma: current status and future directions. Cancer Treat Rev 1988; 15: 1–31. [DOI] [PubMed] [Google Scholar]
- 8. Yeo W, Mok TS, Zee B et al . A randomized phase III study of doxorubicin versus cisplatin/interferon alpha‐2b/doxorubicin/fluorouracil (PIAF) combination chemotherapy for unresectable hepatocellular carcinoma. J Natl Cancer Inst 2005; 97: 1532–8. [DOI] [PubMed] [Google Scholar]
- 9. Folkman J. What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst 1990; 82: 4. [DOI] [PubMed] [Google Scholar]
- 10. O’Reilly MS, Boehm T, Shing Y et al . Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 1997; 88: 277–85. [DOI] [PubMed] [Google Scholar]
- 11. Sudhakar A, Sugimoto H, Yang C, Lively J, Zeisberg M, Kalluri R. Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by alpha v beta 3 and alpha 5 beta 1 integrins. Proc Natl Acad Sci USA 2003; 100: 4766–71. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 12. Yu Y, Moulton KS, Khan MK et al . E‐selectin is required for the antiangiogenic activity of endostatin. Proc Natl Acad Sci USA 2004; 101: 8005–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nyberg P, Heikkila P, Sorsa T et al . Endostatin inhibits human tongue carcinoma cell invasion and intravasation and blocks the activation of matrix metalloprotease‐2‐9, and – 13. J Biol Chem 2003; 278: 22 404–11. [DOI] [PubMed] [Google Scholar]
- 14. Shichiri M, Hirata Y. Antiangiogenic signals by endostatin. FASEB J 2001; 15: 1044–53. [DOI] [PubMed] [Google Scholar]
- 15. Abdollahi A, Hahnfeldt P, Maercker C et al . Endostatin's antiangiogenic signaling network. Mol Cell 2004; 13: 649–63. [DOI] [PubMed] [Google Scholar]
- 16. Folkman J. Antiangiogenesis in cancer therapy‐endostatin and its mechanisms of action. Exp Cell Res 2006; 312: 594–607. [DOI] [PubMed] [Google Scholar]
- 17. Graepler F, Verbeek B, Graeter T et al . Combined endostatin/sFlt‐1 antiangiogenic gene therapy is highly effective in a rat model of HCC. Hepatology 2004; 41: 879–86. [DOI] [PubMed] [Google Scholar]
- 18. Li G, Sham J, Yang J et al . Potent antitumor efficacy of an E1B 55 kDa‐deficient adenovirus carrying murine endostatin in hepatocellular carcinoma. Int J Cancer 2005; 113: 640–8. [DOI] [PubMed] [Google Scholar]
- 19. Fu GF, Li X, Hou YY, Fan YR, Liu WH, Xu GX. Bifidobacterium longum as an oral delivery system of endostatin for gene therapy on solid liver cancer. Cancer Gene Ther 2005; 12: 133–40. [DOI] [PubMed] [Google Scholar]
- 20. Liang ZH, Wu PH, Li L, Xue G, Zeng YX, Huang WL. Inhibition of tumor growth in xenografted nude mice with adenovirus‐mediated endostatin gene comparison with recombinant endostatin protein. Chin Med J (Engl) 2004; 117: 1809–14. [PubMed] [Google Scholar]
- 21. Bertolini F, Fusetti L, Mancuso P et al . Endostatin, an antiangiogenic drug, induces tumor stabilization after chemotherapy or anti‐CD20 therapy in a NOD/SCID mouse model of human high‐grade non‐Hodgkin lymphoma. Blood 2000; 96: 282–7. [PubMed] [Google Scholar]
- 22. Abraham D, Abri S, Hofmann M, Holtl W, Aharinejad S. Low dose carboplatin combined with angiostatic agents prevents metastasis in human testicular germ cell tumor xenografts. J Urol 2003; 170: 1388–93. [DOI] [PubMed] [Google Scholar]
- 23. Velde EA, Vogten JM, Gebbink MFGB, Van Gorp JM, Voest EE, Borel Rinkes IHM. Enhanced antitumor efficacy by combining conventional chemotherapy with angiostatin or endostatin in a liver metastasis model. Br J Surg 2002; 89: 1302–9. [DOI] [PubMed] [Google Scholar]
- 24. Plum SM, Hanson AD, Volker KM et al . Synergistic activity of recombinant human endostatin in combination with adriamycin: analysis of in vitro activity on endothelial cells and in vivo tumor progression in an orthotopic murine mammary carcinoma model. Clin Cancer Res 2003; 9: 4619–26. [PubMed] [Google Scholar]
- 25. Wu Y, Yang L, Hu B et al . Synergistic anti‐tumor effect of recombinant human endostatin adenovirus combined with gemcitabine. Anti-Cancer Drugs 2005; 16: 551–7. [DOI] [PubMed] [Google Scholar]
- 26. Subramanian IV, Nguyen TMB, Truskinovsky AM, Tolar J, Blazar BR, Ramakrishnan S. Adeno‐associated virus‐mediated delivery of a mutant endostatin in combination with carboplatin treatment inhibits orthotopic growth of ovarian cancer and improves long‐term survival. Cancer Res 2006; 66: 4319–28. [DOI] [PubMed] [Google Scholar]
- 27. Sun X, Qiao H, Jiang H et al . Intramuscular delivery of anti‐angiogenic genes suppresses secondary metastases after removal of primary tumors. Cancer Gene Ther 2005; 12: 35–45. [DOI] [PubMed] [Google Scholar]
- 28. Ma L, Luo L, Qiao H et al . Vasostatin synergizes with B7H3‐mediated immunotherapy to eradicate hepatocellular carcinomas. J Hepatol 2006; 46: 98–106. [DOI] [PubMed] [Google Scholar]
- 29. Sun X, Liu M, Wei Y et al . Overexpression of von Hippel–Lindau tumor suppressor protein and antisense HIF‐1alpha eradicates gliomas. Cancer Gene Ther 2006; 13: 428–35. [DOI] [PubMed] [Google Scholar]
- 30. Taraboletti G, Giavazzi R. Modelling approaches for angiogenesis. Eur J Cancer 2004; 40: 881–9. [DOI] [PubMed] [Google Scholar]
- 31. Li J, Tan H, Dong X et al . Antisense integrin αV and β3 gene therapy suppresses subcutaneously implanted hepatocellular carcinomas. Dig Liver Dis 2007; 39: 557–65. [DOI] [PubMed] [Google Scholar]
- 32. Yuan F, Chen Y, Dellian M, Safabakhsh N, Ferrara N, Jain RK. Time‐dependent vascular regression and permeability changes in established human tumor xenografts induced by an anti‐vascular endothelial growth factor/vascular permeability factor antibody. Proc Natl Acad Sci USA 1996; 93: 14 765–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Griscelli F, Li H, Bennaceur‐Griscelli A et al . Angiostatin gene transfer: inhibition of tumor growth in vivo by blockage of endothelial cell proliferation associated with a mitosis arrest. Proc Natl Acad Sci USA 1998; 95: 6367–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nooijen PT, Manusama ER, Eggermont AM et al . Synergistic effects of TNF‐alpha and melphalan in an isolated limb perfusion model of rat sarcoma: a histopathological, immunohistochemical and electron microscopical study. Br J Cancer 1996; 74: 1908–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jain RK. Normalizing tumor vasculature with anti‐angiogenic therapy: a new paradigm for combination therapy. Nat Med 2001; 7: 987–9. [DOI] [PubMed] [Google Scholar]
- 36. Abdollahi A, Hlatky L, Huber PE. Endostatin: the logic of antiangiogenic therapy. Drug Resist Updat 2005; 8: 59–74. [DOI] [PubMed] [Google Scholar]
- 37. Browder T, Butterfield CE, Kraling BM et al . Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug‐resistant cancer. Cancer Res 2000; 60: 1878–86. [PubMed] [Google Scholar]
- 38. Kerbel RS, Klement G, Pritchard KI, Kamen B. Continuous low‐dose anti‐angiogenic/metronomic chemotherapy: from the research laboratory into the oncology clinic. Ann Oncol 2002; 13: 12–15. [DOI] [PubMed] [Google Scholar]
- 39. Pawliuk R, Bachelot T, Zurkiya O, Eriksson A, Cao Y, Leboulch P. Continuous intravascular secretion of endostatin in mice from transduced hematopoietic stem cells. Mol Ther 2002; 5: 345–51. [DOI] [PubMed] [Google Scholar]
- 40. Eisterer W, Jiang X, Bachelot T et al . Unfulfilled promise of endostatin in a gene therapy‐xenotransplant model of human acute lymphocytic leukemia. Mol Ther 2002; 5: 352–9. [DOI] [PubMed] [Google Scholar]
- 41. Hori K, Li HC, Saito S, Sato Y. Increased growth and incidence of lymph node metastases due to the angiogenesis inhibitor AGM‐1470. Br J Cancer 1997; 75: 1730–4. [DOI] [PMC free article] [PubMed] [Google Scholar]