

Abstract
INTRODUCTION:
Bacteria within the gut continuously adapt their gene expression to environmental conditions that are associated with diet, health, and disease. Noninvasive measurements of bacterial gene expression patterns throughout the intestine are important to understand in vivo microbiota physiology and pathophysiology. Current methods do not offer sufficient information about transient or proximal events within the intestine without using indirect or invasive approaches that disturb normal physiology and are inapplicable to clinical practice.
RATIONALE:
Transcriptional recording by CRISPR spacer acquisition from RNA (Record-seq) enables engineered bacteria to continuously record the history of gene expression in a population of bacteria. Over time, snippets of intracellular RNA are converted into DNA and integrated as a historical record of spacer sequences within CRISPR arrays through the action of an integration complex that contains a reverse transcriptase Cas1 fusion protein (RT-Cas1) and Cas2. Here, using a refined Record-seq methodology, we used transcriptional recording Escherichia coli sentinel cells to reveal intestinal and microbiota physiology under different dietary and disease contexts along the length of the unmanipulated mouse intestine.
RESULTS:
We used transcriptional recording sentinel cell technology in the gastrointestinal tract of germ-free and gnotobiotic mice to assess how the DNA record of spacer sequences in fecal samples uncovered distinct transcriptional records during passage from the proximal to the distal intestine, which depended on diet, inflammation, and microbe-microbe interactions.
Sentinel cells retrieved from feces of stably colonized germ-free mice accumulated new spacers over time and during intestinal transit. Fecal Record-seq profiles were distinct for mice on chow diet versus starch or fat diets. Transcriptional records of these diets were pre-served in fecal spacer sequences even 2 weeks after a dietary switch, whereas diet-specific fecal bacterial RNA-sequencing (RNA-seq) profiles were rapidly lost. Direct measurements showed that Record-seq efficiently captured proximal transient transcriptional events. This included evidence of different carbon source preferences and bacterial hexuronate metabolism through the Entner-Doudoroff pathway under conditions of restricted carbon source availability, which was then verified through competitive colonization of wild-type (WT) E. coli and a ΔidnK/ΔgntK mutant defective in hexuronate catabolism. In addition to information about carbon source preference and metabolism, the transcriptome-scale records also provided evidence of an acid-stress response that was associated with a lowered pH in the cecum of starch-fed mice. In a dextran sulfate sodium (DSS) colitis model, sentinel cells recorded transcriptional alterations consistent with reduced anaerobic metabolism, a stringent response, and increased oxidative and membrane stress. Cocolonization with Bacteroides thetaiotaomicron revealed likely cross-feeding of E. coli from records of uptake and metabolism of fiber-derived saccharides liberated by B. thetaiotaomicron glycoside hydrolases.
Record-seq was also able to capture diet-specific signatures in mice colonized with a 12-organism model microbiota. Moreover, by using barcoded CRISPR arrays, we could show that Record-seq can be multiplexed in several strains of the same bacterial species that co-colonize the intestine, thus elucidating the compensatory response of a single-gene mutant to competition with the WT strain.
CONCLUSION:
Transcriptional recording sentinel cells function in vivo in the mouse intestine and record transcriptome-scale information about diet, disease, and microbial interactions integrated along the length of the intestinal tract over time. Transcriptional recording enables noninvasive measurement of the intestinal tract with potential for biomedical research and future biomedical diagnostic applications.
Transcriptional recording by CRISPR spacer acquisition from RNA endows engineered Escherichia coli with synthetic memory, which through Record-seq reveals transcriptome-scale records. Microbial sentinels that traverse the gastrointestinal tract capture a wide range of genes and pathways that describe interactions with the host, including quantitative shifts in the molecular environment that result from alterations in the host diet, induced inflammation, and microbiome complexity. We demonstrate multiplexed recording using barcoded CRISPR arrays, enabling the reconstruction of transcriptional histories of isogenic bacterial strains in vivo. Record-seq therefore provides a scalable, noninvasive platform for interrogating intestinal and microbial physiology throughout the length of the intestine without manipulations to host physiology and can determine how single microbial genetic differences alter the way in which the microbe adapts to the host intestinal environment.
Graphical Abstract
Transcriptional recording sentinel cells noninvasively report interactions with diet, host, other microbes, and pathological environments. Throughout intestinal transit, sentinel cells capture information about transient mRNA expression into plasmid-encoded CRISPR arrays through the action of a reverse transcriptase Cas1-Cas2 complex. This information is retrieved by means of fecal sampling and deep sequencing followed by computational analyses. Barcoded CRISPR arrays enable transcriptional profiling of isogenic bacteria coinhabiting the intestine.
Engineered microorganisms that harbor molecular recording technologies enable the conversion of cellular histories into heritable nucleotide sequence archives encoded in DNA. Current recording tools—based on recombinases, single-stranded DNA recombineering, CRISPR-Cas9, and CRISPR spacer acquisition—are emerging as valuable platforms for encoding diverse biological features into DNA, including environmental metabolite concentration, cell lineage, gene expression, and horizontal gene transfer (1–7). These technologies can illuminate complex dynamic cellular processes but have largely only been deployed in vitro. An unmet need is to lever-age molecular recording to engineer sentinel cells capable of traversing the gastrointestinal tract and report on microbial and host physiology in the otherwise inaccessible intestinal lumen. Current sentinel cell technologies use biosensors to report on single biomolecules (8–13) but fall considerably short of capturing the complexity of the mammalian gastrointestinal tract.
The contents of the mammalian intestine are exposed to and shape dramatically different luminal environments during transit and colonization (14). The microorganisms that inhabit the small intestine and colon adapt their gene expression to environmental changes associated with nutrients and pathological states. Metatranscriptomics from fecal bacteria are largely uninformative about adaptations to the proximal luminal environment (15), sampling of which requires invasive or disruptive procedures or use of devices that cannot preserve transient signals (16). Because metabolites and RNA are short-lived, omics-based measurements of transient stimuli only yield a snapshot of highly dynamic processes. An integrated measure of intestinal function therefore requires a noninvasive system that is able to sample a wide range of conditions and to preserve proximal transient signals in fecal samples.
Our overriding goal in this work was to use molecular recording to establish a scalable, noninvasive sentinel cell system for assessing intestinal and microbial physiology. To achieve this, we used Record-seq (17), which uses the CRISPR spacer adaptation complex of Fusicatenibacter saccharivorans (FsRT-Cas1–Cas2) to acquire snippets of cellular RNAs as DNA within CRISPR arrays that can later be sequenced to reveal memory of past microbial gene expression. Record-seq captures transcriptome-scale information about the intestinal environment, recording the differences in microbia-host interactions according to diet, disease, or alterations of microbiota composition.
Results
Transcriptional recording sentinel cells acquire transcriptional records within the mouse gut
To establish transcriptional recording as a noninvasive recording tool in the mouse intestine, we orally gavaged germ-free C57BL/6(J) mice with Escherichia coli MG1655 carrying an anhydrotetracycline (aTc)–inducible transcriptional recording plasmid (Fig. 1A). Comparisons between Record-seq on sequentially collected fecal samples and contents of the ileum, cecum, and colon revealed that sentinel cells acquired increasing transcriptional records (E. coli genome-aligning spacers) throughout time and position along the intestinal tract (Fig. 1, B and C, and fig. S1, A and B) and according to aTc concentration (fig. S1C). The functional characteristics of FsRT-Cas1–Cas2 (fig. S1, D to H) were consistent with our previous in vitro experiments (17, 18).
Fig. 1. Transcriptional recording sentinel cells acquire transcriptional records within the mouse gut and preserve this information throughout time.
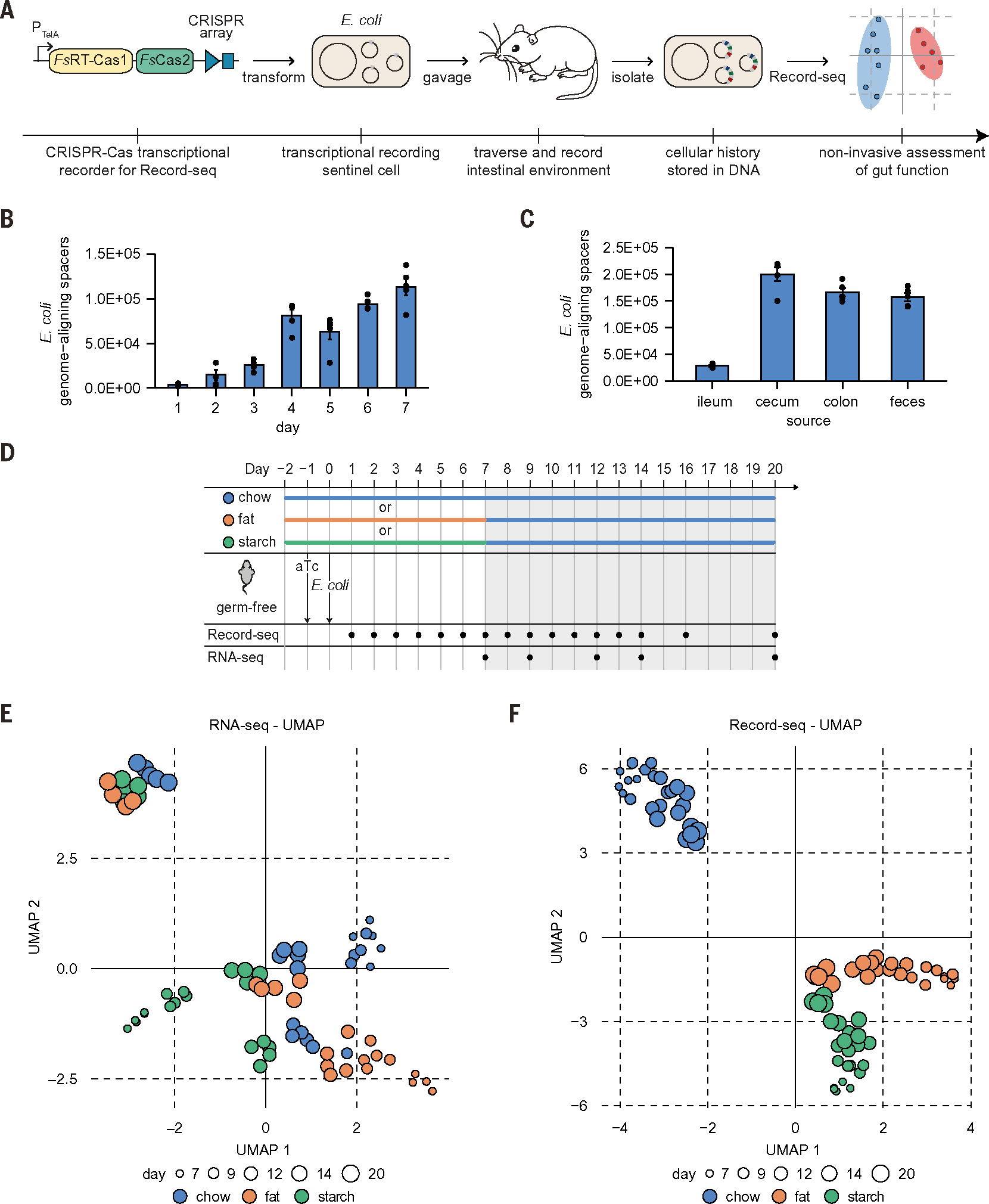
(A) Schematic of experimental workflow for in vivo experiments with transcriptional recording sentinel cells. E. coli with recording plasmid encoding aTc-inducible FsRT-Cas1–Cas2 and a CRISPR array were orally gavaged into mice. Record-seq was performed on feces or intestinal contents. (B and C) Numbers of E. coli genome-aligning spacers (B) obtained from feces on the indicated days and (C) from the indicated intestinal sections on day 20 after gavage. (B) and (C) show mean ± SEM of n = 5 independent biological replicates. (D) Timeline of longitudinal in vivo recording experiment assessing the impact of diet on the intestinal E. coli transcriptome. Germ-free mice were supplied with aTc in the drinking water and gavaged with E. coli MG1655 transformed with the recording plasmid. Mice were fed a chow, fat, or starch diet starting 2 days before gavage until day 7. After day 7, all groups received the chow diet. Fecal RNA/Record-seq sampling is indicated, with day 7 samples being collected before changing the diets. Data for days 1 to 6, 8, 10, 11, 13, and 16 are shown in (B) and figs. S1A and S2C. (E and F) UMAP embedding of (E) RNA-seq or (F) Record-seq data from E. coli under chow (blue), fat (orange), or starch (green) diet on indicated days corresponding to (D). Dot sizes indicate successive time points; n = 5 independent biological replicates. Count thresholds were 104 (Record-seq) and 105 (RNA-seq). Outliers were excluded on the basis of modified z-score and relative deviation from the mean.
Transcriptome-scale recording of complex and dynamic intraluminal environments
We next assessed the capacity of transcriptional recording sentinel cells to record differences in the intestinal environment by varying the animals’ diet. Mice monocolonized with sentinel cells were fed one of three diets: a standard chow or a purified diet based on either starch or fat (referred to as starch or fat diets below). Starting from day 7, all groups received the chow diet (Fig. 1D and table S1). RNA-sequencing (RNA-seq) and Record-seq of fecal samples enabled the characterization of transcriptional changes and the stability of the recorded information. Before the diet switch, both RNA-seq and Record-seq readily distinguished the diet groups (fig. S2, A and B). After the switch to chow, the transcriptional signatures corresponding to starch or fat diets were rapidly lost with RNA-seq but not Record-seq (Fig. 1, E and F, and fig. S2,C to E). Thus, RNA-seq represents a snapshot measurement, whereas Record-seq durably reveals past transcriptional information.
To verify the reproducibility of Record-seq, we replicated the first 14 days in an independent experiment, largely confirming our previous observations (fig. S3, A to H). We also directly compared the two experiments and found a 95% overlap in the regulation of Record-seq differentially expressed genes (DEGs) (fig. S3I and tables S2 and S3). Thus, transcriptional recording sentinel cells reproducibly record cellular transcriptomes and provide an archive of complex and dynamic features of different mammalian intestinal environments in vivo.
Record-seq reveals adaptation of E. coli to intraluminal conditions
Record-seq characterization of the genes and pathways altered in its response to different diets delivered a detailed picture of E. coli’s adaptation to intraluminal environments. DEGs (table S2) readily classified the three diet conditions upon hierarchical clustering (Fig. 2A and table S4), and pairwise comparisons between each diet yielded hundreds of DEGs (Fig. 2B and fig. S4, A and B).
Fig. 2. Record-seq reveals transcriptional changes describing the adaptation of E. coli to diet-dependent intraluminal environments.
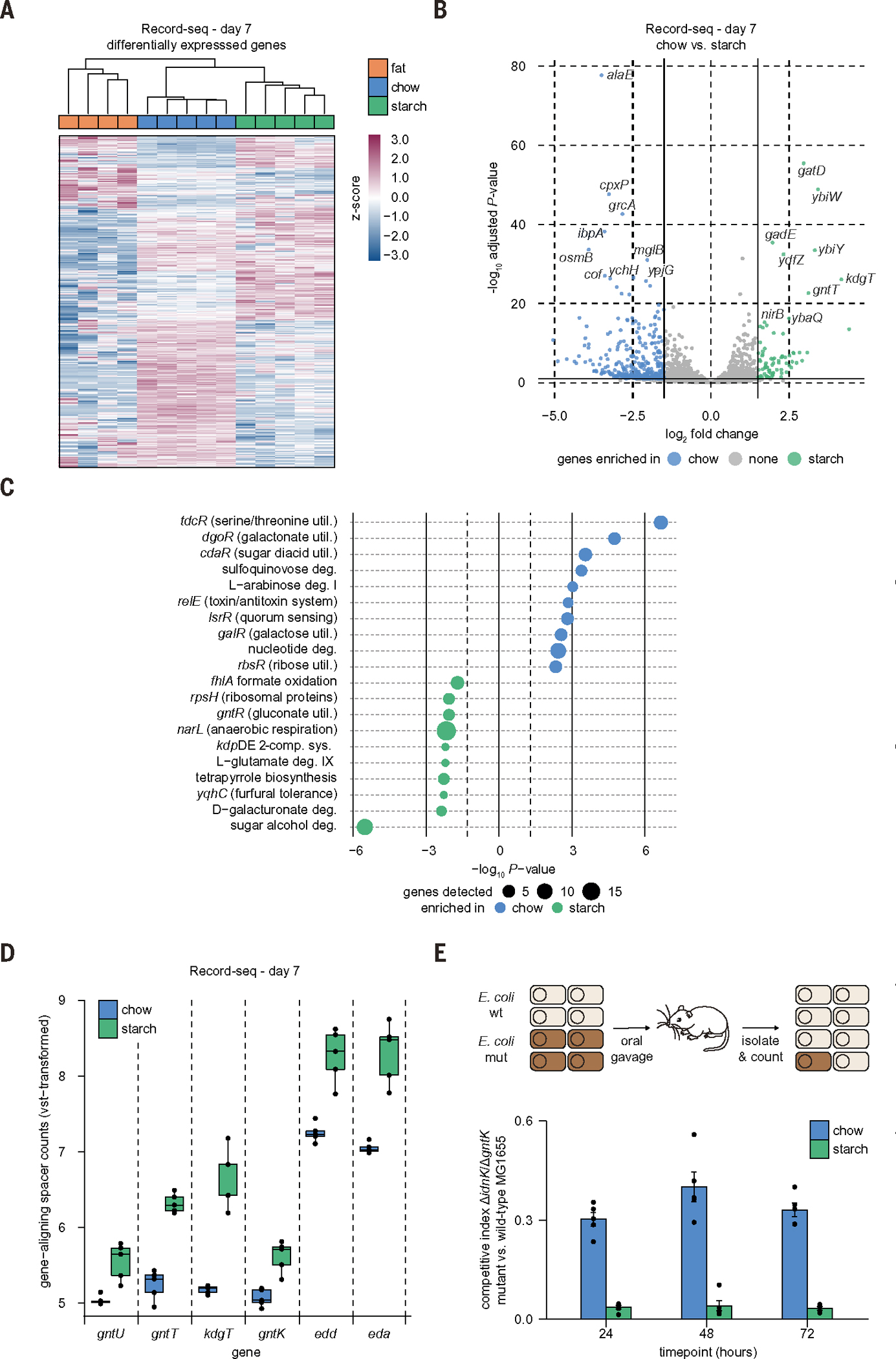
(A to C) Record-seq data from day 7 feces of mice fed a chow (blue), fat (orange), or starch (green) diet. (A) Heatmap showing hierarchical clustering by using 1183 differentially expressed genes (DEGs). z-score standardized gene-aligning spacer counts are shown. (B) Volcano plot with DEGs (Padj < 0.1, log2FC > 1.5) indicated. (C) Pathways and transcriptional and translational regulators enriched per diet group by use of EcoCyc. Dot sizes indicate gene numbers detected as significantly up-regulated for the respective pathway. (D) Box plot showing vst-transformed E. coli genome-aligning spacer counts for selected genes involved in gluconate metabolism corresponding to the indicated diets. (E) Competitive colonization experiment. Germ-free mice fed either a chow or starch diet were orally gavaged with a 1:1 mixture of WT E. coli MG1655 and E. coli MG1655 ΔidnK/ΔgntK (mut). Competitive indices were calculated from fecal recoveries as the ratio of mutant to WT CFU (mean ± SEM, n = 5 independent biological replicates, P = 3.93 × 10−17 likelihood ratio test representative result of two independent experiments). (A) to (D) correspond to Fig. 1D; n = 5 independent biological replicates. Count thresholds were 104 (Record-seq) and 105 (RNA-seq). Outliers were excluded on the basis of modified z-score and relative deviation from the mean.
Pathway enrichment analysis (tables S5 and S6) (19) revealed a number of diet-dependent shifts in a wide range of cellular behaviors among the diet conditions (Fig. 2C and fig. S4, C and D). For further analysis, we focused on a comparison of chow versus starch diets. In starch-fed animals, we found signals indicative of nitrate use as an electron acceptor for anaerobic metabolism (narL), adaptation to low pH (gadC), potassium uptake (kdpDE), and carbohydrate utilization. In chow-fed animals, E. coli used diverse carbon sources as suggested by overrepresentation of utilization pathways for galactonate, glucarate, galactarate, sulfoquinovose, arabinose, galactose, ribose, and fucose (Fig. 2C and fig. S4E). These findings are in line with the diverse composition of the chow diet, which is enriched in plant material (table S1), as well as the previously described adaptation of intestinal symbionts to diverse polysaccharide carbon sources (20, 21).
Record-seq characterization of mice on the starch diet revealed the metabolic adaptation of E. coli in vivo to more restricted carbon source availability. The sugar acids galacturonate and gluconate are present in the host mucus and were shown to be used as carbon sources through the Entner-Doudoroff pathway (EDP) for E. coli colonization in streptomycin-treated mice (22–24). In this study, we could use Record-seq directly in an unperturbed system in which the only manipulation was a diet change, revealing that gluconate and galacturonate are alternative carbon sources in the face of nutritional limitation as shown by enrichment for their catabolic pathways (Fig. 2C and fig. S4F). These changes included key genes of gluconate metabolism, which were strongly up-regulated in E. coli from starch-fed mice compared with chow-fed animals, such as the low- and high-affinity gluconate transporters (gntU and gntT, respectively), the permease for 2-keto-3-deoxygluconate (kdgT), gluconate kinase (gntK), as well as the central enzymes of the EDP (edd and eda) (Fig. 2D).
To confirm gluconate carbon source adaptation directly, we carried out competitive colonizations with wild-type (WT) E. coli MG1655 and mutant E. coli MG1655 ΔidnK/ΔgntK (Fig. 2E). This mutant is unable to catabolize gluconate because it lacks both isoforms of the gluconate kinase, so we expected it to suffer from a substantial competitive disadvantage compared with WT cells. The competitive disadvantage of E. coli MG1655 ΔidnK/ΔgntK over the WT cells was nearly 10-fold greater in starch-versus chow-fed mice (Fig. 2E), suggesting that E. coli becomes more dependent on sugar acids from the host mucus purely because of carbon source changes. Thus, Record-seq delivers a detailed assessment of individual genes and entire pathways altered in E. coli’s adaptation to different diet-dependent intraluminal environments without confounding manipulations.
Sentinel cells capture the intestinal milieu and preserve transient features of the cecum and colon
An important requirement of any intestinal reporting system that avoids potentially confounding manipulations or animal sacrifice is the ability to capture information from the microbial biomass of inaccessible proximal gut sections. Given the ability of Record-seq to preserve information over time, we directly addressed whether the sentinel cells retain information along the longitudinal axis of the gastrointestinal tract by comparing fecal Record-seq with RNA-seq of E. coli from different intestinal segments of mice fed a chow or starch diet. We found that ~46% of the fecal Record-seq DEGs were identified as differentially expressed in the same direction by fecal RNA-seq (Fig. 3A and table S7), highlighting concordance between the two technologies. However, ~54% of the genes detected by fecal Record-seq as up-regulated under the chow or starch diet were found to either be oppositely regulated or show no differences between the diets in fecal RNA-seq (Fig. 3A and table S7).
Fig. 3. Record-seq sentinel cells capture the milieu along the length of the intestine and preserve transient features of proximal large intestinal segments.
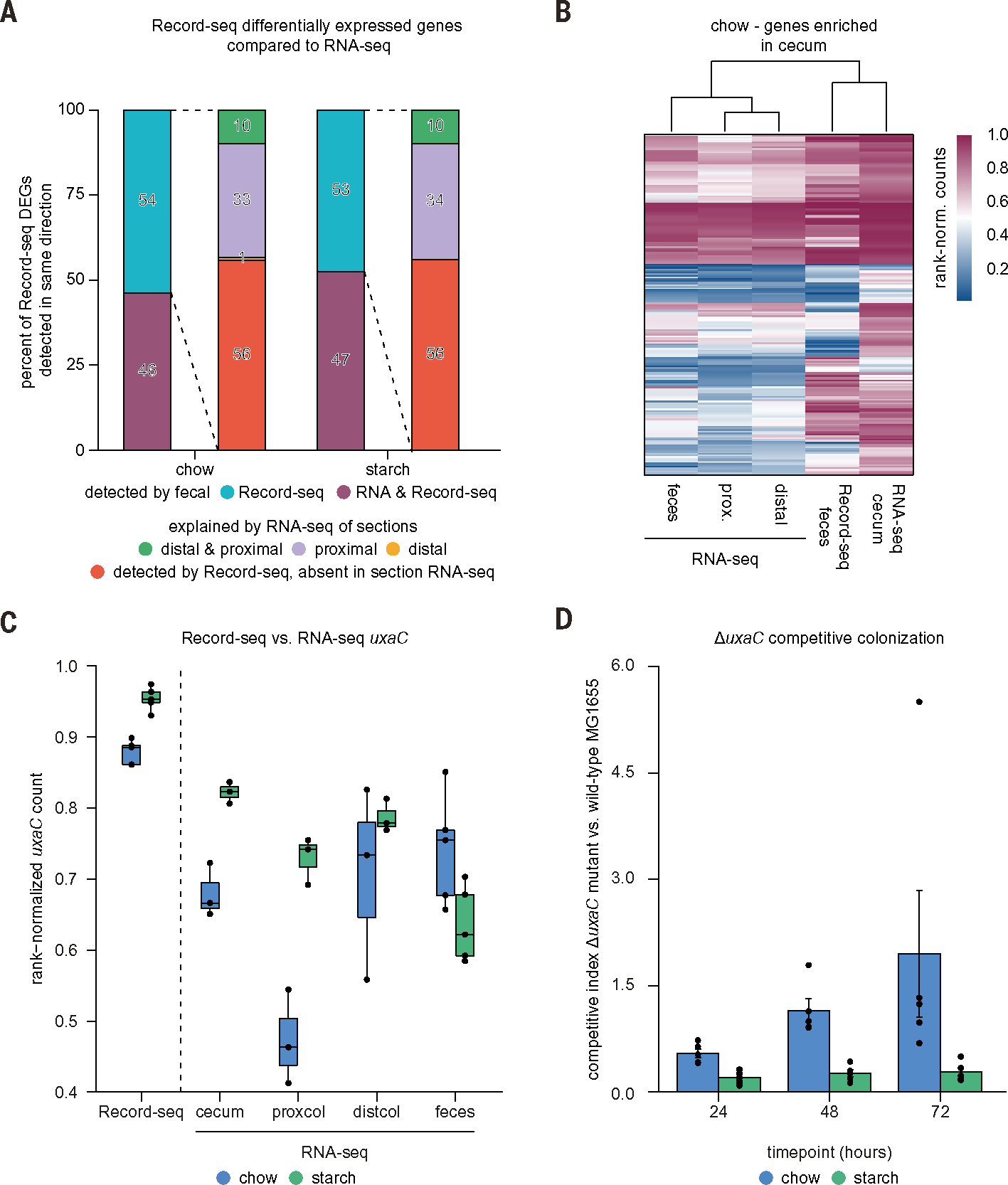
Mice colonized with Record-seq sentinel cells were fed a chow or starch diet for 7 days. (A) Stacked bar plots showing fractions of Record-seq DEGs that were also detected with RNA-seq from feces or intestinal segments as up-regulated under a chow (430 genes) or starch diet (278 genes). Record-seq DEGs not identified with fecal RNA-seq (left bar) are categorized according to differential RNA expression in isolated gut sections (right bar): cecum or proximal colon (“proximal”), distal colon (“distal”), or both proximal and distal sections (“distal and proximal”). (B) Heatmap of cecum signature genes (213 genes overexpressed in the cecum) showing hierarchical clustering of rank-normalized RNA-seq and Record-seq data from the indicated intestinal sections from mice fed a chow diet. (C) Box plot showing E. coli rank-normalized counts of uxaC as determined with Record-seq or RNA-seq from feces, cecum, proximal colon, and distal colon corresponding to the indicated diets on day 7. Data in (A) to (C) are a combined analysis of n = 3 biological replicates per group, each pooled from n = 3 individual mice for gut sections RNA-seq, and n = 5 biological replicates per group for fecal RNA-seq and Record-seq. Count thresholds were 104 (Record-seq) and 105 (RNA-seq). Outliers were excluded on the basis of modified z-score and relative deviation from the mean. (D) Competitive colonization experiment. Germ-free mice fed either a chow or starch diet were orally gavaged with a 1:1 mixture of WT E. coli MG1655 and E. coli MG1655 ΔuxaC. Competitive indices were calculated from fecal ratios of mutant to WT CFU [mean ± SEM, n = 5 independent biological replicates, P = 0.0022 likelihood ratio test (representative result of two independent experiments)].
We considered that fecal Record-seq captures transient events in proximal sections that are absent from fecal RNA-seq. After using RNA-seq to confirm that the transcriptional states of E. coli from different gut segments or feces were distinct (fig. S5, A to D), we addressed the origin of the 54% of genes identified only by Record-seq in the chow condition (Fig. 3A and table S7). A substantial proportion (33%) of these genes were explained by segment-specific RNA-seq signals in the cecum and proximal colon, and a further 10% were explained by expression in the cecum and throughout the colon. Very few (1%) were expressed only in the distal colon. The remaining 56% identified with Record-seq but not RNA-seq were potentially differentially expressed in niches not covered by the sampling performed here (such as the small intestine or colonic mucus) or reflected successive transcriptional events that fell below detection thresholds in RNA-seq. Results from the starch diet were comparable with these findings, although fewer genes were selectively expressed proximally, likely owing to transit effects arising from differences between the fiber content (25) of the diets (Fig. 3A and table S7). Rank-based hierarchical clustering performed on genes overexpressed in the cecum compared with other sections revealed better cecal RNA-seq clustering with Record-seq than with any other RNA-seq dataset, confirming that proximal information is preserved with Record-seq (Fig. 3B and fig. S5E).
To validate a subset of insights regarding proximal gut sections revealed by fecal Record-seq, we followed up on two findings using independent methodologies. First, uxaC—part of the hexuronate catabolism pathway—was distinctly up-regulated in fecal Record-seq and proximal gut RNA-seq under the starch diet (Fig. 3C), suggesting that hexuronates are preferred carbon sources for E. coli under these conditions. In a competitive colonization between WT E. coli and a mutant deficient in hexuronate catabolism (ΔuxaC), the ΔuxaC strain exhibited a significantly greater competitive disadvantage in starch-fed compared with chow-fed mice (Fig. 3D). Second, acid-stress response genes (gadABC and hdeAB) were selectively overexpressed in fecal Record-seq and proximal gut RNA-seq under the starch diet (fig. S5F). Accordingly, the pH of the cecum under the starch diet was significantly reduced compared with that under the chow diet (fig. S5G). Thus, fecal Record-seq reports transcriptional events throughout the length of the intestinal tract and contains information absent from fecal RNA-seq analysis.
Record-seq provides a noninvasive assessment of intestinal inflammation
Another potential application of sentinel cells is their use as noninvasive living diagnostics capable of reporting on gastrointestinal diseases (10). To test this concept, we used the dextran sulfate sodium (DSS)–induced colitis mouse model. After confirming that DSS had negligible direct impact on the E. coli transcriptome in vitro (fig. S6A), we performed Record-seq on sequentially collected fecal samples from mice treated with increasing concentrations of DSS (Fig. 4A). Record-seq not only distinguished treated versus control mice during DSS exposure and withdrawal but could also accurately report on the phenotypic severity of the colitis model (Fig. 4, B and C, and fig. S6, B to E).
Fig. 4. Record-seq provides a noninvasive assessment of DSS-induced intestinal inflammation.
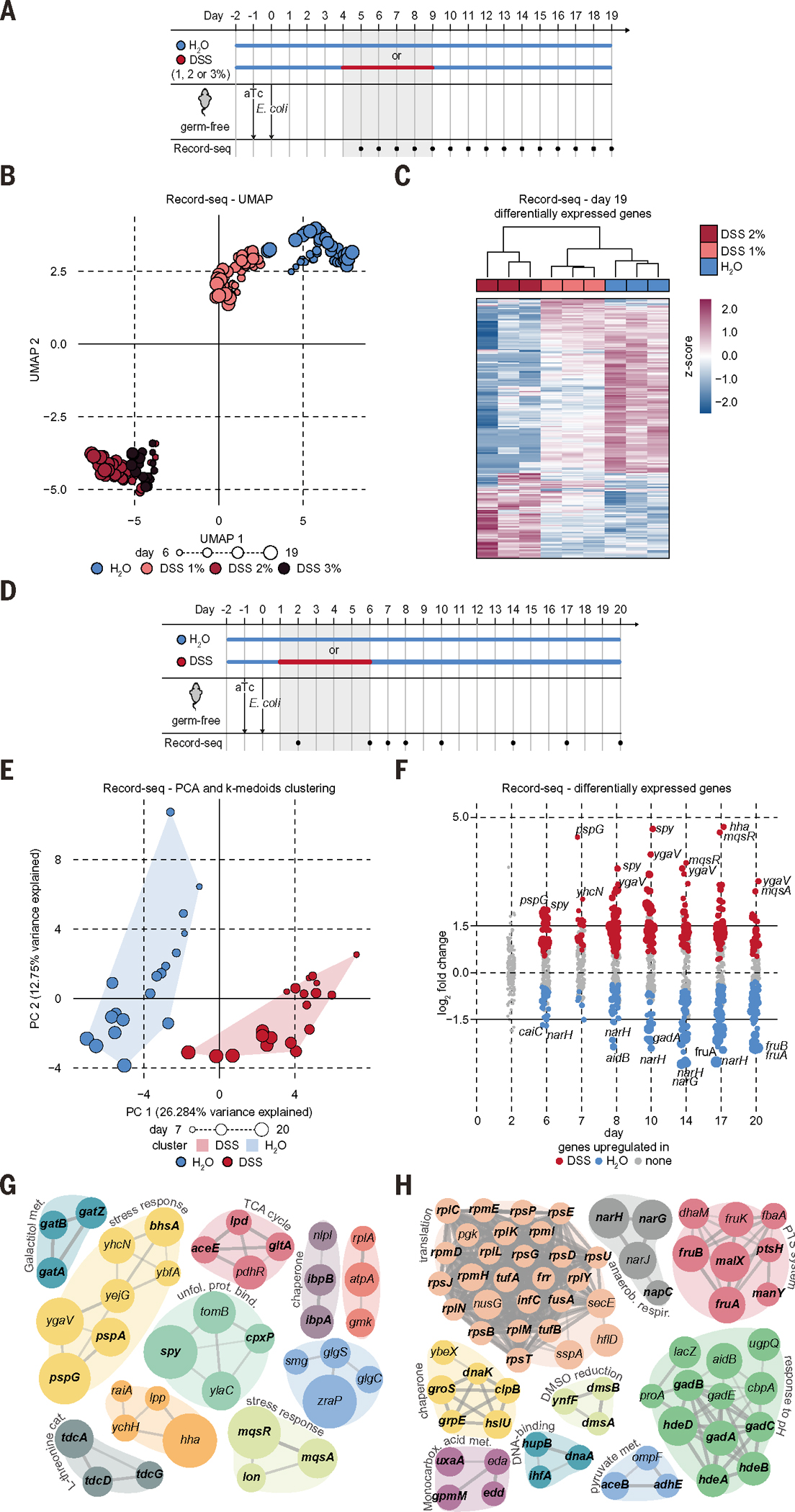
(A) Timeline of DSS colitis recording experiment. Germ-free mice were supplied with aTc in the drinking water; gavaged with E. coli BL21(DE3) sentinel cells; and received 1, 2, or 3% DSS or water as indicated. Fecal Record-seq sampling is indicated. (B) UMAP embedding of Record-seq data for control mice (blue) or mice treated with 1% (salmon), 2% (red), or 3% (black) DSS. Dot sizes indicate successive time points. Mice receiving 3% DSS had to be euthanized on day 13. (C) Heatmap showing hierarchical clustering of Record-seq DEGs from control mice (blue) or mice treated with 1% (salmon) or 2% (red) DSS, day 19. z-score standardized gene-aligning spacer counts are shown. (D) Timeline of DSS colitis recording experiment. Germ-free mice were supplied with aTc in the drinking water, gavaged with E. coli MG1655 sentinel cells, and given 2% DSS or water as indicated. Fecal Record-seq sampling is indicated. (E) PCA-projected Record-seq data on days 7, 8, 10, 14, 17, and 20 for control mice (blue) or mice treated with 2% DSS (red). K-medoids clusters are indicated with convex hulls. Dot sizes indicate successive time points. (F) Dot plot showing log2FC for Record-seq DEGs identified for control mice (blue) or mice treated with 2% DSS (red). Dot sizes increase with significance (Padj range, 9.8 × 10−19 to 1.0). (G and H) STRING analysis of DEGs significantly (G) up-regulated or (H) down-regulated under DSS treatment compared with that of control. Node size increases with log2FC [(G) 0.5 to 4.7; (H) 0.5 to 2.9]. (B) and (C) correspond to the experiment in (A), with n = 3 independent biological replicates. (E) to (H) correspond to the experiment in (D), with n = 3 to 4 independent biological replicates of each condition. Count thresholds were 5× 103 in (B) and (C) and 104 in (E) to (H). Outliers were excluded on the basis of modified z-score and relative deviation from the mean.
Using a longer time course and only the 2% DSS condition, we repeated the in vivo longitudinal recording experiment (Fig. 4D and fig. S6F) to characterize the intraluminal changes that result from DSS-induced inflammation throughout time. We first confirmed that transcriptional records could distinguish DSS-treated and control mice and identify DEGs throughout the duration of the experiment (Fig. 4, E and F; fig. S6, G and H; and tables S4 and S8). Next, we analyzed the DEGs with EcoCyc pathway enrichment (table S9), Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) (table S10), and Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) network tools during and after inflammation and determined that genes required for bacterial membrane integrity (pspG) and post-stress persister cell formation (mqsAR and hha) (26) were up-regulated (Fig. 4, F to H; fig. S6I; and table S8). We also observed the up-regulation of chaperones (spy and cpxP) and heat-shock proteins (ibpA and ibpB) that are induced by oxidative stress (27, 28). By contrast, genes and pathways most prominently down-regulated in DSS-induced colitis included those required for the nitrate and dimethyl sulfoxide (DMSO) electron acceptor pathways (narGH and dmsAB), adaptation to low pH (gad), and the EDP (edd and eda). Down-regulation of the EDP under inflamed conditions likely senses the mucus depletion that characterizes intestinal inflammation (29). These findings also suggest that E. coli shows decreased anaerobic respiration during inflammation but increased levels of envelope and oxidative stress, likely resulting from augmented luminal oxygenation (30). Several ribosomal protein operons were also down-regulated under DSS inflammatory conditions. Transcription of ribosomal proteins is regulated by ppGpp and DksA as part of the stringent response to nutrient limitation (31, 32). Thus, Record-seq sentinel cells can accurately report on DSS-induced colitis disease severity while simultaneously revealing multiple features of the intestinal inflammatory environment.
Record-seq illuminates both host-microbe and microbe-microbe interactions
To assess the performance of Record-seq in the presence of other intestinal microbes, we started by performing longitudinal in vivo recording experiments in mice in the presence of one other prototypical member of the human gut microbiota, Bacteroides thetaiotaomicron (B. theta). Distinct transcriptional archives were obtained from mice either monocolonized with E. coli or cocolonized with E. coli and B. theta (Fig. 5, A to C, and tables S11 and S12). The transcriptional records revealed alterations over a wide range of microbial functions, including a dramatic shift in inferred E. coli carbon source preferences (Fig. 5D; fig. S7, A and B; and tables S13 and S14). In the presence of B. theta, pathways for utilization of xylose, arabinose, sialic acid, amino sugars citrate, rhamnose, maltose, and lactose were significantly up-regulated. Given that B. theta has a far richer content of polysaccharide utilization loci compared with that of E. coli (21, 33) and that mice were fed with chow containing complex plant cell wall carbohydrates (table S1) (25), our data supports that cross-feeding (Fig. 5E) by B. theta liberates usable input nutrients (such as mono- and oligosaccharides) from otherwise unmetabolizable complex diet- or host-derived materials (such as plant pectins and mucus glycans) (34). Supporting the notion that nutrient cross-feeding is beneficial for E. coli, we observed a 3.4-fold increase in E. coli biomass in the presence of B. theta compared with that in monocolonized mice (fig. S7C).
Fig. 5. Record-seq illuminates both host-microbe and microbe-microbe interactions.
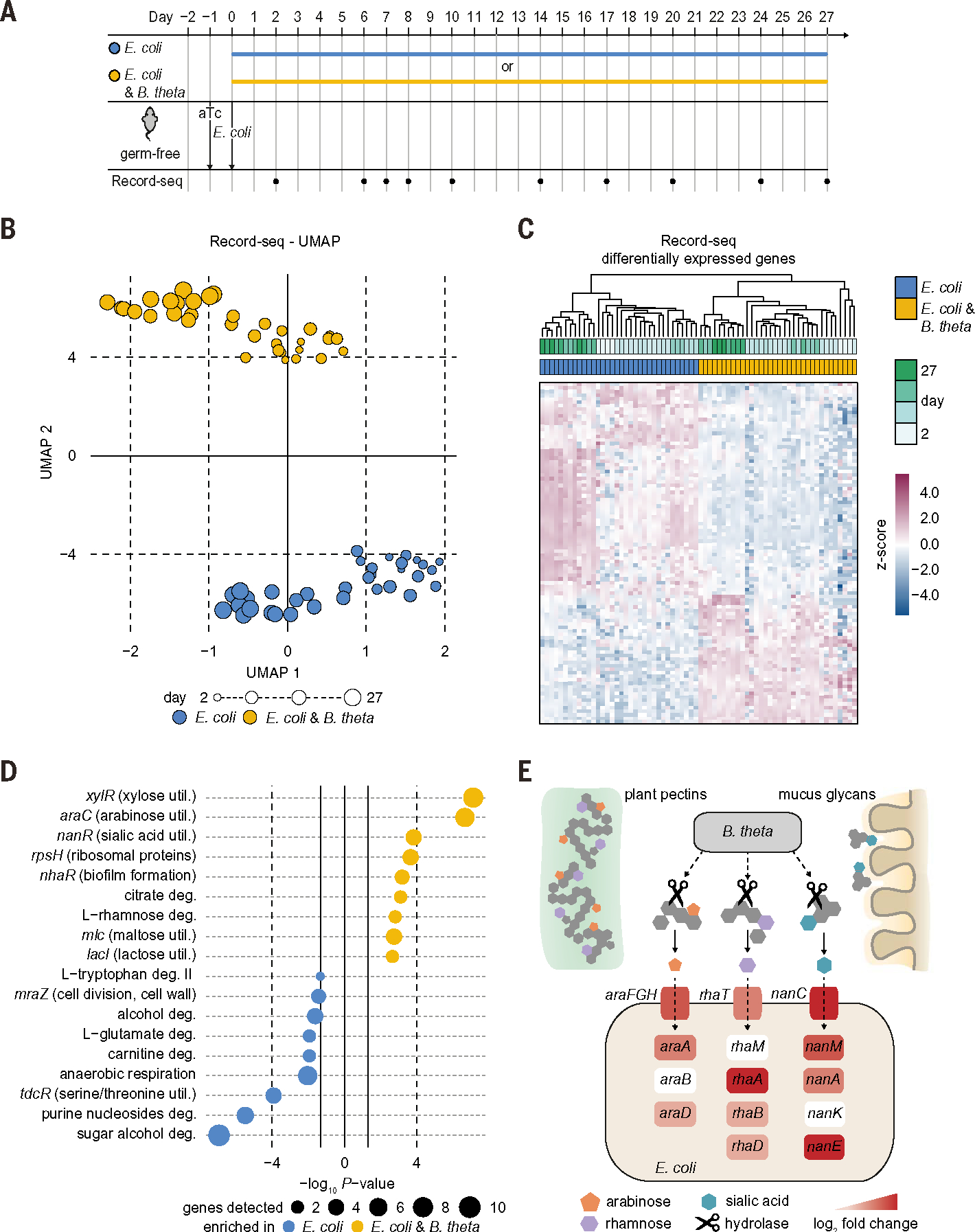
(A) Timeline of longitudinal in vivo recording experiment on interaction of E. coli with B. theta in the gut. Germ-free mice were supplied with aTc in the drinking water and gavaged with E. coli MG1655 sentinel cells alone or together with B. theta. Fecal Record-seq sampling is indicated. (B) UMAP embedding of Record-seq data from E. coli in the presence (yellow) or absence (blue) of B. theta on the indicated days. Dot sizes indicate successive time points. (C) Heatmap showing hierarchical clustering of Record-seq data from E. coli in the presence (yellow) or absence (blue) of B. theta on the indicated days by using identified DEGs. z-score standardized gene-aligning spacer counts are shown. (D) Pathways and transcriptional and translational regulators identified as enriched (P < 0.05) by use of EcoCyc in E. coli in the presence (yellow) or absence (blue) of B. theta on days 2 to 27. Dot sizes indicate gene numbers significantly up-regulated for the respective pathway. (E) Schematic depicting nutrient cross-feeding relationship between E. coli and B. theta inferred by Record-seq. E. coli genes encoding transporters and enzymes are depicted with color codes reflecting their Record-seq–based log2FC of up-regulation in the presence versus absence of B. theta (0 to 5.0). (B) to (E) correspond to (A), with n = 4 independent biological replicates of each condition. Count threshold was 5 × 103. Outliers were excluded on the basis of modified z-score and relative deviation from the mean.
The presence of B. theta also led to down-regulation of E. coli genes involved in metabolism of sugar alcohols, amino acids, fructose, nucleotides, and ethanolamine, which suggests that these secondary carbon sources were not required during bicolonization (Fig. 5D and fig. S7B). Selective expression of ribosomal proteins when E. coli cocolonized with B. theta (Fig. 5D and fig. S7A) is likely indicative of a stringent response on nonpreferred carbon sources when E. coli colonizes alone. These results, which we validated independently (fig. S7, D to G, and tables S12 to S14), demonstrate that Record-seq sentinel cells sense alterations in the intestinal environment in the presence of another taxon and report on the transcriptional adaptations that occur as a result.
We next assessed the capacity of Record-seq to characterize gut function in the presence of a complex microbiota by gavaging sentinel cells into chow- or starch-fed mice colonized by a defined 12-member sDMDMm2 consortium (Fig. 6A) (35). Although colonization resistance caused most gavaged E. coli sentinel cells to rapidly pass through the gastrointestinal tract (fig. S8A), we detected transcriptional records as early as 6 hours after gavage (fig. S8B) and differential expression according to diet from 10 hours after gavage (Fig. 6B). Additionally, the recorded information was sufficient for stratifying the diet groups (fig. S8C) and reproduced in an independent experiment (fig. S8D). By 21 hours, Record-seq detected 220 DEGs in E. coli passing through sDMDMm2-colonized mice on a chow versus starch diet (Fig. 6C). STRING network, KEGG/GO, and EcoCyc pathway enrichment analysis revealed diverse features of E. coli’s adaptation to these ecologically complex environments, including diet-dependent alterations in carbon metabolism (such as galactonate metabolism), anaerobic versus aerobic energy harvesting, and stress responses (Fig. 6D; fig. S8, E to G; and tables S15 to S18). Because none of the bacterial species from the sDMDMm2 microbiota encode a complete DeLey-Doudoroff galactonate degradation pathway, the finding of prominent up-regulation of genes involved in galactonate metabolism in E. coli from chow-fed sDMDMm2 mice indicates that galactonate is likely available as a substrate for E. coli in this in vivo microbial consortium (36). Consistent with our earlier monocolonization results (Fig. 2C), E. coli from starch-fed sDMDMm2 mice overexpressed genes for utilization of mucus-derived sugar acids such as hexuronates and gluconate (fig. S8G), supporting the interpretation that host-derived sugar acids become more important even in the presence of an unmanipulated gnotobiotic microbiota as the dietary nutrient sources become less rich (22, 36). Thus, transcriptional recording sentinel cells function in the context of an intestinal microbiota to reveal a portfolio of host-microbe and microbe-microbe interactions.
Fig. 6. Sentinel cells are deployable within a complex microbiota.
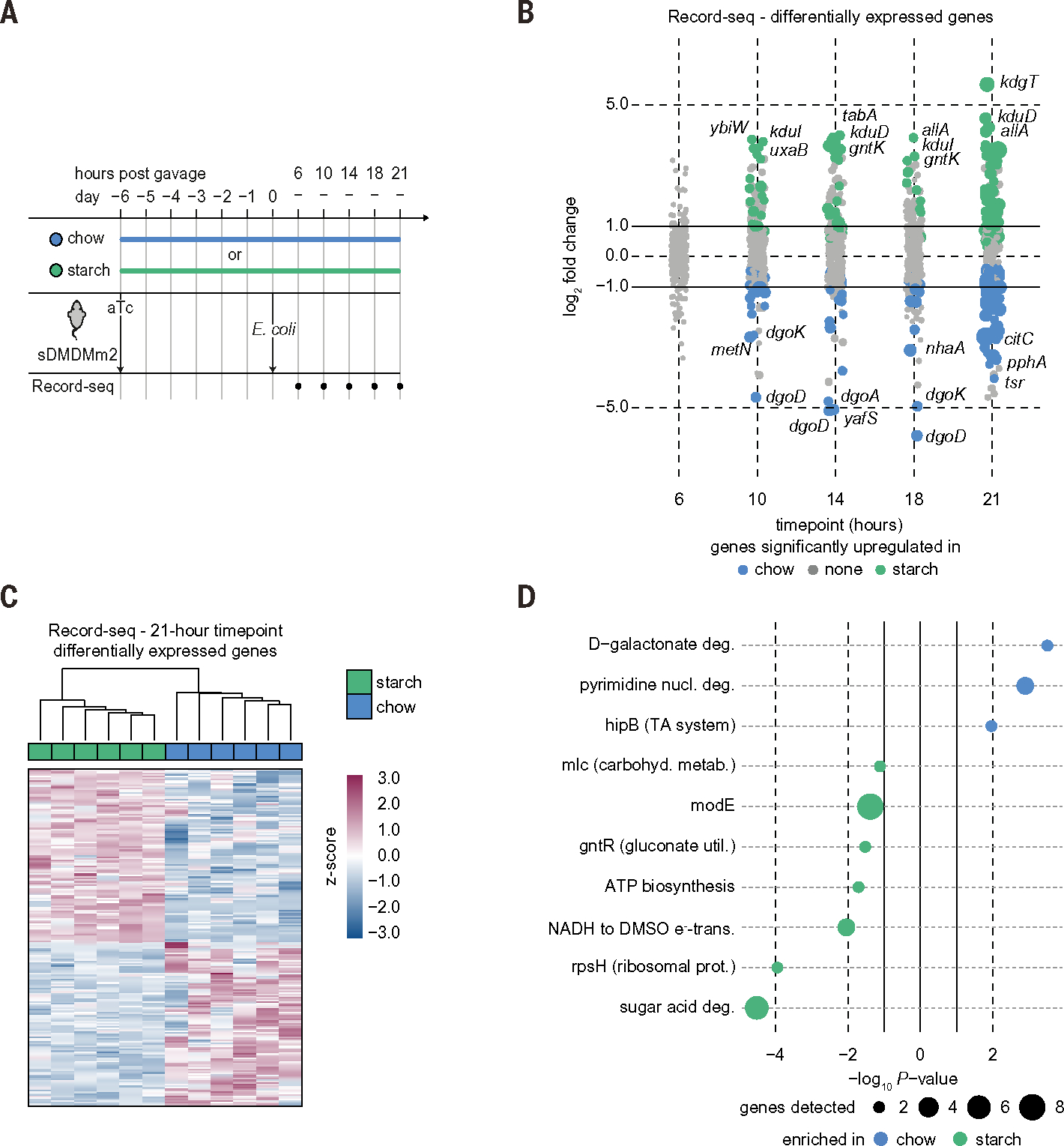
(A) Timeline of complex microbiota recording experiment. Mice harboring a representative 12-member intestinal microbiota (sDMDMm2) were placed on either a chow or starch diet, supplied with aTc in the drinking water, and gavaged with a dose of 7 × 1010 CFU aTc-pretreated E. coli MG1655 as indicated. Fecal Record-seq sampling is indicated. (B) Dot plot showing the log2FC for Record-seq DEGs corresponding to the indicated diet and time. Dot sizes increase with significance (Padj range, 1.9 × 10−17 to 1.0). Colored dots indicate a Padj < 0.1, and gray dots indicate nonsignificant differences. (C) Heatmap showing hierarchical clustering of Record-seq data at 21 hours from mice fed a chow (blue) or starch (green) diet based on identified DEGs. z-score standardized gene-aligning spacer counts are shown. (D) Pathways and transcriptional and translational regulators identified as enriched (P < 0.05) in mice fed chow (blue) or starch (green) by use of EcoCyc. Dot sizes increase with number of genes detected as significantly up-regulated for the respective pathway. (B) to (D) correspond to (A), with n = 6 independent biological replicates for each diet. Count threshold was 5 × 103. Outliers were excluded on the basis of modified z-score and relative deviation from the mean.
Multiplexed Record-seq enables parallel transcriptional profiling of isogenic bacterial strains coinhabiting the mouse intestine
Although genetic polymorphisms between taxa potentially allow RNA-seq to distinguish the transcriptional profiles of different taxa within an intestinal consortium, it is not informative of adaptive transcriptional differences between isogenic strains of the same taxon differing according to one genetic locus (37). We hypothesized that Record-seq could meet this need, allowing a mechanistic understanding of how a particular genetic lesion is functionally compensated within a taxon when two strains are coinhabiting the intestine.
To test this concept, we modified the Record-seq technology. First, we leveraged recent insights revealing that CRISPR spacer acquisition is aided by transcription-coupled repair (38) and introduced a constitutive promoter upstream of the CRISPR array within the recording plasmid, which improved recording efficiency. Second, we developed multiplexed transcriptional recording (Fig. 7A) by using two orthogonal CRISPR arrays with distinct leader and direct repeat (DR) sequences (17), referred to here as DR1 and DR2. After confirming that the barcoded recording constructs facilitated labeling and computational stratification of the transcriptional archives of isogenic E. coli strains in vitro and in vivo (figs. S9 and S10), we investigated whether multiplexed Record-seq could reveal the compensatory mechanism of an isogenic single-gene mutant in a competitive setting in vivo. We prioritized uxaC-deficient E. coli because Record-seq had revealed the importance of uxaC under the starch diet (Fig. 3, C and D). In two independent experiments, germ-free mice cocolonized with WT and uxaC-deficient (ΔuxaC) E. coli MG1655 harbored barcoded recording plasmids in which WT-DR2 was in competition with ΔuxaC-DR1 (group 1) or conversely WT-DR1 competed with ΔuxaC-DR2 (group 2) (fig. S11A). Each isogenic strain revealed robust transcriptional recording activity in vivo, and differences in the transcriptional signatures were driven by genotype (Fig. 7B and figs. S11B and S12), demonstrating multiplexed transcriptional recording of two isogenic E. coli strains inside the same mouse intestine.
Fig. 7. Sentinel cells enable multiplexed transcriptional profiling of isogenic bacterial strains coinhabiting the mouse intestine.
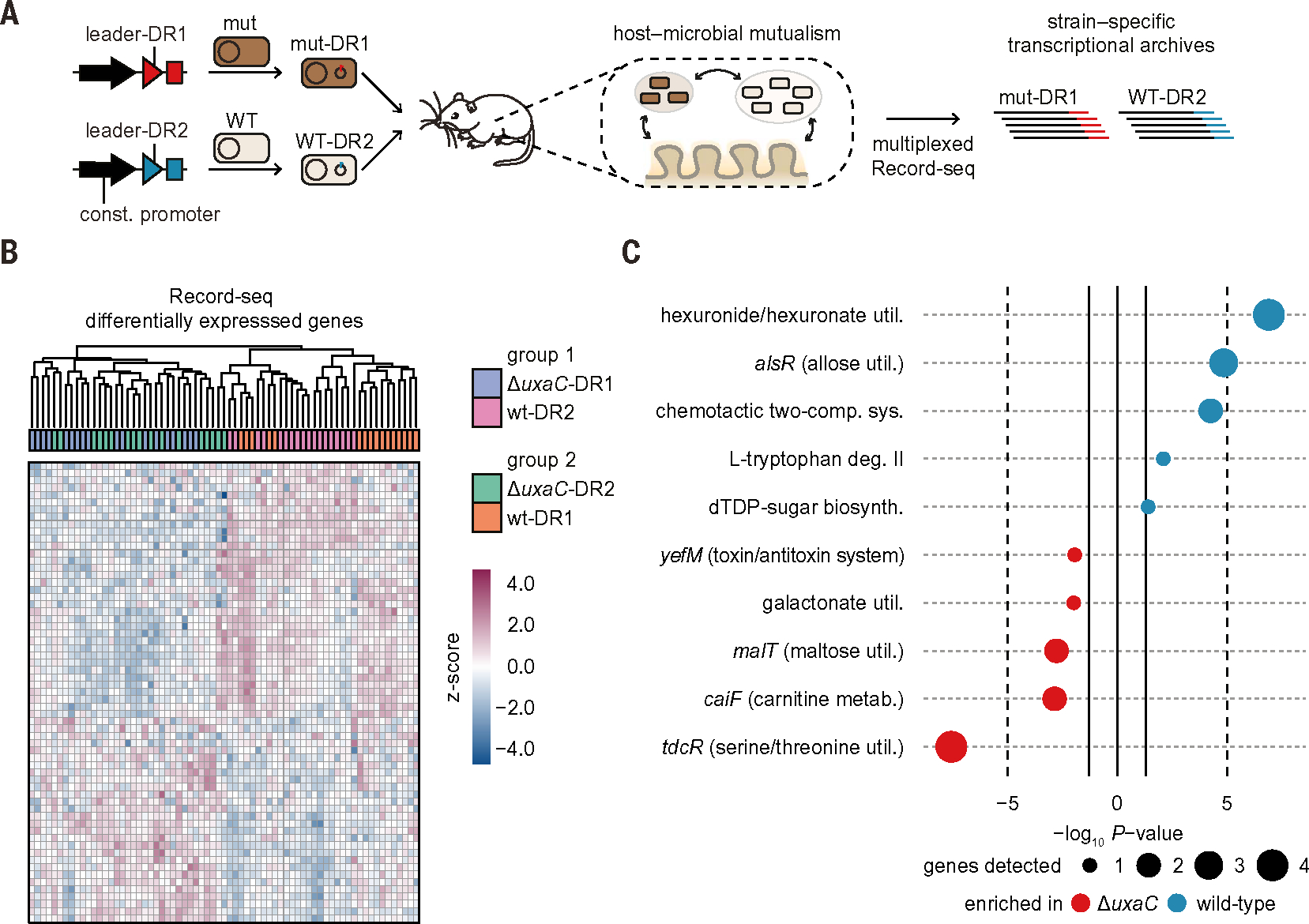
(A) Schematic for multiplexed in vivo recording with sentinel cells barcoded by their CRISPR array. Recording plasmids with either a leader-DR1 or leader-DR2 CRISPR array were transformed into WT or mutant E. coli cells and orally gavaged into mice at a 1:1 ratio. Spacers were assigned to the appropriate strain by using the leader-DR barcode. (B) Germ-free mice on a starch diet were orally gavaged with a 1:1 mixture of WT and uxaC-deficient barcoded E. coli sentinel cells. Two different pairings between genotype (WT or ΔuxaC) and CRISPR array (DR1 or DR2) were used. Group 1 was given ΔuxaC-DR1 (blue) and WT-DR2 (pink). Group 2 was given ΔuxaC-DR2 (green) and WT-DR1 (orange). Fecal Record-seq samples were collected daily from day 7 to day 10, and a combined heatmap is shown. Hierarchical clustering was performed by using the top 50 DEGs detected in both comparisons. z-score standardized gene-aligning spacer counts are shown. (C) Pathways and transcriptional and translational regulators identified as enriched (P < 0.05) by use of EcoCyc on the basis of high-confidence DEGs between ΔuxaC (red) and WT (blue) E. coli. Dot sizes indicate gene number significantly up-regulated for the respective pathway. (B) and (C) correspond to fig. S11A, with n = 5 independent biological replicates of each condition and days 7 to 10. Count threshold was 104. Outliers were excluded on the basis of modified z-score and relative deviation from the mean.
Analysis of DEGs (table S19) and pathways (tables S20 and S21) revealed decreased expression by the uxaC-deficient mutant of other hexuronate utilization genes such as uxaA, uxaB, uxuA, and uxuB in addition to the expected lack of uxaC (Fig. 7C and fig. S12C). Because hexuronate utilization genes are induced by galacturonate and/or glucuronate (39), the WT strain likely displaces the ΔuxaC strain from niches where these sugar acids are available. Furthermore, the ΔuxaC strain appears to compensate for this deficiency through the up-regulation of serine/threonine– and maltose-utilizing gene products. Thus, Record-seq has the distinctive capacity to enable multiplexed transcriptional profiling of two isogenic strains of E. coli in the same mouse gut. This approach reveals compensatory mechanisms in response to intraspecies competition in which RNA-seq–based experiments are uninformative.
Conclusions
We demonstrated that transcriptional recording sentinel cells by using FsRT-Cas1–Cas2 to integrate RNA-derived spacers from the E. coli transcriptome into plasmid DNA-encoded CRISPR arrays are capable of recording complex and dynamic transcriptional changes during the adaptation of E. coli throughout time, transit, and perturbation of the mammalian intestinal tract. This scalable, noninvasive system for assessing intestinal function in vivo archives characteristic microbial signatures of physiological or pathological states. Transcriptome-scale recordings elucidate microbial responses to alterations in the intraluminal environment across nutrition, intestinal inflammation, and microbe-microbe interactions. We have illustrated how carbon preferences can be shown with the tool without confounding manipulations and validated findings of intraluminal microbial adaptation under different dietary conditions.
Record-seq offers multiple advantages compared with contemporary techniques. First, unlike conventional cell-based biosensors, Record-seq does not require a specific biosensor for every biomolecule of interest and can report on a wide range of complex biological features, serving as an unbiased discovery tool. Second, compared with conventional omics-based technologies run on fecal samples, Record-seq integrates information on gut function along the length of the intestine, which is particularly valuable for studying the proximal large intestinal environment that has been largely refractory to detailed studies because of its inaccessible location. Third, multiplexed Record-seq reveals diverse in situ microbe-microbe interactions, even between variants of a single microbial species within the same animal over time, which cannot be readily scaled or implemented in high through-put with conventional methods.
We envision that transcriptional recording sentinel cells and the Record-seq principle may enable understanding intestinal and microbiota physiology under different dietary conditions, disease contexts, or constraints in humans where longitudinal sample collection based on surgical, endoscopic, fecal, ingestible device, or post mortem methods are neither feasible nor sufficient.
Materials and methods
Bacterial strains
Bacterial strains used in this study were Escherichia coli strains MG1655 (ATCC no. 700926) and BL21(DE3) Gold (Agilent no. 230132) and Bacteroides thetaiotaomicron strain VPI-5482 (ATCC no. 29148). E. coli MG1655 StrR, E. coli MG1655 StrR NalR, E. coli MG1655 StrR ΔidnK/ΔgntK, and E. coli MG1655 StrR ΔuxaC were provided by T. Conway and the KanR marker of the ΔuxaC strain was removed using pCP20 recombination as reported previously (40) yielding E. coli MG1655 StrR ΔuxaC ΔKanR. All E. coli strains used in this study are reported in table S22. MG1655 isolates have been sequenced and their genomic sequences are available in the NCBI Assembly database (PRJNA807125). NCBI Reference Sequences U00096.3 and NC_012947.1 were used for MG1655 and BL21(DE3) Gold, respectively. The stable defined moderately diverse mouse microbiota 2 (sDMDMm2) has been described previously (36, 41) and is available through the Deutsche Sammlung für Mikroorganismen und Zellkulturen DSMZ. The constituting taxa were originally isolated from the mouse intestine and comprise Bacteroides I48, Blautia YL58, Akkermansia YL44, Bacteroidales YL27, Ruminococcaceae KB18, Lactobacillus I49, Lachnospiraceae YL32, Erysipelotrichaceae I46, Enterococcus KB1, Flavonifractor YL31, Parasutterella YL45 and Bifidobacterium YL2 (table S23).
Mice
All mouse experiments were performed in accordance with Swiss federal and cantonal regulations under permit numbers BE43/16, BE44/18 and BE107/20. Germ-free C57BL/6(J) mice were born and housed in flexible-film isolators in the Clean Mouse Facility, University of Bern, Switzerland. Unless noted otherwise, mice received a vitamin-fortified rodent chow diet (Kliba Nafag 3307) sterilized by autoclaving for 20 min at 132°C and water ad libitum. Age and sex-matched mice were used at 6 to 15 weeks of age (mostly 8 to 12 weeks). Mice were constantly and independently confirmed to be germ-free within the breeding isolators by culture-dependent methods (liquid cultures in brain-heart infusion (BHI) broth (Thermo Fisher Scientific) aerobically at 37°C at 180 rpm and anaerobically in an anaerobic cabinet (Meintrup DWS) containing 80% N2, 10% H2, and 10% CO2 at 37°C without shaking) and culture-independent methods (i.e., microscopic examination of fecal smears stained with the DNA dye SYTOX green (Thermo Fisher Scientific)).
During experiments, the absence of bacteria other than E. coli (and B. theta in experiments related to Fig. 5 and fig. S7) was constantly confirmed by culturing of fecal suspensions on lysogeny broth (LB) agar aerobically and on BHI agar with 5% defibrinated sheep blood anaerobically. The microbial biomass per gram feces was determined by weighing fecal pellets, homogenizing them in 1 ml of PBS using a Retsch MM400 tissue lyser at 30 Hz for 3 min and streaking serial dilutions in PBS onto LB agar.
One day before gavage, drinking water of the mice was exchanged for water containing 30 μg/ml of anhydrotetracycline (aTc) (Adipogen) and 100 μg/ml of kanamycin sulfate (MP biochemicals) which was prepared by diluting stock solutions of 2 mg/ml of aTc in 95% ethanol and 100 μg/ml of kanamycin sulfate in aqua bidest into sterile tap water.
Plasmid transformation
For plasmid transformation, E. coli BL21 (DE3) Gold (Agilent Technologies) and E. coli MG1655 (ATCC no. 700926) were made chemically competent using the Mix & Go E. coli Transformation Kit & Buffer Set (Zymo Research). For this, strains were streaked to single colonies on LB (Difco) agar (Huberlab) plates without antibiotics followed by growth overnight at 37°C. A single colony of E. coli was inoculated into 50 ml of ZymoBroth (Zymo Research) and grown at 19°C, 220 rpm in an orbital shaker (New Brunswick Innova 40R) to an optical density of OD600 = 0.45. Subsequently, cells were made competent following the manufacturers protocol, dispensed to aliquots of 25 μl, flash-frozen in liquid nitrogen, and stored at −80°C.
Transformation with the recording plasmid pFS_0453 (Addgene #117006) was performed by adding ~60 ng of plasmid DNA to 25 μl of competent cells, followed by heat shock (42°C, 30 s), recovery in 120 μl of S.O.C medium at 37°C, 900 rpm, 30 min and spreading on LB agar plates containing 50 μg/ml of kanamycin sulfate (Biochemica). Glycerol stocks were created by growth of transformants in LB with 50 μg/ml of kanamycin sulfate at 37°C, 180 rpm in bacterial culture tubes followed by mixing of 500 μl of saturated culture with 500 μl of sterile filtered 50% (v/v) glycerol and freezing at −80°C for long-term storage.
Oral gavage
Unless stated otherwise, a saturating gavage dose of 1 × 109 colony forming units (CFU) of E. coli was used to avoid confounding the biological signal reported by our sentinel cells with an initial expansion in the gastrointestinal tract (42). To maintain the recording plasmid and ensure functional stability of the sentinel cells, we added kanamycin sulfate to the drinking water and confirmed that the transformed cells colonized at (7.9 ± 2.6) × 109 CFU/g feces, comparably to what has been reported for the parental strain (42). For oral gavage, E. coli MG1655 or BL21(DE3) each transformed with pFS_0453 was inoculated from freshly grown colonies and cultured overnight under aerobic conditions in LB containing 50 μg/ml of kanamycin sulfate at 37°C, 180 rpm. Upon saturation, cultures were centrifuged at 3480g for 10 min at room temperature and washed twice with the equivalent volume as the LB culture in sterile phosphate-buffered saline (PBS) (8 g per liter of NaCl, 0.2 g per liter of KCl, 1.44 g per liter of Na2HPO4, and 0.24 g per liter of KH2PO4, all from Sigma-Aldrich). The required dose of bacteria was resuspended in PBS (1 × 109 CFU per 500 μl). The bacterial suspension was orally gavaged directly into the mouse duodenum with a 12 gauge straight stainless-steel needle (Provet AG) attached to a 2-ml syringe. The culture volume was chosen according to the number of mice to be gavaged with the equivalent of 3 ml of saturated culture being gavaged into each mouse. Culture vessels were at least twice as large as the culture volume to ensure proper aeration. For example, to gavage n = 15 mice, E. coli was grown in 100 ml of LB broth in a 250-ml bottle, washed twice with 100 ml of PBS, and resuspended in 16.6 ml of PBS from which 500 μl was gavaged into each mouse. Gavage doses and absence of contamination were confirmed by streaking serially diluted suspensions onto LB agar.
Isolation of RNA and RNA-seq
Fecal pellets were homogenized in 1 ml of PBS using a Retsch MM400 tissue lyser at 30 Hz for 3 min. Large particles were pelleted by centrifugation at 200g for 2 min at room temperature. Bacteria in the supernatant were pelleted by centrifugation at 6800g for 3 min at room temperature and lysed in 100 μl of RNA extraction solution containing 95% (v/v) formamide (VWR), 0.025% (w/v) SDS (Bio-Rad), 18 mM EDTA (Merck Millipore), and 1% 2-mercaptoethanol (Merck Millipore) (43) at 95°C, 1200 rpm shaking for 7 min. Following centrifugation at 16,000g for 5 min at room temperature, the RNA in the supernatant was purified with the RNeasy clean-up kit (QIAGEN) following the manufacturer’s instructions, including the optional DNase treatment (15 min at room temperature). RNA was frozen at −80°C for storage and submitted to the Next Generation Sequencing (NGS) Platform Bern for ribosomal RNA (rRNA) depletion using the RiboMinus Transcriptome Isolation Kit, bacteria (Invitrogen), followed by library preparation using the Illumina TruSeq Stranded Total RNA Kit (Illumina) and sequencing on an Illumina NovaSeq platform using the NovaSeq 6000 SP Reagent Kit (100 cycles).
Isolation of plasmid DNA from feces and intestinal contents.
For E. coli monocolonized mice, 50–100 mg of fecal material or intestinal contents were collected and frozen at −20°C. After thawing, feces or intestinal contents were homogenized in 1 ml of PBS using a Retsch MM400 tissue lyser at 30 Hz for 3 min. Large particles were pelleted by centrifugation at 200g for 2 min at room temperature. Bacteria in the supernatant were pelleted by centrifugation at 6800g for 3 min at room temperature and plasmid DNA was isolated using the QIAprep spin Miniprep kit (QIAGEN) with the following modifications: E. coli cells were resuspended in 500 μl of buffer P1 and then lysed by the addition of 500 μl of buffer P2 for 5 min at room temperature. The reaction was neutralized by the addition of 700 μl of buffer N3 and centrifuged at 18,000g for 10 min at room temperature to pellet debris. Supernatant was passed through a spin column on a vacuum manifold (QIAGEN), upon which the column was washed with 500 μl of buffer PB followed by 700 μl of buffer PE. Residual wash buffer was removed by centrifugation at 18,000g for 1 min at room temperature. For cecal contents of monocolonized mice (~200 mg), buffer volumes were increased to 1000 μl of buffer P1, 1000 μl of buffer P2, and 1400 μl of buffer N3, whereas volumes of buffer PB and PE remained the same. Plasmid DNA was eluted from the column by the addition of 50 μl of pre-warmed buffer EB (55°C) followed by incubation at 55°C at 850 rpm for 3 min followed by centrifugation at 10,000g for 1 min. A total of three elution steps with 50 μl were performed yielding 150 μl of eluate. DNA from this eluate was precipitated by the addition of 15 μl of 3 M sodium acetate solution (Sigma-Aldrich) and 150 μl of 2-propanol (Merck-Millipore) followed by incubation at −20°C for 20 min and centrifugation at 20,000g for 20 min at room temperature. Supernatant was carefully removed without disturbing the pellet which was washed with 500 μl of 80% (v/v) ethanol and centrifuged at 20,000g for 15 min at room temperature. Ethanol was removed completely and the pellet was briefly dried (55°C, 30 s) before resuspension in 15 μl of buffer EB and transfer to 96-well PCR plates for storage at −20°C or immediate use in SENECA.
Quantification of isolated plasmid DNA by droplet digital PCR (ddPCR)
For quantification by ddPCR, plasmid DNA was first diluted 100,000-fold in ddPCR dilution buffer. ddPCR dilution buffer consisted of 2 ng/μl of sheared salmon sperm DNA (Sigma Aldrich) and 0.05% Pluronic F-68 (Invitrogen) in UltraPure DNase/RNase-Free distilled water (Thermo Fisher Scientific). Dilution steps were performed in twin.tec PCR plate 96 LoBind (Eppendorf) as follows: dilution 1: 1 μl of plasmid DNA, 49 μl of ddPCR dilution buffer; followed by dilution 2: 1 μl of dilution 1, 49 μl of ddPCR dilution buffer; and finally dilution 3:1 μl of dilution 2, 39 μl of ddPCR dilution buffer. Primer-probe assays targeting FsRT-Cas1 were prepared by mixing of 180 μl of FS_2814 (100 μM), 180 μl FS_2815 (100 μM), and 50 μl of FS_2816 (100 μM) with 590 μl of TE buffer (Sigma Aldrich) (table S24). Aliquots of 100 μl of primer-probe assay were prepared and stored in 1.5-ml amber tubes (Eppendorf) at −20°C. ddPCR was performed by mixing 4.5 μl of dilution 3 template, 1.1 μl of primer-probe assay, 0.25 μl of FastDigest XhoI (Thermo Fisher Scientific), 11 μl of ddPCR Supermix for probes (no dUTP) (Bio-Rad), and 5.4 μl of UltraPure DNase/RNase-Free distilled water per reaction. PCR reactions were dispensed into droplets using the QX100 droplet generator (Bio-Rad) according to the manufacturer’s protocol. PCR amplification was performed in an Eppendorf Mastercycler Gradient (95°C for 10 min, followed by 42 cycles of 95°C for 30 s, 57.1°C for 60 s, 72°C for a 15-s final extension, and a final 98°C 10-min step) with a ramp rate for all steps of 2°C/s as specified by the manufacturer. Readouts were measured using a QX100 droplet reader (Bio-Rad) with a cut-off for positive droplets manually set to 3500.
Selective amplification of expanded CRISPR arrays (SENECA)
The SENECA library preparation method has been extensively described before (18). All DNA oligonucleotides were ordered from IDT (table S25), FastDigest FaqI (ThermoFisher Scientific), T7 DNA Ligase and NEBNext High Fidelity PCR Master Mix, 2X (both New England Biolabs). Due to the low concentration of plasmid DNA extracted from fecal pellets and residual genomic DNA, input DNA was not normalized but instead 3.75 to 7.5 μl of purified plasmid DNA was used for SENECA adapter ligation, volumes are stated in the subsections below corresponding to the respective experiments along with the specific annealed oligonucleotides (carrying library barcodes) for adapter ligation. SENECA first round PCR was performed with 23 cycles instead of 22 cycles and otherwise as described before (18). After second-round PCR, 3 μl of each sample were mixed with 17 μl of UltraPure DNase/RNase-free distilled water (Thermo Fisher Scientific) and loaded on an E-Gel 48 Agarose Gel, 2% along 150 ng of GeneRuler low-range DNA ladder (Thermo Fisher Scientific) in 20 μl for gel-based quantification using the software Bio-Rad Image Lab version 6.0.1. Samples from an experiment were assigned to five bins based on their DNA concentrations and pooled according to these bins. Each sub-pool was purified separately by PCR purification and gel extraction from E-GelEX 2% (Thermo Fisher Scientific) agarose gels as described previously, individually quantified by quantitative PCR (qPCR) using the KAPA Library Quantification Kit for Illumina Platforms (Roche), pooled to achieve equal sequencing depth according to the number of samples in each sub-pool and sequenced on an Illumina NextSeq 500/550 platform as described before. As Record-seq is a population-based measurement requiring many cells to reconstruct a cellular history, and in vivo experiments present a material-limiting environment, we addressed the technical inputs and outputs of our workflow (fig. S1, A and B). Record-seq usually used an input, of approximately (4.7 ± 0.7) × 108 cells per sample (3 to 4 fecal pellets of ~27 mg each per mouse with a biomass of 5.6×109 sentinel cells per gram). After 24 hours of in vivo recording, the Record-seq output was (3.6 ± 0.6) × 103 spacers, which increased by (1.0 ± 0.5) × 104 spacers per day. After 7 days of recording, we found (1.2 ± 0.1) × 105 spacers, which aligned to 91 ± 1% of the 4419 transcripts in the E. coli transcriptome.
Primary analysis of data
The single-end sequencing readout from Record-seq and RNA-seq was processed and analyzed using a two-stage computational pipeline as described before (18). The first step in the primary analysis pipeline involved pre-processing of sequencing reads using FastQC v0.11.4 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) and trimmomatic v0.35 (44). For Record-seq, FASTQ files containing sequencing results were converted to FASTA files using the FASTX-toolkit v0.0.14 (http://hannonlab.cshl.edu/fastx_toolkit). Reads without the library barcode were excluded and acquired unique spacer sequences were identified from the remaining reads using a dedicated python script which incorporates fuzzy string matching (spacerExtractor.py). Identified unique spacer sequences for Record-seq and sequencing reads for RNA-seq were aligned to merged references that contain E. coli genome and plasmid sequences and annotation using Bowtie 2 (45). The following E. coli reference genomes were used: E. coli str. K-12 substr. MG1655 (GenBank U00096.3) and E. coli BL21-Gold(DE3) pLysS AG (Ensembl ASM2366v1). Alignments were then processed and assigned to either the reference E. coli genome or plasmid using Samtools v1.3 (46), and for Record-seq, duplicate alignments were removed using a custom python script (SErmdup.py). This two-step stringent filter for duplicate spacers was incorporated to remove multiple instances of the same spacer arising due to amplification or plasmid replication, and thus obtain a conservative estimate of spacer diversity. Count matrices were generated by quantifying alignments using featureCounts from the Subread package (47). These count matrices contain transcript counts for each RNA-seq sample and transcript-aligning spacer counts for each Record-seq sample. All the steps of this primary analysis pipeline were implemented in Snakemake (48) workflows. The Snakemake workflows for each experiment described in this manuscript, python scripts, reference genomes, and ancillary scripts for primary analysis are available on GitHub (Data and materials availability).
Secondary analysis of data
Secondary analysis was performed on generated count matrices by building upon the previously described recoRdseq package implemented in R (18). This broadly involved unsupervised clustering of samples and identification of classifier genes based on differential expression analysis. Complete analyses for all experiments described in this manuscript are available as R notebooks on GitHub (Data and materials availability). In general, the first step involved filtering count matrices by excluding outlier samples with low cumulative counts () among replicates using an empirically adjusted absolute cumulative counts threshold, as well as a combined threshold for outliers as described below:
include replicate if modified Z-score (, where = cumulative count for replicate = median cumulative count for all replicates), else
if , include replicate if relative deviation from the mean of replicates , where = cumulative count for replicate , and = mean cumulative count for all replicates).
Lowly abundant (or recorded) transcripts, defined as transcripts having a low cumulative count across samples (), were also excluded from the analysis. Further, the first day after gavage (Day 1) generally yielded low spacer counts and noisy data compared to later days in multi-day experiments, hence Day 1 was excluded from all subsequent analyses. The count matrices were then normalized and transformed using the variance-scaling transformation (VST) implemented in the DESeq2 package (49), which rendered the data approximately homoscedastic. For dimensionality reduction and unsupervised cluster discovery, principal component analysis (PCA) using the R base stats package and Uniform Manifold Approximation and Projection (UMAP) (50) using the umap package implemented in R were performed on the vst-transformed count matrices. UMAP parameters and hyperparameters were tuned for each dataset to achieve optimal separation between experimental groups. k-medoids clustering was used to detect clusters in PCA-projected data. Fixed random number seeds were used to ensure reproducibility of clustering algorithms. PCA and UMAP results as well as other illustrative plots were plotted using the ggplot2 package in R. Differential expression analysis was performed using the Wald test (pairwise comparisons) or likelihood ratio test (multiple-group comparisons) implemented in DESeq2 and the quasi-likelihood F-test implemented in the edgeR package in R. DEGs were defined as the intersect of significant genes (, where = Benjamini-Hochberg adjusted P-value detected by these two tools). For time-course datasets, DEGs were independently identified for each timepoint and combined differentially expressed gene lists, ordered by the number of independent timepoints each individual gene was detected on, were generated for downstream pathway analysis. Volcano plots were generated for individual timepoints using the log2-fold change (log2FC) and values calculated by DESeq2. For time-course analysis, log2FC for all genes in the combined differentially expressed gene lists were plotted over time, with point size indicating values. For downstream pathway analyses, the log2FC for each gene (and each comparison) was defined as the maximum log2FC detected for that gene over the time-course. Hierarchical clustering was performed on vst-transformed counts of DEGs after Z-score standardization for each gene, and heatmaps were generated using the pheatmap package in R.
EcoCyc pathway enrichment analysis was performed using the Fisher Exact test for lists of DEGs generated for each experiment, and pathway enrichment plots were created using the top hits. Further, network analysis was performed using the StringApp package (51) in Cytoscape with a confidence score ≥0.4 for these DEGs. MCL clustering using the cluster-Maker2 package (52) was used to generate gene clusters within gene networks detected using StringApp analysis in Cytoscape, and the functional enrichment function was used to annotate these clusters using KEGG pathways, UniProt keywords, NetworkNeighborAL, GO Process, GO Function, GO Component. Nodes contributing to functional enrichment of a cluster were marked by bold font. The size of a node corresponding to each gene in the STRING networks was adjusted to reflect the detected log2FC value for that gene. Over-representation analysis (OA) was performed for differentially expressed gene lists from each experiment based on both the GO resource (53) and Kyoto Encyclopedia of Genes and Genomes (KEGG) (54) using the clusterProfiler package (55) in R. Wherever experiments were replicated, regression analysis was performed to examine the similarity of regulation in the latter experiment for DEGs detected in the initial experiment. The specific analysis parameters used for each experiment and any changes or additions to the workflow are reported in the following sections.
Titration of aTc concentration in drinking water
Germ-free C57BL/6(J) mice were maintained on the chow diet as described above and received water containing 1, 10, or 30 μg/ml of aTc as well as 100 μg/ml of kanamycin sulfate 1 day prior to gavage of 1 × 109 cells of E. coli BL21(DE3) transformed with pFS_0453. Plasmid DNA was extracted and concentrated as described above and 6.25 μl of plasmid DNA was used as an input into SENECA using annealed adapter ligation oligonucleotides FS_0963 and FS_0964 (table S25).
Record-seq comparison of transient chow, fat, and starch-based dietary stimulus
Germ-free C57BL/6(J) mice received the standard chow diet (Kliba Nafag 3307) prior to the experiment. At the beginning of the experiment (2 days before gavage), one group remained on the chow diet, whereas the other two groups received either a starch-based purified diet (Research Diets D12450Jii) or a fat-based (lard-based) purified diet (Research Diets D12492ii), both of which were sterilized by two rounds of irradiation with each 10–20 kGy. Mice received drinking water containing 30 μg/ml of aTc and 100 μg/ml of kanamycin sulfate 1 day before gavage with 1 × 109 cells of E. coli MG1655 transformed with pFS_0453 as described above. After 7 days on different diets, mice from all groups received the chow diet (effectively switching fat- and starch-fed mice to the chow diet). Plasmid DNA from fecal pellets was extracted and concentrated as outlined above. Additionally, intestinal contents were sampled yielding the data presented in fig. S1. The initial, 20-day diet switch experiment described in Fig. 1 and fig. S2 used 3.75 μl of plasmid DNA as an input into SENECA using annealed adapter ligation oligonucleotides FS_2759 and FS_2769 (table S25). The consecutive, 14-day diet switch experiment described in fig. S3 used 7.5 μl of plasmid DNA as an input into SENECA using annealed adapter ligation oligonucleotides FS_2240 and FS_2248 (table S25).
Primary analysis was performed for Record-seq and RNA-seq readout as described above for both experiments. During secondary analysis, the minimum value of cumulative transcript-aligning spacer counts (C) was set to 10,000 for Record-seq samples, and the minimum value of transcript-aligning counts for RNA-seq samples was set to 100,000. PCA projections and UMAP embeddings were generated for the top 500 most variable genes across days and diets. For the initial 20-day experiment, hierarchical clustering and heatmap generation were performed for DEGs detected by multiple testing on day 7 since this was the last day when the mice were fed different diets prior to switching all mice to the chow diet. For both experiments, diet-specific signature genes were defined as the top 500 DEGs detected for day 7.
Hierarchical clustering and heatmap generation were performed for the final day in each experiment using these diet-specific signature genes. For the initial experiment, genes enriched or depleted in each diet pair comparison were identified on day 7 using pairwise DE testing and used for generating volcano plots. EcoCyc pathway (table S5) enrichment was performed using DEGs identified for each diet pair (tables S2 and S3) and the top hits were used for creating pathway enrichment plots. STRING network analysis was performed using Cytoscape using DEG with log2FC ≥ 1.0. The granularity parameter for MCL clustering of STRING networks was set at 2.5 and stringdb:: score was used as array source with an edge cutoff of 0.5. Clusters containing more than four nodes were reported. KEGG- and GO-based OA were performed for DEGs identified for each diet pair using clusterProfiler (table S6). Regression analysis was performed to examine the similarity of regulation in the latter experiment for DEGs in mice fed the chow diet or purified diet based on starch detected in the initial experiment.
E. coli ΔidnK/ΔgntK in vivo competition assay
One group of germ-free C57BL/6(J) mice was switched to the starch-based diet 48 hours before gavage whereas the other remained on the standard chow diet. E. coli MG1655 (WT, NCBI Assembly database PRJNA807125) was grown in 200 ml of LB and E. coli MG1655 StrR ΔidnK/ΔgntK in 250 ml of LB with 30 μg/ml of chloramphenicol (Sigma-Aldrich) at 37°C, 180 rpm overnight. Cultures were pelleted by centrifugation at 3480g for 10 min at room temperature, washed twice with 250 ml of sterile PBS and combined after the first washing step at a 1:1 ratio. The combined E. coli strains were finally resuspended in 7.5 ml of sterile PBS and diluted 1:10 in sterile PBS to gavage ~1 × 109 CFU per mouse in 500 μl. Feces were collected at 24 hours, 48 hours, and 72 hours after gavage and lysed in 1 ml of PBS using a Retsch MM400 tissue lyser at 30 Hz for 3 min. Serial dilutions were streaked onto LB agar and LB agar containing 30 μg/ml of chloramphenicol and the competitive indices were calculated as .
E. coli ΔuxaC in vivo competition assay
The competition experiment using the ΔuxaC mutant was performed as above with the following alterations: E. coli MG1655 StrR ΔuxaC (NCBI Assembly database PRJNA807125) was gavaged into germ-free mice together with either MG1655 WT (NCBI Assembly database PRJNA807125) or with MG1655 StrR (NCBI Assembly database PRJNA807125). Each strain was grown in 100 ml of LB and the ΔuxaC mutant with an additional 50 μg/ml of kanamycin sulfate. Strains were washed with 100 ml of PBS, mixed, and then resuspended in a final volume of 33 ml to achieve a gavage dose of ~1 × 109 CFU per mouse in 500 μl. Serial dilutions of feces were streaked onto LB agar with and without 50 μg/ml of kanamycin sulfate.
Record-seq and RNA-seq assessment of different anatomical sections of the murine gut
One group of germ-free C57BL/6(J) mice was switched to the starch-based diet 48 hours before gavage whereas the other remained on the standard chow diet. Mice received drinking water containing 30 μg/ml of aTc and 100 μg/ml of kanamycin sulfate water and were gavaged with 1 × 109 cells of E. coli MG1655 transformed with pFS_0453 as described above. Record-seq plasmid and/or fecal RNA was extracted from individual mouse feces collected daily. On day 7 after gavage, mice were sacrificed and E. coli RNA was collected from various intestinal sections of individual mice. Cecal contents were mixed in a Petri dish to homogenize them and then ~100 mg was collected for RNA extraction. Proximal colon contents were collected at a 1–2-cm distance from the cecum. Distal colon contents were collected in the terminal 2 cm of the colon. RNA was extracted, sequenced, and analyzed as described above. The experiment was performed twice: once with samples being pooled from each three individual mice, once with sampling from individual mice. Primary analysis was performed for RNA-seq readout as described above for both experiments. During secondary analysis, the minimum value of transcript-aligning counts was set to 100,000. Differential expression analysis between the diet groups was performed for each intestinal section (cecum, proximal colon and distal colon) independently. Record-seq DEGs were overlapped with DEGs identified through RNA-seq proximally (cecum or proximal colon) or distally (distal colon) (). Differential expression analysis between the intestinal sections was performed for each diet group independently. Genes enriched in a particular intestinal section were defined as an overlap of genes identified as up-regulated in that section compared to the other two sections (e.g., genes enriched in the cecum on the chow diet were defined as genes that were up-regulated in the cecum on the chow diet compared to both the proximal and distal colon). Rank-based normalization was used for comparing Record-seq and RNA-seq counts to account for the differences in count distributions.
In vitro exposure of E. coli to DSS
For each replicate, three colonies of E. coli MG1655 transformed with pFS_0453 were inoculated into 2 ml of terrific broth (TB) (24 g per liter of yeast extract, 20 g per liter of tryptone, 4 ml per liter of glycerol, 17 mM KH2PO4, and 72 mM K2HPO4) containing 50 ng/ml of aTc and 0.1, 0.3, 1, 3, or 10% (w/v) of DSS. After overnight culture at 37°C, 220 rpm bacterial cultures were pelleted and plasmid DNA extracted using 500 μl of buffer P1, 500 μl of buffer P2, and 700 μl of buffer N3. Washes were performed as described above. A single elution was performed by adding 60 μl of buffer TE, followed by incubation at 55°C, 850 rpm for 1 min and centrifugation at 20,000g for 1 min at room temperature. Plasmid DNA was quantified as described (18), and input normalized for SENECA using annealed adapter ligation oligonucleotides FS_2108 and FS_2109 (table S25). Primary and secondary analysis of data was performed as described above, with the minimum value of cumulative transcript-aligning spacer counts per sample (C) set to 10,000.
Record-seq assessment of DSS colitis in vivo
Germ-free C57BL/6(J) mice received drinking water containing 30 μg/ml of aTc and 100 μg/ml of kanamycin sulfate water and were gavaged with 1 × 109 cells of E. coli MG1655 or E. coli BL21(DE3) transformed with pFS_0453 as described above. DSS (molecular weight 36 to 50 kDa, Lucerna-Chem) was dissolved in tap water to 1, 2, or 3% (w/v) with aTc and kanamycin sulfate as above. Mice received DSS-containing drinking water for 5 days as illustrated in the experimental outlines. Mice were subsequently switched back to aTc and kanamycin sulfate-containing water without DSS. Mice were monitored daily for signs of distress. Mice treated with 3% DSS (w/v) in the drinking water had to be removed from the study on day 13 of the experiment due to colitis-induced distress and are thus omitted from the analysis for days 14 to 19 in Fig. 4, B and C. Plasmid DNA from fecal pellets was extracted and concentrated as outlined above.
The 2% DSS (w/v) experiment (Fig. 4D) with E. coli MG1655 used 3.75 μl of plasmid DNA as an input to SENECA using annealed adapter ligation oligonucleotides FS_2806 and FS_2807 (table S25). The initial experiment, including different concentrations of DSS (1%, 2%, 3% w/v) with E. coli BL21(DE3) used 7.5 μl of plasmid DNA as an input to SENECA using the annealed adapter ligation oligonucleotides FS_2238 and FS_2246 (table S25).
Primary analysis of the Record-seq sequencing readout for both experiments was performed as described above. For the initial experiment (Fig. 4A) using E. coli BL21(DE3), the minimum value of C was set to 5,000 counts. Hierarchical clustering and heatmap generation using DEGs was performed for day 19. For inferring the trajectory of DSS-induced differences between treatment groups using PCA-projected data from days 5 to 9 (fig. S6D), an approach analogous to Slingshot (56) was employed: clusters were defined using k-medoids clustering with an optimal k value (k = 5) determined by the Elbow method based on minimizing within-cluster sum of squares. Branching of trajectories was inferred based on the assumption that clusters with samples from earlier timepoints branch out into clusters with samples from later timepoints, with the nodes of the plotted trajectory indicating the center of each cluster. For discriminating between treatment groups, Record-seq data on days 6 to 19 (treatment and post-treatment) was randomly split into a 70% training and a 30% test set using a fixed random seed to ensure reproducibility. One-versus-rest classification was performed on the test sets with SVM models trained and tuned on the training sets using leave-one-out cross-validation implemented in the e1071 package in R. Receiver operating characteristic (ROC) curves were generated using the ROCR package in R.
For secondary analysis, in the 2% DSS experiment (E. coli MG1655), the threshold for C was set to 10,000 counts for Record-seq. PCA dimensionality reduction and k-medoids clustering were performed for Record-seq samples collected post DSS treatment. k-medoids cluster identity was encoded by convex hulls in PCA projections. UMAP and time-course log2FC plots was generated and differential expression was performed using days 2 to 20, and identified DEGs were used for EcoCyc pathway enrichment (table S9) and STRING network analysis using Cytoscape. MCL clustering was performed with the granularity parameter set at 2.5 and stringdb::score was used as array source with an edge cutoff of 0.5 to identify groups in the STRING networks. Clusters containing more than two genes were reported. KEGG- and GO-based OA were performed for DEGs using clusterProfiler (table S10).
Record-seq in the presence or absence of B. theta
Germ-free C57BL/6(J) mice received the standard chow diet throughout the entire experiment, drinking water containing 30 μg/ml of aTc and 100 μg/ml of kanamycin sulfate from the day before gavage with 1 × 109 cells of E. coli MG1655 transformed with pFS_0453 as described above. For oral gavage of B. theta, a single colony of B. theta was grown overnight in 140 ml of brain heart infusion broth (Thermo Fisher Scientific) supplemented with 0.5 mg per liter of menadione and 5 mg per liter of hemin (both Sigma-Aldrich) at 37°C under anaerobic conditions without agitation. After two wash steps with 150 ml of sterile anaerobic PBS, B. theta was resuspended and mixed with E. coli suspension to gavage each 1 × 109 CFU E. coli and 1 × 109 CFU B. theta in a total volume of 500 μl. Plasmid DNA was extracted from fecal samples using the procedure outlined above but increasing the buffer volumes to 1000 μl of buffer P1, 1000 μl of buffer P2, and 1400 μl of buffer N3. 2-propanol precipitation and resuspension were performed as described above. The 9-day experiment used 3.75 μl of plasmid DNA as an input into the SENECA using annealed adapter ligation oligonucleotides FS_2762 and FS_2772 (table S25). The 27-day experiment used 3.75 μl of plasmid DNA as an input and annealed adapter ligation oligonucleotides FS_2758 and FS_2768 (table S25).
For measurement of bacterial colonization levels, feces were collected, weighed, and lysed in 1 ml of PBS using a Retsch MM400 tissue lyser at 30 Hz for 3 min. Serial dilutions were streaked onto LB agar plates to determine E. coli CFU and on BHI agar with 5% defibrinated sheep blood to determine B. theta CFU and incubated aerobically or anaerobically, respectively.
Primary analysis of data was performed for Record-seq data as described above for both experiments. For secondary analysis, the minimum value of C was set to 5000. This lower threshold for cumulative transcript-aligning spacer counts was chosen due to fewer observed counts, possibly explained by the difficulty in plasmid DNA extraction from E. coli colonized with B. theta. Hierarchical clustering was performed and heatmaps were generated for the full time-course using DEGs detected on at least 2 days. Combined lists of DEGs identified on any day (table S11 and S12) were used for subsequent EcoCyc pathway enrichment (table S13), STRING network analysis using Cytoscape, and KEGG- and GO-based OA using clusterProfiler (table S14). The granularity parameter for MCL clustering of STRING networks was set at 2.5 and stringdb::score was used as array source with an edge cutoff of 0.5. Clusters containing more than three genes were reported.
Record-seq function in complex microbiomes (sDMDMm2 mice)
C57BL/6(J) mice with the sDMDMm2 microbiota (table S23) received the standard chow diet or the starch-based purified diet (Research Diets D12450Jii) and were switched to drinking water containing 30 μg/ml of aTc but no kanamycin sulfate 6 days before gavage. The gavage procedure was modified as follows: 400 ml of an overnight culture of E. coli MG1655 cells transformed with pFS_0453 in LB with 50 μg/ml of kanamycin sulfate were diluted 1:5 into 2 liters pre-warmed LB with 30 ng/μl of aTc and 50 μg/ml of kanamycin sulfate and cultured for another 2 hours. Bacteria were pelleted by centrifugation at 3480g for 10 min at room temperature, washed twice with 1 liter of PBS and resuspended in 12 ml to gavage 6 × 1010 CFU into each mouse in 500 μl of PBS. Plasmid DNA was extracted from fecal samples using the procedure outlined above but increasing the buffer volumes to 1000 μl of buffer P1, 1000 μl of buffer P2, and 1400 μl of buffer N3. Isopropanol precipitation and resuspension were performed as described above. SENECA adapter ligation was performed using 3.75 μl of plasmid DNA and annealed adapter ligation oligonucleotides FS_2760 and FS_2770 as well as FS_2761 and FS_2771 (table S25) with the following modification compared to the standard protocol: annealed adapter ligation oligonucleotides were diluted 1:10 instead of 1:100 in TE buffer after annealing. E. coli colonization levels were measured by homogenizing and serial dilution of fecal pellets as described for the B. theta experiment above but serial dilutions were spread on MacConkey agar (Thermo Fisher Scientific) with 50 μg/ml of kanamycin sulfate to ensure the selective growth of E. coli sentinel cells.
Primary analysis of data was performed for Record-seq data as described above for both experiments. For secondary analysis, the minimum value of C was set to 5000. Since the genome-aligning spacer counts increased over the sampling time course, data from the final 21-hour timepoint was used for identifying DEGs, hierarchical clustering and heatmap generation, EcoCyc pathway enrichment (table S17), STRING network analysis using Cytoscape, and KEGG- and GO-based OA using clusterProfiler (table S18). The granularity parameter for MCL clustering of STRING networks was set at 2.5 and stringdb::score was used as array source with an edge cutoff of 0.5. Clusters containing at least two genes were reported.
Transcription-stimulated spacer acquisition
Transcription-stimulated recording constructs were generated by designing gBlock FS_3046 designed following the supplementary methods of (38). gBlock FS_3046 (table S25) encodes “T7UUCG(terminator)_rrnBT1(terminator)_T7Term(terminator)_Golden-Gate(BbsI)_FsLeader-CRISPR-02-array-DR2_50bp(stuffer)_EK120029600(terminator_inverted)” and is inserted downstream of FsRT-Cas1–Cas2 in pFS_0453 cloning with XhoI and NotI. This yields a plasmid where oligonucleotides encoding various promoter sequences can be inserted using a Golden Gate reaction with BbsI. Based on pFS_1061, plasmids pFS_1061 to pFS_1067 as well as pFS_1112 to pFS_1114 were generated. These plasmids encode constitutive E. coli promoters of different transcriptional activity upstream of the FsLeader_CRISPR_02_array. These promoters are inserted into pFS_1061 by generating double stranded (dsDNA) fragments encoding the respective promoter and appropriate overhangs through the annealing of oligonucleotides FS_3047 and FS_3048 (for pFS_1062), FS_3049 and FS_3050 (for pFS_1063), FS_3051 and FS_3052 (for pFS_1064), FS_3053 and FS_3054 (for pFS_1065), FS_3055 and FS_3056 (for pFS_1066), FS_3057 and FS_3058 (for pFS_1067), FS_3210 and FS_3211 (for pFS_1112), FS_3212 and FS_3213 (for pFS_1113), FS_3214 and FS_3215 (for pFS_1114). Oligonucleotide sequences are listed in table S25. Oligonucleotides were annealed by mixing 2.5 μl of 100 μl oligonucleotides in buffer TE with 5 μl of NEBuffer 2.0 and 40 μl of Ultrapure H2O (ThermoFisher Scientific) per reaction.
The reaction was heated to 95°C for 5 min in a thermocycler and cooled to 22°C at a rate of 0.5°C/sec. Then the annealed oligonucleotides were diluted 1:200 in Ultrapure H2O. For each target plasmid a Golden Gate reaction was performed containing 40 fmol of pFS_1061, 1 μl of 1:200 diluted, annealed oligonucleotides, 1 μl ofa mixture of ATP and DTT (10 mM each) (ThermoFisher Scientific), 0.25 μl of T7 DNA Ligase (NEB), 0.75 μl of 40% (w/v) PEG8000 (Sigma-Aldrich), 0.75 μl of BpiI (ThermoFisher Scientific), and 1 μl of buffer green (ThermoFisher Scientific). Each reaction was brought up to a 10-μl final volume using Ultrapure H2O (ThermoFisher Scientific).
From each Golden Gate reaction, 0.5 μl was transformed into 5 μl of chemically competent E. coli Stbl3. Individual clones were grown in LB media containing 50 μg/ml of kanamycin sulfate, isolated by plasmid mini-prep and validated by Sanger sequencing (GATC Eurofins). Correct clones were then transformed into chemically competent E. coli MG1655 to assess efficiency of recording. In vitro recording and SENECA reaction was performed as described previously (17, 18). Upon identification of J23103 [iGEM Registry of Standard Biological Parts (http://parts.igem.org)] gBlock FS_3344 encoding “T7UUCG(terminator)_rrnBT1 (terminator)_T7Term(terminator)_J23103_FsLeader-CRISPR-01-array-DR1_50bp(stuffer)_EK120029600(terminator_inverted)” was designed and cloned into pFS_0453 using XhoI and NotI, yielding pFS_1142.
Record-seq for multiplexed recording in different bacterial chassis
For in vivo multiplexed recording experiments, pFS_1113 and pFS_1142 were transformed into chemically competent MG1655 StrRΔuxaC (“ΔuxaC”, NCBI Assembly database PRJNA807125) or MG1655 StrR NalR (“wt”, NCBI Assembly database PRJNA807125) and spread on LB-Agar plates containing 50 μg/ml of kanamycin sulfate. Germ-free C57BL/6(J) mice were switched to the starch-based diet 72 hours before gavage and received drinking water containing 30 μg/ml of aTc and 100 μg/ml of kanamycin sulfate from 24 hours before gavage onwards. All E. coli strains were inoculated into LB medium with 100 μg/ml of streptomycin sulfate and 50 μg/ml of kanamycin sulfate with or without 50 μg/ml of nalidixic acid, respectively and grown overnight at 37°C with 180 rpm shaking. Following washing as described above, the following strains were mixed 1:1 and a total of 1 × 1010 CFU (5 × 109 CFU per strain and mouse) were orally gavaged into recipient mice. Figure 7B, wt-pFS_1113 together with ΔuxaC pFS_1142 and wt-pFS_1142 together with ΔuxaC pFS_1113. Colonization levels of the strains were determined by fecal dilution and plating on LB/Str100/Kan50 (both ΔuxaC and wt) and LB/Str100/Kan50/Nal50 (only wt).
Plasmid DNA was extracted from fecal samples using the procedure outlined above but increasing the buffer volumes to 500 μl of buffer P1, 500 μl of buffer P2, and 700 μl of buffer N3. 2-propanol precipitation and resuspension were performed as described above.
SENECA adapter ligation was performed using 7.5 μl of plasmid DNA and annealed adapter ligation with annealed oligonucleotides FS_3194 and 3204 as well as FS_3316 and 3321 (table S25) with the following modification compared to the standard protocol: annealed adapter ligation oligonucleotides were diluted 1:10 instead of 1:100 in TE buffer after annealing. Upon annealing to dsDNA fragments, oligonucleotides FS_3194 and 3204 form an overhang compatible with FaqI-digested FsDR2 and oligonucleotides FS_3316 and 3321 form an overhang compatible with FaqI-digested FsDR1. Therefore, both sets of annealed oligos were used in a single SENECA adapter ligation reaction to simultaneously read out CRISPR spacer acquisition into pFS_1113 and pFS_1142. Accordingly, SENECA first-round PCR was performed with FS_968, FS_969, FS_970, FS_971, FS_972, FS_973, FS_974 described previousl, which are primers compatible with FsDR2 along with additional primers compatible with FsDR1, namely FS_3325, FS_3326, FS_3327, FS_3328, FS_3329, FS_3330, FS_3331 (table S25) and were each used at a concentration of 0.714 μM. The universal reverse primer FS_911 (18) was used at a concentration of 10 μM. SENECA second-round PCR was performed as described above.
For primary analysis of data, a modified version of the Snakemake workflow described above was used. This workflow requires the additional input of a table with sample-specific entries for the reference plasmid, DR sequence and library barcode. Samples with multiple DR-barcoded E. coli strains, such as those from the in vivo multiplexed recording experiment, resulted in reads containing both DR sequences (FsDR2 and FsDR1). These were stratified into strain-specific reads by identifying the DR-specific library barcode using fuzzy string matching and processed independently. Secondary analysis was performed as described above.
For the in vitro multiplexed recording experiment, the minimum value of cumulative transcript-aligning spacer counts (C) was set to 30,000. For the in vivo multiplexed recording experiment, the minimum value of cumulative transcript-aligning spacer counts (C) was set to 10,000. DEGs were identified between the strains (wt vs ΔuxaC) for days 7 to 10 in both groups of mice independently (group 1: ΔuxaC-DR1, wt-DR2; group 2: wt-DR1, ΔuxaC-DR2; n = 5 in each group). High-confidence DEGs were defined as DEGs identified as regulated in the same direction for at least 3 out of 4 days in both comparisons. These high-confidence DEGs (table S19) were used for EcoCyc pathway enrichment (table S20) and KEGG- and GO-based OA using clusterProfiler (table S21).
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to C. Beisel, E. Burcklen, I. Nissen, K. Eschbach, M. Feldkamp, and T. Schär from the Genomics Facility Basel and T. Leeb and P. Nicholson of the Next-Generation Sequencing Facility of the University of Bern for assistance in Illumina sequencing, as well as H.-M. Kaltenbach, G. Rätsch, and the entire Platt and Macpherson laboratories for discussing results. S. Misztal provided experimental support, and J. Limenitakis undertook E. coli whole-genome sequencing analysis.
Funding:
This work was supported by the European Research Council grant 851021 (F.S., T.T, and R.J.P.); European Union Horizon 2020 Marie Skłodowska-Curie grant 744257 (J.Z.); European Research Council grant 742195 (J.Z and A.J.M.); ETH Zurich grant to Gunnar Rätsch 1–001785-000 (T.T.); Botnar Research Centre for Child Health Multi-Investigator Project Grant (R.F., A.J.M., and R.J.P.); Public Health Service grant GM117324 (T.C.); Swiss National Science Foundation grants CRSII5_177164, CRSII3_154414, and 310030_179479 (A.J.M.); National Centres of Competence – Molecular Systems Engineering 51NF40–182895 (A.J.M. and R.J.P.); Personalized Health and Related Technologies grant 2021–542 (A.J.M. and R.J.P.); and Brain & Behavior Research Foundation grant 26606, Personalized Health and Related Technologies grant PHRT-203, Swiss National Science Foundation grant 31003A_175830, ETH Zurich grant ETH-27 18–2, Swiss Cancer Research Foundation grant KFS-4863–08-2019, Botnar Research Centre for Child Health Fast Track Call grant, EMBO grant 4217, and J. Fickel (R.J.P.). The Clean Mouse Facility is supported by the Genaxen Foundation, Inselspital, and the University of Bern.
Footnotes
Competing interests: F.S., J.Z., T.T., A.J.M., and R.J.P. are co-inventors on patent applications filed by ETH Zurich related to this work. All other authors have no competing interests.
The list of author affiliations is available in the full article online.
Data and materials availability:
Plasmid pFS_0453 (#117006), pFS_1061 (#184738), pFS_1064 (#184739), pFS_1112 (#184740), pFS_1113 (#184741) and pFS_1142 (#184742) used for recording experiments in this study are available from Addgene. Deep sequencing data of RNA-seq and Record-seq experiments is available in the National Center for Biotechnology Information Sequence Read Archive (PRJNA807096). Whole-genome sequencing of E. coli strains used in this study is available in the National Center for Biotechnology Information Assembly database (PRJNA807125). The datasets generated and/or analyzed during the current study are available from the corresponding authors upon reasonable request. Software tools, custom scripts, and R notebooks describing workflows are available on the Platt lab GitHub (https://github.com/plattlab).
REFERENCES AND NOTES
- 1.Sheth RU, Wang HH, DNA-based memory devices for recording cellular events. Nat. Rev. Genet. 19, 718–732 (2018). doi: 10.1038/s41576-018-0052-8; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Farzadfard F, Lu TK, Emerging applications for DNA writers and molecular recorders. Science 361, 870–875 (2018). doi: 10.1126/science.aat9249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schmidt F, Platt RJ, Applications of CRISPR-Cas for synthetic biology and genetic recording. Curr. Opin. Syst. Biol. 5, 9–15 (2017). doi: 10.1016/j.coisb.2017.05.008 [DOI] [Google Scholar]
- 4.Shipman SL, Nivala J, Macklis JD, Church GM, Molecular recordings by directed CRISPR spacer acquisition. Science 353, aaf1175 (2016). doi: 10.1126/science.aaf1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ishiguro S, Mori H, Yachie N, DNA event recorders send past information of cells to the time of observation. Curr. Opin. Chem. Biol. 52, 54–62 (2019). doi: 10.1016/j.cbpa.2019.05.009 [DOI] [PubMed] [Google Scholar]
- 6.Yang L et al. , Permanent genetic memory with >1-byte capacity. Nat. Methods 11, 1261–1266 (2014). doi: 10.1038/nmeth.3147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Munck C, Sheth RU, Freedberg DE, Wang HH, Recording mobile DNA in the gut microbiota using an Escherichia coli CRISPR-Cas spacer acquisition platform. Nat. Commun. 11, 95 (2020). doi: 10.1038/s41467-019-14012-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Riglar DT, Silver PA, Engineering bacteria for diagnostic and therapeutic applications. Nat. Rev. Microbiol. 16, 214–225 (2018). doi: 10.1038/nrmicro.2017.172 [DOI] [PubMed] [Google Scholar]
- 9.Charbonneau MR, Isabella VM, Li N, Kurtz CB, Developing a new class of engineered live bacterial therapeutics to treat human diseases. Nat. Commun. 11, 1738 (2020). doi: 10.1038/s41467-020-15508-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riglar DT et al. , Engineered bacteria can function in the mammalian gut long-term as live diagnostics of inflammation. Nat. Biotechnol. 35, 653–658 (2017). doi: 10.1038/nbt.3879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Archer EJ, Robinson AB, Süel GM, Engineered E coli that detect and respond to gut inflammation through nitric oxide sensing. ACS Synth. Biol. 1, 451–457 (2012). doi: 10.1021/sb3000595 [DOI] [PubMed] [Google Scholar]
- 12.Kotula JW et al. , Programmable bacteria detect and record an environmental signal in the mammalian gut. Proc. Natl. Acad. Sci. U.S.A. 111, 4838–4843 (2014). doi: 10.1073/pnas.1321321111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mimee M, Tucker AC, Voigt CA, Lu TK, Programming a human commensal bacterium, bacteroides thetaiotaomicron, to sense and respond to stimuli in the murine gut microbiota. Cell Syst. 2, 214 (2016). doi: 10.1016/j.cels.2016.03.007 [DOI] [PubMed] [Google Scholar]
- 14.Donaldson GP, Lee SM, Mazmanian SK, Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 14, 20–32 (2016). doi: 10.1038/nrmicro3552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zmora N et al. , Personalized gut mucosal colonization resistance to empiric probiotics is associated with unique host and microbiome features. Cell 174, 1388–1405.e21 (2018). doi: 10.1016/j.cell.2018.08.041 [DOI] [PubMed] [Google Scholar]
- 16.Tang Q et al. , Current sampling methods for gut microbiota: A call for more precise devices. Front. Cell. Infect. Microbiol. 10, 151 (2020). doi: 10.3389/fcimb.2020.00151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmidt F, Cherepkova MY, Platt RJ, Transcriptional recording by CRISPR spacer acquisition from RNA. Nature 562, 380–385 (2018). doi: 10.1038/s41586-018-0569-1 [DOI] [PubMed] [Google Scholar]
- 18.Tanna T, Schmidt F, Cherepkova MY, Okoniewski M, Platt RJ, Recording transcriptional histories using Record-seq. Nat. Protoc. 15, 513–539 (2020). doi: 10.1038/s41596-019-0253-4; pmid: [DOI] [PubMed] [Google Scholar]
- 19.Keseler IM et al. , The EcoCyc database: Reflecting new knowledge about Escherichia coli K-12. Nucleic Acids Res. 45 (D1), D543–D550 (2017). doi: 10.1093/nar/gkw1003; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hooper LV, Gordon JI, Glycans as legislators of host-microbial interactions: Spanning the spectrum from symbiosis to pathogenicity. Glycobiology 11, 1R–10R (2001). doi: 10.1093/glycob/11.2.1R [DOI] [PubMed] [Google Scholar]
- 21.Martens EC, Chiang HC, Gordon JI, Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe 4, 447–457 (2008). doi: 10.1016/j.chom.2008.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sweeney NJ, Laux DC, Cohen PS, Escherichia coli F-18 and E. coli K-12 eda mutants do not colonize the streptomycin-treated mouse large intestine. Infect. Immun. 64, 3504–3511 (1996). doi: 10.1128/iai.64.9.3504-3511.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leatham MP et al. , Mouse intestine selects nonmotile flhDC mutants of Escherichia coli MG1655 with increased colonizing ability and better utilization of carbon sources. Infect. Immun. 73, 8039–8049 (2005). doi: 10.1128/IAI.73.12.8039-8049.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Conway T, Cohen PS, Commensal and pathogenic Escherichia coli metabolism in the gut. Microbiol. Spectr. 3, 3.3.24 (2015). doi: 10.1128/microbiolspec.MBP-0006-2014; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ricci M, Laboratory animal control diets: Very important, often neglected. Lab Anim. (NY) 44, 240–241 (2015). doi: 10.1038/laban.786 [DOI] [Google Scholar]
- 26.Kim Y, Wood TK, Toxins Hha and CspD and small RNA regulator Hfq are involved in persister cell formation through MqsR in Escherichia coli. Biochem. Biophys. Res. Commun. 391, 209–213 (2010). doi: 10.1016/j.bbrc.2009.11.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kitagawa M, Matsumura Y, Tsuchido T, Small heat shock proteins, IbpA and IbpB, are involved in resistances to heat and superoxide stresses in Escherichia coli. FEMS Microbiol. Lett. 184, 165–171 (2000). doi: 10.1111/j.1574-6968.2000.tb09009.x [DOI] [PubMed] [Google Scholar]
- 28.Patwa LG et al. , Chronic intestinal inflammation induces stress-response genes in commensal Escherichia coli. Gastroenterology 141, 1842–51.e1, 10 (2011). doi: 10.1053/j.gastro.2011.06.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johansson ME et al. , Bacteria penetrate the inner mucus layer before inflammation in the dextran sulfate colitis model. PLOS ONE 5, e12238 (2010). doi: 10.1371/journal.pone.0012238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cevallos SA et al. , Increased epithelial oxygenation links colitis to an expansion of tumorigenic bacteria. mBio 10, e02244–19 (2019). doi: 10.1128/mBio.02244-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lemke JJ et al. , Direct regulation of Escherichia coli ribosomal protein promoters by the transcription factors ppGpp and DksA. Proc. Natl. Acad. Sci. U.S.A. 108, 5712–5717 (2011). doi: 10.1073/pnas.1019383108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ng KM et al. , Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 502, 96–99 (2013). doi: 10.1038/nature12503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Desai MS et al. , A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell 167, 1339–1353.e21 (2016). doi: 10.1016/j.cell.2016.10.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marcobal A et al. , Bacteroides in the infant gut consume milk oligosaccharides via mucus-utilization pathways. Cell Host Microbe 10, 507–514 (2011). doi: 10.1016/j.chom.2011.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yilmaz B et al. , Long-term evolution and short-term adaptation of microbiota strains and sub-strains in mice. Cell Host Microbe 29, 650–663.e9 (2021). doi: 10.1016/j.chom.2021.02.001 [DOI] [PubMed] [Google Scholar]
- 36.Brugiroux S et al. , Genome-guided design of a defined mouse microbiota that confers colonization resistance against Salmonella enterica serovar Typhimurium. Nat. Microbiol. 2, 16215 (2016). doi: 10.1038/nmicrobiol.2016.215 [DOI] [PubMed] [Google Scholar]
- 37.Shakya M, Lo CC, Chain PSG, Advances and challenges in metatranscriptomic analysis. Front. Genet. 10, 904 (2019). doi: 10.3389/fgene.2019.00904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Budhathoki JB et al. , Real-time observation of CRISPR spacer acquisition by Cas1-Cas2 integrase. Nat. Struct. Mol. Biol. 27, 489–499 (2020). doi: 10.1038/s41594-020-0415-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nemoz G, Robert-Baudouy J, Stoeber F, Physiological and genetic regulation of the aldohexuronate transport system in Escherichia coli. J. Bacteriol. 127, 706–718 (1976). doi: 10.1128/jb.127.2.706-718.1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Datsenko KA, Wanner BL, One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645 (2000). doi: 10.1073/pnas.120163297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Uchimura Y et al. , Complete genome sequences of 12 species of stable defined moderately diverse mouse microbiota 2. Genome Announc. 4, e00951–16 (2016). doi: 10.1128/genomeA.00951-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H et al. , The outer mucus layer hosts a distinct intestinal microbial niche. Nat. Commun. 6, 8292 (2015). doi: 10.1038/ncomms9292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stead MB et al. , RNAsnap™: A rapid, quantitative and inexpensive, method for isolating total RNA from bacteria. Nucleic Acids Res. 40, e156 (2012). doi: 10.1093/nar/gks680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bolger AM, Lohse M, Usadel B, Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). doi: 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Langmead B, Salzberg SL, Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). doi: 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li H et al. , The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). doi: 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liao Y, Smyth GK, Shi W, featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). doi: 10.1093/bioinformatics/btt656 [DOI] [PubMed] [Google Scholar]
- 48.Köster J, Rahmann S, Snakemake—A scalable bioinformatics workflow engine. Bioinformatics 28, 2520–2522 (2012). doi: 10.1093/bioinformatics/bts480 [DOI] [PubMed] [Google Scholar]
- 49.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). doi: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McInnes L, Healy J, Melville J, UMAP: Uniform Manifold Approximation and Projection for dimension reduction (2018). [Google Scholar]
- 51.Doncheva NT, Morris JH, Gorodkin J, Jensen LJ, Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome Res. 18, 623–632 (2019). doi: 10.1021/acs.jproteome.8b00702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morris JH et al. , clusterMaker: A multi-algorithm clustering plugin for Cytoscape. BMC Bioinformatics 12, 436 (2011). doi: 10.1186/1471-2105-12-436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gene Ontology Consortium, The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 49 (D1), D325–D334 (2021). doi: 10.1093/nar/gkaa1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kanehisa M, Furumichi M, Sato Y, Ishiguro-Watanabe M, Tanabe M, KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 49 (D1), D545–D551 (2021). doi: 10.1093/nar/gkaa970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu G, Wang LG, Han Y, He QY, clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012). doi: 10.1089/omi.2011.0118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Street K et al. , Slingshot: Cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19, 477 (2018). doi: 10.1186/s12864-018-4772-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Plasmid pFS_0453 (#117006), pFS_1061 (#184738), pFS_1064 (#184739), pFS_1112 (#184740), pFS_1113 (#184741) and pFS_1142 (#184742) used for recording experiments in this study are available from Addgene. Deep sequencing data of RNA-seq and Record-seq experiments is available in the National Center for Biotechnology Information Sequence Read Archive (PRJNA807096). Whole-genome sequencing of E. coli strains used in this study is available in the National Center for Biotechnology Information Assembly database (PRJNA807125). The datasets generated and/or analyzed during the current study are available from the corresponding authors upon reasonable request. Software tools, custom scripts, and R notebooks describing workflows are available on the Platt lab GitHub (https://github.com/plattlab).