

Abstract
Leishmaniasis is a neglected tropical disease that is estimated to afflict over 12 million people. Current drugs for leishmaniasis suffer from serious deficiencies, including toxicity, high cost, modest efficacy, primarily parenteral delivery, and emergence of widespread resistance. We have discovered and developed a natural product inspired tambjamine chemotype, known to be effective against Plasmodium spp, as a novel class of antileishmanial agents. Herein, we report in-vitro, and in-vivo antileishmanial activities, detailed structure-activity relationships and metabolic/pharmacokinetic profiles of a large library of tambjamines. A number of tambjamines exhibited excellent potency against both Leishmania mexicana and Leishmania donovani parasites with good safety and metabolic profiles. Notably, tambjamine 110 offered excellent potency and provided partial protection to leishmania-infected mice at 40 and/or 60 mg/kg/10 days of oral treatment. This study presents the first account of antileishmanial activity in the tambjamine family, and paves the way for the generation of new oral antileishmanial drugs.
Graphical Abstract
INTRODUCTION
Leishmaniasis is a spectrum of diseases caused by parasitic protists of the genus Leishmania, with about 20 species responsible for human disease.1 Infection is initiated by a bite of a parasite-carrying sand fly, and animals often serve as reservoirs for transmission of infection between animals and humans. The pathology is dependent upon the species of parasite and the host immune response and can range from self-healing but potentially disfiguring cutaneous disease, to highly disfiguring mucocutaneous leishmaniasis, and to fatal visceral leishmaniasis where the liver, spleen, and bone marrow are colonized.2 This latter and most serious disease is usually caused by Leishmania donovani or Leishmania infantum. Two parasites that cause cutaneous leishmaniasis, Leishmania major and Leishmania mexicana, have been employed widely to study the basic cell and molecular biology of these protozoa but are also agents of widespread disease. The principal cells infected by the parasite are those of the reticuloendothelial system such as macrophages and dendritic cells. Within the sand fly vector, Leishmania parasites exist as various developmental forms of the elongated, flagellated, motile, extracellular promastigotes.3 When an infected sand fly bites a vertebrate host, it injects infectious metacyclic promastigotes that invade macrophages, where they are incorporated into phagolysosomes that mature into acidic parasitophorous vacuoles. Within these vacuoles, the promastigotes transform into nonmotile oval-shaped amastigotes that are adapted for survival and replication within the vacuole and that ultimately lyse the infected host cell, releasing free amastigotes that reinvade other macrophages.
Disease burden globally is estimated to involve ~12 million cases with 1 million new cases of cutaneous leishmaniasis and ~300,000 cases of visceral leishmaniasis per year.1 Furthermore, asymptomatic leishmaniasis doubtlessly significantly increases the number of infected individuals well beyond the numbers cited above.4 Treatment options are poor, with current drugs such as pentavalent antimonials, pentamidine, amphotericin B, and paromomycin suffering from toxicity, high cost, and development of widespread resistance.5 Only one drug, miltefosine5-7 is orally deliverable but exhibits severe toxicity and high cost. Hence, there is a pressing need for novel drugs with improved efficacy and safety profiles that can be employed to combat this burdensome but neglected disease that afflicts largely tropical and subtropical regions of the globe.
One obstacle to the discovery of novel drugs for leishmaniasis is the limited number of validated targets. Hence, phenotypic screening has played an important role the in recent discovery of drug candidates. These include nitroimidazooxazoles,8 the benzoxaborale DNDI-6148,9 and the nitroimidazooxazine DNDI-0690,10, 11 and other nitro compounds have activity against Leishmania parasites.9, 12 In addition, the Genomics Institute of the Novartis Foundation discovered the dimethyl oxazole-carboxamide derivative GNF6702 and a related compound LXE40813 as drug leads, and using genetic screens for resistance these compounds were determined to be inhibitors of the parasite proteasome. Cytochrome bc1 has been identified as a promiscuous drug target by phenotypic screening.14 However, the pyrollopyrimidine DNDI-6174, which targets the Qi site of Cytochrome b, was highly effective in decreasing the burden of L. infantum in the livers of infected mice.15 A screen of an ~200,000 compound library identified 15 chemotypes with demonstrated activity against L. major, one of which was tested for efficacy in a mouse model of cutaneous leishmaniasis.16 Screening of an ~600,000 compound library against L. mexicana identified 9 chemotypes with potential for drug development and two of these were shown to have partial efficacy in an animal model.17 In addition, a recent study established pyrazolopyrrolidinones as a new antileishmanial chemotype.18 Despite these notable advances, there is still a need for discovery of additional antileishmanial oral drug leads to maintain a robust pipeline in the face of inevitable failures on the way to clinical approval.
As a part of an ongoing interest in the discovery of a novel class of antiparasitic agents with novel mode(s) of action (MoA) to overcome the emerging drug resistance, recently we have discovered and developed a class of intriguing natural and synthetic products that has exhibited strong potency against the malaria parasite Plasmodium falciparum, including the prodiginines (PGs) and the structurally related tambjamines (TAs)19-23 (Figure 1). PGs and TAs contain 3 and 2 pyrrole rings, respectively, and share 4-methoxy-2,2'-bipyrrole-5-carboxaldehyde (MBC, 1; Figure 1) as a common precursor in their biosynthetic pathways.24-26 In addition, these natural and synthetic compounds have been extensively investigated for various therapeutic applications, including antimicrobial,27-29 anticancer,30-35 antitumor,27, 29, 36 and immunosuppressive37 activities with various MoA.38-41 Indeed, a recent study demonstrated the effectiveness of both TAs and PGs against the kinetoplastid parasites such as Trypanosoma cruzi,42 the causative agent of Chagas disease. To the best of our knowledge, the exact MoA are unknown for these novel TA chemotypes; however, they likely induce mitochondrial swelling and lysosomal dysfunction leading to autophagy and necrotic cell death in cancer cells.43
Figure 1.

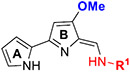
Biologically active natural PGs (2 and 3), and TAs (4 and 5), and their common biosynthetic precursor MBC (1).
To date, antileishmanial activities of these promising PG and TA compounds have not been investigated. Although Plasmodium and Leishmania parasites belong to different orders of protozoa, apicomplexa and kinetoplastida, respectively, the efficacy of PGs and TAs against the former genus suggested that they might be active against Leishmania as well. To test this possibility, we exposed intracellular amastigotes of both L. mexicana and L. donovani to various PGs and TAs and found that a subset of TA compounds had inhibitory activity for growth of the parasites with EC50 values in the nanomolar (nM) range while exhibiting significantly less toxicity toward the host macrophages. Exploration of a large library of TA analogues allowed assignment of structure-activity relationships (SAR). Furthermore, TAs with potent in vitro activity were tested for in vivo activity, via oral administration in a mouse model of cutaneous leishmaniasis induced by infection with L. mexicana, and they were shown to have partial efficacy in controlling disease. In addition, we also report the in vitro metabolic and in vivo pharmacokinetic (PK) profiles of the most potent TAs. These results establish TAs as a promising chemotype for the development of novel antileishmanial oral drugs and define several early lead compounds.
RESULTS AND DISCUSSION
Chemistry.
To explore the effects of TAs on L. mexicana and L. donovani intracellular amastigotes, and understand the SARs, we relied upon TAs both previously synthesized in our studies on P. falciparum19 and a large library of novel TAs recently synthesized with various structural features (Tables 1-5). The key 2,2'-bipyrrole-5-carboxaldehyde intermediates 1 and 6–24 required in the synthesis of various A- and B-ring functionalized TAs are depicted in Figure 2. Using the reported methodology,44 MBC (1) was prepared from commercially available starting materials in two steps, and the intermediates 6–19 were synthesized by our recently reported synthetic routes19, 20, 22, 45 in good to excellent yields. The synthetic routes of aryloxy- and aryl-substituted 2,2'-bipyrrole-5-carboxaldehyde analogues 20–24 are shown in Scheme 1.
Table 1.
In Vitro Antileishmanial Activity of TAs (42–63) with Modifications at Terminal Amine.
|
survival (%) at 0.1 μMa vs |
EC50 (nM)a vs |
||||
|---|---|---|---|---|---|---|
| compound | R1 |
L. mexicana amastigote |
J774A.1 macrophage |
L. mexicana amastigote |
J774A.1 macrophage |
TI |
| 42 | n-C6H13 | 11.1 ± 4.91 | 127 ± 71.0 | 52.5 ± 0.71 | > 1000 ± 0.0 | > 19 |
| 43 | n-C11H23 | 11.9 ± 3.80 | 112 ± 72.7 | 47.5 ± 0.71 | > 1000 ± 0.0 | > 21 |
| 44 |
|
7.33 ± 0.96 | 88.9 ± 25.1 | 40.5 ± 4.95 | > 1000 ± 0.0 | > 25 |
| 45 |
|
4.84 ± 1.19 | 99.6 ± 59.1 | 32.5 ± 2.12 | 550 ± 630 | 17 |
| 46 |
|
5.30 ± 2.33 | 72.2 ± 25.9 | 48.5 ± 3.53 | > 1000 ± 0.0 | > 21 |
| 47 |
|
12.4 ± 1.90 | 66.6 ± 12.5 | 40.0 ± 0.0 | 696 ± 734 | 17 |
| 48 |
|
9.48 ± 1.73 | 74.4 ± 20.7 | 46.7 ± 17.8 | 1098 ± 1218 | 24 |
| 49 |
|
28.3 ± 3.0 | 51.1 ± 13.6 | 87.7 ± 41.9 | 2986 ± 0.0 | 34 |
| 50 |
|
24.6 ± 12.0 | 124 ± 78.6 | 79.5 ± 10.6 | 625 ± 530 | 7.9 |
| 51 |
|
14.3 ± 2.81 | 72.8 ± 23.7 | 73.0 ± 2.83 | 1041 ± 1262 | 14 |
| 52 |
|
5.49 ± 2.59 | 50.8 ± 14.8 | 41.0 ± 2.83 | 171 ± 165 | 4.2 |
| 53 |
|
9.85 ± 0.69 | 95.4 ± 31.9 | 47.0 ± 9.89 | > 1000 ± 0.0 | > 21 |
| 54 |
|
18.9 ± 10.8 | 89.6 ± 61.5 | 72.5 ± 29.0 | > 1000 ± 0.0 | > 14 |
| 55 |
|
26.0 ± 11.9 | 69.8 ± 33.6 | 93.0 ± 3.61 | 2741 ± 0.0 | 29 |
| 56 |
|
8.65 ± 2.03 | 50.2 ± 14.6 | 66.0 ± 4.24 | > 1000 ± 0.0 | > 15 |
| 57 |
|
13.6 ± 2.55 | 65.3 ± 6.27 | 91.5 ± 14.8 | 570 ± 610 | 6.2 |
| 58 |
|
11.4 ± 1.01 | 83.7 ± 13.1 | 54.0 ± 4.24 | 760 ± 340 | 14 |
| 59 |
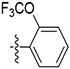
|
69.9 ± 13.8 | 81.0 ± 22.9 | ND | ND | ND |
| 60 |
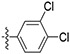
|
23.8 ± 9.67 | 67.5 ± 25.7 | 100 ± 7.07 | > 1000 ± 0.0 | > 10 |
| 61 |
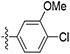
|
77.6 ± 23.3 | 57.3 ± 12.9 | ND | ND | ND |
| 62 |
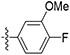
|
98.3 ± 20.7 | 58.7 ± 14.9 | ND | ND | ND |
| 63 |
|
9.03 ± 4.12 | 77.3 ± 26.2 | 32.0 ± 7.07 | > 1000 ± 0.0 | > 31 |
| miltefosine | - | 0.020 ± 0.010 | 39.2 ± 5.1 | 3460 ± 781 | >100000 ± 0.0 | > 29 |
Data represent the mean ± standard deviation of results of at least two independent experiments; an error of 0.0 indicates identical values were obtained for each replicate; TI = Therapeutic Index ((J774A.1 EC50/L. mexicana EC50); ND, not determined.
Figure 2.

Key 2,2'-bipyrrole-5-carboxaldehydes 1 and 6–24 for the synthesis of A- and B-ring functionalized TAs.
Scheme 1.

Synthesis of Aryloxy and Aryl Substituted 2,2'-Bipyrrole-5-carboxaldehydes (20–24).
Synthesis of 4-Aryloxy- and 4-Aryl-2,2'-bipyrrole-5-carboxaldehydes (20–23).
Coupling of Boc-glycine (25) with Meldrum’s acid in the presence of 4-(N,N-dimethylamino)pyridine (DMAP) and isopropyl chloroformate led to the acylated Meldrums’s acid, which when refluxed in ethyl acetate furnished the 4-hydroxy-1,5-dihydro-pyrrol-2-one (26) in a 64% yield.46 The hydroxy intermediate 26 was then treated with p-toluenesulfonyl chloride in the presence of N,N-diisopropylethylamine (DIPEA) to give the tosylated intermediate 27 in a 94% yield. According to Marchal’s procedure,47 27 was coupled with substituted phenols 28a–c in the presence of 1,4-diazabicyclo[2.2.2]octane (DABCO) at room temperature for 16 h to furnish the corresponding aryloxy intermediates 29a–c (Scheme 1, top panel). Conversely, a standard Suzuki cross-coupling reaction19, 45 between intermediate 27 and 4-(trifluoromethoxy)phenylboronic acid (33) furnished the desired intermediate 34. Upon treatment with trifluoroacetic acid (TFA), 29a–c and 34 smoothly delivered the corresponding free-NH intermediates 30a–c and 35, respectively, in excellent yields. Subjecting 30a–c and 35 to a standard Vilsmeier formylation44 led to the bromo intermediates 31a–c and 36, which when further subjected to Suzuki cross-coupling with N-Boc-2-pyrroleboronic acid (32) followed by deprotection of the N-Boc group gave the desired 4-aryloxy-2,2'-bipyrrole-5-carboxaldehydes 20–22 and 4-aryl-2,2'-bipyrrole-5-carboxaldehyde 23, respectively, in good yields (Scheme 1, top panel).
Synthesis of 5'-Aryl-2,2'-bipyrrole-5-carboxaldehyde (24).
By employing a similar strategy to the synthesis of 20–23, compound 24 was obtained in good yield from readily available 4-bromoanisole (37) and 4-methoxy-3-pyrrolin-2-one (40) via the appropriate intermediates 39 and 41 (Scheme 1, bottom panel). The pyrroleboronic acid intermediate 39 was easily synthesized from 37 via a two-step sequence of Suzuki cross-coupling with 32 followed by treatment of lithium diisopropylamide (LDA) and trimethyl borate.
Synthesis of Tambjamine Analogues (42–124).
The acid-catalyzed condensation19 of the key 2,2'-bipyrrole-5-carboxaldehydes 1 and 6–24 with various alkyl/cycloalkyl/arylamines provided the desired TA analogues 42–48 and 50–124 (Tables 1-5) in good to excellent yields (Scheme 2, top panel). Conversely, compound 49 was synthesized in excellent yield under the neutral conditions22 (Na2SO4, dichloromethane, 60 °C, in a sealed tube) to avoid deprotection of the N-Boc group, from 1 and tert-butyl ((1r,4r)-4-aminocyclohexyl)carbamate (Scheme 2, bottom panel). All of the intermediates and final target compounds were structurally characterized using NMR and HRMS analyses.
Scheme 2.

Synthesis of Tambjamine Analogues 42–124.
Biological Activity.
Growth Inhibitory Activity of Intracellular Amastigotes.
To measure the growth inhibitory activity of our target TA analogues against Leishmania parasites, we infected J774A.1 murine macrophage-like cells with stationary phase L. mexicana or L. donovani promastigotes, expressing red-shifted firefly (Photinus pyralis) luciferase,48 (RS-LUC) from an integrated rRNA site, at a multiplicity of 10:1. After 24 h, parasites that had not been phagocytosed were removed by washing, test compounds were added over a range of concentrations, and luciferase activity representing viable intracellular amastigotes was measured following 96 h incubation in minimal essential medium (MEM) plus 10% heat inactivated fetal bovine serum. Initially, all target TA analogues 42–124 were tested at 1 μM (data not shown) and 0.1 μM (Tables 1-5), and those that exhibited pronounced inhibition (typically ≥ 50% at 0.1 μM) were subsequently tested in a dose-response format to determine the EC50 value as a measure of potency (Tables 1-6). Conversely, a series of PG analogues21, 22 were also tested in this assay at 1 and 0.1 μM, however, they have shown only moderate potency (data not shown) and were thus not further pursued. In parallel, cytotoxicity against the uninfected J774A.1 macrophages was determined in a similar format, except that viability was determined using SYBR Green II fluorescence as a measure of DNA content.48 The therapeutic index (TI) was calculated as the ratio of the EC50 value for J774A.1 macrophages over the EC50 value for L. mexicana and/or L. donovani amastigotes (TI = EC50(J774A.1)/EC50(L. mexicana or L. donovani)). All of these results are summarized in Tables 1-6, and discussed in the following sections.
In Vitro Antileishmanial Activity and SAR of TAs (42–63).
Initially, a series of TAs 42–63 containing various alkyl, cycloalkyl, and aryl moieties at the right-hand-side terminal amine and OMe group at the 4-position of ring-B (as observed in natural TAs, Figure 1), were evaluated for their in vitro antileishmanial activity against L. mexicana amastigotes and cytotoxicity against J774A.1 macrophages, and the results are shown in Table 1. TAs 42 and 43, containing n-alkyl moieties at terminal amine, exhibited excellent potency against L. mexicana amastigotes with low EC50 values of 52.5 and 47.5 nM, respectively. Notably, these two compounds were much less inhibitory to the growth of the J774A.1 macrophage line (EC50 ≥ 1000 nM), indicating a feasible TI (Table 1). Interestingly, the replacement of n-alkyl groups of 42 and 43 at terminal amine with cycloalkyl moieties as in 44–48, slightly improved the potency (EC50 = 32.5–48.5 nM vs L. mexicana). Approximately, a 2-fold antileishmanial potency loss was observed when the cyclohexyl group of 44 was replaced with N-substituted cycloalkyl moieties as in 49 and 50 (49 EC50 = 87.7 nM and 50 EC50 = 79.5 nM vs 44 EC50 = 40.5 nM vs L. mexicana). Next, we tested a series of TAs containing various mono-substituted aryl moieties on terminal amine (51–59). All of these mono-substituted aryl TAs, with the exception of 59 (~30% inhibition at 0.1 μM vs L. mexicana), displayed excellent antileishmanial potency (51–58 EC50 = 41.0–93.0 nM vs L. mexicana) that was comparable with the alkyl/cycloalkyl TAs (42–50 EC50 = 32.5–87.7 nM vs L. mexicana). These findings suggested that the OCF3 group at ortho-position of aryl moiety on terminal amine of 59 is detrimental to antileishmanial potency. Introduction of an additional Cl substitution on the aryl moiety at terminal amine as in 60 led to a two-fold loss of potency (60 EC50 = 100 nM vs 53 EC50 = 47.0 nM vs L. mexicana). A significant loss of potency was observed for 61 and 62 containing a combination of halogen (Cl/F) and OMe groups on the aromatic ring at terminal amine (~22% inhibition for 61, and ~2% inhibition for 62 at 0.1 μM). Interestingly, the potency was retained after the introduction of a cyclic-diether moiety on the aryl ring (63 EC50 = 32.0 nM vs L. mexicana) (Table 1). Taken together, in vitro potency and SAR analyses of TAs 42–63 demonstrated that the OMe group at the 4-position of ring-B and n-alkyl, cycloalkyl, and mono-substituted aryl moieties at terminal amine is best tolerated.
In Vitro Antileishmanial Activity and SAR of TAs (64–81).
To probe the importance of OMe group at the 4-position of ring-B of TAs, a series of new TAs (64–81), in which the OMe group is replaced by various substituted aryloxy and/or aryl moieties, was generated and evaluated for their in vitro antileishmanial activity against L. mexicana amastigotes (Table 2). In vitro antileishmanial activity of TAs 64–75 containing 4-(trifluoromethoxy)phenoxy and/or 3,5-difluorophenoxy moieties at the 4-position of ring-B and n-alkyl, and cycloalkyl moieties at terminal amine, demonstrated a 2.5- to 6-fold diminished potency when compared to the corresponding OMe group containing TAs (Table 2 vs Table 1). Conversely, TAs 76–78, which contain a 4-chlorophenoxy moiety at the 4-position of ring-B and cycloalkyl moieties at terminal amine, retained the potency (76–78 EC50 = 38.0–72.3 nM), suggesting that the aryloxy moiety with a Cl substitution is well tolerated on ring-B. Replacement of the 4-(trifluoromethoxy)phenoxy moiety at the 4-position of ring-B with 4-(trifluoromethoxy)phenyl moiety as in 79–81 resulted in a decrease in antileishmanial potency, suggesting that the oxygen (O) atom at the 4-position of ring-B (i.e., OAr or OMe) plays a pivotal role in the enhancement of potency. Notably, all 1-adamantyl moiety containing TAs 70 (EC50 = 41.0 nM), 74 (EC50 = 79.0 nM), 77 (EC50 = 38.3 nM) and 80 (EC50 = 90.5 nM) exhibited excellent potency regardless of the substitutions at the 4-position of ring-B. These results demonstrated that the substitutions at the 4-position of ring-B have an important role in potent antileishmanial activity, and the OMe group can be replaced by aryloxy and/or aryl moieties when 1-adamantyl moiety exists at the terminal amine of the TA scaffold.
Table 2.
In Vitro Antileishmanial Activity of TAs (64–81) with Modifications on Ring-B and Terminal Amine.
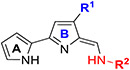
|
survival (%) at 0.1 μMa vs |
EC50 (nM)a vs |
|||||
|---|---|---|---|---|---|---|---|
| compound | R1 | R2 |
L. mexicana amastigote |
J774A.1 macrophage |
L. mexicana amastigote |
J774A.1 macrophage |
TI |
| 64 |
|
n-C6H13 | 46.1 ± 13.3 | 110 ± 66.5 | 303 ± 102 | 1104 ± 288 | 3.6 |
| 65 |
|
n-C11H23 | 58.0 ± 18.9 | 96.7 ± 39.4 | ND | ND | ND |
| 66 |
|
|
52.7 ± 9.40 | 81.3 ± 25.7 | 84.0 ± 11.3 | 982 ± 557 | 11.7 |
| 67 |
|
|
43.0 ± 11.2 | 82.8 ± 33.4 | 155 ± 2.12 | 642 ± 250 | 4.1 |
| 68 |
|
|
37.6 ± 6.85 | 92.9 ± 32.9 | 99.0 ± 15.6 | 982 ± 430 | 9.9 |
| 69 |
|
|
42.9 ± 9.92 | 97.6 ± 39.3 | 167 ± 27.6 | 717 ± 378 | 4.3 |
| 70 |
|
|
35.7 ± 7.48 | 80.7 ± 28.2 | 41.0 ± 2.82 | 353 ± 196 | 8.6 |
| 71 |
|
|
79.9 ± 3.06 | 112 ± 39.0 | ND | ND | ND |
| 72 |
|
|
33.2 ± 1.10 | 108 ± 11.2 | 88.7 ± 42.5 | 852 ± 298 | 9.6 |
| 73 |
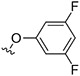
|
|
87.2 ± 4.48 | 65.8 ± 10.4 | ND | ND | ND |
| 74 |
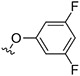
|
|
27.3 ± 4.39 | 59.3 ± 1.66 | 79.0 ± 29.5 | 680 ± 233 | 8.6 |
| 75 |
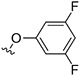
|
|
96.2 ± 1.86 | 55.3 ± 7.14 | ND | ND | ND |
| 76 |
|
|
19.8 ± 1.30 | 100 ± 17.0 | 63.7 ± 20.3 | 367 ± 54 | 5.8 |
| 77 |
|
|
7.16 ± 1.96 | 67.1 ± 18.0 | 38.3 ± 26.8 | 567 ± 273 | 15 |
| 78 |
|
|
27.5± 5.72 | 80.1 ± 17.1 | 72.3 ± 45.2 | 321 ± 341 | 4.4 |
| 79 |
|
|
77.9 ± 18.4 | 95.8 ±15.7 | ND | ND | ND |
| 80 |
|
|
39.0 ± 3.94 | 70.6 ± 16.2 | 90.5 ± 32.7 | 908 ± 112 | 10 |
| 81 |
|
|
81.6 ± 20.5 | 116 ± 19.5 | ND | ND | ND |
| miltefosine | - | - | 0.020 ± 0.010 | 39.2 ± 5.1 | 3460 ± 781 | >100000 ± 0.0 | > 29 |
Data represent the mean ± standard deviation of results of at least two independent experiments; an error of 0.0 indicates identical values were obtained for each replicate; TI = Therapeutic Index (J774A.1 EC50/L. mexicana EC50); ND, not determined.
In Vitro Antileishmanial Activity and SAR of TAs (82–109).
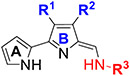
Having established the importance of the OMe, and aryloxy/aryl moieties at the 4-position of ring-B and the n-alkyl, cycloalkyl and aryl moieties at terminal amine as optimal, we then investigated the effects of the alkyl groups either at the 3-position or 4-position or both the 3-and 4-positions on ring-B (Table 3). The potency was slightly diminished when OMe at the 4-position of ring-B of 45 (Table 1) was replaced by short alkyl groups (methyl and ethyl) as in 82 and 83 (82: ~75% inhibition; 83: ~60% inhibition vs 45 >95% inhibition at 0.1 μM vs L. mexicana), suggesting that the OMe group on ring-B is playing a crucial role on antileishmanial activity. The loss of potency was also observed when the alkyl groups shifted from the 4-position to the 3-position with the compounds 84–87 (Table 3), whereas the TAs containing 1-adamatyl moiety at terminal amine and short alkyl groups at the 3-position of ring-B (88 and 89) showed excellent potency with low EC50 values of 48.0 nM and 37.5 nM, respectively. Again, these results are consistent and support the findings that the 1-adamantyl moiety at terminal amine is very important for potent antileishmanial activity. We next investigated whether substitutions at both the 3- and 4-positions are tolerated. Interestingly, TAs with 3,4-dialkyl substitutions on ring-B and cyclcoalkyl moieties at terminal amine (90–95 and 101–104), exhibited better potency compared with those of the corresponding 3- and 4-monoalkyl substituted TAs (Table 3). The greatest loss of potency was observed in TAs 96–100 and 105–109, in which cycloalkyl moiety is replaced by substituted aryl moieties, suggesting that the aryl functionality is detrimental to potency when ring-B is substituted with alkyl groups. Together, from the 3-, and 4-monoalkylated and 3,4-dialkylated TAs the data showed that the combination of dialkyl substitutions on ring-B and cycloalkyl moieties at terminal amine were well tolerated (Table 3).
Table 3.
In Vitro Antileishmanial Activity of TAs (82–109) with Modifications on Ring-B and Terminal Amine.
|
survival (%) at 0.1 μMa vs |
EC50 (nM)a vs |
||||||
|---|---|---|---|---|---|---|---|---|
| compound | R1 | R2 | R3 |
L. mexicana amastigote |
J774A.1 macrophage |
L. mexicana amastigote |
J774A.1 macrophage |
TI |
| 82 | H | Me |
|
26.3 ± 3.29 | 88.4 ± 29.4 | ND | ND | ND |
| 83 | H | Et |
|
42.1 ± 3.13 | 88.7 ± 22.0 | ND | ND | ND |
| 84 | Me | H |
|
89.0 ± 17.6 | 127 ± 83.3 | ND | ND | ND |
| 85 | Me | H |
|
48.6 ± 9.03 | 150 ± 60.4 | ND | ND | ND |
| 86 | Et | H |
|
33.9 ± 10.5 | 32.2 ± 9.96 | ND | ND | ND |
| 87 | Et | H |
|
34.6 ± 5.87 | 35.8 ± 11.9 | ND | ND | ND |
| 88 | Me | H |
|
15.4 ± 3.98 | 86.9 ± 27.5 | 48.0 ± 4.24 | 1131 ± 185 | 24 |
| 89 | Et | H |
|
11.3 ± 2.09 | 50.0 ± 22.1 | 37.5 ± 2.12 | 884 ± 163 | 24 |
| 90 | Me | Me |
|
18.2 ± 1.57 | 62.8 ± 7.71 | 55.0 ± 1.41 | 908 ± 159 | 16 |
| 91 | Me | Me |
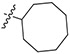
|
33.5 ± 7.06 | 43.5 ± 14.4 | ND | ND | ND |
| 92 | Me | Me |
|
14.2 ± 1.0 | 53.2 ± 22.2 | 91.7 ± 48.3 | 1600 ± 900 | 17 |
| 93 | Et | Et |
|
27.5±3.37 | 26.7±5.21 | ND | ND | ND |
| 94 | Et | Et |
|
17.0±2.70 | 29.5±9.59 | ND | ND | ND |
| 95 | Et | Et |
|
17.1 ± 5.6 | 87.8 ± 10.7 | 73.3 ± 0.0 | 1424 ± 0.0 | 19 |
| 96 | Me | Me |
|
74.0 ± 7.9 | 54.2 ± 7.9 | ND | ND | ND |
| 97 | Me | Me |
|
92.2 ± 18.6 | 88.1 ± 23.4 | ND | ND | ND |
| 98 | Me | Me |
|
102 ± 25 | 77.2 ± 18.1 | ND | ND | ND |
| 99 | Me | Me |
|
108 ± 32 | 65.9 ± 3.7 | ND | ND | ND |
| 100 | Me | Me |
|
111 ± 30 | 66.2 ± 20.6 | ND | ND | ND |
| 101 |
|
|
40.9 ± 7.6 | 60.3 ± 13.4 | 130 ± 0.0 | 2934 ± 0.0 | 23 | |
| 102 |
|
|
24.7 ± 12.0 | 80.0 ± 6.71 | 74.0 ± 9.54 | 252±0.0 | 3.4 | |
| 103 |
|
|
33.4 ± 13.5 | 75.8 ± 25.7 | ND | ND | ND | |
| 104 |
|
|
16.1 ± 4.5 | 75.0 ± 16.9 | ND | ND | ND | |
| 105 |
|
|
112 ± 23 | 95.1 ± 16.9 | ND | ND | ND | |
| 106 |
|
|
103 ± 6 | 67.2 ± 19.5 | ND | ND | ND | |
| 107 |
|
|
84.9 ± 10.7 | 92.7 ± 51.2 | ND | ND | ND | |
| 108 |
|
|
89.8 ± 19.4 | 62.5 ± 19.1 | ND | ND | ND | |
| 109 |
|
|
102 ± 24 | 58.2 ± 9.5 | ND | ND | ND | |
| miltefosine | - | - | 0.020±0.010 | 39.2 ± 5.1 | 3460 ± 781 | >100000±0.0 | >29 | |
Data represent the mean ± standard deviation of results of at least two independent experiments; an error of 0.0 indicates identical values were obtained for each replicate; TI = Therapeutic Index (J774A.1 EC50/L. mexicana EC50); ND, not determined.
In Vitro Antileishmanial Activity and SAR of TAs (110–116).
After establishing the substitution pattern at the 3-, and 4-positon of ring-B, we investigated the importance of alkyl substitutions at different positions on ring-A by keeping cycloalkyl moieties at terminal amine as an active pharmacophore for all TAs 110–116 (Table 4). Interestingly, compounds with alkyl groups (ethyl and iso-butyl) at the 5'-position of ring-A and cycloheptyl and/or cyclooctyl moiety at terminal amine (110–112) showed superior potency (>96% inhibition at 0.1 μM vs L. mexicana) to that of the corresponding TAs (90 and 91) that are lacking the substitutions on ring-A (<80% inhibition at 0.1 μM vs L. mexicana; Table 3). In subsequent dose response studies, 110–112 also exhibited excellent potency with low EC50 values (EC50 = 31.4–44.3 nM) and enhanced TI (Table 4). Surprisingly, shifting the ethyl group of 110 from the 5'-position to 4'-position as in 113 led to a 3-fold diminished antileishmanial potency (113 EC50 = 117 nM vs 110 EC50 = 40.9 nM vs L. mexicana). Approximately a 5-fold less potency was observed with the compound 114, in which ring-A was substituted with methyl groups at both the 3'- and 5'-positions, as compared to the 110 (114 EC50 = 182 nM vs 110 EC50 = 40.9 nM vs L. mexicana). It is noteworthy that the antileishmanial potency was retained after replacing the cycloheptyl moiety by 1-adamantyl moiety at terminal amine in 115 (115 EC50 = 46.5 nM vs 114 EC50 = 182 nM vs L. mexicana). Conversely, the compound 116 containing tri-alkyl substitutions on ring-A and 1-adamatnyl moiety at terminal amine exhibited great potency (116 EC50 = 54.0 nM vs L. mexicana). These results demonstrated that the positions of alkyl substitutions on ring-A play a crucial role in altering the potency, specifically, alkyl substitutions at the 5'-position play a crucial role in enhancement of the antileishmanial potency and metabolic stability.
Table 4.
In Vitro Antileishmanial Activity of TAs (110–116) with Modifications on Ring-A and Terminal Amine.
| survival (%) at 0.1 μMa vs |
EC50 (nM)a vs |
|||||
|---|---|---|---|---|---|---|
| compound | structure |
L.mexicana amastigote |
J774A.1 macrophage |
L.mexicana amastigote |
J774A.1 macrophage |
TI |
| 110 |
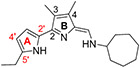
|
3.7 ± 1.0 | 71.2 ± 20.5 | 40.9 ± 19.6 | 1140 ± 498 | 28 |
| 111 |
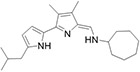
|
2.3 ± 0.1 | 74.5 ± 15.7 | 44.3 ± 24.7 | 1237 ± 130 | 28 |
| 112 |
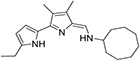
|
1.3 ± 0.4 | 53.6 ± 14.8 | 31.4 ± 12.6 | 573 ± 63.4 | 18 |
| 113 |
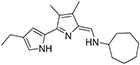
|
23.8 ± 4.3 | 63.3 ± 26.0 | 117 ± 47.4 | 1328 ± 73.5 | 11 |
| 114 |
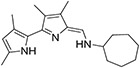
|
43.8 ± 7.5 | 56.8 ± 15.9 | 182 ± 80.7 | ND | ND |
| 115 |
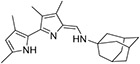
|
12.5 ± 3.00 | 93.2 ± 50.4 | 46.5 ± 12.0 | 667 ± 473 | 14 |
| 116 |
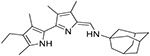
|
10.7 ± 5.4 | 63.5 ± 16.2 | 54.0 ± 15.5 | 675 ± 459 | 12 |
| miltefosine | - | 0.020 ± 0.010 | 39.2 ± 5.1 | 3460 ± 781 | >100000 ± 0.0 | > 29 |
Data represent the mean ± standard deviation of results of at least two independent experiments; an error of 0.0 indicates identical values were obtained for each replicate; TI = Therapeutic Index (J774A.1 EC50/L. mexicana EC50); ND, not determined.
In Vitro Antileishmanial Activity and SAR of TAs (117–124).
Our detailed SAR investigations around ring-A, ring-B and terminal amine of the TA scaffold led us to valuable insight into the structural features that are required for potent antileishmanial activity. Lastly, we sought to explore whether the substitution at the 5'-position on ring-A is tolerated when ring-B is substituted with the OMe group at the 4-position (as observed in natural TAs, Figure 1). To that end, we generated a series of TAs containing alkyl, aryl and benzyl substitutions at the 5'-positon on ring-A and cycloalkyl, and aryl moieties at terminal amine (117–124) and evaluated these compounds for in vitro antileishmanial activity (Table 5). Interestingly, TAs 117–123 containing cycloalkyl moiety at terminal amine substantially inhibited the growth of L. mexicana amastigotes at 0.1 μM concentration, whereas the compound 124 with an aryl moiety at terminal amine showed poorest activity (~7% inhibition at 0.1 μM vs L. mexicana). It is noteworthy that the inhibitory percentage of 117–123 is highest (Table 5) than any of the tested TA analogues in this study (Tables 1-4). In our subsequent dose-response evaluation, these compounds 117–123 showed the greatest potency (EC50 = 11.3–37.1 nM vs L. mexicana) with a feasible TI (14–88). Overall, the detailed SAR of the TA analogues suggested that various substitutions at the 5'-positon on ring-A, OMe and short alkyl groups on ring-B and cycloalkyl (including 1-adamantyl) moieties at terminal amine play key roles in enhancing in vitro antileishmanial potency with reduced toxicity toward the host macrophages. These latter potent compounds were developed subsequent to initiating the in vivo experiments discussed below and reported in Figured 3 for TAs 95, 101, and 110. However, future investigations of in vivo efficacy for some of TAs 117–123 would be a priority.
Table 5.
In Vitro Antileishmanial Activity of TAs (117–124) with Modifications on Ring-A and Terminal Amine.
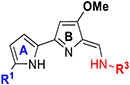
|
survival (%) at 0.1 μMa vs |
EC50 (nM)a vs |
|||||
|---|---|---|---|---|---|---|---|
| compound | R1 | R2 |
L. mexicana amastigote |
J774A.1 macrophage |
L. mexicana amastigote |
J774A.1 macrophage |
TI |
| 117 | Et |
|
0.3 ± 0.1 | 52.5 ± 16.6 | 37.1 ± 1.82 | 512 ± 0.0 | 14 |
| 118 |
|
|
−0.02 ± 0.03 | 77.3 ± 45.3 | 18.8 ± 9.70 | 450 ± 20.1 | 24 |
| 119 |
|
|
1.40 ± 0.20 | 86.6 ± 51.4 | 23.8 ± 15.9 | 717 ± 0.0 | 30 |
| 120 |
|
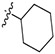
|
0.010 ± 0.10 | 84.6 ± 46.6 | 22.6 ± 13.9 | 946 ± 687 | 42 |
| 121 |
|
|
−0.11 ± 0.02 | 84.6 ± 47.8 | 16.2 ± 6.24 | 959 ± 0.0 | 59 |
| 122 |
|
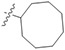
|
−0.11 ± 0.04 | 84.8 ± 63.4 | 15.8 ± 4.38 | 547 ± 0.0 | 35 |
| 123 |
|
|
−0.05 ± 0.04 | 59.6 ± 35.6 | 11.3 ± 2.04 | > 1000 ± 0.0 | > 88 |
| 124 |
|
|
93.5 ± 26.6 | 101 ± 46 | ND | ND | ND |
| miltefosine | - | - | 0.020 ± 0.010 | 39.2 ± 5.1 | 3460 ± 781 | >100000 ± 0.0 | > 29 |
Data represent the mean ± standard deviation of results of at least two independent experiments; an error of 0.0 indicates identical values were obtained for each replicate; TI = Therapeutic Index (J774A.1 EC50/L. mexicana EC50); ND, not determined. Negative values for % survival represent luminescence measurements that were somewhat below that of background and probably reflect error in measurements of very low intensities. In some instances, e.g., 117 and 119, the rank order of % survival does not correspond to that of the EC50 values. The % survival for such compounds is also very low, and the EC50 values are likely more accurate, as they entail multiple measurements over a range of concentrations and luminescence values.
In Vitro Antileishmanial Activity against L. donovani of Selected TAs.
We next investigated a subset of the potent TAs (Table 6) against the L. donovani amastigotes (causes fatal visceral leishmaniasis) to identify TAs that are broadly active against these clinically important Leishmania species. Notably all of these tested TAs exhibited activity against L. donovani amastigotes with low EC50 values (Table 6). Furthermore, the TI values for these TAs, compared to inhibition of growth of HepG2 cells, were quite robust ranging, for all but 55, from 26 to 300. However, the potency against L. donovani amastigotes was typically ~2- to 3-fold lower than for L. mexicana.
Table 6.
In Vitro Antileishmanial Activity against L. donovani Amastigotes and In Vitro Cytotoxicity of Selected TAs.
| in vitro activity EC50 (nM)a vs |
cytotoxicity IC50 (nM)b vs |
||
|---|---|---|---|
| compound | L. donovani | HepG2 cells | TI |
| 42 | 118 ± 16 | 26700 | 226 |
| 45 | 139 ± 61 | 10100 | 73 |
| 46 | 88.0 ± 11 | 6058 | 69 |
| 47 | 122 ± 35 | 3425 | 28 |
| 48 | 198 ± 61 | 5120 | 26 |
| 51 | 226 ± 106 | ND | ND |
| 55 | 285 ± 57 | 1734 | 6.0 |
| 74 | 184 ± 88 | 59400 | 323 |
| 77 | 181 ± 32 | 14529 | 80 |
| 80 | 398 ± 127 | 85513 | 215 |
| 88 | 100 ± 39 | 30000 | 300 |
| 89 | 90.5 ± 6.3 | 15200 | 168 |
| 90 | 79.5 ± 16 | 21300 | 268 |
| 95 | ND | 15933 | ND |
| 101 | ND | 8208 | ND |
| 102 | 101 ± 53 | 6200 | 61 |
| 110 | ND | 2994 | ND |
| 116 | 81.2 ± 8.7 | 21323 | 263 |
Data represent the mean ± standard deviation of results of at least two independent experiments
IC50 values (nM) are the average of at least three determinations, each carried out in triplicate (±10%); ND, not determined; TI = Therapeutic Index (HepG2 IC50/L. donovani EC50).
In Vitro Cytotoxicity.
The preliminary in vitro general cytotoxicity was tested for the selected TAs using human hepatic HepG2 cells.19, 49 Inhibition of mammalian cells only occurred at relatively very high concentrations (IC50 >10000 nM against HepG2 cells) with many of these TAs (Table 6).
In Vitro Metabolic Stability.
A number of antileishmanial TAs, selected for their potency (EC50 < 100 nM) and to represent a range of structurally diverse analogues, were also assessed for in vitro metabolic stability using human liver microsomes (HLM) and mouse liver microsomes (MLM) with well-established methods.50, 51 Notably, many of the most active TAs appear to be stable for up to 60 minutes or longer in both HLM and MLM (Table 7).
Table 7.
In Vitro Metabolic Stability of Selected TA Analogues against Liver Microsomes.
| metabolic stability (t1/2, min) vs | ||
|---|---|---|
| compound | HLM | MLM |
| 42 | 17.1 | 10.2 |
| 45 | 52.1 | 8.20 |
| 46 | 18.6 | 12.3 |
| 47 | 17.3 | 11.4 |
| 48 | 13.0 | 15.4 |
| 49 | 8.45 | 9.71 |
| 52 | 30.0 | 28.9 |
| 54 | > 60.0 | 45.1 |
| 55 | > 60.0 | 46.9 |
| 56 | > 60.0 | 30.1 |
| 57 | > 60.0 | 42.0 |
| 58 | > 60.0 | 15.8 |
| 60 | 56.2 | 46.7 |
| 61 | > 60.0 | > 60.0 |
| 62 | > 60.0 | 19.1 |
| 63 | > 60.0 | 15.8 |
| 70 | > 60.0 | > 60.0 |
| 74 | > 60.0 | > 60.0 |
| 77 | > 60 | > 60 |
| 80 | > 60.0 | > 60.0 |
| 88 | > 60.0 | 17.6 |
| 90 | 45.1 | 11.2 |
| 95 | 23.3 | 37.5 |
| 101 | 26.7 | 33.0 |
| 102 | > 60.0 | 9.71 |
| 104 | > 60.0 | > 60.0 |
| 105 | 42.1 | 31.2 |
| 108 | > 60.0 | > 60.0 |
| 109 | > 60.0 | > 60.0 |
| 110 | > 60.0 | > 60.0 |
| 111 | > 60.0 | > 60.0 |
| 112 | > 60.0 | > 60.0 |
| 113 | > 60.0 | 11.0 |
| 116 | > 60.0 | 29.0 |
| 117 | > 60.0 | 7.98 |
| 118 | > 60.0 | > 60.0 |
| 119 | 11.7 | 10.4 |
| 120 | 19.7 | 13.8 |
| 121 | 25.6 | 14.8 |
| 122 | 50.8 | 16.5 |
| 123 | > 60.0 | > 60.0 |
HLM = human liver microsomes; MLM = mouse live microsomes.
In Vivo Antileishmanial Efficacy of TAs.
Given the promising antileishmanial potency of a number of TAs against L. mexicana and L. donovani along with a favorable TI against J774A.1 macrophage lines, and a favorable combination of good physicochemical properties, an in vivo proof of concept study in a well-established murine model of cutaneous leishmaniasis using BALB/c mice infected by footpad injection52 was undertaken with 3 promising and representative TAs 95, 101, and 110. These compounds were chosen because they had a combination of properties suggesting potential in vivo efficacy, including good in vitro potency, strong TI, and acceptable in vitro metabolic stability, with 110 being the most promising by these criteria. Importantly, these three compounds had also shown strong efficacy in curing Plasmodium yoelli infections in mice (unpublished results), thus ensuring that they must have sufficient in vivo metabolic stability and bioavailability to evaluate their efficacy against L. mexicana in vivo. However, it should be noted that in general there is not strong correlation between the in vitro potencies of TAs between L. mexicana and Plasmodium parasites, in which they were first investigated.19
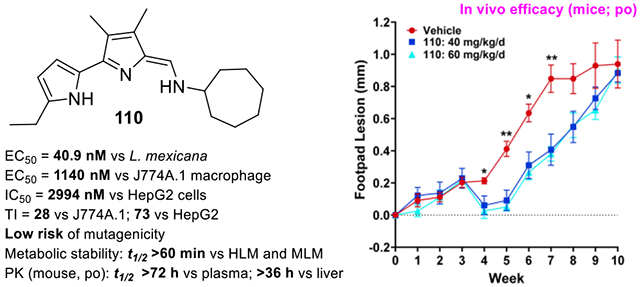
Twenty days post-infection (pi) with L. mexicana promastigotes expressing red-shifted luciferase, animals were treated with 95, 101, and 110 daily for 10 days by oral gavage at 40–60 mg/kg/d. Lesion growth was monitored in two independent experiments measuring either the width of the footpad lesion (Figure 3A) or the luminescence signal over the infected footpad (Figure 3B). For both experiments, TAs 95, 101, and 110 provided retardation of lesion progression compared to the animals treated with vehicle alone, particularly between weeks 5–7 pi, demonstrating partial efficacy in controlling the disease. In particular, compound 110 provided greater efficacy than both 95 and 101. However, the effect was transient, as the lesion sizes or luminescence signal (not shown) reached that of vehicle-treated animals by week 10 pi. These results imply that in vitro potency does translate to in vivo oral efficacy, although the “drug-like” properties of these TAs would need to be improved to exhibit a curative effect. Several potent TAs with a feasible TI and good metabolic stability that emerged later in this study, including 118 and 123, will be tested in vivo in future studies to identify potentially more promising antileishmanial TA lead compounds.
Figure 3.

In vivo efficacy of 95, 101, and 110 in mice. (A) mice (initially 5 per treatment) infected with L. mexicana were treated with vehicle, 101 and 110 and footpad lesion thickness was measured. p Values were determined by two-tailed paired t-test; (B) Mice infected with RS-LUC-expressing L. mexicana were treated (days 20–29) with vehicle, 95, 101 and 110 and imaged at 6 weeks post-infection. The ratio of Relative Luminescence Units (RLU) for each mouse was calculated by dividing the RLU at day 45 post-infection (pi) by the RLU at day 19, just before treatment was begun. This ratio quantifies the increase in RLU between day 19 and day 45. Statistical significance was determined using ANOVA and Dunnett’s post-test. *** p<0.001, ** p<0.01, * p<0.05.
In Vivo Pharmacokinetic Analysis.
Preliminary in vivo PK analysis of lead compound 110 was conducted following a single intragastric (po) administration in mice at 40 mg/kg with blood and liver samples taken at the following time points: 0, 0.5, 1, 2, 4, 8, 24, 30, 48, 54, and 72 h.53, 54 Key PK parameters in both plasma and liver are summarized in Table 8. Significantly, compound 110 showed long half-life (t1/2 = > 72 h for plasma, and > 36 h for liver) and rapid absorption in both plasma and liver (Tmax = 0.0 h for plasma and 2.0 h for liver). It is noteworthy that 110 is highly concentrated in the liver versus plasma (3667 ng/mL for liver vs 35 ng/mL for plasma), a potentially significant advantage for the treatment of visceral leishmaniasis, where the liver is a major site of parasite colonization. The PK data of 110 indicates the observed oral dose efficacy might be the result of a combination of high concentration (3667 ng/mL) and long elimination half-life (t1/2 > 36 h) of 110 in the liver (Table 8). Indeed, these favorable PK properties were one of the reasons we chose to test 110 in vivo.
Table 8.
Key PK Parameters of 110 in Plasma and Liver Following Single Oral Dose of 40 mg/kg Administrations in Mice.
| matrix | Cmax (ng/mL) |
Tmax (h) |
t1/2 (h) |
AUClast (ng.h/mL) |
AUCinf (ng·h/mL) |
AUCextrap (%) |
|---|---|---|---|---|---|---|
| liver | 3667 | 2.0 | > 36 | 22982 | 23043 | 0.262 |
| plasma | 35.0 | 0.0 | > 72 | 2421 | 206250 | 98.8 |
Cmax, maximum plasma or hepatic concentration; Tmax, time to Cmax; t1/2, apparent elimination half-life; AUClast, area under the concentration-time curve from 0 up to the last sampling time at which a quantifiable concentration is found; AUCinf, area under the concentration-time curve from 0 up to infinity; AUCextrap, percentage of the AUC extrapolated from the last observed time point.
In Vitro Mutagenicity.
Given the promising in vitro and in vivo antileishmanial activities of TA analogues, lead compound 110 was investigated for the potential of mutagenicity risk using the Ames assay55, 56 (EPBI Inc.) at concentrations up to 10 μM, with and without S9 metabolic activation, against two Salmonella typhimurium TA100 and T98 strains. Results were negative: there was no increase over the background reversion rate with 110, suggesting low risk of mutagenicity with this TA class of compounds.
CONCLUSIONS
In summary, we have discovered and developed a natural product inspired tambjamine chemotype as a novel class of antileishmanial agents. In this work, we have generated and investigated a large library of TA analogues with various structural features on ring-A, ring-B, and terminal amine of the TA scaffold, against two Leishmania parasites, cutaneous leishmaniasis causing L. mexicana, and fatal visceral leishmaniasis causing L. donovani. Several strengths of the TA chemotype are summarized here. The robust SARs identified a number of novel TA analogues with excellent in vitro potency with low nanomolar EC50 values against both L. mexicana and L. donovani parasites, while demonstrating great TI against both the J774A.1 macrophages and human hepatic HepG2 cells. It is noteworthy that the TAs demonstrated superior in vitro antileishmanial activity than the control drug, miltefosine (TAs EC50s < 100 nM vs miltefosine EC50 = 3460 nM), and in general the low nM potency of many TAs makes them potentially superior to currently available antileishmanial therapies. Furthermore, selected lead TAs possess good in vitro metabolic stability in both human and mouse liver microsomes and in vivo PK profiles in mice with long half-life and rapid absorption in both plasma and liver. In addition, these TAs have shown moderate in vivo efficacy in mice after oral administration. Specifically, lead compound 110 provided retardation of lesion progression compared to animals treated with vehicle alone, particularly between weeks 5 and 7 pi, demonstrating partial efficacy in controlling disease. The partial in vivo efficacy indicates that this chemotype has not yet been sufficiently optimized to qualify as a late lead. Our further structural optimizations focus on the identification of novel TAs that retain or increase antileishmanial potency, including some of those listed in Table 5, while improving efficacy in animals and “drug-like” properties such as favorable PK and oral bioavailability, with mitigated toxicity.
Another objective for future studies is to address the potential targets and MoA of TAs in Leishmania parasites. Currently, targets and MoA are not known, be it for Plasmodium parasites, T. cruzi, or L. mexicana. One approach that has been employed successfully for other anti-trypanosomatid chemotypes is to generate resistant mutants followed by whole genome sequencing to identify genetic modifications.13 Alternatively, analogs that incorporate photoactivatable probes can be employed to identify proteins with which TAs interact.57 Recently, proteome-wide approaches have been developed to identify proteins that interact with specific compounds, inducing a shift in thermal denaturation.58 In addition, phenotypic alterations in TA-treated parasites might reveal important cellular pathways whereby such compounds act. While such comprehensive studies are beyond the scope of the current initial demonstration of antileishmanial potency, they provide potential strategies for elucidation of the mechanisms whereby this chemotype targets Leishmania intracellular amastigotes.
EXPERIMENTAL SECTION
General.
NMR spectra were recorded on Bruker AMX-400 and AMX-600, spectrometers at 400 and 600 MHz, respectively. NMR experiments were recorded in CDCl3 and/or DMSO-d6 at 25 °C. Chemical shifts are given in parts per million (ppm) downfield from internal standard tetramethylsilane (TMS). High-resolution mass spectra (HRMS) (electrospray ionization (ESI)) were recorded on a vanquish ultrahigh-performance liquid chromatography/high-performance liquid chromatography (UHPLC/HPLC) system coupled with a high-resolution (350000) Q Exactive Orbitrap mass spectrometer. Unless otherwise stated, all reagents and solvents were purchased from commercial supplies and used without further purification. Reactions that required the use of anhydrous, inert atmosphere techniques were carried out under an atmosphere of argon/nitrogen. Chromatography was executed on a CombiFlash instrument using silica gel (230–400 mesh) as stationary phase and mixtures of ethyl acetate and hexanes as eluents. Analytical HPLC analysis was performed on an Agilent 1260 Infinity II LC System using C18 column (3.0 × 50 mm) with a linear elution gradient of water/methanol (containing 10 mM ammonium acetate) ranging from 50:50 to 0:100 for 6.0 min at a flow rate of 0.75 mL/min, at 254 nm. A purity of >95% has been established for all final compounds.
Synthesis of tert-Butyl 4-Hydroxy-2-oxo-2,5-dihydro-1H-pyrrole-1-carboxylate (26).
To a stirred solution of (tert-butoxycarbonyl)glycine (25, 10 g, 57.1 mmol) in 200 mL of anhydrous dichloromethane (DCM) were added Meldrum’s acid (9.87 g, 68.6 mmol) and DMAP (17.4 g, 143 mmol) under argon atmosphere at 0 °C. A solution of isopropyl chloroformate (86 mL, 85.7 mmol, 1 N in toluene) was added dropwise, and the reaction mixture was stirred for 4 h at 0 °C. The reaction mixture was diluted with DCM (200 mL), washed with 15% KHSO4 (2 × 100 mL), and organic layer was dried over anhydrous Na2SO4, and the solvent was evaporated under reduced pressure to give the acylated Meldrum’s acid. This material was then refluxed in ethyl acetate (1 L) for 1 h, and the solvent was evaporated under reduced pressure, and the product was recrystallized from ethyl acetate to give the desired product 26 (7.28 g, 64%) as a white crystalline solid. 1H NMR (DMSO-d6, 400 MHz) δ 12.1 (br s, 1H), 4.88 (s, 1H), 4.14 (s, 2H), 1.44 (s, 9H); HRMS (ESI) calcd for C9H13NNaO4 (M + Na)+ 222.0737; found 222.0729.
Synthesis of tert-Butyl 2-Oxo-4-(tosyloxy)-2,5-dihydro-1H-pyrrole-1-carboxylate (27).
To a stirred solution of 26 (5.0 g, 25.1 mmol) in anhydrous DCM (100 mL) were added p-toluenesulfonyl chloride (4.77 g, 25.1 mmol) and DIPEA (6.48 g, 50.2 mmol). The resulting reaction mixture was stirred for 6 h at room temperature. Then the reaction mixture was washed with 5% HCl (2 × 40 mL), and brine and dried over anhydrous Na2SO4. The organic solvent was removed under reduced pressure and the product was chromatographed on silica gel, with ethyl acetate/hexanes as eluent, to afford 27 (8.34 g, 94%) as a white solid. 1H NMR (CDCl3, 400 MHz) δ 7.86 (d, J = 8.4 Hz, 2H), 7.42 (d, J = 8.4 Hz, 2H), 5.75 (s, 1H), 4.22 (d, J = 1.2 Hz, 2H), 2.50 (s, 3H), 1.52 (s, 9H); HRMS (ESI) calcd for C16H19NNaO6S (M + Na)+ 376.0825; found 376.0831.
Synthesis of tert-Butyl 2-Oxo-4-(4-(trifluoromethoxy)phenoxy)-2,5-dihydro-1H-pyrrole-1-carboxylate (29a).
To a stirred solution of 27 (4.0 g, 11.3 mmol) in anhydrous DCM (150 mL) were added 4-(trifluoromethoxy)phenol (28a; 4.0 g, 22.7 mmol) and DABCO (2.79 g, 24.9 mmol). The resulting reaction mixture was stirred for 16 h at room temperature. Then the reaction mixture was washed with 5% HCl (2 × 75 mL), and 10% NaOH solution (150 mL) and dried over anhydrous Na2SO4. The organic solvent was removed under reduced pressure and the product was chromatographed on silica gel, with ethyl acetate/hexanes as eluent, to afford 29a (2.03 g, 50%) as a white solid. 1H NMR (CDCl3, 400 MHz) δ 7.31 (d, J = 9.1 Hz, 2H), 7.20 (d, J = 9.1 Hz, 2H), 4.98 (s, 1H), 4.43 (s, 2H), 1.57 (s, 9H); HRMS (ESI) calcd for C16H16F3NNaO5 (M + Na)+ 382.0873; found 382.0860.
Synthesis of Intermediates 29b and 29c.
Compounds 29b and 29c were synthesized and purified by the same procedure as described for 29a from 27, and 28b and 28c, respectively.
tert-Butyl 4-(4-Chlorophenoxy)-2-oxo-2,5-dihydro-1H-pyrrole-1-carboxylate (29b).
Yield: 1.61 g (45%) white solid; 1H NMR (CDCl3, 400 MHz) δ 7.40 (d, J = 8.7 Hz, 2H), 7.10 (d, J = 8.7 Hz, 2H), 4.95 (s, 1H), 4.40 (d, J = 0.9 Hz, 2H), 1.55 (s, 9H); HRMS (ESI) calcd for C15H16ClNNaO4 (M + Na)+ 332.0660; found 332.0650.
tert-Butyl 4-(3,5-difluorophenoxy)-2-oxo-2,5-dihydro-1H-pyrrole-1-carboxylate (29c).
Yield: 2.21 g (44%) white solid; 1H NMR (CDCl3, 400 MHz) δ 7.8–6.72 (m, 3H), 5.09 (s, 1H), 4.44 (s, 2H), 1.56 (s, 9H); HRMS (ESI) calcd for C15H15F2NaNO4 (M + Na)+ 334.0861; found 334.0850.
Synthesis of 4-(4-(Trifluoromethoxy)phenoxy)-1,5-dihydro-2H-pyrrol-2-one (30a).
To a stirred solution of 29a (2.0 g, 5.57 mmol) in anhydrous DCM (50 mL) was added dropwise TFA (2.54 g, 22.3 mmol). The resulting reaction mixture was stirred for an additional hour at room temperature. The solvent was evaporated under reduced pressure, and the crude material was then dissolved in ethyl acetate (200 mL). The organic layer was washed with 5% NaHCO3 (100 mL) and brine and dried over anhydrous Na2SO4. The organic solvent was removed under reduced pressure and the solid product was washed with DCM (50 mL) to afford the pure product 30a (1.32 g, 92%) as a white solid. 1H NMR (DMSO-d6, 400 MHz) δ 7.67 (br s, 1H), 7.48 (d, J = 9.2 Hz, 2H), 7.42 (d, J = 9.2 Hz, 2H), 4.85 (d, J = 1.3 Hz, 1H), 4.08 (s, 2H); HRMS (ESI) calcd for C11H9F3NO3 (M + H)+ 260.0529; found 260.0520.
Synthesis of Intermediates 30b and 30c.
Compounds 30b and 30c were synthesized and purified by the same procedure as described for 30a from 29b and 29c, respectively.
4-(4-Chlorophenoxy)-1,5-dihydro-2H-pyrrol-2-one (30b).
Yield: 1.13 g (95%) white solid; 1H NMR (DMSO-d6, 400 MHz) δ 7.66 (br s, 1H), 7.52 (d, J = 8.6 Hz, 2H), 7.32 (d, J = 8.6 Hz, 2H), 4.84 (s, 1H), 4.06 (s, 2H); HRMS (ESI) calcd for C10H9ClNO2 (M + H)+ 210.0316; found 210.0310.
4-(3,5-Difluorophenoxy)-1,5-dihydro-2H-pyrrol-2-one (30c).
Yield: 1.34 g (90%) white solid; 1H NMR (DMSO-d6, 400 MHz) δ 7.72 (br s, 1H), 7.72–7.13 (m, 3H), 5.06 (d, J = 0.89 Hz, 1H), 4.06 (s, 2H); HRMS (ESI) calcd for C10H8F2NO2 (M + H)+ 212.0518; found 212.0511.
Synthesis of (Z)-N-((5-Bromo-3-(4-(trifluoromethoxy)phenoxy)-2H-pyrrol-2-ylidene)methyl)-N-ethylethanamine (31a).
To a stirred solution of diethylformamide (1.52 g, 15.0 mmol) in anhydrous DCM (10 mL) was added dropwise a solution of phosphorus oxybromide (POBr3; 3.58 g, 12.5 mmol) in DCM (10 mL) under an argon atmosphere at 0 °C. The resulting thick suspension was stirred at 0 °C for an hour to obtain the Vilsmeier complex as a white solid. Then, the solid was crushed with a spatula, DCM (50 mL) was added, and the mixture was cooled to 0 °C. A solution of 30a (1.3 g, 5.0 mmol) in DCM (10 mL) was added dropwise, and the mixture was warmed to room temperature and then heated at 50 °C for 6 h. The mixture was poured onto ice water, and the pH was adjusted to 8 by treating with 5 N NaOH solution, and then stirred for an additional 30 min. The organic layer was separated and the aqueous layer was extracted with DCM (3 × 50 mL). The combined organic extracts were washed with water and brine, and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure, and the crude product was chromatographed on silica gel, with ethyl acetate/hexanes as eluent, to afford the title compound 31a as a tan solid (1.3 g, 64%). 1H NMR (CDCl3, 400 MHz) δ 7.23–7.16 (m, 5H), 5.64 (s, 1H), 4.24 (q, J = 7.1 Hz, 2H), 3.48 (q, J = 7.1 Hz, 2H), 1.38–1.33 (m, 6H); HRMS (ESI) calcd for C16H17BrF3N2O2 (M + H)+ 405.0420; found 405.0406 and 407.0383 (+2, isotope).
Synthesis of Intermediates 31b and 31c.
Compounds 31b and 31c were synthesized and purified by the same procedure as described for 31a from 30b and 30c, respectively.
(Z)-N-((5-Bromo-3-(4-chlorophenoxy)-2H-pyrrol-2-ylidene)methyl)-N-ethylethanamine (31b).
Yield: 1.21 g (65%) pale-brown solid; 1H NMR (CDCl3, 400 MHz) δ 7.21 (d, J = 8.8 Hz, 2H), 7.06 (s, 1H), 7.01 (d, J = 8.8 Hz, 2H), 5.51 (s, 1H), 4.13 (q, J = 6.9 Hz, 2H), 3.38 (q, J = 7.2 Hz, 2H), 1.28–1.24 (m, 6H); HRMS (ESI) calcd for C15H17BrClN2O (M + H)+ 355.0207; found 355.0194 and 357.0169 (+2, isotope).
(Z)-N-((5-Bromo-3-(3,5-difluorophenoxy)-2H-pyrrol-2-ylidene)methyl)-N-ethylethanamine (31c).
Yield: 1.46 g (72%) pale-brown solid; 1H NMR (CDCl3, 400 MHz) δ 7.07 (s, 2H), 6.69–6.50 (m, 3H), 5.78 (s, 1H), 4.22 (q, J = 7.2 Hz, 2H), 3.46 (q, J = 7.3 Hz, 2H), 1.36–1.31 (m, 6H); HRMS (ESI) calcd for C15H16BrF2N2O (M + H)+ 357.0409; found 357.0394 and 359.0371 (+2, isotope).
Synthesis of 4-(4-(Trifluoromethoxy)phenoxy)-1H,1'H-[2,2'-bipyrrole]-5-carbaldehyde (20).
To a degassed solution of 31a (1.3 g, 3.21 mmol) and 2-pyrroleboronic acid (32, 1.0 g, 4.82 mmol) in 10% water/1,4-dioxane (70 mL) were added Pd(PPh3)4 (0.186 g, 0.16 mmol) and Na2CO3 (0.682 g, 6.43 mmol). The reaction mixture was stirred for 4 h at 100 °C. The solvents were evaporated under reduced pressure, and the obtained crude product was then treated with 10% NaOH solution at 90 °C for 20 min. Then the crude product was extracted with ethyl acetate (3 × 50 mL). The combined organic layers were washed with brine and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure, and the crude product was then chromatographed on silica gel, with ethyl acetate/hexanes as eluent, to afford the title compound 20 as a pale-yellow solid (0.886 g, 82%). 1H NMR (CDCl3, 400 MHz) δ 11.96 (br s, 1H), 11.66 (br s, 1H), 9.39 (s, 1H), 7.30–7.25 (m, 4H), 6.98 (m, 1H), 6.62 (m, 1H), 6.29 (m, 1H), 5.99 (d, J = 2.6 Hz, 1H); HRMS (ESI) calcd for C16H12F3N2O3 (M + H)+ 337.0795; found 337.0781.
Synthesis of MBC Analogues 21 and 22.
Compounds 21 and 22 were synthesized and purified by the same procedure as described for 20 from 31b and 31c, respectively.
4-(4-Chlorophenoxy)-1H,1'H-[2,2'-bipyrrole]-5-carbaldehyde (21).
Yield: 0.687 g (85%) pale-yellow solid; 1H NMR (DMSO-d6, 400 MHz) δ 11.8 (br s, 1H), 11.2 (br s, 1H), 9.41 (s, 1H), 7.47 (d, J = 8.8 Hz, 2H), 7.24 (d, J = 8.8 Hz, 2H), 6.89 (m, 1H), 6.80 (m, 1H), 6.16 (d, J = 1.9 Hz, 1H), 6.11 (m, 1H); HRMS (ESI) calcd for C15H12ClN2O2 (M + H)+ 287.0582; found 287.0574.
4-(3,5-Difluorophenoxy)-1H,1'H-[2,2'-bipyrrole]-5-carbaldehyde (22).
Yield: 0.501 g (62%) pale-yellow solid; 1H NMR (DMSO-d6, 600 MHz) δ 11.9 (br s, 1H), 11.2 (br s, 1H), 9.40 (s, 1H), 7.06 (tt, J = 9.3, 2.0 Hz, 1H), 6.98 (dd J = 8.3, 1.8 Hz, 2H), 6.91 (m, 1H), 6.81 (br s, 1H), 6.30 (d, J = 2.0 Hz, 1H), 6.13 (m, 1H); HRMS (ESI) calcd for C15H11F2N2O2 (M + H)+ 289.0783; found 289.0775.
Synthesis of tert-Butyl 2-Oxo-4-(4-(trifluoromethoxy)phenyl)-2,5-dihydro-1H-pyrrole-1-carboxylate (34).
To a degassed solution of 27 (3.9 g, 11.0 mmol) and (4-(trifluoromethoxy)phenyl)boronic acid (33, 3.4 g, 16.6 mmol) in 10% water/THF (100 mL) were added Pd(dppf)Cl2 (0.403 g, 0.55 mmol) and Cs2CO3 (10.8 g, 33.1 mmol). The reaction mixture was stirred for 6 h at 85 °C and then filtered through Celite. The filtrate was evaporated under reduced pressure, and the obtained crude product was then chromatographed on silica gel, with ethyl acetate/hexanes as eluent, to afford the title compound 34 as a white solid (3.0 g, 79%). 1H NMR (CDCl3, 400 MHz) δ 7.61 (d, J = 8.6 Hz, 2H), 7.32 (d, J = 8.6 Hz, 2H), 6.43 (s, 1H), 4.70 (s, 2H), 1.61 (s, 9H); HRMS (ESI) calcd for C16H16F3NNaO4 (M + Na)+ 366.0924; found 366.0913.
Synthesis of 4-(4-(Trifluoromethoxy)phenyl)-1,5-dihydro-2H-pyrrol-2-one (35).
Compound 35 was synthesized and purified by the same procedure as described for 30a from 34. Yield: 1.91 g (90%) white solid; 1H NMR (DMSO-d6, 400 MHz) δ 8.24 (br s, 1H), 7.80 (d, J = 8.8 Hz, 2H), 7.44 (d, J = 8.8 Hz, 2H), 6.58 (s, 1H), 4.39 (s, 2H); HRMS (ESI) calcd for C11H9F3NO2 (M + H)+ 244.0580; found 244.0572.
Synthesis of (Z)-N-((5-Bromo-3-(4-(trifluoromethoxy)phenyl)-2H-pyrrol-2-ylidene)methyl)-N-ethylethanamine (36).
Compound 36 was synthesized and purified by the same procedure as described for 31a from 35. Yield:1.71 g (63%) pale-brown solid; 1H NMR (CDCl3, 400 MHz) δ 7.39–7.25 (m, 4H), 6.94 (s, 1H), 6.39 (s, 1H), 4.35 (m, 2H), 3.47 (m, 2H), 1.41 (t, J = 7.1 Hz, 3H), 1.33 (t, J = 7.2 Hz, 3H); HRMS (ESI) calcd for C16H17BrF3N2O (M + H)+ 389.0471; found 389.0455 and 391.0431 (+2, isotope).
Synthesis of 4-(4-(Trifluoromethoxy)phenyl)-1H,1'H-[2,2'-bipyrrole]-5-carbaldehyde (23).
Compound 23 was synthesized and purified by the same procedure as described for 20 from 32 and 36. Yield: 0.800 g (63%) pale-yellow solid; 1H NMR (DMSO-d6, 600 MHz) δ 12.1 (br s, 1H), 11.3 (br s, 1H), 9.46 (s, 1H), 7.70 (d, J = 8.7 Hz, 2H), 7.45 (d, J = 8.7 Hz, 2H), 6.93 (d, J = 1.1 Hz, 1H), 6.80 (d, J = 1.6 Hz, 1H), 6.73 (s, 1H), 6.14 (m, 1H); HRMS (ESI) calcd for C16H12F3N2O2 (M + H)+ 321.0845; found 321.0837.
Synthesis of tert-Butyl 2-(4-Methoxyphenyl)-1H-pyrrole-1-carboxylate (38).
Compound 38 was synthesized and purified by the same procedure as described for 20 from 32 and 37, with the exception of 10% NaOH solution treatment. Yield: 4.5 g (62%) clear syrup; 1H NMR (CDCl3, 400 MHz) δ 7.35 (m, 1H), 7.30 (d, J = 8.6 Hz, 2H), 6.92 (d, J = 8.6 Hz, 2H), 6.23 (t, J = 3.3 Hz, 1H), 6.16 (m, 1H), 3.85 (s, 3H), 1.42 (s, 9H); HRMS (ESI) calcd for C16H19NNaO3 (M + Na)+ 296.1259; found 296.1248.
Synthesis of (1-(tert-Butoxycarbonyl)-5-(4-methoxyphenyl)-1H-pyrrol-2-yl)boronic Acid (39).
To a stirred solution of 38 (3.5 g, 12.8 mmol) in 75 mL of anhydrous THF was added dropwise lithium diisopropylamide (LDA, 2 M; 19.3 mL, 63.6 mmol) under an argon atmosphere at −78 °C. The reaction mixture was stirred for 2 h at −78 °C, then trimethyl borate (6.60 g, 64.1 mmol) was added, and the resulting solution was allowed to react at ambient temperature overnight. The reaction was quenched with saturated NH4Cl solution, filtered through Celite, and extracted with ethyl acetate (3 × 50 mL). The combined organic phases were dried over anhydrous Na2SO4, and concentrated under reduced pressure. The crude product was further washed with hexanes, to afford the pure product 39 as a white solid (3.82 g, 94%). 1H NMR (CDCl3, 400 MHz) δ 7.23 (d, J = 8.8 Hz, 2H), 7.11 (d, J = 3.4 Hz, 1H), 6.91 (br s, 2H), 6.93 (d, J = 8.8 Hz, 2H), 6.22 (d, J = 3.4 Hz, 1H), 3.86 (s, 3H), 1.22 (s, 9H); HRMS (ESI) calcd for C16H20BNNaO5 (M + Na)+ 340.1330; found 340.1313.
Synthesis of (Z)-N-((5-Bromo-3-methoxy-2H-pyrrol-2-ylidene)methyl)-N-ethylethanamine (41).
Compound 41 was synthesized and purified by the same procedure as described for 31a from 40. Yield: 7.53 g (66%) tan solid; 1H NMR (CDCl3, 600 MHz) δ 7.01 (s, 1H), 5.61 (s, 1H), 4.14 (q, J = 7.4 Hz, 2H), 3.78 (s, 3H), 3.41 (q, J = 7.2 Hz, 2H), 1.31 (t, J = 7.4 Hz, 3H), 1.30 (t, J = 7.2 Hz, 3H); HRMS (ESI) calcd for C10H16BrN2O (M + H)+ 259.0441; found 259.0434 and 259.0412 (+2, isotope).
Synthesis of 4-Methoxy-5'-(4-methoxyphenyl)-1H,1'H-[2,2'-bipyrrole]-5-carbaldehyde (24).
Compound 24 was synthesized and purified by the same procedure as described for 20 from 39 and 41. Yield: 1.72 g (75%) tan solid; 1H NMR (DMSO-d6, 400 MHz) δ 11.4 (br s, 1H), 11.3 (br s, 1H), 9.31 (s, 1H), 7.61 (d, J = 8.8 Hz, 2H), 6.99 (d, J = 8.8 Hz, 2H), 6.77 (t, J = 2.3 Hz, 1H), 6.51 (dd, J = 2.3, 1.0 Hz, 1H), 6.40 (d, J = 2.6 Hz, 1H), 3.86 (s, 3H), 3.79 (m, 3H); HRMS (ESI) calcd for C17H17N2O3 (M + H)+ 297.1234; found 297.1224.
Synthesis of (Z)-1-Cycloheptyl-N-((4'-methoxy-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)-methanamine Hydrochloride (46).
To a stirred solution of 1 (0.100 g, 0.52 mmol) and cycloheptylmethanamine (0.134 g, 1.05 mmol) in anhydrous methanol (10 mL) were added a few drops of methanolic HCl (2 N, catalytic amount) under an argon atmosphere at room temperature. The resulting pale yellow colored solution was stirred at refluxing temperature for 5 h (in some cases between 2 and10 h). On completion of the reaction, methanol was evaporated under reduced pressure and the obtained crude product was then dissolved in ethyl acetate (50 mL) and washed with 2 N HCl (2 × 30 mL). The organic layer was washed with brine and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure, and the crude product was then chromatographed on silica gel, with ethyl acetate/hexanes as eluent, to afford the desired TA analogue 46 with hydrogen chloride salt (0.153 g, 87% ) as a yellow solid. 1H NMR (CDCl3, 400 MHz) δ 13.73 (br s, 1H), 10.64 (br s, 1H), 9.44 (br s, 1H), 7.31 (d, J = 14.9 Hz, 1H), 7.07 (m, 1H), 6.75 (m, 1H), 6.25 (m, 1H), 5.96 (d, J = 1.8 Hz, 1H), 3.94 (s, 3H), 3.33 (t, J = 6.4 Hz, 2H), 1.94–1.89 (m, 3H), 1.86–1.80 (m, 2H), 1.75–1.45 (m, 6H), 1.32–1.23 (m, 2H); HRMS (ESI) calcd for C18H26N3O (M + H)+ 300.2070; found 300.2058.
Synthesis of tert-Butyl ((1r,4r)-4-(((Z)-(4'-Methoxy-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)amino)cyclohexyl)carbamate (49).
Compound 1 (0.100 g, 0.52 mmol), tert-butyl ((1r,4r)-4-aminocyclohexyl)carbamate (0.225 g, 1.05 mmol), anhydrous Na2SO4 (10 g), and anhydrous DCM (50 mL) were placed in a sealed tube with a Teflon-lined cap under an argon atmosphere at room temperature. The reaction mixture was stirred and heated for 72 h at 60 °C. The mixture was filtered by a sintered funnel and washed with DCM (50 mL). The filtrate was concentrated under reduced pressure to give a crude residue, which was further chromatographed on silica gel, with ethyl acetate/hexanes as eluent, to afford the titled compound 49 in excellent yield (0.175 g, 86%) as a yellow solid. 1H NMR (CDCl3, 400 MHz) δ 13.70 (br s, 1H), 10.59 (br s, 1H), 9.60 (br s, 1H), 7.36 (d, J = 14.5 Hz, 1H), 7.07 (br s, 1H), 6.74 (br s, 1H), 6.28 (dd, J = 5.3, 2.5 Hz, 1H), 5.93 (br s, 1H), 3.92 (s, 3H), 3.62–3.51 (m, 4H), 3.30 (m, 1H), 2.20 (br s, 1H), 2.07–1.88 (m, 3H), 1.70 (m, 1H), 1.47 (s, 9H); HRMS (ESI) calcd for C21H31N4O3 (M + H)+ 387.2391; found 387.2377.
Synthesis of Target TA Analogues.
Synthesis and structural characterization of 42–45, 50, 53, 82–91, 93, 94, 102, 103, 115 and 116 were reported in our previous publication19 that describes the antiplasmodial activity of early-stage TAs. The rest of the new TAs were synthesized and purified by the same procedure as described for 46 from 1, 6–24 (100 mg scale, 1.0 equivalent) and appropriate amines (2.0 equivalents). Note: Majority of the final TA analogues are reported in HCl salt form. Hence, an additional proton, which protonates the nitrogen of ring-B, appears at downfield region in the 1H NMR spectra.
(1r,4r)-N-((Z)-(4'-Methoxy-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)-4-methylcyclohexan-1-amine Hydrochloride (47).
Yield: 142 mg (84%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 13.65 (s, 1H), 10.62 (s, 1H), 9.57 (d, J = 15.1 Hz, 1H), 7.42 (d, J = 15.1 Hz, 1H), 7.08 (m, 1H), 6.74 (m, 1H), 6.29 (m, 1H), 5.95 (d, J = 2.3 Hz, 1H), 3.94 (s, 3H), 3.34 (m, 1H), 2.11 (m, 2H), 1.85 (m, 2H), 1.72–1.60 (m, 2H), 1.49–1.41 (m, 1H), 1.09–0.98 (m, 2H), 0.95 (d, J = 6.4 Hz, 3H); HRMS (ESI) calcd for C17H24N3O (M + H)+ 286.1914; found 286.1904.
(1r,4r)-N-((Z)-(4'-Methoxy-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)-4-(trifluoromethyl)cyclohexan-1-amine Hydrochloride (48).
Yield: 176 mg (89%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 13.98 (br s, 1H), 10.73 (br s, 1H), 9.51 (d, J = 14.8 Hz, 1H), 7.38 (d, J = 14.8 Hz, 1H), 7.10 (m, 1H), 6.77 (m, 1H), 6.30 (m, 1H), 5.97 (d, J = 2.3 Hz, 1H), 3.95 (s, 3H), 3.79 (m, 1H), 2.19–1.97 (m, 5H), 1.89–1.77 (m, 4H); HRMS (ESI) calcd for C17H21F3N3O (M + H)+ 340.1631; found 340.1621.
(Z)-N-((4'-Methoxy-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)aniline Hydrochloride (51).
Yield: 144 mg (91%) brownish red solid; 1H NMR (CDCl3, 400 MHz) δ 14.02 (br s, 1H), 11.26 (d, J = 14.5 Hz, 1H), 10.74 (br s, 1H), 7.82 (d, J = 14.5 Hz, 1H), 7.41 (m, 4H), 7.22 (m, 1H), 7.17 (m, 1H), 6.86 (m, 1H), 6.35 (m, 1H), 6.03 (d, J = 2.2 Hz, 1H), 4.01 (s, 3H); HRMS (ESI) calcd for C16H16N3O (M + H)+ 266.1288; found 266.1278.
(Z)-N-((4'-Methoxy-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)-4-methylaniline Hydrochloride (52).
Yield: 156 mg (94%) brownish red solid; 1H NMR (CDCl3, 400 MHz) δ 13.95 (br s, 1H), 11.25 (d, J = 14.6 Hz, 1H), 10.70 (br s, 1H), 7.79 (d, J = 14.6 Hz, 1H), 7.32 (d, J = 8.2 Hz, 2H), 7.21 (d, J = 8.2 Hz, 2H), 7.15 (br s, 1H), 6.83 (br s, 1H), 6.34 (d, J = 2.1 Hz, 1H), 6.01 (d, J = 1.7 Hz, 1H), 4.00 (s, 3H), 2.36 (s, 3H); HRMS (ESI) calcd for C17H18N3O (M + H)+ 280.1444; found 280.1434.
(Z)-N-((4'-Methoxy-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)-4-(trifluoromethoxy)aniline Hydrochloride (54).
Yield: 192 mg (95%) brownish red solid; 1H NMR (DMSO-d6, 400 MHz) δ 13.25 (br s, 1H), 12.60 (d, J = 13.7 Hz, 1H), 12.09 (br s, 1H), 8.28 (d, J = 13.7 Hz, 1H), 7.73 (d, J = 8.5 Hz, 2H), 7.46 (d, J = 8.5 Hz, 2H), 7.29 (d, J = 6.2 Hz, 2H), 6.62 (s, 1H), 6.36 (s, 1H), 4.02 (s, 3H); HRMS (ESI) calcd for C17H15F3N3O2 (M + H)+ 350.1111; found 350.1099.
(Z)-N-((4'-Methoxy-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)-4-(p-tolyloxy)aniline (55).
Yield: 162 mg (83%) brownish red solid; 1H NMR (CDCl3, 400 MHz) δ 7.79 (s, 1H), 7.34 (d, J = 8.8 Hz, 2H), 7.17 (d, J = 8.4 Hz, 2H), 7.12 (dd, J = 2.4, 1.2 Hz, 1H), 7.02 (d, J = 8.8 Hz, 2H), 6.94 (d, J = 8.4 Hz, 2H), 6.80 (dd, J = 3.7, 1.2 Hz, 1H), 6.34 (dd, J = 2.7, 0.97 Hz, 1H), 6.02 (s, 1H), 3.99 (s, 3H), 2.36 (s, 3H); HRMS (ESI) calcd for C23H22N3O2 (M + H)+ 372.1707; found 372.1695.
(Z)-N-((4'-Methoxy-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)-4-(4-methoxyphenoxy)aniline (56).
Yield: 165 mg (81%) brownish red solid; 1H NMR (CDCl3, 400 MHz) δ 7.77 (s, 1H), 7.34 (d, J = 8.8 Hz, 2H), 7.13 (dd, J = 2.4, 1.3 Hz, 1H), 7.78 (d, J = 9.0 Hz, 2H), 6.98 (d, J = 8.8 Hz, 2H), 6.91 (d, J = 9.0 Hz, 2H), 6.80 (dd, J = 3.7, 1.1 Hz, 1H), 6.34 (dd, J = 3.7, 1.1 Hz, 1H), 6.01 (s, 1H), 3.99 (s, 3H), 3.83 (s, 3H); HRMS (ESI) calcd for C23H22N3O3 (M + H)+ 388.1656; found 388.1643.
(Z)-3-Chloro-N-((4'-methoxy-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)aniline Hydrochloride (57).
Yield: 155 mg (88%) brownish red solid; 1H NMR (DMSO-d6, 400 MHz) δ 13.21 (br s, 1H), 12.52 (d, J = 13.9 Hz, 1H), 12.10 (br s, 1H), 8.35 (d, J = 13.9 Hz, 1H), 7.78 (s, 1H), 7.58 (d, J = 8.2 Hz, 1H), 7.46 (t, J = 8.2 Hz, 1H), 7.30–7.25 (m, 3H), 6.62 (d, J = 1.9 Hz, 1H), 6.38 (dd, J = 5.2, 2.3 Hz, 1H), 4.03 (s, 3H); HRMS (ESI) calcd for C16H15ClN3O (M + H)+ 300.0898; found 300.0887.
(Z)-N-((4'-Methoxy-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)-3-(trifluoromethoxy)aniline Hydrochloride (58).
Yield: 184 mg (91%) brownish red solid; 1H NMR (DMSO-d6, 400 MHz) δ 13.24 (br s, 1H), 12.60 (br s, 1H), 12.12 (br s, 1H), 8.36 (br s, 1H), 7.71–7.19 (m, 6H), 6.63 (br s, 1H), 6.38 (br s, 1H), 4.03 (s, 3H); HRMS (ESI) calcd for C17H15F3N3O2 (M + H)+ 350.1111; found 350.1098.
(Z)-N-((4'-Methoxy-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)-2-(trifluoromethoxy)aniline Hydrochloride (59).
Yield: 188 mg (93%) brownish red solid; 1H NMR (DMSO-d6, 400 MHz) δ 13.55 (br s, 1H), 12.15 (br s, 1H), 11.75 (d, J = 13.4 Hz, 1H), 8.07 (d, J = 13.4 Hz, 1H), 7.79 (d, J = 7.7 Hz, 1H), 7.53 (d, J = 9.2 Hz, 1H), 7.49 (d, J = 7.7 Hz, 1H), 7.39 (t, J = 7.7 Hz, 1H), 7.31 (s, 1H), 7.27 (s, 1H), 6.65 (s, 1H), 6.39 (br s, 1H), 4.03 (s, 3H); HRMS (ESI) calcd for C17H15F3N3O2 (M + H)+ 350.1111; found 350.1099.
(Z)-3,4-Dichloro-N-((4'-methoxy-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)aniline Hydrochloride (60).
Yield: 165 mg (85%) brownish red solid; 1H NMR (DMSO-d6, 400 MHz) δ 13.16 (br s, 1H), 12.53 (d, J = 13.9 Hz, 1H), 12.12 (br s, 1H), 8.35 (d, J = 13.9 Hz, 1H), 7.98 (s, 1H), 7.69 (d, J = 8.7 Hz, 1H), 7.60 (d, J = 8.7 Hz, 1H), 7.30 (s, 2H), 6.62 (s, 1H), 6.38 (s, 1H), 4.03 (s, 3H); HRMS (ESI) calcd for C16H14Cl2N3O (M + H)+ 334.0508; found 334.0495.
(Z)-4-Chloro-3-methoxy-N-((4'-methoxy-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)aniline Hydrochloride (61).
Yield: 175 mg (91%) brownish red solid; 1H NMR (DMSO-d6, 400 MHz) δ 13.24 (br s, 1H), 12.51 (d, J = 14.0 Hz, 1H), 12.07 (br s, 1H), 8.40 (d, J = 14.0 Hz, 1H), 7.46 (d, J = 8.4 Hz, 1H), 7.44 (d, J = 9.0 Hz, 1H), 7.29 (d, J = 10.0 Hz, 2H), 7.18 (d, J = 8.4 Hz, 1H), 6.62 (s, 1H), 6.36 (s, 1H), 4.02 (s, 3H), 3.94 (s, 3H); HRMS (ESI) calcd for C17H17ClN3O2 (M + H)+ 330.1004; found 330.0993.
(Z)-4-Fluoro-3-methoxy-N-((4'-methoxy-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)aniline Hydrochloride (62).
Yield: 162 mg (88%) brownish red solid; 1H NMR (DMSO-d6, 400 MHz) δ 13.21 (br s, 1H), 12.54 (br s, 1H), 12.05 (br s, 1H), 8.37 (s, 1H), 7.47–7.16 (m, 5H), 6.61 (s, 1H), 6.35 (s, 1H), 4.01 (s, 3H), 3.93 (s, 3H); HRMS (ESI) calcd for C17H17FN3O2 (M + H)+ 314.1299; found 314.1286.
(Z)-N-((4'-Methoxy-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)-2,3-dihydrobenzo[b][1,4]dioxin-6-amine Hydrochloride (63).
Yield: 132 mg (93%) brownish red solid; 1H NMR (DMSO-d6, 400 MHz) δ 13.10 (s, 1H), 12.45 (d, J = 14.3 Hz, 1H), 11.98 (s, 1H), 8.21 (d, J = 14.3 Hz, 1H), 7.22 (br s, 3H), 7.11 (dd, J = 6.5, 2.3 Hz, 1H), 6.93 (d, J = 8.6 Hz, 1H), 6.57 (d, J = 1.0 Hz, 1H), 6.34 (d, J = 1.0 Hz, 1H), 4.28 (dd, J = 5.1, 3.1 Hz, 4H), 4.00 (s, 3H); HRMS (ESI) calcd for C18H18N3O3 (M + H)+ 324.1343; found 324.1328.
(Z)-N-((4'-(4-(Trifluoromethoxy)phenoxy)-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)hexan-1-amine (64).
Yield: 106 mg (85%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 7.55 (s, 1H), 7.31 (d, J = 9.0 Hz, 2H), 7.24 (d, J = 9.0 Hz, 2H), 7.06 (br s, 1H), 6.65 (br s, 1H), 6.27 (d, J = 2.5 Hz, 1H), 5.82 (s, 1H), 3.57 (t, J = 7.2 Hz, 2H), 1.82 (m, 2H), 1.47–1.42 (m, 2H), 1.37–1.35 (m, 4H), 0.92 (t, J = 6.9 Hz, 3H); HRMS (ESI) calcd for C22H25F3N3O2 (M + H)+ 420.1893; found 420.1879.
(Z)-N-((4'-(4-(Trifluoromethoxy)phenoxy)-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)undecan-1-amine Hydrochloride (65).
Yield: 126 mg (81%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 13.98 (br s, 1H), 10.56 (br s, 1H), 10.05 (br s, 1H), 7.56 (d, J = 12.2 Hz, 1H), 7.31 (d, J = 8.9 Hz, 2H), 7.24 (d, J = 8.9 Hz, 2H), 7.07 (br s, 1H), 6.66 (br s, 1H), 6.27 (m, 1H), 5.81 (s, 1H), 3.57 (t, J = 6.7 Hz, 2H), 1.83 (m, 2H), 1.47–1.42 (m, 2H), 1.33–1.28 (m, 14H), 0.89 (t, J = 6.4 Hz, 3H); HRMS (ESI) calcd for C27H35F3N3O2 (M + H)+ 490.2676; found 490.2663.
(Z)-N-((4'-(4-(Trifluoromethoxy)phenoxy)-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)cyclohexanamine Hydrochloride (66).
Yield: 117 mg (87%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 13.95 (br s, 1H), 10.56 (br s, 1H), 10.10 (br s, 1H), 7.62 (d, J = 15.3 Hz, 1H), 7.31 (d, J = 8.8 Hz, 2H), 7.24 (d, J = 8.8 Hz, 2H), 7.08 (m, 1H), 6.65 (m, 1H), 6.27 (m, 1H), 5.82 (d, J = 2.4 Hz, 1H), 3.49 (m, 1H), 2.14 (m, 2H), 1.96–1.92 (m, 2H), 1.74–1.64 (m, 2H), 1.43–1.29 (m, 4H); HRMS (ESI) calcd for C22H23F3N3O2 (M + H)+ 418.1737; found 418.1724.
(Z)-N-((4'-(4-(Trifluoromethoxy)phenoxy)-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)cycloheptanamine Hydrochloride (67).
Yield: 124 mg (89%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 13.98 (br s, 1H), 10.57 (br s, 1H), 10.14 (m, 1H), 7.60 (d, J = 15.2 Hz, 1H), 7.31 (d, J = 8.8 Hz, 2H), 7.25–7.22 (m, 2H), 7.08 (m, 1H), 6.64 (m, 1H), 6.27 (m, 1H), 5.81 (d, J = 2.2 Hz, 1H), 3.68 (m, 1H), 2.19–2.11 (m, 2H), 1.99–1.92 (m, 2H), 1.90–1.83 (m, 2H), 1.68–1.63 (m, 4H), 1.59–1.51 (m, 2H); HRMS (ESI) calcd for C23H25F3N3O2 (M + H)+ 432.1893; found 432.1882.
(Z)-N-((4'-(4-(Trifluoromethoxy)phenoxy)-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)cyclooctanamine Hydrochloride (68).
Yield: 132 mg (92%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 14.00 (s, 1H), 10.58 (br s, 1H), 10.08 (m, 1H), 7.60 (d, J = 15.2 Hz, 1H), 7.31 (d, J = 8.8 Hz, 2H), 7.23 (m, 2H), 7.08 (d, J = 1.2 Hz, 1H), 6.64 (m, 1H), 6.27 (m, 1H), 5.81 (d, J = 2.2 Hz, 1H), 3.72 (m, 1H), 2.09–2.01 (m, 4H), 1.92–1.84 (m, 2H), 1.70–1.54 (m, 8H); HRMS (ESI) calcd for C24H27F3N3O2 (M + H)+ 446.2050; found 446.2037.
(Z)-1-Cycloheptyl-N-((4'-(4-(trifluoromethoxy)phenoxy)-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)methanamine Hydrochloride (69).
Yield: 123 mg (86%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 14.03 (s, 1H), 10.56 (s, 1H), 9.98 (m, 1H), 7.52 (d, J = 15.0 Hz, 1H), 7.32 (d, J = 8.8 Hz, 2H), 7.26–7.23 (m, 2H), 7.08 (m, 1H), 6.66 (m, 1H), 6.27 (m, 1H), 5.82 (d, J = 2.3 Hz, 1H), 3.42 (t, J = 6.4 Hz, 2H), 1.99 (m, 1H), 1.90–1.84 (m, 2H), 1.76–1.70 (m, 2H), 1.67–1.61 (m, 2H), 1.58–1.50 (m, 4H), 1.37–1.30 (m, 2H); HRMS (ESI) calcd for C24H27F3N3O2 (M + H)+ 446.2050; found 446.2038.
(3s,5s,7s)-N-((Z)-(4'-(4-(Trifluoromethoxy)phenoxy)-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)adamantan-1-amine Hydrochloride (70).
Yield: 123 mg (82%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 13.95 (br s, 1H), 10.60 (br s, 1H), 10.22 (d, J = 15.7 Hz, 1H), 7.65 (d, J = 15.7 Hz, 1H), 7.32 (d, J = 8.8 Hz, 2H), 7.27–7.23 (m, 2H), 7.08 (m, 1H), 6.63 (m, 1H), 6.26 (m, 1H), 5.82 (d, J = 2.2 Hz, 1H), 2.27 (br s, 3H), 2.07 (d, J = 2.5 Hz, 6H), 1.80–1.72 (m, 6H); HRMS (ESI) calcd for C26H27F3N3O2 (M + H)+ 470.2050; found 470.2039.
(Z)-4-methyl-N-((4'-(4-(Trifluoromethoxy)phenoxy)-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)cyclohexan-1-amine Hydrochloride (71) (mixture of two isomers).
Yield: 126 mg (91%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 14.19 (br s, 0.6H), 13.92 (br s, 0.4H), 10.73 (br s, 0.6H), 10.54 (br s, 0.4H), 10.13 (m, 0.6H), 9.90 (m, 0.4H), 7.64–7.59 (m, 1H), 7.32 (d, J = 9.0 Hz, 2H), 7.26–7.21 (m, 2H), 7.08 (m, 1H), 6.64 (m, 1H), 6.27 (m, 1H), 5.81 (m, 1H), 3.74 (m, 0.6H), 3.43 (m, 0.4H), 2.17 (m, 1H), 2.04 (m, 1H), 1.85–1.81 (m, 3H), 1.77–1.65 (m, 2H), 1.51–1.44 (m, 0.7H), 1.11–1.00 (m, 3H), 0.98 (m, 1H); HRMS (ESI) calcd for C23H25F3N3O2 (M + H)+ 432.1893; found 432.1882.
(1r,4r)-N-((Z)-(4'-(4-(Trifluoromethoxy)phenoxy)-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)-4-(trifluoromethyl)cyclohexan-1-amine Hydrochloride (72).
Yield: 135 mg (87%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 14.27 (s, 1H), 10.66 (br s, 1H), 10.03 (d, J = 10.6 Hz, 1H), 7.59 (d, J = 14.9 Hz, 1H), 7.33 (d, J = 8.7 Hz, 2H), 7.26–7.24 (m, 2H), 7.10 (m, 1H), 6.67 (m, 1H), 6.28 (m, 1H), 5.81 (d, J = 2.1 Hz, 1H), 3.88 (m, 1H), 2.23–2.14 (m, 3H), 2.12–2.03 (m, 2H), 1.93–1.82 (m, 4H); HRMS (ESI) calcd for C23H22F6N3O2 (M + H)+ 486.1611; found 486.1597.
(Z)-N-((4'-(3,5-Difluorophenoxy)-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)cycloheptanamine Hydrochloride (73).
Yield: 128 mg (88%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 14.09 (br s, 1H), 10.56 (br s, 1H), 10.29 (br s, 1H), 7.58 (d, J = 13.2 Hz, 1H), 7.09 (br s, 1H), 6.79–6.68 (m, 4H), 6.29 (m, 1H), 5.96 (s, 1H), 3.67 (m, 1H), 2.18–2.12 (m, 2H), 1.99–1.92 (m, 2H), 1.90–1.84 (m, 2H), 1.68–1.63 (m, 4H), 1.57–1.51 (m, 2H); HRMS (ESI) calcd for C22H24F2N3O (M + H)+ 384.1882; found 384.1869.
(3s,5s,7s)-N-((Z)-(4'-(3,5-Difluorophenoxy)-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)adamantan-1-amine Hydrochloride (74).
Yield: 133 mg (84%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 14.06 (br s, 1H), 10.59 (br s, 1H), 10.36 (d, J = 15.5 Hz, 1H), 7.62 (d, J = 15.5 Hz, 1H), 7.09 (br s, 1H), 6.79–6.71 (m, 3H), 6.67 (br s, 1H), 6.28 (m, 1H), 5.96 (d, J = 1.8 Hz, 1H), 2.27 (br s, 3H). 2.06 (br s, 6H), 1.80–1.72 (m, 6H); HRMS (ESI) calcd for C25H26F2N3O (M + H)+ 422.2038; found 422.2025.
(1r,4r)-N-((Z)-(4'-(3,5-Difluorophenoxy)-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)-4-methylcyclohexan-1-amine Hydrochloride (75).
Yield: 121 mg (83%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 14.04 (br s, 1H), 10.52 (br s, 1H), 10.28 (br s, 1H), 7.59 (d, J = 12.9 Hz, 1H), 7.09 (br s, 1H), 6.78–6.68 (m, 4H), 6.29 (m, 1H), 5.96 (s, 1H), 3.46 (m, 1H), 2.16–2.12 (m, 2H), 1.89–1.86 (m, 2H), 1.77–1.66 (m, 2H), 1.48 (m, 1H), 1.10–1.00 (m, 2H), 0.95 (t, J = 6.4 Hz, 3H); HRMS (ESI) calcd for C22H24F2N3O (M + H)+ 384.1882; found 384.1869.
(Z)-N-((4'-(4-Chlorophenoxy)-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)cycloheptanamine Hydrochloride (76).
Yield: 130 mg (89%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 13.95 (br s, 1H), 10.57 (br s, 1H), 10.10 (br s, 1H), 7.60 (d, J = 15.0 Hz, 1H), 7.42 (d, J = 8.9 Hz, 2H), 7.15 (d, J = 8.9 Hz, 2H), 7.07 (br s, 1H), 6.62 (br s, 1H), 6.26 (m, 1H), 5.78 (d, J = 2.3 Hz, 1H), 3.67 (m, 1H), 2.19–2.11 (m, 2H), 1.98–1.83 (m, 4H), 1.67–1.64 (m, 4H), 1.57–1.51 (m, 2H); HRMS (ESI) calcd for C22H25ClN3O (M + H)+ 382.1681; found 382.1670.
(3s,5s,7s)-N-((Z)-(4'-(4-Chlorophenoxy)-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)adamantan-1-amine Hydrochloride (77).
Yield: 134 mg (84%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 13.91 (br s, 1H), 10.60 (br s, 1H), 10.18 (d, J = 15.8 Hz, 1H), 7.65 (d, J = 15.8 Hz, 1H), 7.42 (d, J = 8.6 Hz, 2H), 7.17 (d, J = 8.6 Hz, 2H), 7.07 (br s, 1H), 6.61 (br s, 1H), 6.26 (m, 1H), 5.78 (d, J = 2.1 Hz, 1H), 2.26 (br s, 3H), 2.06 (s, 6H), 1.80–1.72 (m, 6H); HRMS (ESI) calcd for C25H27ClN3O (M + H)+ 420.1837; found 420.1825.
(1r,4r)-N-((Z)-(4'-(4-Chlorophenoxy)-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)-4-methylcyclohexan-1-amine Hydrochloride (78).
Yield: 121 mg (83%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 13.90 (s, 1H), 10.54 (br s, 1H), 10.08 (m, 1H), 7.62 (d, J = 15.3 Hz, 1H), 7.42 (d, J = 8.8 Hz, 2H), 7.15 (d, J = 8.8 Hz, 2H), 7.07 (br s, 1H), 6.63 (br s, 1H), 6.27 (m, 1H), 5.78 (d, J = 2.0 Hz, 1H), 3.42 (m, 1H), 2.17–2.12 (m, 2H), 1.89–1.86 (m, 2H), 1.76–1.66 (m, 2H), 1.51 (m, 1H), 1.11–1.00 (m, 2H), 0.95 (d, J = 6.5 Hz, 3H); HRMS (ESI) calcd for C22H25ClN3O (M + H)+ 382.1681; found 382.1669.
(Z)-N-((4'-(4-(Trifluoromethoxy)phenyl)-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)cycloheptanamine Hydrochloride (79).
Yield: 128 mg (91%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 14.40 (br s, 1H), 10.76 (br s, 1H), 10.68 (br s, 1H), 7.46 (d, J = 8.4 Hz, 2H), 7.38–7.35 (m, 3H), 7.11 (br s, 1H), 6.81 (br s, 1H), 6.65 (s, 1H), 6.32 (m, 1H), 3.66 (m, 1H), 2.16–2.11 (m, 2H), 2.01–1.92 (m, 2H), 1.89–1.84 (m, 2H), 1.66–1.64 (m, 4H), 1.54–1.52 (m, 2H); HRMS (ESI) calcd for C23H25F3N3O (M + H)+ 416.1944; found 416.1931.
(3s,5s,7s)-N-((Z)-(4'-(4-(Trifluoromethoxy)phenyl)-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)adamantan-1-amine Hydrochloride (80).
Yield: 133 mg (87%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 14.38 (br s, 1H), 10.81 (d, J = 15.1 Hz, 1H), 10.71 (br s, 1H), 7.47–7.45 (m, 3H), 7.38 (d, J = 8.1 Hz, 2H), 7.11 (br s, 1H), 6.81 (br s, 1H), 6.65 (d, J = 1.7 Hz, 1H), 6.32 (m, 1H), 2.27 (br s, 3H), 2.05 (m, 6H), 1.80–1.71 (m, 6H); HRMS (ESI) calcd for C26H27F3N3O (M + H)+ 454.2101; found 454.2086.
(1r,4r)-4-Methyl-N-((Z)-(4'-(4-(trifluoromethoxy)phenyl)-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)cyclohexan-1-amine Hydrochloride (81).
Yield: 120 mg (85%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 14.34 (br s, 1H), 10.74 (br s, 1H), 10.64 (m, 1H), 7.46 (d, J = 8.4 Hz, 2H), 7.41–7.35 (m, 3H), 7.11 (br s, 1H), 6.81 (br s, 1H), 6.65 (s, 1H), 6.32 (m, 1H), 3.42 (m, 1H), 2.13–2.10 (m, 2H), 1.88–1.85 (m, 2H), 1.80–1.70 (m, 2H), 1.48 (m, 1H), 1.09–0.98 (m, 2H), 0.94 (d, J = 6.4 Hz, 3H); HRMS (ESI) calcd for C23H25F3N3O (M + H)+ 416.1944; found 416.1931.
(Z)-1-Cycloheptyl-N-((3',4'-dimethyl-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)methanamine Hydrochloride (92).
Yield: 158 mg (89%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 13.73 (br s, 1H), 10.80 (br s, 1H), 10.12 (m, 1H), 7.25 (d, J = 15.1 Hz, 1H), 7.09 (m, 1H), 6.79 (m, 1H), 6.31 (m, 1H), 3.38 (t, J = 6.5 Hz, 2H), 2.19 (s, 3H), 2.18 (s, 3H), 1.95 (m, 1H), 1.86–1.80 (m, 2H), 1.73–1.66 (m, 2H), 1.64–1.58 (m, 2H), 1.55–1.47 (m, 4H), 1.32–1.23 (m, 2H); HRMS (ESI) calcd for C19H28N3 (M + H)+ 298.2278; found 298.2266.
(1r,4r)-N-((Z)-(3',4'-Diethyl-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)-4-methylcyclohexan-1-amine Hydrochloride (95).
Yield: 148 mg (92%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 13.59 (br s, 1H), 10.87 (d, J = 15.0 Hz, 1H), 10.33 (br s, 1H), 7.39 (br s, 1H), 7.09–7.08 (m, 1H), 6.79 (m, 1H), 6.30 (m, 1H), 3.39 (m, 1H), 2.68 (q, J = 7.5 Hz, 2H), 2.59 (q, J = 7.6 Hz, 2H), 2.11–2.08 (m, 2H), 1.85–1.82 (m, 2H), 1.72–1.65 (m, 2H), 1.46–1.41 (m, 1H), 1.22–1.17 (m, 6H), 1.07–0.96 (m, 2H), 0.92 (d, J = 6.5 Hz, 3H); HRMS (ESI) calcd for C20H30N3 (M + H)+ 312.2434; found 312.2423.
(Z)-N-((3',4'-Dimethyl-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)-4-methylaniline Hydrochloride (96).
Yield: 155 mg (93%) brownish red solid; 1H NMR (CDCl3, 400 MHz) δ 13.86 (br s, 1H), 11.69 (d, J = 14.3 Hz, 1H), 10.89 (m, 1H), 7.74 (d, J = 14.3 Hz, 1H), 7.31 (d, J = 8.1 Hz, 2H), 7.17 (br s, 1H), 7.13 (d, J = 8.1 Hz, 2H), 6.85 (br s, 1H), 6.35 (m, 1H), 2.31 (s, 3H), 2.26 (s, 3H), 2.16 (s, 3H); HRMS (ESI) calcd for C18H20N3 (M + H)+ 278.1652; found 278.1643.
(Z)-4-Chloro-N-((3',4'-dimethyl-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)aniline Hydrochloride (97).
Yield: 166 mg (94%) brownish red solid; 1H NMR (CDCl3, 400 MHz) δ 13.82 (br s, 1H), 11.64 (d, J = 14.3 Hz, 1H), 10.86 (br s, 1H), 7.71 (d, J = 14.3 Hz, 1H), 7.33 (d, J = 8.8 Hz, 2H), 7.23 (d, J = 8.8 Hz, 2H), 7.19 (br s, 1H), 6.89 (br s, 1H), 6.37 (m, 1H), 2.25 (s, 3H), 2.13 (s, 3H); HRMS (ESI) calcd for C17H17ClN3 (M + H)+ 298.1106; found 298.1097.
(Z)-N-((3',4'-Dimethyl-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)-4-(trifluoromethoxy)aniline Hydrochloride (98).
Yield: 193 mg (95%) brownish red solid; 1H NMR (CDCl3, 400 MHz) δ 13.93 (br s, 1H), 11.66 (d, J = 14.1 Hz, 1H), 10.90 (br s, 1H), 7.68 (d, J = 14.1 Hz, 1H), 7.44 (d, J = 9.0 Hz, 2H), 7.20–7.18 (m, 3H), 6.92 (br s, 1H), 6.38 (m, 1H), 2.25 (s, 3H), 2.16 (s, 3H); HRMS (ESI) calcd for C18H17F3N3O (M + H)+ 348.1318; found 348.1306.
(Z)-N-((3',4'-Dimethyl-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)-4-(4-methoxyphenoxy)aniline Hydrochloride (99).
Yield: 199 mg (89%) brownish red solid; 1H NMR (CDCl3, 400 MHz) δ 13.86 (br s, 1H), 11.80 (d, J = 14.6 Hz, 1H), 10.86 (br s, 1H), 7.67 (d, J = 14.6 Hz, 1H), 7.38 (d, J = 8.9 Hz, 2H), 7.16 (br s, 1H), 6.98–6.87 (m, 7H), 6.36 (m, 1H), 3.81 (s, 3H), 2.24 (s, 3H), 2.16 (s, 3H); HRMS (ESI) calcd for C24H24N3O2 (M + H)+ 386.1863; found 386.1850.
(Z)-N-((3',4'-Dimethyl-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)-4-(p-tolyloxy)aniline Hydrochloride (100).
Yield: 187 mg (87%) brownish red solid; 1H NMR (CDCl3, 400 MHz) δ 13.92 (br s, 1H), 11.85 (d, J = 14.6 Hz, 1H), 10.88 (br s, 1H), 7.67 (d, J = 14.6 Hz, 1H), 7.40 (d, J = 8.9 Hz, 2H), 7.18 (s, 1H), 7.15 (d, J = 8.4 Hz, 2H), 6.99 (d, J = 8.9 Hz, 2H), 6.91 (d, J = 8.4 Hz, 2H), 6.88 (br s, 1H), 6.36 (m, 1H), 2.34 (s, 3H), 2.26 (s, 3H), 2.19 (s, 3H); HRMS (ESI) calcd for C24H24N3O (M + H)+ 370.1914; found 370.1902.
(Z)-N-((3-(1H-Pyrrol-2-yl)-4,5,6,7-tetrahydro-1H-isoindol-1-ylidene)methyl)cyclohexanamine Hydrochloride (101).
Yield: 141 mg (91%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 13.72 (br s, 1H), 10.75 (br s, 1H), 9.99 (br s, 1H), 7.29 (d, J = 14.6 Hz, 1H), 7.10 (br s, 1H), 6.71 (br s, 1H), 6.29 (m, 1H), 3.34 (m, 1H), 2.65–2.63 (m, 4H), 2.80–2.04 (m, 2H), 1.92–1.79 (m, 6H), 1.70–1.60 (m, 3H), 1.39–1.24 (m, 3H); HRMS (ESI) calcd for C19H26N3 (M + H)+ 296.2121; found 296.2111.
(Z)-1-(3-(1H-Pyrrol-2-yl)-4,5,6,7-tetrahydro-1H-isoindol-1-ylidene)-N-(cycloheptylmethyl)methanamine Hydrochloride (104).
Yield: 141 mg (84%) brownish red solid; 1H NMR (CDCl3, 400 MHz) δ 13.79 (br s, 1H), 10.75 (br s, 1H), 9.89 (br s, 1H), 7.17 (d, J = 14.5 Hz, 1H), 7.10 (m, 1H), 6.72 (m, 1H), 6.30 (m, 1H), 3.35 (t, J = 6.3 Hz, 2H), 2.65–2.62 (m, 4H), 1.92 (m, 1H), 1.85–1.79 (m, 6H), 1.72–1.65 (m, 2H), 1.59–1.57 (m, 2H), 1.53–1.46 (m, 4H), 1.37–1.22 (m, 2H); HRMS (ESI) calcd for C21H30N3 (M + H)+ 324.2434; found 324.2423.
(Z)-N-((3-(1H-Pyrrol-2-yl)-4,5,6,7-tetrahydro-1H-isoindol-1-ylidene)methyl)-4-methylaniline Hydrochloride (105).
Yield: 147 mg (93%) brownish red solid; 1H NMR (CDCl3, 400 MHz) δ 13.87 (br s, 1H), 11.47 (d, J = 14.6 Hz, 1H), 10.81 (br s, 1H), 7.69 (d, J = 14.6 Hz, 1H), 7.27 (d, J = 8.5 Hz, 2H), 7.16 (br s, 1H), 7.07 (d, J = 8.5 Hz, 2H), 6.77 (br s, 1H), 6.34 (d, J = 3.0 Hz, 1H), 2.74 (t, J = 5.6 Hz, 2H), 2.59 (t, J = 5.6 Hz, 2H), 2.28 (s, 3H), 2.14–1.82 (m, 4H); HRMS (ESI) calcd for C20H22N3 (M + H)+ 304.1808; found 304.1797.
(Z)-N-((3-(1H-Pyrrol-2-yl)-4,5,6,7-tetrahydro-1H-isoindol-1-ylidene)methyl)-4-chloroaniline Hydrochloride (106).
Yield: 151 mg (90%) brownish red solid; 1H NMR (DMSO-d6, 400 MHz) δ 13.42 (br s, 1H), 12.53 (d, J = 13.9 Hz, 1H), 11.19 (br s, 1H), 8.45 (d, J = 13.9 Hz, 1H), 7.70 (d, J = 8.7 Hz, 2H), 7.55 (d, J = 8.7 Hz, 2H), 7.27 (br s, 1H), 7.11 (br s, 1H), 6.37 (br s, 1H), 2.84 (t, J = 6.2 Hz, 2H), 2.69 (t, J = 6.2 Hz, 2H), 1.81–1.79 (m, 4H); HRMS (ESI) calcd for C19H19ClN3 (M + H)+ 324.1262; found 324.1251.
(Z)-N-((3-(1H-Pyrrol-2-yl)-4,5,6,7-tetrahydro-1H-isoindol-1-ylidene)methyl)-4-(trifluoromethoxy)aniline Hydrochloride (107).
Yield: 174 mg (91%) brownish red solid; 1H NMR (CDCl3, 400 MHz) δ 14.01 (br s, 1H), 11.59 (d, J = 14.3 Hz, 1H), 10.84 (br s, 1H), 7.60 (d, J = 14.3 Hz, 1H), 7.42 (d, J = 9.1 Hz, 2H), 7.20 (s, 1H), 7.19 (d, J = 9.1 Hz, 2H), 6.86 (br s, 1H), 6.36 (m, 1H), 2.74 (t, J = 5.4 Hz, 2H), 2.63 (t, J = 5.4 Hz, 2H), 1.84–1.83 (m, 4H); HRMS (ESI) calcd for C20H19F3N3O (M + H)+ 374.1475; found 374.1465.
(Z)-N-((3-(1H-Pyrrol-2-yl)-4,5,6,7-tetrahydro-1H-isoindol-1-ylidene)methyl)-4-(4-methoxyphenoxy)aniline Hydrochloride (108).
Yield: 182 mg (87%) brownish red solid; 1H NMR (CDCl3, 400 MHz) δ 13.97 (br s, 1H), 11.66 (d, J = 14.6 Hz, 1H), 10.81 (br s, 1H), 7.59 (d, J = 14.6 Hz, 1H), 7.36 (d, J = 9.1 Hz, 2H), 7.17 (br s, 1H), 6.98–6.94 (m, 4H), 6.81 (d, J = 9.1 Hz, 2H), 6.82 (br s, 1H), 6.35 (m, 1H), 3.81 (s, 3H), 2.73 (t, J = 5.2 Hz, 2H), 2.66 (t, J = 5.2 Hz, 2H), 1.87–1.56 (m, 4H); HRMS (ESI) calcd for C26H26N3O2 (M + H)+ 412.2020; found 412.2007.
(Z)-N-((3-(1H-Pyrrol-2-yl)-4,5,6,7-tetrahydro-1H-isoindol-1-ylidene)methyl)-4-(p-tolyloxy)aniline Hydrochloride (109).
Yield: 173 mg (86%) brownish red solid; 1H NMR (CDCl3, 400 MHz) δ 13.93 (br s, 1H), 11.64 (d, J = 14.6 Hz, 1H), 10.80 (br s, 1H), 7.60 (d, J = 14.6 Hz, 1H), 7.36 (d, J = 8.8 Hz, 2H), 7.16 (br s, 1H), 7.14 (d, J = 8.4 Hz, 2H), 6.96 (d, J = 8.9 Hz, 2H), 6.89 (d, J = 8.4 Hz, 2H), 6.81 (br s, 1H), 6.35 (m, 1H), 2.73 (t, J = 5.9 Hz, 2H), 2.64 (t, J = 5.4 Hz, 2H), 2.34 (s, 3H), 1.86–1.82 (m, 4H); HRMS (ESI) calcd for C26H26N3O (M + H)+ 396.2070; found 396.2059.
(Z)-N-((5-Ethyl-3',4'-dimethyl-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)cycloheptanamine Hydrochloride (110).
Yield: 148 mg (92%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 13.59 (br s, 1H), 10.80 (br s, 1H), 10.03 (br s, 1H), 7.28 (t, J = 14.9 Hz, 1H), 6.71 (t, J = 3.3 Hz, 1H), 6.05 (br s, 1H), 3.64 (m, 1H), 2.81 (q, J = 7.6 Hz, 2H), 2.18 (s, 3H), 2.17 (s, 3H), 2.14–2.08 (m, 2H), 1.98–1.83 (m, 4H), 1.65–1.64 (m, 4H), 1.56–1.48 (m, 2H), 1.37 (t, J = 7.6 Hz, 3H); HRMS (ESI) calcd for C20H30N3 (M + H)+ 312.2434; found 312.2424.
(Z)-N-((5-Isobutyl-3',4'-dimethyl-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)cycloheptanamine Hydrochloride (111).
Yield: 144 mg (94%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 13.61 (br s, 1H), 10.80 (br s, 1H), 10.03 (m, 1H), 7.27 (d, J = 14.8 Hz, 1H), 6.72 (t, J = 3.0 Hz, 1H), 6.01 (br s, 1H), 3.64 (m, 1H), 2.62 (d, J = 7.6 Hz, 2H), 2.18 (s, 3H), 2.17 (s, 3H), 2.14–2.06 (m, 3H), 1.98–1.83 (m, 4H), 1.66–1.60 (m, 4H), 1.56–1.50 (m, 2H), 0.97 (d, J = 6.7 Hz, 6H); HRMS (ESI) calcd for C22H34N3 (M + H)+ 340.2747; found 340.2737.
(Z)-N-((5-Ethyl-3',4'-dimethyl-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)cyclooctanamine Hydrochloride (112).
Yield: 159 mg (95%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 13.62 (br s, 1H), 10.82 (br s, 1H), 10.01 (br s, 1H), 7.28 (d, J = 14.9 Hz, 1H), 6.71 (t, J = 3.0 Hz, 1H), 6.05 (br s, 1H), 3.66 (m, 1H), 2.78 (q, J = 7.6 Hz, 2H), 2.19 (s, 3H), 2.17 (s, 3H), 2.05–1.99 (m, 4H), 1.93–1.84 (m, 2H), 1.71–1.52 (m, 8H), 1.37 (t, J = 7.6 Hz, 3H); HRMS (ESI) calcd for C21H32N3 (M + H)+ 326.2591; found 326.2579.
(Z)-N-((4-Ethyl-3',4'-dimethyl-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)cycloheptanamine Hydrochloride (113).
Yield: 140 mg (87%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 13.61 (br s, 1H), 10.56 (br s, 1H), 10.21 (br s, 1H), 7.33 (d, J = 15.0 Hz, 1H), 6.91 (br s, 1H), 6.64 (br s, 1H), 3.63 (m, 1H), 2.55 (q, J = 7.6 Hz, 2H), 2.19 (s, 3H), 2.18 (s, 3H), 2.16–2.10 (m, 2H), 1.98–1.82 (m, 4H), 1.64–1.63 (m, 4H), 1.55–1.49 (m, 2H), 1.23 (t, J = 7.6 Hz, 3H); HRMS (ESI) calcd for C20H30N3 (M + H)+ 312.2434; found 312.2423.
(Z)-N-((3,3',4',5-Tetramethyl-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)cycloheptanamine Hydrochloride (114).
Yield: 144 mg (90%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 12.86 (br s, 1H), 11.11 (br s, 1H), 9.50 (m, 1H), 7.39 (d, J = 15.0 Hz, 1H), 5.83 (br s, 1H), 3.64 (m, 1H), 2.33 (s, 3H), 2.22 (s, 3H), 2.20 (s, 3H), 2.14–2.06 (m, 5H), 2.01–1.94 (m, 2H), 1.89–1.84 (m, 2H), 1.64–1.63 (m, 4H), 1.55–1.49 (m, 2H); HRMS (ESI) calcd for C20H30N3 (M + H)+ 312.2434; found 312.2422.
(Z)-N-((5-Ethyl-4'-methoxy-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)cycloheptanamine Hydrochloride (117).
Yield: 147 mg (92%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 13.55 (br s, 1H), 10.62 (br s, 1H), 9.33 (br s, 1H), 7.33 (d, J = 14.7 Hz, 1H), 6.65 (t, J = 3.0 Hz, 1H), 6.01 (br s, 1H), 5.89 (d, J = 1.8 Hz, 1H), 3.92 (s, 3H), 3.58 (m, 1H), 2.76 (q, J = 7.6 Hz, 2H), 2.11–2.05 (m, 2H), 1.92–1.80 (m, 4H), 1.66–1.63 (m, 4H), 1.55–1.48 (m, 2H), 1.37 (t, J = 7.6 Hz, 3H); HRMS (ESI) calcd for C19H28N3O (M + H)+ 314.2227; found 314.2214.
(Z)-N-((4'-Methoxy-5-(4-methoxyphenyl)-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)cycloheptanamine Hydrochloride (118).
Yield: 120 mg (83%) yellow solid; 1H NMR (CDCl3, 400 MHz) δ 13.67 (br s, 1H), 11.07 (br s, 1H), 9.22 (m, 1H), 7.74 (d, J = 8.2 Hz, 2H), 7.27 (d, J = 15.1 Hz, 1H), 6.81 (d, J = 8.2 Hz, 2H), 6.68 (br s, 1H), 6.44 (br s, 1H), 5.86 (br s, 1H), 3.85 (br s, 3H), 3.76 (br s, 3H), 3.55 (m, 1H), 2.01–1.97 (m, 2H), 1.86–1.75 (m, 4H), 1.50–1.45 (m, 6H); HRMS (ESI) calcd for C24H30N3O2 (M + H)+ 392.2333; found 392.2319.
(Z)-N-((4'-Methoxy-5-(4-methoxybenzyl)-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)cyclopentanamine Hydrochloride (119).
Yield: 124 mg (93%) yellow solid; 1H NMR (CDCl3, 600 MHz) δ 13.58 (br s, 1H), 10.62 (br s, 1H), 9.37 (m, 1H), 7.33 (d, J = 15.0 Hz, 1H), 7.27 (d, J = 8.5 Hz, 2H), 6.86 (d, J = 8.5 Hz, 2H), 6.63 (t, J = 3.2 Hz, 1H), 5.96 (t, J = 2.8 Hz, 1H), 5.87 (t, J = 2.2 Hz, 1H), 3.98 (s, 2H), 3.93 (br s, 4H), 3.79 (s, 3H), 2.07–2.03 (m, 2H), 1.97–1.87 (m, 4H), 1.71–1.67 (m, 2H); HRMS (ESI) calcd for C23H28N3O2 (M + H)+ 378.2176; found 378.2164.
(Z)-N-((4'-Methoxy-5-(4-methoxybenzyl)-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)cyclohexanamine Hydrochloride (120).
Yield: 127 mg (92%) yellow solid; 1H NMR (CDCl3, 600 MHz) δ 13.58 (br s, 1H), 10.64 (br s, 1H), 9.36 (m, 1H), 7.35 (d, J = 15.1 Hz, 1H), 7.28 (d, J = 8.6 Hz, 2H), 6.85 (d, J = 8.6 Hz, 2H), 6.63 (t, J = 3.2 Hz, 1H), 5.96 (t, J = 3.2 Hz, 1H), 5.87 (t, J = 2.2 Hz, 1H), 3.98 (s, 2H), 3.91 (s, 3H), 3.79 (s, 3H), 3.38 (m, 1H), 2.07–2.04 (m, 2H), 1.91–1.89 (m, 2H), 1.66–1.59 (m, 4H), 1.39–1.28 (m, 2H); HRMS (ESI) calcd for C24H30N3O2 (M + H)+ 392.2333; found 392.2320.
(Z)-N-((4'-Methoxy-5-(4-methoxybenzyl)-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)cycloheptanamine Hydrochloride (121).
Yield: 135 mg (95%) yellow solid; 1H NMR (CDCl3, 600 MHz) δ 13.59 (br s, 1H), 10.66 (br s, 1H), 9.38 (m, 1H), 7.33 (d, J = 15.3 Hz, 1H), 7.27 (d, J = 8.5 Hz, 2H), 6.86 (d, J = 8.5 Hz, 2H), 6.62 (t, J = 2.9 Hz, 1H), 5.96 (t, J = 2.8 Hz, 1H), 5.87 (d, J = 2.1 Hz, 1H), 3.98 (s, 2H), 3.91 (s, 3H), 3.78 (s, 3H), 3.57 (m, 1H), 2.10–2.06 (m, 2H), 1.90–1.80 (m, 4H), 1.66–1.63 (m, 4H), 1.53–1.49 (m, 2H); HRMS (ESI) calcd for C25H32N3O2 (M + H)+ 406.2489; found 406.2475.
(Z)-N-((4'-Methoxy-5-(4-methoxybenzyl)-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)cyclooctanamine Hydrochloride (122).
Yield: 132 mg (90%) yellow solid; 1H NMR (CDCl3, 600 MHz) δ 13.62 (br s, 1H), 10.68 (br s, 1H), 9.35 (m, 1H), 7.33 (d, J = 15.0 Hz, 1H), 7.27 (d, J = 8.5 Hz, 2H), 6.86 (d, J = 8.5 Hz, 2H), 6.62 (t, J = 2.9 Hz, 1H), 5.96 (t, J = 2.8 Hz, 1H), 5.87 (d, J = 2.2 Hz, 1H), 3.98 (s, 2H), 3.91 (s, 3H), 3.79 (s, 3H), 3.60 (m, 1H), 2.04–1.93 (m, 4H), 1.88–1.83 (m, 2H), 1.67–1.51 (m, 8H); HRMS (ESI) calcd for C26H34N3O2 (M + H)+ 420.2646; found 420.2631.
(3s,5s,7s)-N-((Z)-(4'-Methoxy-5-(4-methoxybenzyl)-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)adamantan-1-amine Hydrochloride (123).
Yield: 137 mg (89%) yellow solid; 1H NMR (CDCl3, 600 MHz) δ 13.57 (br s, 1H), 10.68 (br s, 1H), 9.49 (d, J = 14.9 Hz, 1H), 7.41 (d, J = 15.5 Hz, 1H), 7.28 (d, J = 8.5 Hz, 2H), 6.86 (d, J = 8.5 Hz, 2H), 6.61 (t, J = 2.9 Hz, 1H), 5.96 (t, J = 2.9 Hz, 1H), 5.87 (d, J = 2.5 Hz, 1H), 3.98 (s, 2H), 3.91 (s, 3H), 3.78 (s, 3H), 2.22 (br s, 3H), 2.00 (d, J = 2.4 Hz, 6H), 1.76–1.70 (m, 6H); HRMS (ESI) calcd for C28H34N3O2 (M + H)+ 444.2646; found 444.2632.
(Z)-4-Chloro-N-((4'-methoxy-5-(4-methoxybenzyl)-1H,5'H-[2,2'-bipyrrol]-5'-ylidene)methyl)aniline Hydrochloride (124).
Yield: 138 mg (94%) brownish red solid; 1H NMR (CDCl3, 400 MHz) δ 13.85 (br s, 1H), 11.11 (d, J = 14.3 Hz, 1H), 10.65 (br s, 1H), 7.64 (d, J = 14.3 Hz, 1H), 7.37–7.28 (m, 6H), 6.89 (d, J = 8.5 Hz, 2H), 6.77 (d, J = 2.8 Hz, 1H), 6.05 (d, J = 2.8 Hz, 1H), 5.94 (br s, 1H), 4.03 (s, 2H), 3.78 (s, 3H), 3.80 (s, 3H); HRMS (ESI) calcd for C24H23ClN3O2 (M + H)+ 420.1473; found 420.1462.
BIOLOGICAL EXEPRIMENTS
Infection of J774A.1 Macrophages.
Macrophages (from ATCC) were grown in tissue culture flasks in DMEM plus 10% heat inactivated bovine fetal bovine serum until confluence. L. mexicana (strain MNYC/B2/62/M379, ATTC) expressing red-shifted firefly luciferase from an integrated ribosomal (28S) RNA locus48 (see section below on mouse infections for details of strain construction) were inoculated into RPMI plus 10% heat inactivated fetal bovine serum (FBS) containing 5 μg/mL hemin and 0.1 mM xanthine at a density of 2 × 105 cells/mL and grown at 26 °C for 8 days to stationary phase, ~5–6 × 107 cells/mL. They were then examined microscopically for the presence of metacyclic promastigotes with long flagella and short cell bodies. The parasites were pelleted and resuspended in a volume of PBS that is sufficient to cover the macrophages. Macrophages were infected at a 10:1 ratio of parasites to macrophages by removing medium from the macrophages and adding the parasites in PBS. They were incubated for 10 min at 37 °C in a 5% CO2 atmosphere. Minimal Essential Media (GIBCO) plus 10% heat inactivated FBS was added at half the volume of the parasite suspension in PBS, and the flask was incubated at 37 °C in 5% CO2 atmosphere overnight (~16 h). The medium was removed from the infected macrophages and the monolayer was washed 3× with PBS at 37 °C to remove parasites that has not been internalized. Macrophages were then resuspended in a small volume of warm PBS using a cell scraper, counted, and adjusted to 5 × 104 cells/mL with MEM 10% HI-FBS. A 200 μL aliquot of the cell suspension (1 × 104 cells) was pipetted into each well of a 96-well plate and incubated for 3 h at 37 °C to reattach macrophages to the plates.
Dose-Response Curves for Test Compounds.
For measurement of sensitivity of intracellular amastigotes to each compound, 2 μL of compound at the appropriate concentration in DMSO was added to the well, and the opaque plate was incubated at 37 °C in 5% CO2 for 96 h. Medium was removed from each well, and a 50 μL aliquot of 1:100 firefly luciferase substrate in assay buffer (Promega Firefly Luciferase Assay System) was added and the luminescence signal was read immediately in a Wallac Victor2 1420 Multilabel Counter, using the instrument luminescence program and 1 sec reads. Typically, compounds were applied in serial 3-fold dilutions to cover a range of 10 μM to 1 nM. Data were log transformed and EC50 values were determined using GraphPad Prism 8 (GraphPad Software) and the sigmoidal dose-response with variable slope fitting.
The SYBR Green assay was used for the measurement of sensitivity macrophages. Uninfected macrophages were diluted to 5 × 104 cells/mL in DMEM 10% HI-FBS. A 200 μL aliquot of the cell suspension (1 × 104 cells) was pipetted into each well of a 96-well black plate (Greiner Bio-One, 655086) and incubated for 3 h at 37 °C to reattach macrophages to the plates. Then, compounds were added. After 96 h the medium was removed, the stock SYBR Green II (10,000×) was diluted to 1:1000× in 1% Triton in PBS, and 100 μL was added. Samples were incubated in the dark at room temperature for 1 h with moderate shaking. The plate was then read on the plate reader using Ex 485 nM, Em 535 using a 0.1 s exposure.
Animals and Ethical Statements (Oregon Health & Science University).
All animal work was carried out under the approved Institutional Animal Care and Use Committee protocol IBC-03-07 at the Oregon Health & Sciences University. The facility is accredited by AAALAC International, has a Public Health Service Animal Welfare Assurance, and complies with the Animal Welfare Act Regulations.
In Vivo Antileishmanial Efficacy in a Murine Model.
L. mexicana and L. donovani promastigotes expressing red-shifted firefly luciferase were prepared as follows. The 1647 bp red-shifted firefly luciferase gene PpyRE9H59 was cloned into the Leishmania expression vector pRP-H60 containing a neomycin resistance gene. Following linearization by SwaI restriction digestion, this construct, which contains an rRNA promoter and 28S rRNA sequence flanking the luciferase and neomycin resistance genes, was transfected into the parasites and selected with G418 to achieve integration into the 28S rRNA locus. For in vivo studies, the L. mexicana parasites were inoculated into RPMI plus 10% FBS plus 0.5 μg/mL hemin and 0.1 mM xanthine as above at a density of 2 × 105 cells/mL. Parasites were grown at 26 °C for 8 days to stationary phase ~5–6 × 107 cells/mL, and parasites were examined microscopically for the presence of metacyclics. Parasites were pelleted, washed twice with sterile PBS, and resuspended to a density of 5 × 107 cells/mL in sterile PBS. Female BALB/c mice weighing about 20 g were injected in one hind footpad with 20 μL of resuspended parasites (1 × 106 parasites). Footpad widths were measured with calipers weekly. At 20 days pi, mice were treated with the relevant compound by resuspending at the relevant dose in PEG-400 and delivering 100 μL by oral gavage. Compounds were delivered daily for 10 days. Lesion sizes were determined by subtracting the footpad width at day 19 (just before treatment) from the width at the subsequent time of measurement.
For in vivo imaging system (IVIS), mice infected with L. mexicana expressing red-shifted firefly luciferase were injected intraperitoneally with D-luciferin (PerkinElmer Health Science, Inc.) reconstituted at a concentration of 30 mg/mL in Dulbecco’s PBS without Mg2+ or Ca2+ and filtered using a 0.2 μm filter unit. The luciferin solution was warmed to 37 °C and injected at a dose of 300 mg/mL. Mice were anesthetized with 5% isoflurane and maintained in a sedated state with 2–5–3.5% isoflurane administered by nose cone. Images were taken 10 min after injection with luciferin using an IVIS Spectrum instrument (Revvity Inc., Small Animal Research Imaging Core, Oregon Health & Science University) with 1 min scans. Data settings included the following: F-stop 1, no emission filter, 22 cm field of view, CCD exposure time automatically determined by instrument for optimal signal, data binning 8. For data analysis, Living Image software version 4.7.3.20616 (Revvity, Inc.) was employed. Areas of interest surrounding the infected and uninfected hind footpads were quantified, and the luminescence units of the uninfected footpad were subtracted from those of the infected footpad to determine relative luminescence units for each mouse.
In Vitro Cytotoxicity Assessments.
To identify toxicity to mammalian cells, a dimethylthiazol-diphenyltetrazolium bromide (MTT) assay using human hepatocarcinoma cells (HepG2) was employed. HepG2 cells were cultured in complete minimal essential medium (MEM; Gibco-Invitrogen, No. 11090-099) prepared by supplementing MEM with 0.19% sodium bicarbonate (Gibco-BRL No. 25080-094), 10% heat-inactivated fetal bovine serum (FBS; Gibco-Invitrogen No. 16000-036), 2 mM L-glutamine (Gibco-Invitrogen, No. 25030-081), 0.1 mM MEM nonessential amino acids (Gibco-Invitrogen, No. 11140-050), 0.009 mg/mL insulin (Sigma No. I1882), and 1.76 mg/mL bovine serum albumin (Sigma No. A1470). Cell viability was assessed using trypan blue. 96-well plates were seeded with 2.5 × 104 cells in 170 μL of culture medium per well and incubated at 37 °C overnight in a humidified 5% CO2 atmosphere. Test compound plates were prepared with a Biomek 4000 automated laboratory station and 96-well plates containing 11 duplicate 1.6-fold serial dilutions of each test compound suspended in dimethyl sulfoxide (DMSO). A 30 μL sample of the diluted test compound was then added to 170 μL of medium per well. Cells were exposed to test compound concentrations ranging from 10 to 0.15 μg/mL for 48 h. After incubation, 30 μL of a 1.5 mg/mL solution of MTT diluted in complete MEM media was added to each well; all plates were subsequently incubated in the dark for 1 h at room temperature. Next, the media and test compounds in each well were removed and plates were then left to dry in a hood for 15 min. 60 μL of acidified isopropyl alcohol was added to dissolve the formazan dye crystals created by the reduction of MTT. Plates were placed on a three-dimensional (3-D) rotator for 15–30 min. Absorbance was determined with the Perkin Elmer EnSight plate reader. All experiments were run in triplicates, and the IC50 values were determined using GraphPad Prism software using the nonlinear regression equation (sigmoidal dose–response, variable slope).
In Vitro Metabolic Stability.
The half-lives of lead TAs in mouse and human liver microsomes were determined, as described previously.50, 51 All samples were tested in human and mouse liver microsomal preparations. Sample stocks at 10 or 20 μM (depending on solubility) in DMSO were diluted to a final concentration of 1 μM with a mixture containing 0.5 mg/mL prewarmed pooled human or mouse liver microsomes (BD Gentest), 1.3 mM NADP (Sigma), 3.3 mM MgCl2 (Sigma), and 100 mM potassium phosphate buffer (pH 7.4) using a TECAN Genesis robotic liquid handler. The reaction was started with the addition of 1 U/mL glucose-6-phosphate dehydrogenase (G6PD). The mixture was incubated on a shaking platform at 37 °C, and aliquots were taken and quenched with the addition of an equal volume of cold acetonitrile at 0, 10, 20, 30, and 60 minutes. Samples were centrifuged at 3700 rpm for 10 min at 20 °C to remove debris. Sample quantification was carried out by LC/MS, and the metabolic half-life was calculated by log plots of the total ion chromatography area remaining.
Animals and Ethical Statements (Walter Reed Army Institute of Research).
Male ICR-CD1 mice (Charles River Laboratories. Inc. Raleigh, NC) were used in this study. All animals were assigned a study number with an individual ear tag. Cards attached to each animal cage were also used to identify the study groups. All animals were quarantined for stabilization for 7 days prior to infection. Animals were housed in a designated room with food and water supplied ad libitum and in 12:12 light/dark cycle, in a room with temperature ranging from 64–79 °F, and 34–68% relative humidity.
Research was conducted under the approved Institutional Animal Care and Use Committee. The facility is accredited by AAALAC International, has a Public Health Service Animal Welfare Assurance, and complies with the Animal Welfare Act Regulations and other federal statutes relating to research animals.
In Vivo Pharmacokinetic (PK) Studies.
PK studies were conducted using the previously established methods.53, 54 Briefly, male ICR-CD1 mice weighing 23 to 28 g (Charles River Laboratories. Inc. Raleigh, NC) were used for the PK evaluations. Compound 110 was formulated in 50% PEG-400:50% sterile water and given orally using intragastric (IG) administration. A single 40 mg/kg dose of experimental compound 110 was administered to three mice in each experimental endpoint. At each time point, a terminal whole blood sample was collected by cardiac puncture using heparinized syringes (Heparin, Hospira, Lake Forest, IL). Following the separation of appropriate aliquots, plasma was obtained from the whole blood via centrifugation at 16,200 rpm for 10 minutes at 4 °C. Liver samples were also collected and weighed at each experimental time point. All tissue samples were immediately preserved on dry ice and later stored at −80 °C until analytical work was performed. Drug concentrations were determined for each sample taken from animals dosed with test compounds. PK parameters such as time to maximum plasma concentration (Tmax), maximum plasma concentration (Cmax), the elimination half-life (t1/2), area under the curve (AUC), etc. were obtained using noncompartmental analysis via the Phoenix-WinNonlin software package (version 6.4; Pharsight Crop., Mountain View, CA).
AMES Assay.
Mutagenicity evaluation was assessed using the Ames assay55, 56 (EBPI, Ontario, Canada) against S. typhimurium TA100 and TA98, with and without S9 activation. Tester strain S. typhimurium (TA100 and TA98) cultures were inoculated from the lyophilized pellets and grown in nutrient broth provided by EBPI. Each inoculated flask (125- or 150-mL volume) was placed overnight in the incubator at 37 °C. The positive controls for the assay were 5 μg/mL 2-aminoanthracene for incubations with an S9 mix and a mixture of 1 μg/mL 4-nitroquinoline N-oxide and 2 μg/mL 2-nitrofluoroene for incubations without the S9 mix. The tester strains (2.5 mL) were mixed with exposure medium and, where appropriate, 2.0 mL of S9 to give a final volume of 18 mL. An aliquot (0.2 mL) of the mixture was then added to each well of a 96-well plate. The test compound (10 μL/well) was then added to each well. The 96-well plates were covered and sealed in zip-lock bags and incubated for 96 h at 37 °C. Plates were scored visually, and the positive wells against background mutations were recorded.
Supplementary Material
ACKNOWLEDGMENT
This work was supported from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under the award numbers R21AI160670, R01AI141972, and R01AI175444. The National Science Foundation is acknowledged for support of the BioAnalytical Mass Spectrometry Facility at Portland State University (MRI1828573).
ABBREVIATIONS
- EC50
half maximal effective concentration
- IC50
half-maximal inhibitory concentration
- HLM
human liver microsomes
- MBC
4-methoxy-2,2'-bipyrrole-5-carboxaldehyde
- MEM
minimal essential medium
- MLM
mouse liver microsomes
- PK
pharmacokinetics
- PG
prodigiosin or prodiginine
- SAR
structure-activity relationship
- TA
tambjamine
- TAs
tambjamines
- TI
therapeutic index
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
1H NMR spectra and HRMS of all final compounds and HPLC chromatograms of the key compounds (PDF).
Molecular formula strings and in vitro biological data (CSV).
The authors declare no competing financial interest.
The material has been reviewed by the Walter Reed Army Institute of Research (WRAIR). There is no objection to its presentation and/or publication. The opinions or assertions contained herein are the private views of the authors and are not to be constructed as official or as reflecting the true views of the Department of the Army or the Department of Defense. The research was conducted under an IACUC-approved animal use protocol in an AAALAC International - accredited facility with a Public Health Services Animal Welfare Assurance and in compliance with the Animal Welfare Act and other federal statutes and regulations relating to laboratory animals.
This work was prepared while D.O., was employed at Oregon Health & Science University. The opinions expressed in this article are the author’s own and do not reflect the view of the National Institutes of Health, Department of Health and Human Services, or the United States government.
REFERENCES
- (1).Georgiadou SP; Makaritsis KP; Dalekos GN Leishmaniasis revisited: Current aspects on epidemiology, diagnosis and treatment. J. Transl. Int. Med 2015, 3, 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Burza S; Croft SL; Boelaert M Leishmaniasis. Lancet 2018, 392, 951–970. [DOI] [PubMed] [Google Scholar]
- (3).Sunter J; Gull K Shape, form, function and Leishmania pathogenicity: from textbook descriptions to biological understanding. Open Biol. 2017, 7, 170165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Bañuls AL; Bastien P; Pomares C; Arevalo J; Fisa R; Hide M Clinical pleiomorphism in human leishmaniases, with special mention of asymptomatic infection. Clin. Microbiol. Infect 2011, 17, 1451–1461. [DOI] [PubMed] [Google Scholar]
- (5).Nagle AS; Khare S; Kumar AB; Supek F; Buchynskyy A; Mathison CJ; Chennamaneni NK; Pendem N; Buckner FS; Gelb MH; Molteni V Recent developments in drug discovery for leishmaniasis and human African trypanosomiasis. Chem. Rev 2014, 114, 11305–11347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Sindermann H; Engel J Development of miltefosine as an oral treatment for leishmaniasis. Trans R. Soc. Trop. Med. Hyg 2006, 100 Suppl 1, S17–20. [DOI] [PubMed] [Google Scholar]
- (7).Sunyoto T; Potet J; Boelaert M Why miltefosine-a life-saving drug for leishmaniasis-is unavailable to people who need it the most. BMJ Glob. Health 2018, 3, e000709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Gupta S; Yardley V; Vishwakarma P; Shivahare R; Sharma B; Launay D; Martin D; Puri SK Nitroimidazo-oxazole compound DNDI-VL-2098: An orally effective preclinical drug candidate for the treatment of visceral leishmaniasis. J. Antimicrob. Chemother 2015, 70, 518–527. [DOI] [PubMed] [Google Scholar]
- (9).Van Bocxlaer K; Caridha D; Black C; Vesely B; Leed S; Sciotti RJ; Wijnant GJ; Yardley V; Braillard S; Mowbray CE; Ioset JR; Croft SL Novel benzoxaborole, nitroimidazole and aminopyrazoles with activity against experimental cutaneous leishmaniasis. Int. J. Parasitol. Drugs Drug Resist 2019, 11, 129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Thompson AM; O'Connor PD; Marshall AJ; Yardley V; Maes L; Gupta S; Launay D; Braillard S; Chatelain E; Franzblau SG; Wan B; Wang Y; Ma Z; Cooper CB; Denny WA 7-Substituted 2-nitro-5,6-dihydroimidazo[2,1-b][1,3]oxazines: Novel antitubercular agents lead to a new preclinical candidate for visceral leishmaniasis. J. Med. Chem 2017, 60, 4212–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Thompson AM; O'Connor PD; Marshall AJ; Blaser A; Yardley V; Maes L; Gupta S; Launay D; Braillard S; Chatelain E; Wan B; Franzblau SG; Ma Z; Cooper CB; Denny WA Development of (6R)-2-nitro-6-[4-(trifluoromethoxy)phenoxy]-6,7-dihydro-5 H-imidazo[2,1- b][1,3]oxazine (DNDI-8219): A new lead for visceral leishmaniasis. J. Med. Chem 2018, 61, 2329–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Patterson S; Fairlamb AH Current and future prospects of nitro-compounds as drugs for Trypanosomiasis and Leishmaniasis. Curr. Med. Chem 2019, 26, 4454–4475. [DOI] [PubMed] [Google Scholar]
- (13).Khare S; Nagle AS; Biggart A; Lai YH; Liang F; Davis LC; Barnes SW; Mathison CJ; Myburgh E; Gao MY; Gillespie JR; Liu X; Tan JL; Stinson M; Rivera IC; Ballard J; Yeh V; Groessl T; Federe G; Koh HX; Venable JD; Bursulaya B; Shapiro M; Mishra PK; Spraggon G; Brock A; Mottram JC; Buckner FS; Rao SP; Wen BG; Walker JR; Tuntland T; Molteni V; Glynne RJ; Supek F Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature 2016, 537, 229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wall RJ; Carvalho S; Milne R; Bueren-Calabuig JA; Moniz S; Cantizani-Perez J; MacLean L; Kessler A; Cotillo I; Sastry L; Manthri S; Patterson S; Zuccotto F; Thompson S; Martin J; Marco M; Miles TJ; De Rycker M; Thomas MG; Fairlamb AH; Gilbert IH; Wyllie S The Q(i) site of cytochrome b is a promiscuous drug target in Trypanosoma cruzi and Leishmania donovani. ACS Infect. Dis 2020, 6, 515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Braillard S; Keenan M; Breese KJ; Heppell J; Abbott M; Islam R; Shackleford DM; Katneni K; Crighton E; Chen G; Patil R; Lee G; White KL; Carvalho S; Wall RJ; Chemi G; Zuccotto F; González S; Marco M; Deakyne J; Standing D;, Brunori G; Lyon JJ; Castañeda-Casado P; Camino I; Martinez Martinez MS; Zulfiqar B; Avery VM; Feijens PB; Van Pelt N; Matheeussen A; Hendrickx S; Maes L; Caljon G; Yardley V; Wyllie S; Charman SA; Chatelain E DNDI-6174 is a preclinical candidate for visceral leishmaniasis that targets the cytochrome bc(1). Sci. Transl. Med 2023, 15, eadh9902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Sharlow ER; Close D; Shun T; Leimgruber S; Reed R; Mustata G; Wipf P; Johnson J; O'Neil M; Grögl M; Magill AJ; Lazo JS Identification of potent chemotypes targeting Leishmania major using a high-throughput, low-stringency, computationally enhanced, small molecule screen. PLoS Negl. Trop. Dis 2009, 3, e540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ortiz D; Guiguemde WA; Hammill JT; Carrillo AK; Chen Y; Connelly M; Stalheim K; Elya C; Johnson A; Min J; Shelat A; Smithson DC; Yang L; Zhu F; Guy RK; Landfear SM Discovery of novel, orally bioavailable, antileishmanial compounds using phenotypic screening. PLoS Negl. Trop. Dis 2017, 11, e0006157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kavouris JA; McCall LI; Giardini MA; De Muylder G; Thomas D; Garcia-Pérez A; Cantizani J; Cotillo I; Fiandor JM; McKerrow JH; De Oliveira CI; Siqueira-Neto JL; González S; Brown LE; Schaus SE Discovery of pyrazolopyrrolidinones as potent, broad-spectrum inhibitors of Leishmania infection. Front. Trop. Dis 2023, 3, 1011124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kancharla P; Kelly JX; Reynolds KA Synthesis and structure-activity relationships of tambjamines and B-ring functionalized prodiginines as potent antimalarials. J. Med. Chem 2015, 58, 7286–7309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kancharla P; Lu W; Salem SM; Kelly JX; Reynolds KA Stereospecific synthesis of 23-hydroxyundecylprodiginines and analogues and conversion to antimalarial premarineosins via a Rieske oxygenase catalyzed bicyclization. J. Org. Chem 2014, 79, 11674–11689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Papireddy K; Smilkstein M; Kelly JX; Shweta; Salem SM; Alhamadsheh M; Haynes SW; Challis GL; Reynolds KA Antimalarial activity of natural and synthetic prodiginines. J. Med. Chem 2011, 54, 5296–5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kancharla P; Li Y; Yeluguri M; Dodean RA; Reynolds KA; Kelly JX Total synthesis and antimalarial activity of 2-(p-hydroxybenzyl)-prodigiosins, isoheptylprodigiosin, and geometric isomers of tambjamine MYP1 isolated from marine bacteria. J. Med. Chem 2021, 64, 8739–8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Salem SM; Kancharla P; Florova G; Gupta S; Lu W; Reynolds KA Elucidation of final steps of the marineosins biosynthetic pathway through identification and characterization of the corresponding gene cluster. J. Am. Chem. Soc 2014, 136, 4565–4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Hu DX; Withall DM; Challis GL; Thomson RJ Structure, chemical synthesis, and biosynthesis of prodiginine natural products. Chem. Rev 2016, 116, 7818–7853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Burke C; Thomas T; Egan S; Kjelleberg S The use of functional genomics for the identification of a gene cluster encoding for the biosynthesis of an antifungal tambjamine in the marine bacterium Pseudoalteromonas tunicata. Environ. Microbiol 2007, 9, 814–818. [DOI] [PubMed] [Google Scholar]
- (26).Lu W; Kancharla P; Reynolds KA MarH, a bifunctional enzyme involved in the condensation and hydroxylation steps of the marineosin biosynthetic pathway. Org. Lett 2017, 19, 1298–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Boger DL; Patel M Total synthesis of prodigiosin, prodigiosene, and desmethoxy-prodigiosin: Diels-Alder reactions of heterocyclic azadienes and development of an effective palladium(II)-promoted 2,2'-bipyrrole coupling procedure. J. Org. Chem 1988, 53, 1405–1415. [Google Scholar]
- (28).Marchal E; Uddin MI; Smithen DA; Hawco CLA; Lanteigne M; Overy DP; Kerr RG; Thompson A Antimicrobial activity of non-natural prodigiosenes. Rsc Advances 2013, 3, 22967–22971. [Google Scholar]
- (29).Kojiri K; Nakajima S; Suzuki H; Okura A; Suda H A new antitumor substance, BE-18591, produced by a streptomycete. I. Fermentation, isolation, physico-chemical and biological properties. J. Antibiot. (Tokyo) 1993, 46, 1799–1803. [DOI] [PubMed] [Google Scholar]
- (30).Regourd J; Ali AA; Thompson A Synthesis and anti-cancer activity of C-ring-functionalized prodigiosin analogues. J. Med. Chem 2007, 50, 1528–1536. [DOI] [PubMed] [Google Scholar]
- (31).Aldrich LN; Stoops SL; Crews BC; Marnett LJ; Lindsley CW Total synthesis and biological evaluation of tambjamine K and a library of unnatural analogs. Bioorg. Med. Chem. Lett 2010, 20, 5207–5211. [DOI] [PubMed] [Google Scholar]
- (32).Diaz de Grenu B; Iglesias Hernandez P; Espona M; Quinonero D; Light ME; Torroba T; Perez-Tomas R; Quesada R Synthetic prodiginine obatoclax (GX15-070) and related analogues: anion binding, transmembrane transport, and cytotoxicity properties. Chemistry 2011, 17, 14074–14083. [DOI] [PubMed] [Google Scholar]
- (33).Trudel S; Li ZH; Rauw J; Tiedemann RE; Wen XY; Stewart AK Preclinical studies of the pan-Bcl inhibitor obatoclax (GX015-070) in multiple myeloma. Blood 2007, 109, 5430–5438. [DOI] [PubMed] [Google Scholar]
- (34).Nguyen M; Marcellus RC; Roulston A; Watson M; Serfass L; Murthy Madiraju SR; Goulet D; Viallet J; Belec L; Billot X; Acoca S; Purisima E; Wiegmans A; Cluse L; Johnstone RW; Beauparlant P; Shore GC Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 19512–19517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Parikh SA; Kantarjian H; Schimmer A; Walsh W; Asatiani E; El-Shami K; Winton E; Verstovsek S Phase II study of obatoclax mesylate (GX15-070), a small-molecule BCL-2 family antagonist, for patients with myelofibrosis. Clin. Lymphoma Myeloma Leuk. 2010, 10, 285–289. [DOI] [PubMed] [Google Scholar]
- (36).Mukherjee PP; Goldschmidt ME; Williams RP Enzymic formation of prodigiosin analog by a cell-free preparation from Serratia marcescens. Biochim. Biophys. Acta 1967, 136, 182–184. [DOI] [PubMed] [Google Scholar]
- (37).D'Alessio R; Bargiotti A; Carlini O; Colotta F; Ferrari M; Gnocchi P; Isetta A; Mongelli N; Motta P; Rossi A; Rossi M; Tibolla M; Vanotti E Synthesis and immunosuppressive activity of novel prodigiosin derivatives. J. Med. Chem 2000, 43, 2557–2565. [DOI] [PubMed] [Google Scholar]
- (38).Furstner A. Chemistry and biology of roseophilin and the prodigiosin alkaloids: a survey of the last 2500 years. Angew. Chem. Int. Ed. Engl 2003, 42, 3582–3603. [DOI] [PubMed] [Google Scholar]
- (39).Rastogi S; Marchal E; Uddin I; Groves B; Colpitts J; McFarland SA; Davis JT; Thompson A Synthetic prodigiosenes and the influence of C-ring substitution on DNA cleavage, transmembrane chloride transport and basicity. Org. Biomol. Chem 2013, 11, 3834–3845. [DOI] [PubMed] [Google Scholar]
- (40).Melvin MS; Ferguson DC; Lindquist N; Manderville RA DNA binding by 4-methoxypyrrolic natural products. Preference for intercalation at AT sites by tambjamine E and prodigiosin. J. Org. Chem 1999, 64, 6861–6869. [DOI] [PubMed] [Google Scholar]
- (41).Cavalcanti BC; Junior HV; Seleghim MH; Berlinck RG; Cunha GM; Moraes MO; Pessoa C Cytotoxic and genotoxic effects of tambjamine D, an alkaloid isolated from the nudibranch Tambja eliora, on Chinese hamster lung fibroblasts. Chem. Biol. Interact 2008, 174, 155–162. [DOI] [PubMed] [Google Scholar]
- (42).Herráez R; Quesada R; Dahdah N; Viñas M; Vinuesa T Tambjamines and prodiginines: biocidal activity against Trypanosoma cruzi. Pharmaceutics 2021, 13, 705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Rodilla AM; Korrodi-Gregório L; Hernando E; Manuel-Manresa P; Quesada R; Pérez-Tomás R; Soto-Cerrato V Synthetic tambjamine analogues induce mitochondrial swelling and lysosomal dysfunction leading to autophagy blockade and necrotic cell death in lung cancer. Biochem. Pharmacol 2017, 126, 23–33. [DOI] [PubMed] [Google Scholar]
- (44).Dairi K; Tripathy S; Attardo G; Lavallee JF Two-step synthesis of the bipyrrole precursor of prodigiosins. Tetrahedron Lett. 2006, 47, 2605–2606. [Google Scholar]
- (45).Kancharla P; Reynolds KA Synthesis of 2,2'-bipyrrole-5-carboxaldehydes and their application in the synthesis of B-ring functionalized prodiginines and tambjamines. Tetrahedron 2013, 69, 8375–8385. [Google Scholar]
- (46).Li WR; Lin ST; Hsu NM; Chern MS Efficient total synthesis of pulchellalactam, a CD45 protein tyrosine phosphatase inhibitor. J. Org. Chem 2002, 67, 4702–4706. [DOI] [PubMed] [Google Scholar]
- (47).Marchal E; Rastogi S; Thompson A; Davis JT Influence of B-ring modifications on proton affinity, transmembrane anion transport and anti-cancer properties of synthetic prodigiosenes. Org. Biomol. Chem 2014, 12, 7515–7522. [DOI] [PubMed] [Google Scholar]
- (48).Ortiz D; Forquer I; Boitz J; Soysa R; Elya C; Fulwiler A; Nilsen A; Polley T; Riscoe MK; Ullman B; Landfear SM Targeting the cytochrome bc1 complex of leishmania parasites for discovery of novel drugs. Antimicrob. Agents Chemother 2016, 60, 4972–4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Ferrari M; Fornasiero MC; Isetta AM MTT colorimetric assay for testing macrophage cytotoxic activity in vitro. J. Immunol. Methods 1990, 131, 165–172. [DOI] [PubMed] [Google Scholar]
- (50).Korotchenko V; Sathunuru R; Gerena L; Caridha D; Li Q; Kreishman-Deitrick M; Smith PL; Lin AJ Antimalarial activity of 4-amidinoquinoline and 10-amidinobenzonaphthyridine derivatives. J. Med. Chem 2015, 58, 3411–3431. [DOI] [PubMed] [Google Scholar]
- (51).Zhang X; Qu S Proximity of transmembrane segments 5 and 8 of the glutamate transporter GLT-1 inferred from paired cysteine mutagenesis. PLoS One 2011, 6, e21288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Hammill JT; Sviripa VM; Kril LM; Ortiz D; Fargo CM; Kim HS; Chen Y; Rector J; Rice AL; Domagalska MA; Begley KL; Liu C; Rangnekar VM; Dujardin JC; Watt DS; Landfear SM; Guy RK Amino-substituted 3-aryl- and 3-heteroarylquinolines as potential antileishmanial agents. J. Med. Chem 2021, 64, 12152–12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Li Q; Cantilena LR; Leary KJ; Saviolakis GA; Miller RS; Melendez V; Weina PJ Pharmacokinetic profiles of artesunate after single intravenous doses at 0.5, 1, 2, 4, and 8 mg/kg in healthy volunteers: a phase I study. Am. J. Trop. Med. Hyg 2009, 81, 615–621. [DOI] [PubMed] [Google Scholar]
- (54).Li Q; O'Neil M; Xie L; Caridha D; Zeng Q; Zhang J; Pybus B; Hickman M; Melendez V Assessment of the prophylactic activity and pharmacokinetic profile of oral tafenoquine compared to primaquine for inhibition of liver stage malaria infections. Malar. J 2014, 13, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Ames BN; Mccann J; Yamasaki E Methods for detecting carcinogens and mutagens with the Salmonella/mammalian-microsome mutagenicity test. Mutat. Res 1975, 31, 347–364. [DOI] [PubMed] [Google Scholar]
- (56).Pant K. Ames II™ and Ames liquid format mutagenicity screening assays. In high-throughput screening methods in toxicity testing; John Wiley and Sons, Inc., 2013. [Google Scholar]
- (57).Mackinnon AL; Taunton J Target identification by diazirine photo-cross-linking and click chemistry. Curr. Protoc. Chem. Biol 2009, 1, 55–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Gaetani M; Sabatier P; Saei AA; Beusch CM; Yang Z; Lundström SL; Zubarev RA Proteome integral solubility alteration: A high-throughput proteomics assay for target deconvolution. J. Proteome Res 2019, 18, 4027–4037. [DOI] [PubMed] [Google Scholar]
- (59).Branchini BR; Southworth TL; Khattak NF; Michelini E; Roda A Red- and green-emitting firefly luciferase mutants for bioluminescent reporter applications. Anal. Biochem 2005, 345, 140–148. [DOI] [PubMed] [Google Scholar]
- (60).Soysa R; Tran KD; Ullman B; Yates PA Integrating ribosomal promoter vectors that offer a choice of constitutive expression profiles in Leishmania donovani. Mol. Biochem. Parasitol 2015, 204, 89–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.