
Figure 1: Molecular mechanisms of BIPN and sites of therapeutic intervention based on results obtained from rodent models.
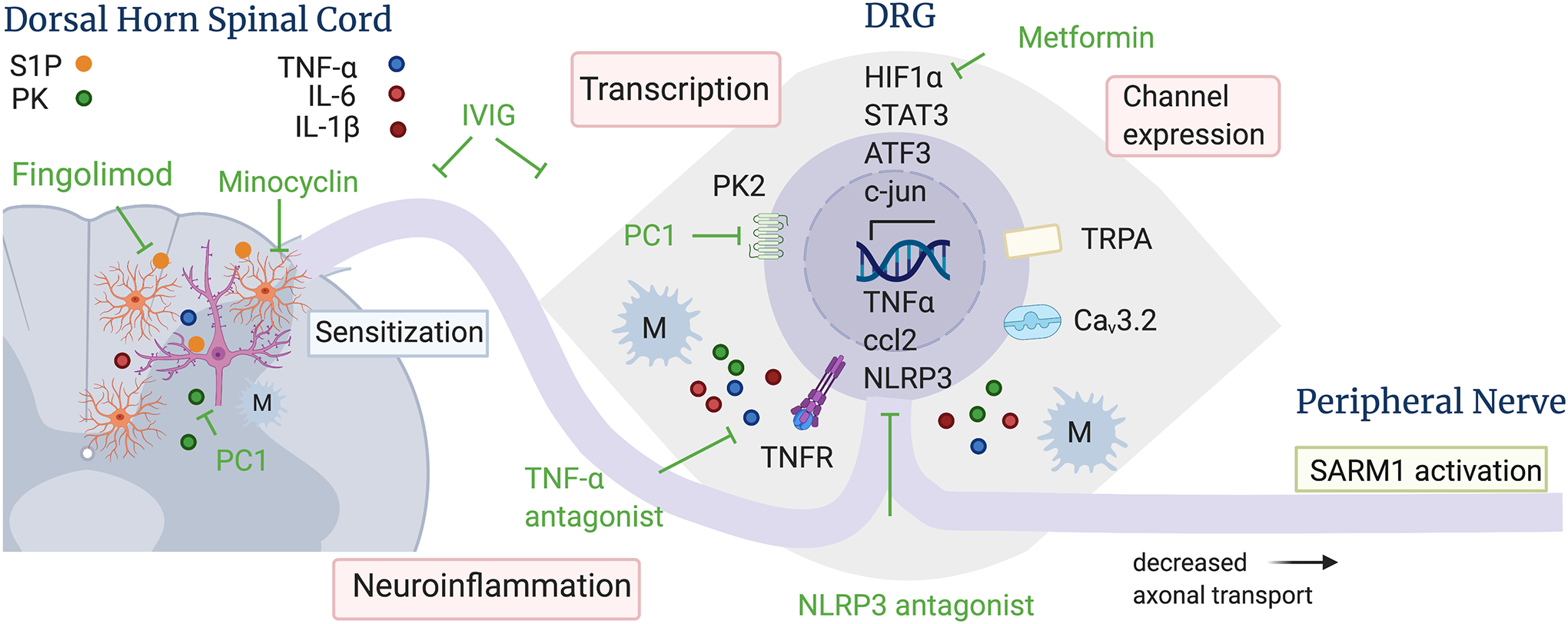
BTZ leads to activation of transcriptional programs and channel overexpression in DRG neurons, which results in neuroinflammation and increased neuronal firing. In the dorsal horn of the spinal cord, BTZ induces astrocyte, but not microglia, activation, dysregulation of sphingolipid metabolism and neuroinflammation, which is associated with increased firing of wide dynamic range neurons and pain sensitization. Several pharmacological interventions indicated in green have been shown to reduce BTZ-induced hyperalgesia in rodent models, including TNFalpha and NLRP3 antagonists, Fingolimod (blocks sphingosine-1-phosphate receptor signaling in astrocytes), PC1, (inhibits prokineticin receptors), metformin (blocks the binding of HIF1alpha to the HIF1-alpha responsive binding element), and IVIg (blocks neuroinflammation). Mechanisms that lead to BTZ-induced axon degeneration and treatment strategies are shown in more detail in figure 2. Abbreviations: ATF3 – activating transcription factor 3; Ccl2 – chemokine ligand 2; Cav3.2 – voltage gated calcium channel 3.2; HIF1alpha – Hypoxia inducible factor 1 subunit alpha; IL – interleukin; M – macrophage; NLRP3 – nod like receptor protein 3; PC1 – prokineticin receptor antagonist; PK – prokineticin; S1P – sphingosine 1 phosphate, STAT3 – signal transducer and activator of transcription 3; TNF-alpha – tumor necrosis factor alpha; TNFR – TNF receptor; TRPA - transient receptor potential ankyrin 1 ion channel.