

Abstract
When a replicative DNA polymerase encounters a lesion on the template strand and stalls, it is replaced with another polymerase(s) with low processivity that bypasses the lesion to continue DNA synthesis. This phenomenon is known as translesion replication or replicative bypass. Failing this, the cell is increasingly likely to undergo apoptosis. In this study, we found that proteasome inhibitors prevent translesion replication in human cancer cells but not in normal cells. Three proteasome inhibitors, MG‐132, lactacystin, and MG‐262, inhibited UV‐induced translesion replication in a wide range of cancer cell lines, including HeLa, HGC‐27, MCF‐7, HepG2, WiDr, a malignant melanoma, an acute lymphoblastic leukemia, and a multiple myeloma cell line; irrespective of cell origin, histological type, or p53 status. In contrast, these inhibitors had little or no influence on normal fibroblasts (NB1RGB and TIG‐1) or a normal liver mesenchymal (LI90) cell line. Among the DNA‐damaging antineoplastic agents, cisplatin caused a UV‐type translesion reaction; the proteasome inhibitors delayed cisplatin‐induced translesion replication in cancer cell lines but had only a weak effect on normal cell lines. Therefore, translesion replication would be an effective target of proteasome inhibitors for cancer chemotherapy by which cancer cells can be efficiently sensitized to DNA‐damaging antineoplastic agents, such as cisplatin. (Cancer Sci 2008; 99: 863–871)
Abbreviations:
- ASDG
alkaline sucrose density gradient centrifugation
- CPT
camptothecin
- Da
Dalton
- D'MEM
Dulbecco's modified Eagle's medium
- DMSO
dimethyl sulfoxide
- FCS
fetal calf serum
- Mb
megabase
- MMS
methyl methanesulfonate
- PBS
Ca2+‐ and Mg2+‐free phosphate‐buffered saline
- PCNA
proliferating cell nuclear antigen
- XP‐V
xeroderma pigmentosum variant.
Patients with the autosomal recessive disorder XP‐V are predisposed to skin cancer, and XP‐V cells demonstrate hyper‐mutability after UV irradiation (for reviews, see( 1 , 2 )). The defective gene in XP‐V encodes one of the Y‐family DNA polymerases (polη), which catalyzes translesion synthesis past the cyclobutane dimer of UV‐lesions instead of the replicative polymerase(s) that have stalled just before the lesion. Polη then incorporates nucleotides relatively accurately in a manner similar to AA opposite the cis‐syn TT‐dimer (error‐free bypass)( 3 ) as proved by in vitro lesion‐bypass assays. By pulse‐labeling cells with [14C]thymidine and using a modified protocol of ASDG,( 4 ) we precisely detected translesion replication in vivo, which indicated that it is delayed in XP‐V cells but not completely abolished.( 5 ) Taking the above results together, error‐prone (= mutagenic) and inefficient polymerase(s) are plausibly involved in the UV‐mutagenesis in XP‐V cells devoid of polη (for a review, see( 6 )). To learn more about the in vivo function of the polymerase(s), specific DNA polymerase inhibitors were added. Butylphenyldeoxyguanosine (BuPGdR) inhibited the elongation of pulse‐labeled replication products in the UV‐irradiated XP‐V cells( 5 ) suggesting that the ‘mismatch extender’( 7 ) polζ, is involved in this polη‐independent bypass. Evidence that polζ causes mutagenesis is being accumulated (for a review, see( 8 )).
Caffeine at concentrations of the order of mM significantly prevents elongation in UV‐exposed XP‐V cells but not in normal cells,( 9 ) indicating that polη‐independent and error‐prone bypass is caffeine‐sensitive and is regulated differently from error‐free bypass. Caffeine has pleiotropic effects on DNA metabolism or cell‐cycle checkpoint.( 10 ) Although this inhibitory mechanism remains unexplained, the target of the inhibition must be a Ser/Thr kinase, which functions during the DNA damage response. Even at 10 mM, caffeine did not delay the bypass in normal cells (Fig. 1c), indicating that error‐free UV‐bypass predominates exclusively in normal cells.
Figure 1.

Alkaline sucrose density gradient centrifugation (ASDG) profiles of replication products in UV‐irradiated (a,c) NB1RGB (normal) and (b,d) HeLa cells. (a,b) Time‐course of elongation, (c,d) effect of caffeine. Cells synchronized in the mid‐S phase were UV (10 J/m2)‐irradiated, incubated in normal medium for 30 min, pulse‐labeled with 10 µCi/mL of [14C]thymidine for 1 h, washed twice with phosphate‐buffered saline (PBS), and incubated for 1–5 h at 37°C in a normal medium containing (c,d) 1.25–10 mM caffeine, or (a,b) no inhibitor. (c) Some of these profiles overlap. Sedimentation is from right to left. The arrow indicates the position of the T4 phage DNA (166 kb, i.e. approximately 5.5 × 107 Da/single strand). Labeled E. coli DNA (approximately 4 Mb) sedimented near the bottom (fractions 3–6).( 4 ) The average fragment length (in Mb) of each profile is shown in square brackets. cpm, counts per minute.
Error‐free bypass is reportedly initiated by RAD6/RAD18, a protein complex that conjugates monoubiquitins to PCNA (a DNA polymerase sliding clamp) arrested at the lesion‐site and recruits polη.( 11 , 12 ) Polη is also recruited to the site via direct physical interaction with RAD18.( 12 ) Monoubiquitinated PCNA was observed in UV‐irradiated XP‐V cells( 11 ) suggesting that unaccomplished error‐free bypass is later switched to polη‐independent (error‐prone) bypass via unknown mechanisms. Several models have been proposed for the ‘polymerase switching’ (for reviews, see( 13 , 14 )).
In this study, we detected UV‐induced translesion replication in cancer cell lines. Similar to that observed in XP‐V cells, the translesion in cancer cells was considerably slower than that in normal cells, and it was sensitive to caffeine, indicating that UV‐bypass in cancer cells is predominantly of the polη‐independent type.
Additionally, we found that proteasome inhibitors prevent the UV‐bypass in cancer cells. Three different types of proteasome inhibitors (for a review, see( 15 )) were used. MG‐132 is of the peptide‐aldehyde type, and it blocks the proteolytic activity of the 26S proteasome but also inhibits lysosomal cysteine proteases and calpains. Lactacystin is a natural product derived from actinomycetes, and it differs in structure from peptide aldehydes; it is substantially more specific to the proteasome. The very potent MG‐262 (Cbz‐LLL‐B(OH)2) has an inhibitory boronate group attached to the peptide moiety; it does not inhibit any cellular proteases and is therefore highly specific.
Usually, translesion replication is detected using the ASDG technique. After UV irradiation, the pulse‐labeled replication products in XP‐V cells are smaller than those in non‐irradiated cells, but on prolonged incubation, they eventually attain a high molecular weight similar to that in non‐irradiated cells. This conversion is interpreted that DNA synthesis is temporarily retarded by a pyrimidine dimer and then continues beyond the dimer, leaving a gap that is subsequently sealed.( 9 ) The initial size of the newly synthesized DNA is approximately equal to the average distance between dimers in the template strands.( 16 ) This means that the gaps in the newly synthesized DNA are opposite the dimers (for a review, see( 17 )). Therefore, the mechanism by which the gaps are sealed (by translesion or other postreplication repair mechanisms) can be observed by assessing the molecular weight of the labeled DNA. Using the ASDG technique, we actually detected postreplication repair other than translesion in camptothecin‐treated RAD18‐/‐ chicken DT40 cells.( 18 )
Postreplication repair was designated to be the mechanisms by which the stalling of the replication fork is avoided, including translesion replication and template switching. The latter is also known as ‘strand displacement and branch migration’;( 19 ) however, its real nature is yet unclear. By analogy with postreplication repair mechanisms in E. coli, homologous recombination (HR)‐mediated mechanisms have long been considered.
In order to determine whether the gaps in the newly synthesized strands are sealed by a recombinational exchange or translesion synthesis, A.R. Lehmann used a bromodeoxyuridine (BrUdR)‐313 nm light photolysis method( 16 ) in UV‐irradiated L5178Y cells. The results indicated that the gaps in the newly synthesized strands opposite the pyrimidine dimers are sealed by translesion. (Reportedly, post‐UV conversion of replication products in the mouse lymphoma L5178Y cell is slow in kinetics and is caffeine‐sensitive,( 17 ) similar to that in human cancer cells observed in this study.)
We also tried to detect these recombinational exchanges after UV‐irradiation using BrUdR‐density labeling followed by isopycnic sedimentation analysis. Human cells are labeled together with 14C‐cytidine and BrUdR after UV irradiation; their DNAs are isolated, sheared, and then analyzed by neutral CsCl equilibrium density gradient. If the strand exchange occurs between sister chromatids in the vicinity of UV‐lesion, two newly synthesized strands ought to be paired there. The resulting peak will be observed in the full heavy position (HH, i.e. both strands are density‐labeled). But such an HH peak could be seen neither in XP‐V cells nor in HeLa cells (unpublished observations). Therefore, the conversion of post‐UV replication products that we observed is exclusively the result of translesion.
Additionally, many groups have reported a mutation spectrum after UV irradiation not only in normal cells, but also in XP‐V cells or cancer cells including HeLa. They agreed that the hot spot of mutation is the dipyrimidine sequence. This implies that translesion is mainly executed in UV‐irradiated human cells to resume the stalled replication, because HR‐mediated postreplication repair is considered to be error‐free (this has been confirmed in yeast at least).
Materials and Methods
Cell lines and cell cultures. NB1RGB and HGC‐27 cells (Table 1) were purchased from Riken Gene Bank (Tsukuba, Japan). TIG‐1, LI90, WiDr, HepG2, MCF‐7, CCRF‐CEM, Mewo, and RPMI8226 cells were obtained from Health Science Research Resources Bank (Sennan, Osaka, Japan). The CCRF‐CEM and RPMI8226 cells were cultured in RPMI‐1640 medium supplemented with 10% FCS. The remaining cell lines were maintained in monolayers in D'MEM supplemented with 10% FCS (‘normal’ medium).
Table 1.
Cancer or normal cell lines used †
| Origin | Histological type | p53 status | ||
|---|---|---|---|---|
| Cancer cell line | ||||
| HeLa | uterine cervix | squamous cell carcinoma | WT( 20 ) | |
| HGC‐27 | stomach, lymph‐node meta | undifferentiated carcinoma | transplantable in nude mice | |
| WiDr (= HT‐29) | colon | adenocarcinoma | mt( 20 ) | |
| MCF‐7 | breast | adenocarcinoma | WT( 21 ) | |
| HepG2 | liver | hepatocellular carcinoma | WT( 22 ) | |
| CCRF‐CEM | hemo‐lymphocytic | acute T lymphoblastic leukemia | mt( 20 ) | |
| Mewo | malignant melanoma | mt( 20 ) | ||
| RPMI8226 | hemo‐lymphocytic | multiple myeloma | mt( 20 ) | |
| Normal cell line | ||||
| NB1RGB | skin | normal fibroblasts | WT( 23 ) | |
| LI90 | liver | Ito cells( 24 ) | limited life span | |
| TIG‐1 | fetal lung | normal diploid fibroblasts | WT( 20 ) | |
Sources are listed in the references. mt, mutant; WT, wild type.
UV irradiation and translesion replication.( 5 ) Log‐phase adherent cells were trypsinized and seeded into culture dishes (1.3~3 × 105 cells/φ60 mm dish). The following day, the cell phase was synchronized with 1 mM hydroxyurea for 16 h. The cells were further incubated for 3 h in normal medium. Then, thus treated mid‐S phase cells were exposed to UV light (10 J/m2) from a germicidal lamp (GL15; Toshiba, Tokyo, Japan) at 0.6 J/m2/s. After 30 min in culture, the medium was changed to a labeling medium consisting of D'MEM supplemented with 10% FCS and 10 µCi/mL of [U‐14C]thymidine (Moravek MC267, 470 mCi/mmol). The UV‐irradiated cells were pulse‐labeled for 1 h, while the non‐irradiated cells were labeled for 30 min. The medium was then changed to a normal medium with or without the proteasome inhibitors, MG‐132 (Sigma), lactacystin (Wako, Pure Chemical, Osaku, Japan), or MG‐262 (Calbiochem or Wako), or caffeine (Sigma), and the cells were chased for 1–8 h. These cells were harvested by trypsinization and examined using ASDG.
Suspension‐cultured cells were re‐suspended in a small volume of Ca2+‐ and Mg2+‐free PBS containing 1% FCS, placed at the center of a φ100 mm dish, and exposed to UV light (10 J/m2) as described above. The normal medium was added to the dish. After 20 min in culture, the medium was changed to a labeling medium consisting of an RPMI‐1640 medium supplemented with 10% FCS and 10 µCi/mL of [U‐14C]thymidine (<3.2 × 106 cells/mL). Pulse‐labeling was conducted for 30 min. The hot medium was replaced with a normal medium with or without one of the proteasome inhibitors or caffeine, and incubation was continued for 3 h. These cells were harvested and examined using ASDG.
Cisplatin or MMS treatment. Cisplatin (Sigma) was dissolved with DMSO to 20 mM and added to the culture medium (final concentration, 200 µM). After 1 h in culture, the cisplatin‐containing medium was replaced with normal medium, and the cells were incubated for another 30 min. The medium was then changed to the above‐mentioned D'MEM‐based labeling medium. Pulse‐labeling was conducted for 30 min. The rest of the protocol was identical to that used for UV treatment. MMS (Aldrich) was diluted to 4 mg/mL with double‐distilled water and added to the medium (final concentration, 75 µg/mL). After 30 min in culture, the MMS medium was replaced with normal medium, and the cells were incubated for another 40 min. The medium was then changed to the D'MEM‐based labeling medium, and pulse‐labeling was conducted for 1 h. The rest of the protocol was identical to that used for UV treatment.
Camptothecin treatment and X‐ray irradiation. To place camptothecin‐lesions just ahead of the ongoing replication forks, cells were pulse‐labeled in the D'MEM‐based labeling medium for 30 min, prior to camptothecin treatment. Camptothecin (Sigma) was dissolved in DMSO to 0.1 mM and added to the culture medium (0.4 µM). After 15 min in culture, the camptothecin‐containing medium was replaced with normal medium with or without one of the proteasome inhibitors or caffeine, and the cells were further incubated for 2.5 h. They were then collected and examined using ASDG. For X‐ray irradiation, the culture medium was removed, and the dishes were floated on ice‐water and irradiated using an OM‐150R soft X‐ray generator (Ohmic, Tokyo, Japan) at 3.6 Gy/min (100 kV, 10 mA, with a 0.2 mm Al filter). Pulse‐labeling was conducted for 1 h. The rest of the protocol was identical to that used for UV treatment.
ASDG.( 4, 5 ) Cells (approximately 1 × 105 in 50 µL of PBS) were gently layered onto 50 µL of 1% sucrose in PBS that was overlaid on 100 µL of lysis solution (0.6 M KOH, 2.0 M KCl, 10 mM EDTA, and 1% N‐lauroylsarcosine) placed on a 4.35 mL alkaline 5–20% sucrose gradient (0.3 M KOH, 2.0 M KCl, 1 mM EDTA, and 0.1% N‐lauroylsarcosine) with 0.4 mL of alkaline 80% sucrose as a cushion at the bottom. The gradient was centrifuged at 6000 rpm for 15.6 h at 15°C in a Beckman SW 50.1 rotor. The gradient was fractionated onto 30 circles of No. 17 paper (Whatman). The paper circles were dried, immersed in cold 5% trichloroacetic acid for 10 min, washed three times with ethanol and once with acetone, and dried; the radioactivity was then measured. As a molecular weight marker, [14C]‐labeled phage T4 DNA was placed on the lysis layer in the form of phage particles and sedimented in a parallel run. The approximate fragment length of each fraction was estimated based on the position of this T4 DNA marker and that of E. coli DNA,( 4 ) and it was adjusted using a sucrose density gradient curve.( 4 ) The average fragment length (in megabases [Mb]) of each profile is shown in the figures as a fragment length of the median fraction. (The median fraction is the middle fraction that separates the greater half of the profile from the lower half.)
Viability assay of UV‐irradiated or cisplatin‐treated cells. Cell viability was measured by the CellTiter‐Blue Assay (Promega, Madison, WI, US) based on the notion that viable cells retain the ability to reduce resazurin into resorufin, which is highly fluorescent, while non‐viable cells do not. NB1RGB or HeLa cells were seeded into culture dishes (1.1 × 105 cells/φ35 mm dish). The following day, the cells were exposed to various doses of UV light as described above. The cells were cultured in normal medium with 3.0 mM caffeine or 0.33 µM MG‐262 for 5 h. They were then rinsed with PBS and cultured in the normal medium for 1 day. Next, 200 µL of resazurin solution was added to the medium, and the cells were incubated for 1 h. The medium was recovered, and resorufin fluorescence was measured using an MTP‐650FA microplate reader (Corona Electric, Ibaraki, Japan). Cisplatin treatment was performed by incubating the cells in a normal medium containing various doses of cisplatin for 1 h. The rest of the protocol was identical to that used for UV treatment.
Results
Detection of UV‐induced translesion replication in cancer cells. HeLa cells synchronized at the mid‐S phase were irradiated with UV (10 J/m2) and further incubated for 30 min. These cells were pulse‐labeled with [14C]thymidine for 1 h. The replication products after UV irradiation sedimented as a sharp peak and were slightly larger than the marker T4 phage DNA (Fig. 1b). When the cells were chased in the normal medium for 1, 3, or 5 h, the products gradually joined to form larger DNA with lengths of the order of megabases (Mb). The conversion (i.e. joining of the fragments into chromosomal size) was very slow, as compared to that in NB1RGB cells (Fig. 1a).
The conversion after UV irradiation was also very slow in other cancer cell lines (3, 4). The conversion observed in undamaged HeLa or HGC‐27 cells was, however, as fast as that in normal, NB1RGB or LI90 cells (upper panels of Fig. 3).
Figure 3.
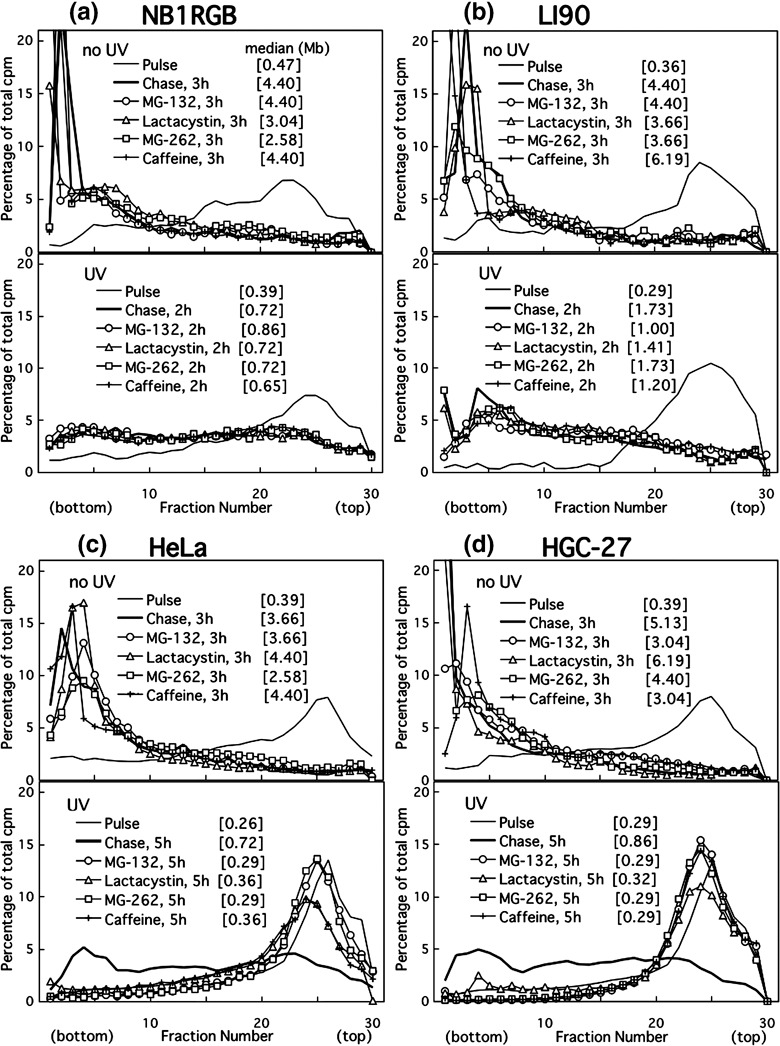
Alkaline sucrose density gradient centrifugation (ASDG) profiles of replication products in (a) NB1RGB (normal), (b) LI90 (normal), (c) HeLa, and (d) HGC‐27 (effects of proteasome inhibitors or caffeine.) Cells synchronized in the mid‐S phase were not irradiated, and were pulse‐labeled with 10 µCi/mL of [14C]thymidine for 30 min, washed twice with phosphate‐buffered saline (PBS), and incubated at 37°C in a normal medium containing 50 µM MG‐132, 200 µM lactacystin, 5.0 µM MG‐262, or 5 mM caffeine for 3 h (upper panel). Similarly synchronized cells were UV (10 J/m2)‐irradiated, incubated in a normal medium for 30 min, pulse‐labeled with the same concentration of [14C]thymidine for 1 h, washed twice with PBS, and incubated for 2 or 5 h at 37°C in normal medium containing 50 µM MG‐132, 200 µM lactacystin, 5.0 µM MG‐262, or 5 mM caffeine (lower panel). (a–d) Some of these profiles overlap. Sedimentation is from right to left. The average fragment length (in Mb) of each profile is shown in square brackets. cpm, counts per minute.
Figure 4.
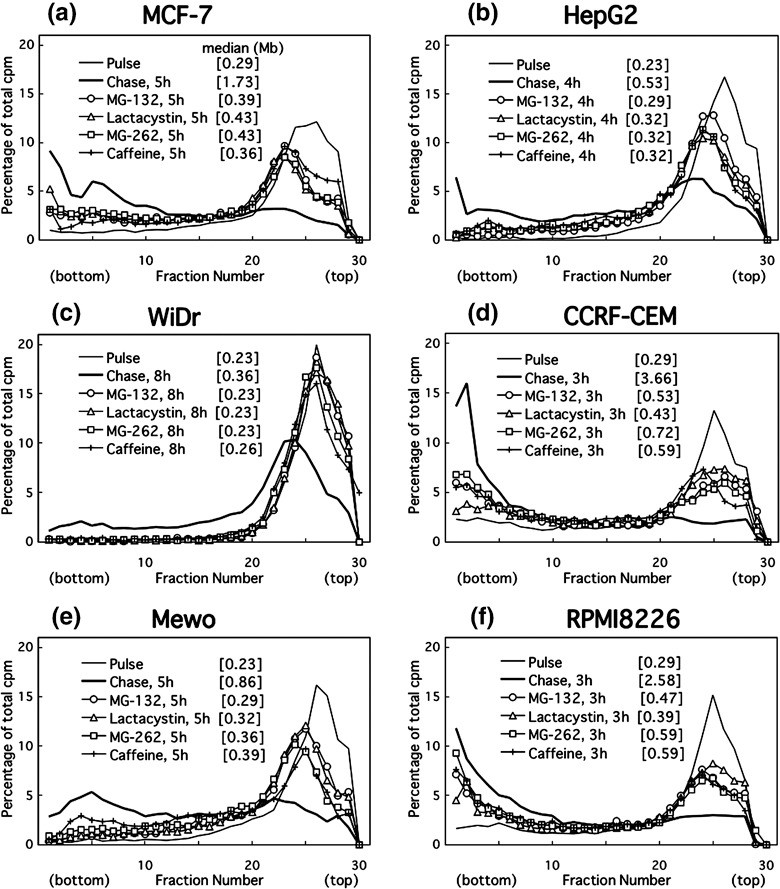
Alkaline sucrose density gradient centrifugation (ASDG) profiles of replication products after UV irradiation in (a) MCF‐7, (b) HepG2, (c) WiDr (= HT‐29), (d) CCRF‐CEM, (e) Mewo, or (f) RPMI8226 cells (effects of proteasome inhibitors or caffeine.) Adherent cells synchronized in the mid‐S phase were UV (10 J/m2)‐irradiated, incubated in the normal medium for 30 min, pulse‐labeled with 10 µCi/mL of [14C]thymidine for 1 h, washed twice with phosphate‐buffered saline (PBS), and incubated for 4–8 h at 37°C in a normal medium containing 50 µM MG‐132, 200 µM lactacystin, 5.0 µM MG‐262, or 5 mM caffeine. CCRF‐CEM and RPMI8226 cells were not synchronized, and pulse‐labeling was conducted for 30 min. Sedimentation is from right to left. The average fragment length (in Mb) of each profile is shown in square brackets. cpm, counts per minute.
Caffeine delays UV‐induced translesion replication in cancer cells. Cells were pulse‐labeled as described above in the absence of caffeine and then chased in a medium containing various concentrations of caffeine. Caffeine inhibited the conversion in UV‐irradiated HeLa (Fig. 1d) and HGC‐27 cells (lower panel of Fig. 3d), as well as in other cancer cell lines (Fig. 4). In contrast, it had no effect on either NB1RGB (1, 3), LI90 cells (Fig. 3b) or non‐damaged controls for HeLa or HGC‐27 cells (upper panels of Fig. 3c, d, respectively). These results suggest that the caffeine‐sensitive polη‐independent pathway is the major pathway to bypass UV‐lesions in cancer cells, although in HeLa cells, functional and intact polη is abundantly expressed( 25 ) which is different from XP‐V cells. The polη‐independent bypass probably displaces the error‐free bypass via an unidentified mechanism.
Effect of proteasome inhibitors on UV‐induced translesion replication in cancer cells. Even at a lower concentration (3.3 µM), MG‐132 delayed the joining of the replication products in UV‐irradiated HeLa cells (partial inhibition) (Fig. 2a). Raising the dose increased the extent of the delay. At 50 µM, the drug caused complete inhibition (Fig. 2a). Similarly, lactacystin partly prevented the joining at 20 µM, prevented it further at 66 µM, and achieved maximal inhibition at 180 or 200 µM (Fig. 2b). In addition, MG‐262 exhibited a partial effect at 0.1 µM but induced complete inhibition at 0.33–5 µM (Fig. 2c). The dose of each inhibitor was selected based on these results (MG‐132, 50 µM; lactacystin, 200 µM; MG‐262, 5 µM).
Figure 2.
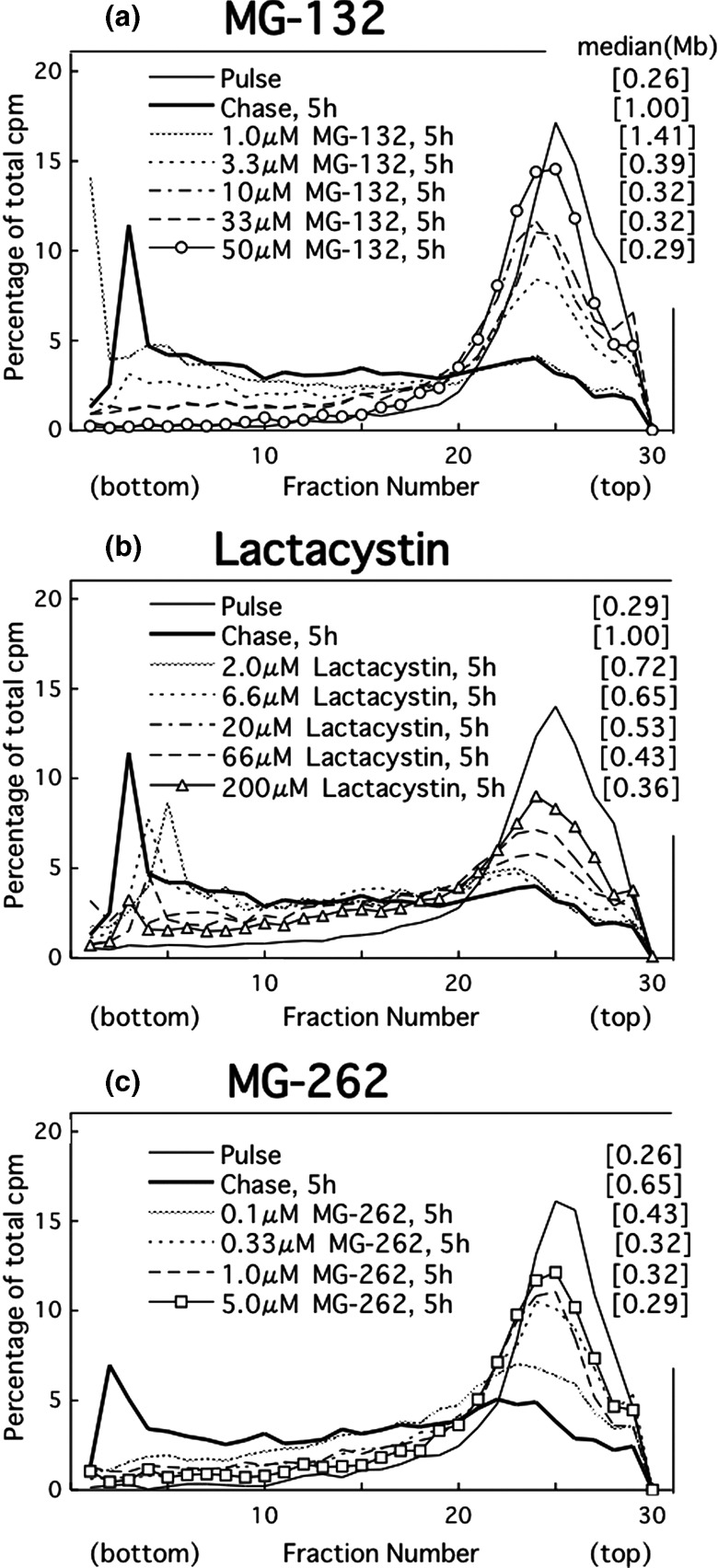
Alkaline sucrose density gradient centrifugation (ASDG) profiles of replication products in UV‐irradiated HeLa cells (effects of proteasome inhibitors.) (a) Effect of MG‐132, (b) lactacystin, and (c) MG‐262. HeLa cells synchronized in the mid‐S phase were UV (10 J/m2)‐irradiated, incubated in a normal medium for 30 min, pulse‐labeled with 10 µCi/mL of [14C]thymidine for 1 h, washed twice with phosphate‐buffered saline (PBS), and incubated for 5 h at 37°C in a normal medium containing (a) 1.0–50 µM MG‐132, (b) 2.0–200 µM lactacystin, or (c) 0.1–5.0 µM MG‐262. Sedimentation is from right to left. The average fragment length (in Mb) of each profile is shown in square brackets. cpm, counts per minute.
These three proteasome inhibitors prevented the joining of post‐UV replication products not only in HeLa but also in HGC‐27 cells (lower panels of Fig. 3c,d). In the undamaged controls, however, the inhibitors had no effect (upper panels of Fig. 3c,d). Moreover, the inhibitors had little or no effect on normal cells, such as NB1RGB (Fig. 3a), TIG‐1 (data not shown), and LI90 cells (Fig. 3b), irrespective of whether the cells were exposed to UV.
Proteasome inhibitors effectively prevent UV‐bypass in various cancer cell lines, irrespective of cell origin, histological type, or p53 status (3, 4; Table 1). Thus, the inhibitors remarkably prevented caffeine‐sensitive bypass in UV‐irradiated cancer cells but not error‐free bypass in normal cells.
We examined the effect of the inhibitors on nucleotide excision repair using ASDG. MG‐132 did not change the profiles of incision or repair‐synthesis in UV‐exposed HeLa cells (unpublished observation). Further, these inhibitors did not nick cellular DNA when added alone (unpublished observation).
Proteasome inhibitors prevent translesion replication across cisplatin‐lesions in cancer cells. The joining of replication products after cisplatin treatment was inhibited by both caffeine and the three proteasome inhibitors in HeLa (Fig. 5b) and HGC‐27 cells (not shown), but it was only weakly inhibited in NB1RGB cells (Fig. 5a). Cisplatin forms three kinds of diadducts in DNA (for a review, see ( 26 )): 65% d(GpG) or 25% d(ApG) intrastrand crosslinks, and 5–8% interstrand crosslinks between the guanines in the sequence d(GpC). Probably because both are dinucleotide lesions and because base‐stacking is similar between cyclobutane pyrimidine dimers (CPDs) and cisplatin intrastrand crosslinks, post‐UV and post‐cisplatin translesion replication have common features.
Figure 5.
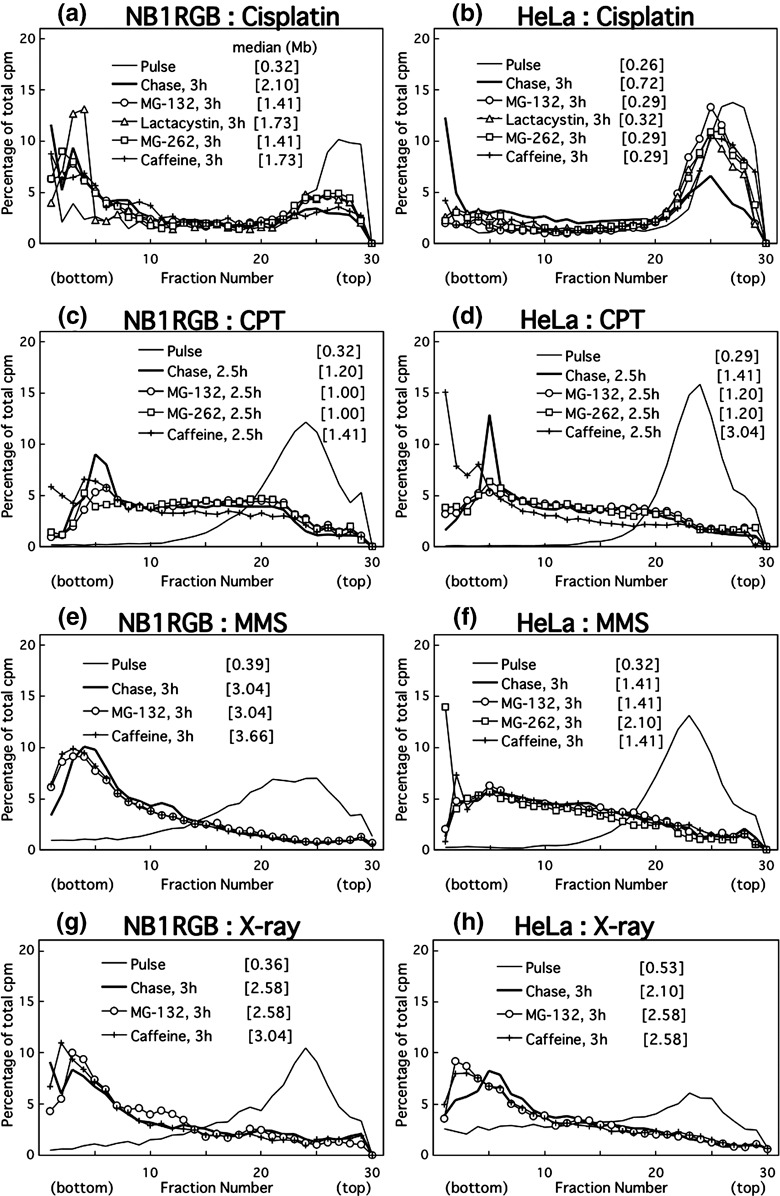
Alkaline sucrose density gradient centrifugation (ASDG) profiles of replication products in (a,b) cisplatin‐, (c,d) camptothecin (CPT)‐, (e,f) methyl methanesulfonate (MMS)‐, or (g,h) X‐ray‐treated (a,c,e,g) NB1RGB (normal) or (b,d,f,h) HeLa cells (effects of proteasome inhibitors or caffeine). Cells synchronized in the mid‐S phase were treated with cisplatin (200 µM, 1 h), MMS (75 µg/mL, 30 min), or X‐rays (10 Gy); incubated in normal medium for 30 or 40 min, and pulse‐labeled with 10 µCi/mL of [14C]thymidine for 0.5 or 1 h. The cells treated with camptothecin (CPT) (0.4 µM, 15 min) were pulse‐labeled for 30 min beforehand. The pulse‐labeled cells were then washed twice with phosphate‐buffered saline (PBS) and incubated at 37°C in a normal medium containing 50 µM MG‐132, 200 µM lactacystin, 5.0 µM MG‐262, or 5 mM caffeine for 2.5 or 3 h. Sedimentation is from right to left. The average fragment length (in Mb) of each profile is shown in square brackets. cpm, counts per minute.
In contrast, neither caffeine nor the proteasome inhibitors had inhibitory effects on the joining of replication products in camptothecin‐ (Fig. 5c,d), MMS‐ (Fig. 5e,f), or X‐ray‐treated NB1RGB or HeLa cells (Fig. 5g,h), suggesting that the replication fork arrest caused by these lesions is overcome by mechanisms distinct from translesion replication.
Caffeine and MG‐262 markedly reduce the viability of UV‐irradiated or cisplatin‐treated HeLa cells. The viability of NB1RGB or HeLa cells after UV irradiation or cisplatin treatment was measured. Caffeine (3 mM) had little or no effect on UV‐exposed or cisplatin‐treated NB1RGB cells (Fig. 6a,c). In contrast, caffeine significantly reduced the viability of similarly treated HeLa cells (Fig. 6b,d).
Figure 6.
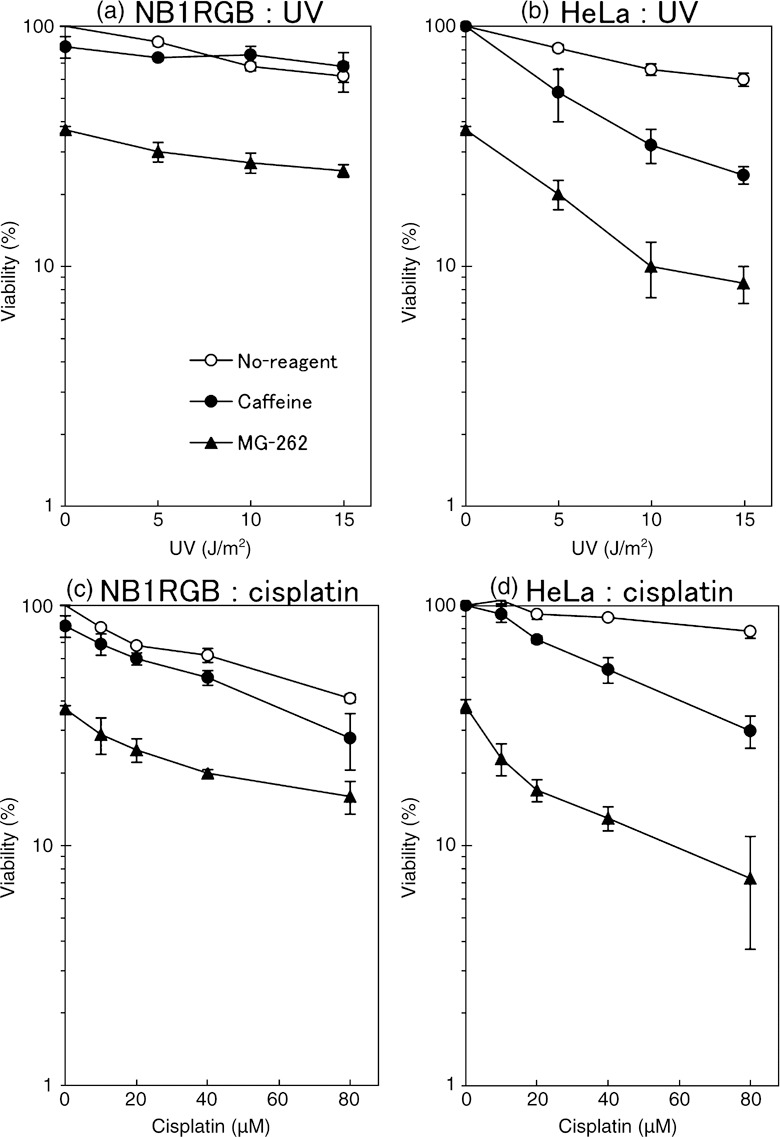
(a,b) Viability of UV‐exposed or (c,d) cisplatin‐treated (a,c) NB1RGB (normal) or (b,d) HeLa cells (effects of MG‐262 or caffeine.) UV‐exposed cells were cultured for 5 h in a normal medium containing 3.0 mM caffeine (closed circle) or 0.33 µM MG‐262 (closed triangle) or without either (open circle). They were rinsed with phosphate‐buffered saline (PBS) and cultured in the normal medium for 1 day. Then, 200 µL of resazurin solution was added to the medium and the cells were incubated for a further 1 h. The medium was recovered, and the resorufin fluorescence was measured. Cisplatin treatment was performed by incubating the cells in a normal medium containing various doses of cisplatin for 1 h, instead of UV. Points, the means of three independent experiments; bars, SE.
MG‐262 (0.33 µM for 5 h) itself is cytotoxic, and therefore the viability of undamaged cells was decreased to approximately 37%. An additive effect was observed in UV‐exposed or cisplatin‐treated NB1RGB cells (Fig. 6a,c), whereas a synergistic effect was observed in HeLa cells (Fig. 6b,d), where the viability reduced rapidly, similar to the effect of caffeine. Thus, caffeine or MG‐262 sensitizes HeLa cells to UV‐ or cisplatin‐induced DNA damages. These results are consistent with those obtained from ASDG experiments.
Discussion
We found that translesion replication after UV irradiation was very slow in cancer cells, compared to that in normal cells. In addition, caffeine delayed UV‐induced translesion replication in cancer cells as well as in XP‐V cells.( 5 ) We also revealed that proteasome inhibitors prevent UV‐bypass not only in XP‐V cells (manuscript in preparation) but also in cancer cells. In contrast, these agents have no effect on error‐free bypass in normal cells. These results imply that the polη‐independent (error‐prone) pathway of translesion reaction is executed in cancer cells.
Our results clearly suggest the involvement of the 26S proteasome in translesion replication. By conducting an epistatic analysis, Podlaska et al. found a link between 20S proteasomal activity and postreplication repair in the yeast Saccharomyces cerevisiae. ( 27 , 28 ) However, the target protein that is probably signaled by polyubiquitins and degraded by the proteasome remains to be identified.
In yeast, genotypes in mutants of postreplication repair are classified into the RAD6 epistasis group (for a review, see( 29 )). In this group, two sets of ubiquitination complex exist: RAD6/RAD18 and UBC13/MMS2/RAD5. RAD6/RAD18 catalyzes the monoubiquitination of PCNA at lysine 164( 30 ) following DNA damage, such as that resulting from UV and MMS. The alternate ubiquitination complex, UBC13/MMS2/RAD5( 31 , 32 ) is inferred to further extend the single ubiquitin moiety with a polyubiquitin chain( 30 , 33 ) in a non‐canonical lysine 63‐linked fashion.( 34 ) The polyubiquitinated PCNA is postulated to function in error‐free ‘template switching’ (for reviews, see( 35 , 36 )), not in proteolysis.( 34 ) Accordingly, no candidate targets of proteasomal disruption have thus far been presented in yeast.
Polyubiquitinated PCNA is rarely detected in human cells.( 11 ) RAD18 protein is autopolyubiquitinated, but this is for the regulation of the protein itself by proteolysis; besides, a mutant that lacks autoubiquitination has normal sensitivity to UV.( 37 ) Less information is available about other candidate targets, for example, the small or large subunit of polδ, RPA, and topoisomerases. The mechanism by which proteasome inhibitors inhibit error‐prone translesion replication remains to be elucidated.
In this paper, we presented evidence that translesion replication is the most effective target of proteasome inhibitors for cancer chemotherapy, with cancer cells being efficiently attacked with minimal damage to normal tissues. Translesion replication was particularly prevented in an undifferentiated gastric carcinoma cell line (HGC‐27), a malignant melanoma cell line (Mewo), and a multiple myeloma cell line (RPMI8226); patients with these cancers are difficult to convalesce. Although UV is rarely used in medicine, cisplatin, one of the most frequently used antineoplastic agents, has been revealed to induce a UV‐type translesion reaction, which proteasome inhibitors prevent in cancer cells.
Bortezomib (Velcade; Millennium Pharmaceuticals, MA, US) is the first proteasome inhibitor in antineoplastic use (for a review, see( 38 )). This agent has recently been approved for use in the United States for the treatment of multiple myeloma, and it has demonstrated promising results. Furthermore, phase II studies of the agent are underway in patients with other hematologic malignancies and solid tumors, in combination with other agents. Bortezomib has a modified peptidyl boronic acid structure, similar to MG‐262. Our findings should contribute to the application of bortezomib and similar drugs to clinical chemotherapy, thereby relieving unendurable pain for a large number of patients.
Proteasome inhibitors may be considerably cytotoxic and non‐specific to translesion replication because the 26S proteasome is involved in the recycling of various unfolded or damaged proteins. Nonetheless, our results are worth reporting. The finding that proteasomal disruption is necessary for translesion replication in cancer cells, which differs from that in normal cells, provides a novel clue regarding the development of chemotherapy with fewer side‐effects.
Acknowledgments
This work was supported in part by grants for nuclear research and scientific research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, and a grant from the Mishima Kaiun Memorial Foundation. We have no financial conflict of interest.
References
- 1. Cleaver JE, Kraemer KH. Xeroderma pigmentosum and cockayne syndrome. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Basis of Inherited Disease, 7th edn, Vol. 3. New York: McGraw‐Hill, 1995; 4393–419. [Google Scholar]
- 2. Friedberg EC, Walker GC, Siede W. DNA Repair and Mutagenesis. Washington D.C.: ASM Press, 1995. [Google Scholar]
- 3. Masutani C, Kusumoto R, Iwai S, Hanaoka F. Mechanisms of accurate translesion synthesis by human DNA polymerase η. EMBO J 2000; 19: 3100–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yamada K, Kameyama Y, Inoue S. An improved method of alkaline sucrose density gradient sedimentation to detect less than one lesion per 1 Mb DNA. Mutat Res 1996; 364: 125–31. [DOI] [PubMed] [Google Scholar]
- 5. Yamada K, Takezawa J, Ezaki O. Translesion replication in cisplatin‐treated xeroderma pigmentosum variant cells is also caffeine‐sensitive: features of the error‐prone DNA polymerase(s) involved in UV‐mutagenesis. DNA Repair 2003; 2: 909–24. [DOI] [PubMed] [Google Scholar]
- 6. Kannouche P, Stary A. Xeroderma pigmentosum variant and error‐prone DNA polymerases. Biochimie 2003; 85: 1123–32. [DOI] [PubMed] [Google Scholar]
- 7. Johnson RE, Washington MT, Haracska L, Prakash S, Prakash L. Eukaryotic polymerase ι and ζ act sequentially to bypass DNA lesions. Nature 2000; 406: 1015–9. [DOI] [PubMed] [Google Scholar]
- 8. Lawrence CW. Cellular roles of DNA polymerase ζ and Rev1 protein. DNA Repair 2002; 1: 425–35. [DOI] [PubMed] [Google Scholar]
- 9. Lehmann AR, Kirk‐bell S, Arlett CF et al . Xeroderma pigmentosum cells with normal levels of excision repair have a defect in DNA synthesis after UV‐irradiation. Proc Natl Acad Sci USA 1975; 72: 219–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kaufmann WK, Heffernan TP, Beaulieu LM et al . Caffeine and human DNA metabolism: the magic and the mystery. Mutat Res 2003; 532: 85–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kannouche PL, Wing J, Lehmann AR. Interaction of human DNA polymerase η with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell 2004; 14: 491–500. [DOI] [PubMed] [Google Scholar]
- 12. Watanabe K, Tateishi S, Kawasuji M, Tsurimoto T, Inoue H, Yamaizumi M. Rad18 guides polη to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J 2004; 23: 3886–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fischhaber PL, Friedberg EC. How are specialized (low‐fidelity) eukaryotic polymerases selected and switched with high‐fidelity polymerases during translesion DNA synthesis? DNA Repair 2005; 4: 279–83. [DOI] [PubMed] [Google Scholar]
- 14. Friedberg EC, Lehmann AR, Fuchs RPP. Trading places: How do DNA polymerases switch during translesion DNA synthesis? Mol Cell 2005; 18: 499–505. [DOI] [PubMed] [Google Scholar]
- 15. Lee DH, Goldberg AL. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol 1998; 8: 397–403. [DOI] [PubMed] [Google Scholar]
- 16. Lehmann AR. Postreplication repair of DNA in ultraviolet‐irradiated mammalian cells. J Mol Biol 1972; 66: 319–37. [DOI] [PubMed] [Google Scholar]
- 17. Lehmann AR. Postreplication repair of DNA in mammalian cells. Life Sci 1974; 15: 2005–16. [DOI] [PubMed] [Google Scholar]
- 18. Yoshimura A, Nishino K, Takezawa J et al . A novel Rad18 function involved in protection of the vertebrate genome after exposure to camptothecin. DNA Repair 2006; 5: 1307–16. [DOI] [PubMed] [Google Scholar]
- 19. Higgins NP, Kato K, Strauss B. A model for replication repair in mammalian cells. J Mol Biol 1976; 101: 417–25. [DOI] [PubMed] [Google Scholar]
- 20. Jia L‐G, Osada M, Ishioka C et al . Study of p53 status of cultured cell lines maintained mainly in the JCRB Cell Bank. Mol Carcinogenesis 1997; 19: 243–53. [DOI] [PubMed] [Google Scholar]
- 21. Casey G, Lo‐Hsueh M, Lopez ME, Vogelstein B, Stanbridge EJ. Growth suppression of human breast cancer cells by the introduction of a wild‐type p53 gene. Oncogene 1991; 6: 1791–7. [PubMed] [Google Scholar]
- 22. Hosono S, Lee C‐S, Chou M‐G, Yang C‐S, Shih C. Molecular analysis of the p53 alleles in primary hepatocellular carcinomas and cell lines. Oncogene 1991; 6: 237–43. [PubMed] [Google Scholar]
- 23. Tsuboi K, Tsuchida Y, Nose T, Ando K. Cytotoxic effect of accelerated carbon beams on glioblastoma cell lines with p53 mutation: clonogenic survival and cell‐cycle analysis. Int J Radiat Biol 1998; 74: 71–9. [DOI] [PubMed] [Google Scholar]
- 24. Murakami K, Abe T, Miyazawa M et al . Establishment of a new human cell line, LI90, exhibiting characteristics of hepatic Ito (Fat‐Storing) Cells. Laboratory Invest 1995; 72: 731–9. [PubMed] [Google Scholar]
- 25. Yuasa M, Masutani C, Eki T, Hanaoka F. Genomic structure, chromosomal localization and identification of mutations in the xeroderma pigumentosum variant (XP‐V) gene. Oncogene 2000; 19: 4721–8. [DOI] [PubMed] [Google Scholar]
- 26. Dronkert MLG, Kanaar R. Repair of DNA interstrand cross‐links. Mutat Res 2001; 486: 217–47. [DOI] [PubMed] [Google Scholar]
- 27. Podlaska A, McIntyre J, Skoneczna A, Sledziewska‐Gojska E. The link between 20S proteasome activity and post‐replication DNA repair in Saccharomyces cerevisiae . Mol Microbiol 2003; 49: 1321–32. [DOI] [PubMed] [Google Scholar]
- 28. McIntyre J, Podlaska A, Skoneczna A, Halas A, Sledziewska‐Gojska E. Analysis of the spontaneous mutator phenotype associated with 20S proteasome deficiency in S. cerevisiae . Mutat Res 2006; 593: 153–63. [DOI] [PubMed] [Google Scholar]
- 29. Barbour L, Xiao W. Regulation of alternative replication bypass pathways at stalled replication forks and its effects on genome stability: a yeast model. Mutat Res 2003; 523: 137–55. [DOI] [PubMed] [Google Scholar]
- 30. Hoege C, Pfander B, Moldovan G‐L, Pyrowolakis G, Jentsch S. RAD6‐dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002; 419: 135–41. [DOI] [PubMed] [Google Scholar]
- 31. Torres‐Ramos CA, Prakash S, Prakash L. Requirement of RAD5 and MMS2 for postreplication repair of UV‐damaged DNA in Saccharomyces cerevisiae . Mol Cell Biol 2002; 22: 2419–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hofmann RM, Pickart CM. Noncanonical MMS2‐encoded ubiquitin conjugating enzyme functions in assembly of novel polyubiquitin chains for DNA repair. Cell 1999; 96: 645–53. [DOI] [PubMed] [Google Scholar]
- 33. Ulrich HD, Jentsch S. Two RING finger proteins mediate cooperation between ubiquitin‐conjugating enzymes in DNA repair. EMBO J 2000; 19: 3388–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Spence J, Sadis S, Haas AL, Finley D. A ubiquitin mutant specific defects in DNA repair and multiubiquitination. Mol Cell Biol 1995; 15: 1265–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lehmann AR, Replication of UV‐damaged DNA. new insights into links between DNA polymerases, mutagenesis and human disease. Gene 2000; 253: 1–12. [DOI] [PubMed] [Google Scholar]
- 36. Broomfield S, Hryciw T, Xiao W. DNA postreplication repair and mutagenesis in Saccharomyces cerevisiae . Mutat Res 2001; 486: 167–84. [DOI] [PubMed] [Google Scholar]
- 37. Miyase S, Tateishi S, Watanabe K et al . Differential regulation of Rad18 through Rad6‐dependent mono‐ and polyubiquitination. J Biol Chem 2005; 280: 515–24. [DOI] [PubMed] [Google Scholar]
- 38. Richardson PG, Hideshima T, Anderson KC. Bortezomib (PS‐341): a novel, first‐in‐class proteasome inhibitor for the treatment of multiple myeloma and other cancers. Cancer Control 2003; 10: 361–9. [DOI] [PubMed] [Google Scholar]