

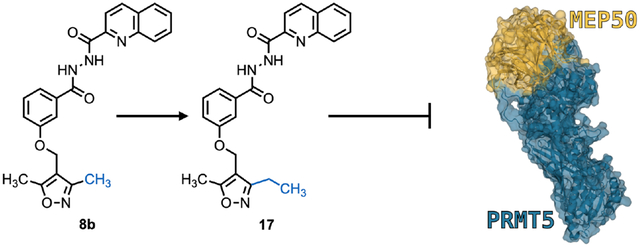
Abstract
Protein arginine methyltransferase 5 (PRMT5) is a master epigenetic regulator and an extensively validated therapeutic target in multiple cancers. Notably, PRMT5 is the only PRMT that requires an obligate cofactor, methylosome protein 50 (MEP50), to function. We developed compound 17, a novel small molecule PRMT5:MEP50 protein:protein interaction (PPI) inhibitor, after initial virtual screen hit identification and analog refinement. Molecular docking indicated that compound 17 targets PRMT5:MEP50 PPI by displacing MEP50 W54 burial into a hydrophobic pocket of PRMT5 TIM barrel. In vitro analysis indicates IC50 <500 nM for prostate and lung cancer cells with selective, specific inhibition of PRMT5:MEP50 substrate methylation and target gene expression, and RNA-seq analysis suggests that compound 17 may dysregulate TGF-β signaling. Compound 17 provides a proof of concept in targeting PRMT5:MEP50 PPI, as opposed to catalytic targeting, as a novel mechanism of action and supports further preclinical development of inhibitors in this class.
Keywords: PRMT5, MEP50, WDR77, Prostate Cancer, methyltransferase inhibitor, protein:protein interaction (PPI)
Graphical Abstract
INTRODUCTION
Protein arginine methyltransferase 5 (PRMT5) is one of nine members of the PRMT family of methyltransferases and is responsible for the majority of symmetric dimethylation of arginine residues in cells.1–3 Through post-translational modification of signaling proteins such as p53, E2F and EGFR as well as epigenetic regulation of target gene expression via symmetric dimethylation of histones (H4R3, H3R2, H3R8 and H2AR3), PRMT5 is required for many cellular processes including cell proliferation, differentiation, survival, DNA damage response, and RNA splicing.4,5 PRMT5 is significantly dysregulated or overexpressed in multiple cancers, and its overexpression appears to correlate with cancer progression and poor clinical outcomes.4,6 One of the major mechanisms to account for its putative oncogenic role is epigenetic repression of tumor suppressors such as Rb1, ST7, PTEN, and p53.6–10 Structural studies have demonstrated that PRMT5 forms a complex with methylosome protein 50 (MEP50) for biological enzymatic function as well as formation of higher order complexes.11–13 Based on in vitro biochemical assays, the presence of MEP50 increases the enzymatic activity of PRMT5 by 100-fold, suggesting that MEP50 is an obligate cofactor.14,15 In addition, multiple PRMT5 interacting proteins appear to serve as adaptors to specifically recruit various substrates or dictate biological activity.16–18 Along these lines, we have recently demonstrated that PRMT5 can also promote prostate cancer cell growth and confer therapy resistance through transcriptional activation of the androgen receptor (AR) in both hormone naïve prostate cancer (HNPC) and castration resistance prostate cancer (CRPC) through interaction with cofactor pICln.19,20 In addition to AR reactivation, we have also demonstrated that PRMT5 mediates transcription of genes involved in DNA damage response pathway following fractionating ionizing radiation, providing two distinct mechanisms for PRMT5 to mediate therapy resistance for prostate cancer cells at two separate stages of disease.19 These data suggest that targeting PRMT5 and its interacting proteins including substrate adaptors such as pICln, COPR5 and RIOK1 may offer a unique and potentially specific approach to target PRMT5 in a context specific manner by exploiting this unique cofactor dependency. Indeed, at the time of manuscript preparation, three recent reports establish a PRMT5 substrate adapter binding motif and subsequent development of an inhibitor targeting said motif to disrupt PRMT5:RIOK1 interaction21–23, though the clinical implication of these inhibitors remain unclear. Because PRMT5 represents a valuable therapeutic target with several Phase I clinical trials currently underway in solid and blood cancers, and because PRMT5 is the only PRMT of 9 family members that requires a cofactor (MEP50) and/or other factors for function, targeting the PRMT5:MEP50 protein:protein interaction (PPI) may offer a specific approach as opposed to the catalytic or pan-MT inhibitors.24–26
Support for this hypothesis also comes from our recent findings that PRMT5 can regulate target gene expression in both MEP50-independent and MEP50-dependent manners.19,20 Thus, development of such inhibitors targeting the PRMT5:MEP50 interaction would not only avoid potential non-specific targeting of other methyltransferases that utilize SAM as a cofactor but also enable selection of a specific type or stage of cancer that is dependent on the function of PRMT5:MEP50. Taking advantage of the existing structure of PRMT5:MEP50, we conducted a virtual screen in conjunction with a bimolecular fluorescence complementation (BiFC)-based screen in cells to discover the first-in-class small molecule inhibitors of the PRMT5:MEP50 interaction27,28. We further improved potency in a second generation of synthesis based on computational docking and BiFC screens. Biological characterization of the lead compound 17 confirmed that the identified compounds can specifically inhibit the target gene expression of PRMT5:MEP50 but not the complexes that involve other cofactors or adaptors. Further, treatment of prostate cancer cells with compound 17 resulted in similar transcriptional response as PRMT5 or MEP50 knockdown, dysregulation of critical cancer cell survival pathways, and cell death with sub-micromolar IC50 in prostate and lung cancer cells.
RESULTS AND DISCUSSION
PRMT5:MEP50 PPI Interface as a Druggable Target
Clinical and in vitro data demonstrate that PRMT5 is frequently overexpressed in cancers and that its overexpression correlates with poor clinical outcome (Fig. S1A). Further, PRMT5 and MEP50 expression correlate positively in patient samples as well as cell lines derived from normal and cancerous tissue. In an analysis of over 1300 cell lines, PRMT5 correlated strongly with MEP50/WDR77 gene expression via the Cancer Dependency Map (Fig S1B,C)29–32. To evaluate a novel mechanism of PRMT5 inhibition through the PPI between PRMT5 and MEP50, we first analyzed PRMT5:MEP50 crystallographic structure 4GQB from Protein DataBank (PDB).13 PRMT5 forms a complex with MEP50 through its N-terminal TIM barrel domain (residues 1–292), and the interaction involves an interface completely occupying the bottom surface (with the top surface defined as the surface following directionality of the innermost beta strand) of the MEP50 protein (Fig. 1A). To evaluate if the interface between PRMT5 and MEP50 is suitable for development of PPI disruptors, we analyzed the PPI interface and observed that electrostatic interactions present in the PPI interface likely contribute significantly to orientation and binding. We identified five residues that may play a role in mediating the interaction. PRMT5 R49 extends from the TIM barrel and closely interacts with MEP50 D99 in the 2nd β-propeller of MEP50 (Fig. 1B,C). PRMT5 R49 also forms contacts with three co-crystallized water molecules as well as 2 contacts with MEP50 D99 and one contact each with MEP50 V83, S47 (Fig. S2A). MEP50 W54 is buried into a pocket of PRMT5, also in the TIM barrel, and appears to be involved in a stacking interaction with PRMT5 H47 (Fig. 1C, S2A). Additionally, MEP50 R52 lies solvent exposed between two alpha helices in the PRMT5 TIM barrel, even though it does not participate in any hydrogen bonding. Adjacent to MEP50 D99, residue D126, located in a loop between the 2nd and 3rd β-propellers of MEP50, appeared to be involved in a hydrogen bond with PRMT5 N21. Collectively, these residues represent potential electrostatic interactions that may be functionally evaluated to assess importance for the PRMT5:MEP50 PPI. Utilizing FastContact binding energy prediction software33, we identified 11 residues each on PRMT5 and MEP50 that likely contribute to binding energy (Fig. S2B–E), and identified additional key residues to include in the BiFC mutant screen. MEP50 D126 (contacts with PRMT5 N21 and co-crystallized water), MEP50 R191 (2 contacts with PRMT5 E161), MEP50 K201 (2 contacts with PRMT5 D166), MEP50 D298 (3 contacts with PRMT5 R62), and MEP50 E276 (one contact with PRMT5 K51 and one with co-crystallized water) were added based on this prediction. We then deployed BiFC assay to assess the feasibility of targeting the interaction via this interface. BiFC is based on the proximity of two interacting proteins and has been used for visualization of PPIs in live cells and animals as well as for screening of PPIs.28,34 We fused the VN (the N-terminal Venus fluorescent protein residues 1–154) to the N-terminal PRMT5 TIM domain and VC (the C-terminal Venus fluorescent protein residues 155–238) to the N-terminal end of MEP50 (Fig. 1D,E). We then introduced mutations described above (PRMT5 residue R49A/D/G as well as MEP50 residues R52D, W54A/D/G, ΔS50-W54 deletion mutant, D99A/R/G, D126R, R191D, K201D, E276R, D298R), confirmed mutant expression, and quantified their interaction via BiFC efficiency28,34 (Fig. 1D–G, S3A,B). PRMT5 mutations R49A/G/D all resulted in suppressed BiFC efficiency (Fig. 1F), indicating decreased interaction between PRMT5 and MEP50. Furthermore, seven MEP50 substitutions (ΔS50-W54, D99A/R/G, D126R, E276R, D298R) also resulted in a significant reduction of the BiFC efficiency (Fig. 1G). Mutations MEP50 D99A/R/G or PRMT5 R49A/G/D resulted in the strongest decrease of PRMT5:MEP50 interaction with over 70% decreased interaction. This is consistent with disruption of a three-residue (MEP50 D99 and PRMT5 R49, in addition to PRMT5 H47) bridge mediated by at least four hydrogen bonds at the PRMT5:MEP50 PPI interface, creating 7 angstrom wide pocket between the bridge and PRMT5 R68 in which MEP50 W54 inserts (Fig. 1C). Further, MEP50 W54 (insertion residue) mutants W54A and W54D resulted in increased variability in BiFC assay, indicating that substitution may make MEP50 marginally more stable, but that MEP50 W54 burial into the PRMT5 TIM barrel pocket is important to mediate PRMT5:MEP50 interaction. Individually, W54 and R52 mutants were unable to decrease binding, but deletion of first β-propeller loop residues S50-W54 reduced interaction by almost 50% (Fig. 1G). Western blotting confirmed that these mutations did not alter the expression and stability of BiFC fusion proteins (Fig. S3). FastContact binding energy prediction also supported live cell BiFC data with MEP50 D99 and PRMT5 R49 contributing significant binding energy to the PPI (Fig. 1H). These encouraging results suggest that targeting the electrostatic interactions at the interface of PRMT5:MEP50 PPI may be achievable. Collectively, in silico prediction of binding energy and in vitro live-cell BiFC analysis with PRMT5 and MEP50 mutants were in agreement and supported development of small molecules targeting PPI, particularly the binding interface via MEP50 W50-W54/D99 and PRMT5 R49.
Figure 1. PRMT5:MEP50 PPI Interface as a Druggable Target.
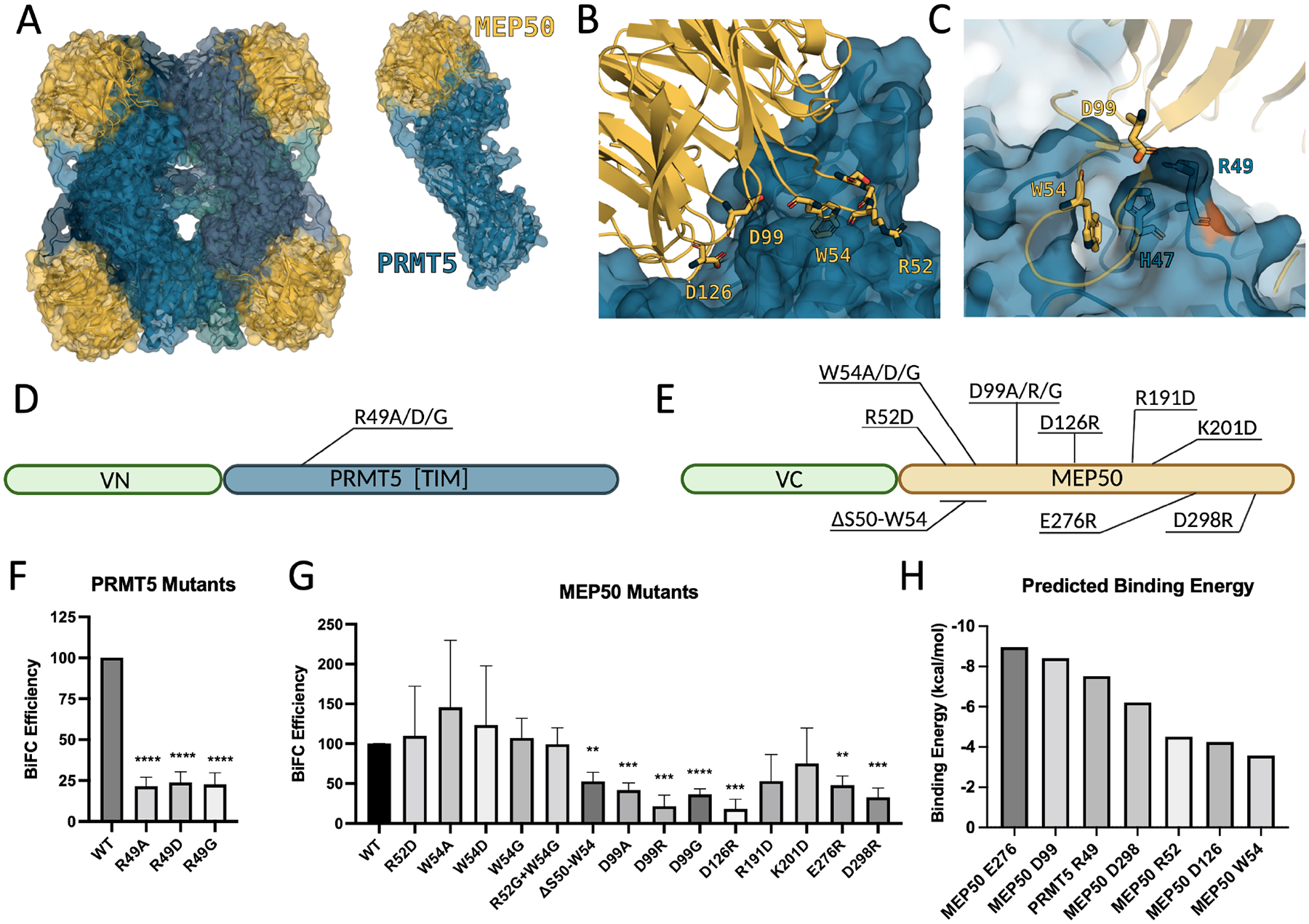
(A) Crystal structure 4GQB of PRMT5:MEP50 showing heterooctameric 4:4 organization (left) as well as single PRMT5:MEP50 heterodimer (right). (B) MEP50 (Shown in cartoon, gold) residues shown are in close interaction with the surface (R52, D99, D126) or buried inside (W54) the TIM barrel of PRMT5 (cartoon with surface, blue). (C) MEP50 (cartoon, gold) residues D99 and W54 occupy 7-angstrom wide pocket in TIM barrel of PRMT5 formed by a ridge consisting of PRMT5 H47 and R49. (D, E) Schematic representation of BiFC constructs and mutations designed to study electrostatic interactions of PRMT5:MEP50 PPI. (VC, venus C-terminus; VN, venus N-terminus). (F) Quantified BiFC efficiency of mutations in PRMT5. (G) Quantified BiFC efficiency of mutations in MEP50. (H) FastContact Binding Energy Prediction of PRMT5 and MEP50 residues used in mutant screen. BiFC efficiency means are average of at least three biological replicates. **p<0.01, ***p<0.001, ****p<0.0001
Identification of Compound 8 as a PPI Inhibitor by Virtual Screen
Next, we performed a virtual screen of close to 30 million small molecules from the ZINC database with the goal of disrupting the PRMT5:MEP50 interaction using ZINCPharmer35. We built a variety of pharmacophore models around the hydrophobic pocket of MEP50 W54 that shows a stacking interaction with PRMT5 H47, as well as addressing hydrogen bonding patterns from crystal waters 811, 840, 935, 985 and 1002 in PDB 4GQB. Compounds that fit the pocket were further minimized using SMINA36, and those that remained in the pocket were selected for testing in vitro (Fig. 2A). We purchased 12 commercially available molecules identified in the SMINA screen and performed BiFC-based screen at 10 μM concentration. Compounds 12, 4, 3, and 10 showed the greatest inhibition at 24–35% (Fig. 2B). However, we noticed that treatment of cells with compound 8 (Cpd 8) at 10 μM resulted in significant cell death, which is suboptimal for the BiFC assay and can prevent successful quantitation, suggesting the use of lower drug concentrations.27,37,38 To address this, we performed a dose-response BiFC assay and identified that compound 8 indeed inhibited the BiFC efficiency in a dose-dependent manner (Fig. 2C). At 0.25 μM, compound 8 inhibited 41% of the BiFC efficiency (Fig. 2D). Thus, we identified compound 8 as a promising hit via initial screen. Molecular docking indicated that compound 8 inhibits interaction of the W54 residue of MEP50 in a small pocket formed by TIM barrel loops 1 (C22-P24) and 2 (P44-H47 and T67-S69), in which the quinoline ring of compound 8 occupies the cavity mediating interaction with MEP50 W54 while the methyl-substituted isoxazole ring is exposed to solvent (in the absence of MEP50), occluding binding of MEP50 W54 into the TIM pocket (Fig. 2E). Additionally, docking suggests: 1) stacking interaction with PRMT5 H47, 2) hydrogen bonding with P44, R68, and I26 mediated by the hydrazide bond, and 3) hydrogen bonding with R49 mediated by the oxygen atom of the isoxazole ring (Fig. 2E, right). Collectively, these data demonstrate successful identification of hit compound 8 due to interaction of the quinoline group with hydrophobic pocket of PRMT5 TIM barrel and solvent-exposed isoxazole ring participating in interaction with PRMT5 R49, together inhibiting MEP50 W54, D99 and PRMT5 R49 from contributing to PPI interaction, supporting further refinement and synthesis of analog compounds.
Figure 2. Identification of Compound 8 as a PPI inhibitor by virtual screen.
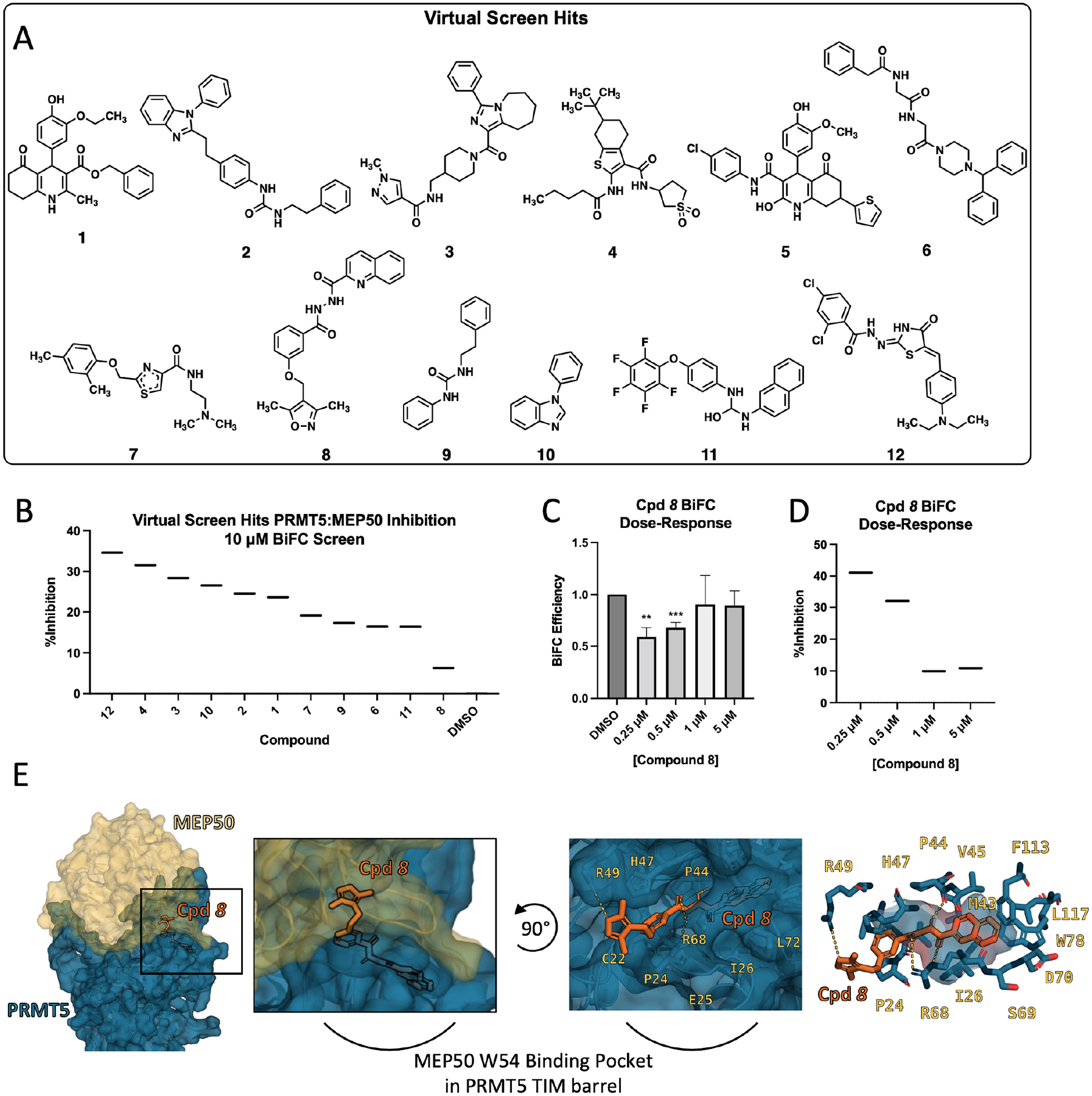
(A) Compounds identified in ZINCPharmer/SMINA virtual screen and utilized in accompanying BiFC-based interaction screen. (B) Ranked-order inhibition in BiFC screen showing percent inhibition of the PRMT5:MEP50 interaction of DMSO control and compounds 1–12 in COS-1 cells following 18-hour treatment. (C) Dose-response of BiFC efficiency showing compound 8 (Cpd 8) inhibition of PRMT5:MEP50 interaction at 0.25 μM − 5 μM doses. (D) Ranked-order inhibition in BiFC screen of compound 8 dose response as depicted in c. Data presented in D and C are mean of three biological replicates but depicted similarly to BiFC screen data to facilitate direct comparison to B. (E) Computational docking of compound Cpd 8 into the TIM barrel of PRMT5 where MEP50 W54 residue normally occupies. Left, PRMT5 (blue) and MEP50 (gold) shown with compound 8 occupying the MEP50 W54 binding pocket in PRMT5 TIM barrel; Middle left, expanded view; Middle right, rotated view with MEP50 removed, compound 8 docking position shows isoxazole ring solvent exposed and hydrogen bonding to PRMT5 R49, R68, and P44 backbone; Right, quinoline ring buried inside MEP50 W54 binding pocket of PRMT5 TIM barrel. BiFC Screens are single replicate. BiFC Efficiency means are average of at least three biological replicates. **p<0.01, ***p<0.001
Synthesis of Compound 8 and Its Analogs
Utilizing the docking data, we hypothesized that either adding bulk to the quinoline group or extending the length of the methyl groups in the oxazole ring may facilitate greater affinity or occlusion of the binding site. We first re-synthesized compound 8 (resynthesized compound is named as 8b; original compound 8 purchased from MolPort is listed as compound 8a) (Scheme 1). Its synthesis started from commercial starting material 27. Reduction of the aldehyde of 27 followed by bromination gave bromide 29 in high yield. Alkylation of phenol 30 with 29 afforded 31, which further reacted with hydrazine to provide hydrazide 32. Acylation of 32 with five different acyl chlorides (33) gave the 8b and four of its analogs (13–16) with structural variation at the original quinoline group. Analogs with extended alkyl chain (17–19) or a phenyl group (20) at the C3 or C5 position of the isoxazole ring to facilitate hydrophobic interactions in the PRMT5 TIM barrel were synthesized as well by following a similar synthetic sequence.
Scheme 1.
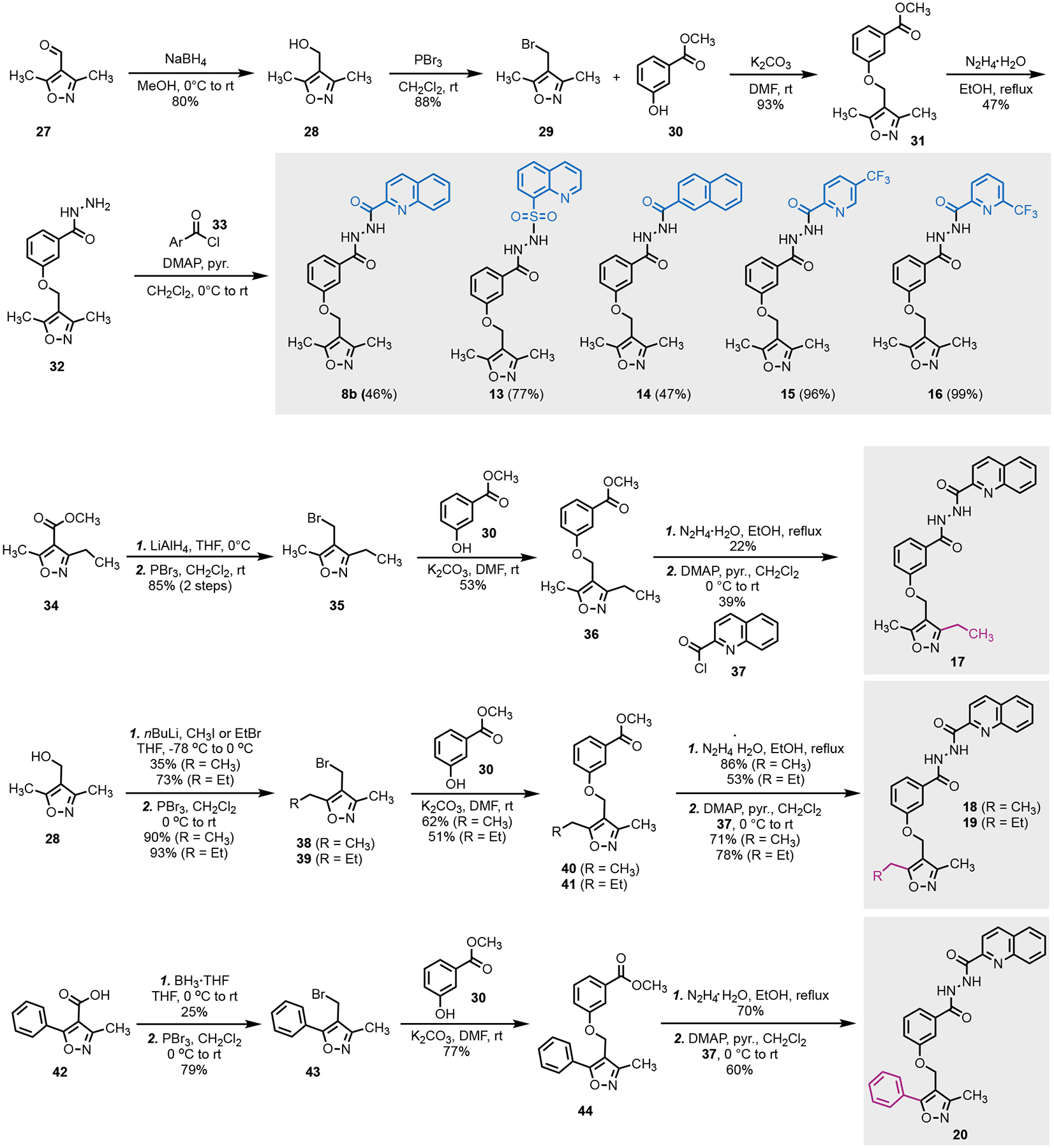
Synthesis of 8b and its hydrazide analogs
Identification of Compound 17 as potent analog of Compound 8
We performed another BiFC screen as described above and observed that the top four inhibitors at 500 nM concentration were 8b, 17, 15, and 8a (Fig. 3A). These results suggested that the bulky groups (trifluorotolyl or quinyl) are desirable for binding and that an ethyl substitution at the C3 position on the isoxazole ring is preferred. With this knowledge, we synthesized a second round of inhibitors by altering the hydrazide linker to imide (24, Scheme 2) or amide with various length (21–23, 25, 26) to avoid potential later downstream PK/PD issues with the hydrazide linker. We then performed third BiFC screen at a lower concentration (250 nM) and determined that compound 17 was still the most effective at inhibiting PRMT5:MEP50 interaction (Fig. 3B). To assess functional PPI inhibition in live cells following treatment with compound 17, we performed co-immunoprecipitation (Co-IP) western blot and identified a 65.4% decrease in amount of MEP50 co-immunoprecipitated with PRMT5 bait across three independent biological replicates (Fig. 3C, D). When molecular docking simulation was performed on compound 17, as with compound 8, both compounds bound to the same pocket of PRMT5, but with the ethyl group of compound 17 extending further into the PRMT5:MEP50 PPI (Fig. 3E). Interestingly, of all compounds synthesized, the most potent compound was highly similar to the initial hit, highlighting importance of the A) hydrogen bond between PRMT5 R49 and oxygen of the isoxazole ring, B) hydrophobic interaction of quinoline group with PRMT5 TIM barrel pocket, and C) electrostatic/hydrogen bonding interactions of the hydrazide linker. Because live cell imaging (BiFC) demonstrated improved potency of compound 17, and Co-IP from cell lysates also suggested successful target engagement, we elected to pursue biological characterization and functional confirmation in vitro utilizing prostate cancer cells due to their dependence on PRMT5:MEP50 and prior validation of PRMT5 as a therapeutic target.
Figure 3. Identification of Compound 17 as potent analog of Compound 8.
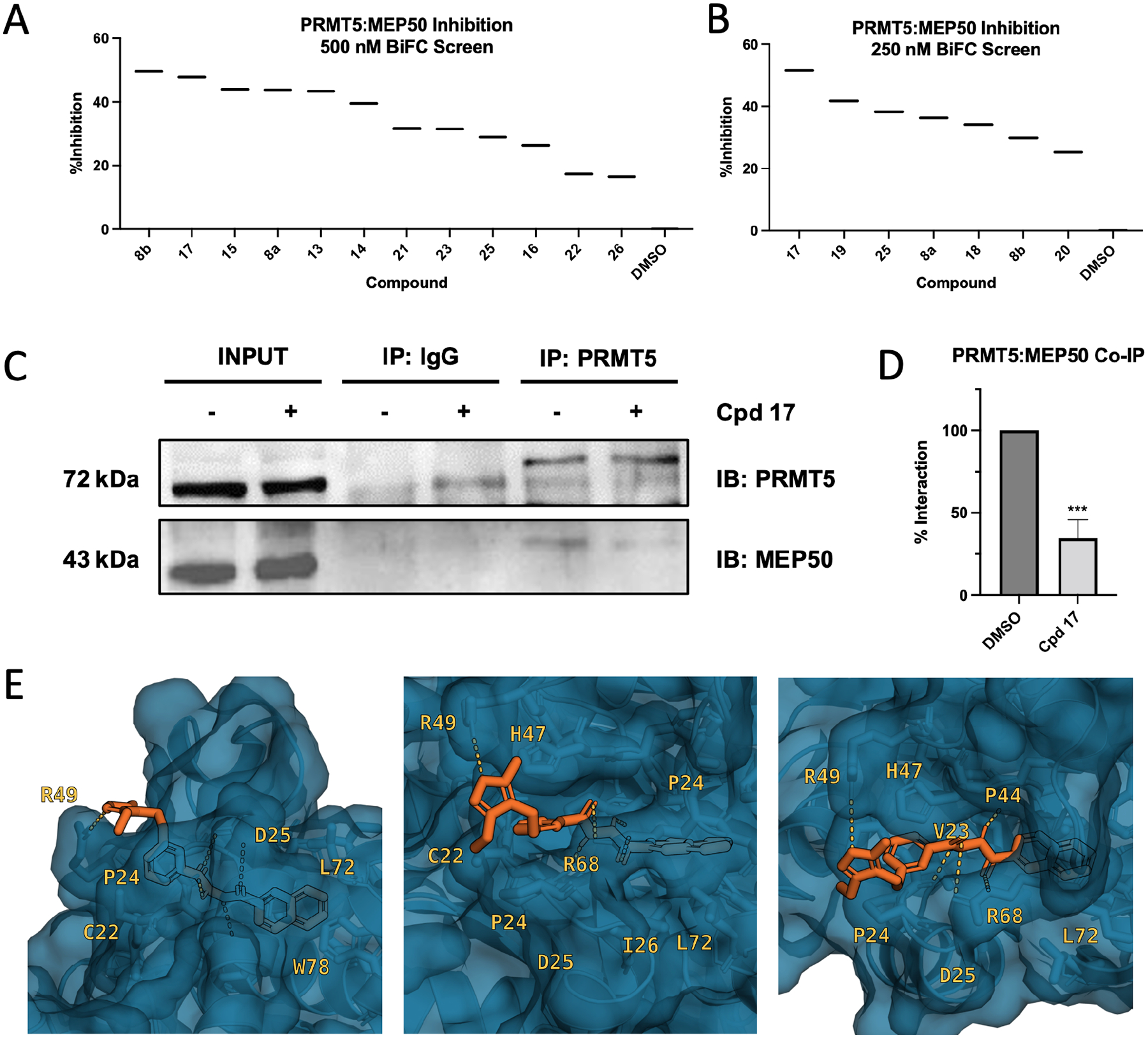
(A) BiFC screen at 500 nM treatment in COS-1 cells indicating %Inhibition of the PRMT5:MEP50 PPI. (B) BiFC screen at 250 nM treatment in COS-1 cells indicating %Inhibition of the PRMT5:MEP50 PPI (C) Western blot of coimmunoprecipitation (Co-IP) of PRMT5 protein in LNCaP cell lysate following treatment with either DMSO or compound 17 representative blot. (D) Integrated density of western blot Co-IP data from C across three biological replicates. (E) Computational docking of compound 8 derivative compound 17 (orange), occupying same binding site along TIM barrel of PRMT5 as MEP50 W54 residue. BiFC screens are single replicate. Co-immunoprecipitation quantitation means are average of three biological replicates. n.s., non-significant, ***p<0.001
Scheme 2.
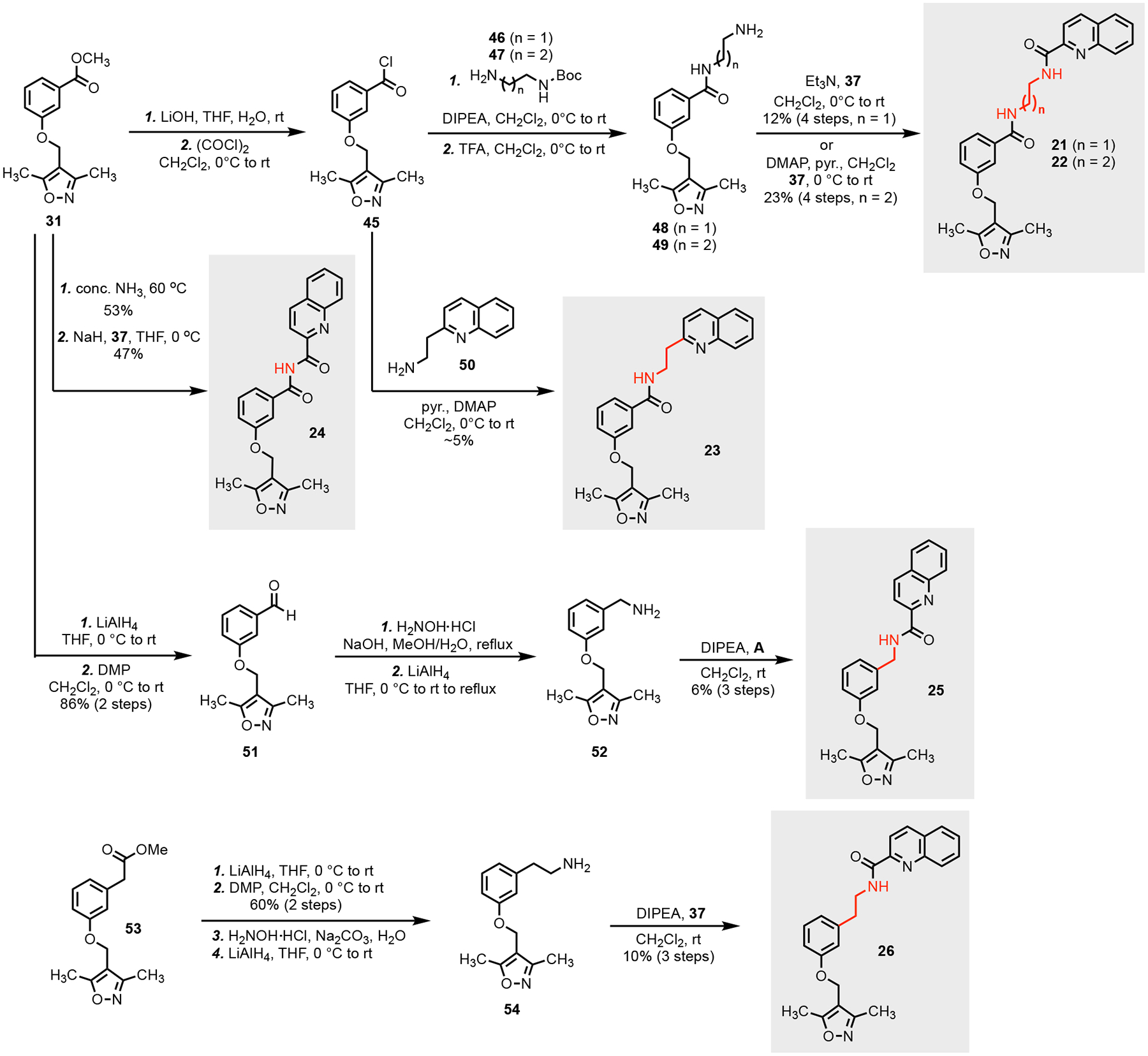
Synthesis of analogs without the hydrazide linker
Compound 17 selectively inhibits PRMT5:MEP50 biological function in prostate cancer cells
To evaluate the biological effects of compound 17 in cells, we utilized prostate cancer cells as an in vitro model system, as we have extensively interrogated the roles of both PRMT5 and MEP50 previously.5,19,20,39 Treatment of hormone naïve LNCaP cells with compound 17 at 250 nM, 500 nM, and 1000 nM for 72 h resulted in both suppression of growth and induction of cell death in a dose dependent manner (Fig. 4A). IC50 of compound 17 was also calculated in LNCaP cells to be 430 nM when treated over 72 h, compared with 1658 nM for parent compound 8b, a roughly 4-fold improved potency (Fig. 4B). As PRMT5:MEP50 are responsible for symmetric dimethylation of arginine 3 residue of Histone H4 (H4R3me2s), we then examined global level of H4R3me2s in LNCaP cells after treatment with compound 17 over 72 h (Fig. 4C). We observed that global H4R3me2s decreased by 65% after treatment with compound 17, and that global H4R3me2s levels decreased in a dose-dependent manner (Fig. 4D). PRMT5:MEP50 occupies the promoter region of the Involucrine (IVL) gene and represses its transcription in LNCaP19 and other40 cells, while PRMT5 and pICln (notably not MEP50) activate AR transcription.20 Thus, the combination of IVL de-repression with unaltered AR expression serve as an ideal model system to evaluate the target engagement and selectivity of compound 17. Toward this end, we treated LNCaP cells with compound 17 for 72 h and performed quantitative reverse transcription polymerase chain reaction (RT-qPCR) to quantify the expression of both IVL and AR. Consistent with target engagement suggested by Co-IP result from LNCaP cells (Fig. 3C), compound 17 significantly de-represses PRMT5:MEP50-regulated IVL gene (as a positive control) without significantly altering expression of PRMT5:pICln-regulated AR gene (as a negative control) in LNCaP cells over 72 h (Fig. 4E/F), supporting the selective effect of compound 17 in cells in inhibition of PRMT5:MEP50 target gene IVL but no inhibition of PRMT5:pICln target gene AR. As it has been reported that PRMT5 regulates multiple tumor suppressor genes, including TP53, PTEN, and RB18,9, we evaluated if treatment with compound 17 de-represses the transcription of these tumor suppressors in prostate cancer cells. Indeed, we observed that compound 17 treatment caused upregulation of both PTEN and RB1 and to some extent TP53, albeit statistically insignificant (Fig. 4G). Loss of histone methylation and de-repression of PRMT5:MEP50 target gene IVL support biologically on-target functional consequence of PRMT5:MEP50 PPI Inhibition. Compound 17 was similarly effective against non-small cell lung cancer (NSCLC) cell line A549 with IC50 447 nM (Fig. S4). Functionally, no change was detected in AR expression, which we have demonstrated is regulated not by PRMT5:MEP50, but by PRMT5:pICln41. Collectively, these biologically functional data suggest that compound 17 is a potent and selective inhibitor of the PRMT5:MEP50 interaction.
Figure 4. Compound 17 selectively inhibits PRMT5:MEP50 biological function in prostate cancer cells.
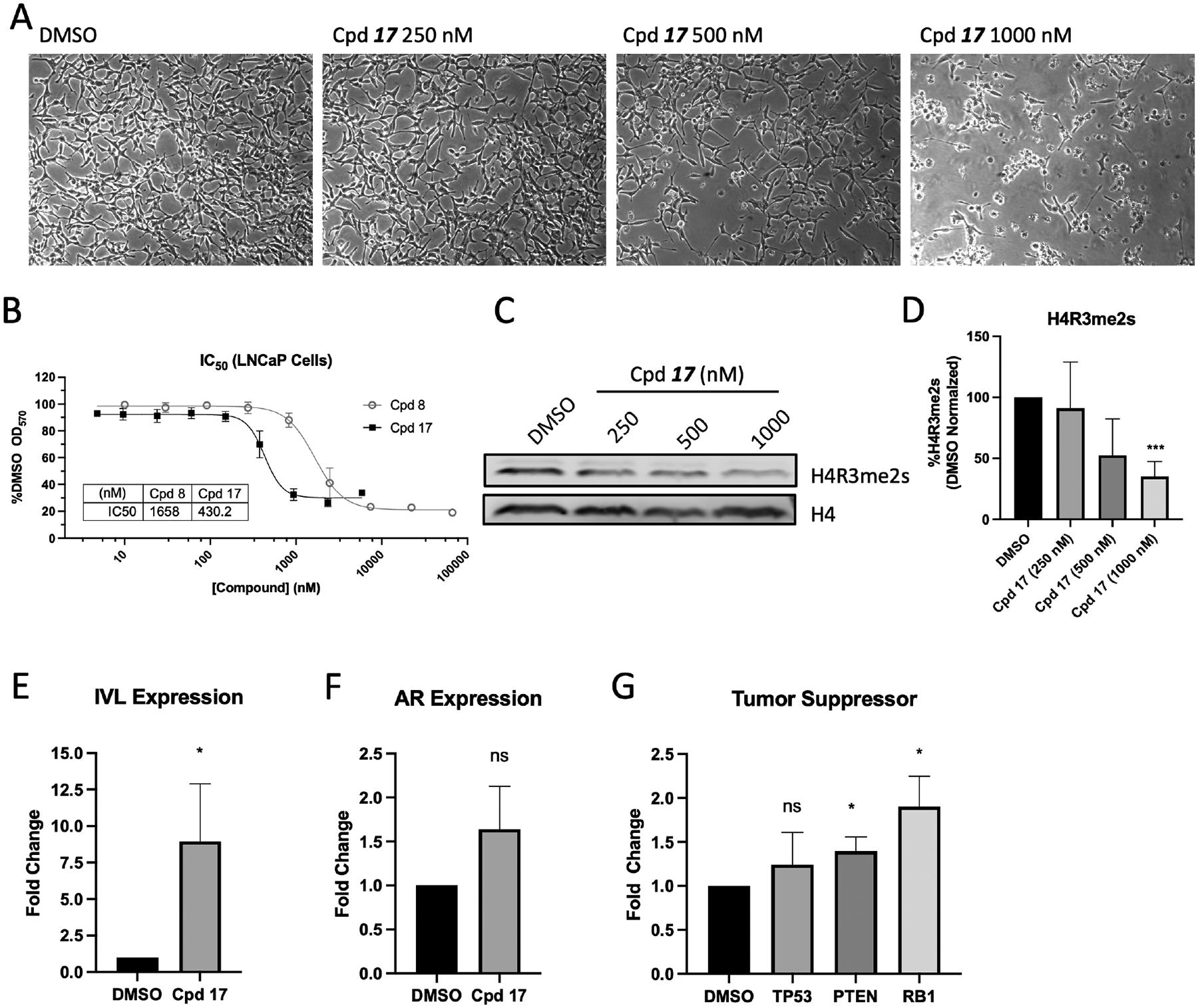
(A) LNCaP cells treated with compound 17 at indicated dose over 72 h period. (B) IC50 measurement of original compound 8b and second-generation derivative compound 17 in LNCaP cells over 72 h treatment. (C) Global histone H4R3 and H4R3me2s western blot from LNCaP cells treated at indicated compound 17 dose over 72 h. (D) Quantified densitometry from bands in C. (E-G) Expression analysis of LNCaP cells treated with 500 nM compound 17 for 72 h followed by RNA isolation and RT-qPCR for PRMT5:MEP50-regulated IVL (E), PRMT5:pICln-regulated AR (F), and tumor suppressor genes TP53, PTEN, and RB1 (G). Means for IC50, western blot, and RT-qPCR are average of three biological replicates. n.s., non-significant, *p<0.05, ***p<0.001
Compound 17 treatment targets PRMT5:MEP50-mediated cellular functions
Because compound 17 targets PRMT5:MEP50 PPI, we hypothesized that we would identify similar dysregulation of genes between PRMT5 knockdown, MEP50 knockdown, and compound 17 treatment. To experimentally address this, we performed RNA-seq in LNCaP cells treated with compound 17 over 72 h. Overall, 1493 differentially expressed genes (DEGs) were identified between compound 17 treatment and DMSO control (Fig. 5A). Consistent with RNA-seq performed on samples with knockdown (KD) of PRMT5 or MEP50 in LNCaP cells, compound 17 treatment did display a small degree of overlap with a core set of up- and down-regulated genes as was observed in PRMT5 or MEP50 KD alone (140 and 112, respectively) (Fig. 5B, C). However, the degree of overlap between DEGs identified in compound 17 treatment versus PRMT5 or MEP50 KD was unexpectedly a minority of the 1493 total genes identified in the compound 17 treatment (911 up- and 582 down-regulated genes). To evaluate the biological consequence of these overlapping genes, we performed a comparative analysis between compound 17 treated LNCaP cells, PRMT5 / MEP50 KD in LNCaP cells, as well as PRMT5 / MEP50 KD in A549 lung cancer cells by analyzing a publicly available dataset in which A549 NSCLC cell lines were also subjected to PRMT5 and MEP50 knockdown followed by RNA-seq.42 After performing differential expression analysis on each of the three datasets, we then performed GO enrichment for the up- or down-regulated genes within each treatment (PRMT5 KD, MEP 50 KD, or compound 17 treatment). As PRMT5:MEP50 mediate multiple pathways in the cell, it was not surprising that certain GO terms were enriched in both up- and down- regulated genes. For this reason, we combined all enriched GO terms agnostic of differential expression directionality and identified enriched terms common to all data sets (compound 17 treatment in LNCaP cells, PRMT5/MEP50 KD in LNCaP cells, and PRMT5/MEP50 KD in A549 cells (Fig. 5D). Broadly, compound 17 treatment and PRMT5/MEP50 knockdown showed commonly enriched pathways in three major pathways significant to the hallmarks of cancer, including differentiation/proliferation, kinase/phosphatase activity, and multiple signaling/survival pathways43 (Fig. 5D top, middle, bottom panels). Significantly, we detected enrichment of TGF-β signaling in all three datasets and wnt signaling shared between compound 17 and PRMT5/MEP50 knockdown in A549 cells. Collectively, we identified 27 enriched GO terms shared between all three datasets and 142 terms shared between compound 17 and at least one other dataset, with all GO terms of Fold Enrichment >2 (Fig. 5D). As complementary approach to GO enrichment, we also utilized gene set enrichment analysis (GSEA) to include all genes identified in the compound 17 RNAseq data set. Predictably, we saw overlap of similar pathways as observed in the GO enrichment, particularly with PRMT5-mediated TP53 signaling, TGF-β signaling, kinase/phosphatase signaling, as well as epithelial cell development/differentiation (Fig. 5E).
Figure 5. Compound 17 treatment targets PRMT5:MEP50-mediated cellular functions.
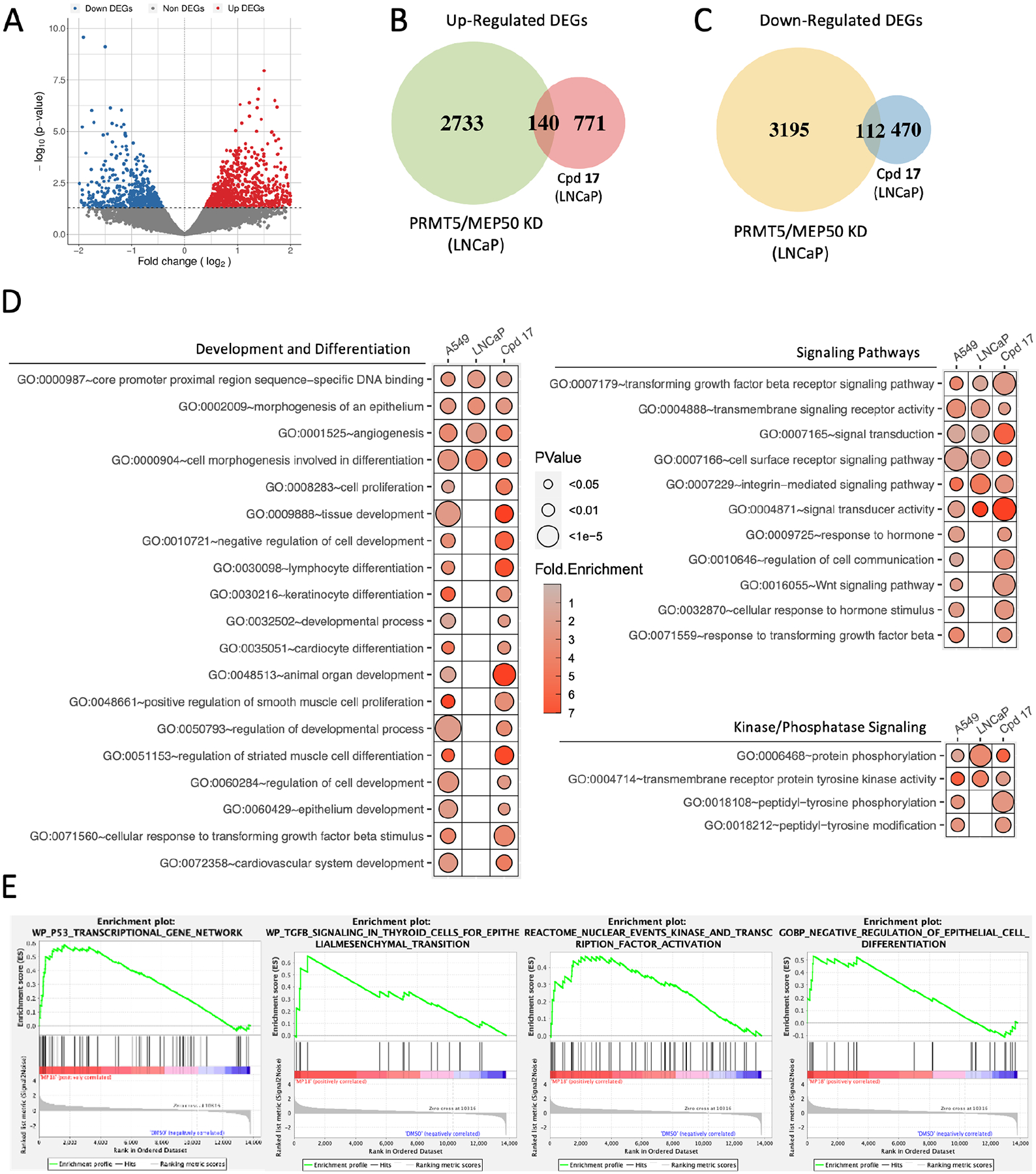
(A) Volcano plot of differentially expressed genes identified in LNCaP cells treated 72 h with compound 17. (B) Venn diagram of up-regulated genes common between either PRMT5 or MEP50 KD in LNCaP cells and compound 17 treatment. (C) Venn diagram of down-regulated genes common between either PRMT5 or MEP50 KD in LNCaP cells and compound 17 treatment. (D) Gene Ontology enrichment of PRMT5-mediated pathways following compound 17 treatment. GO terms shown are shared between compound 17 treatment and one or both of PRMT5/MEP50 knockdown in A549 cells and LNCaP cells. Fold enrichment shown as heat map, P value shown as circle diameter. (E) Gene Set Enrichment Analysis of PRMT5-mediated pathways based on total gene expression in compound 17 compared to DMSO samples.
We utilized a comprehensive approach involving 1) differential expression and GO enrichment of compound 17 treated LNCaP cells, 2) comparative analysis of differential expression across PRMT5/MEP50 KD in LNCaP and A549 cells, and 3) whole-transcriptome analysis utilizing GSEA of compound 17 treated LNCaP cells. Compound 17 treatment resulted in dysregulation of multiple processes in which PRMT5 has been extensively characterized including chromatin structure and epigenetic regulation41,44,45, proliferation/differentiationI4,46–48, MAPK/ERK signaling46,49, and apoptosis/TP53 regulation50,51, and shows significant overlap between PRMT5 and MEP50 knockdown in two independent cell lines. Prostate cancer cell line LNCaP and NCSLC cancer cell line A549 showed similar IC50 < 450 nM (Fig. S4). Most significantly, our data specifically implicated TGF-β signaling present in each of the PRMT5/MEP50 LNCaP knockdown, PRMT5/MEP50 A549 knockdown, and compound 17 LNCaP treatment datasets. Together, these data strongly suggest that A) treatment with compound 17, a PRMT5:MEP50 PPI inhibitor, results in similar biological functional consequence as knockdown of PRMT5 or MEP50 in multiple cell lines, B) compound 17 treatment is able to inhibit multiple PRMT5-regulated pathways critical to the survival and proliferation of lung and prostate cancer cells, and C) PRMT5:MEP50 PPI inhibition via compound 17 may potentially inhibit the TGF-β signaling axis, which has been extensively characterized as a key driver in multiple solid tumor cancers and leukemias/lymphomas. Collectively, our data support further refinement of lead compound 17 as a potential therapeutic inhibitor with specificity to PRMT5:MEP50-regualted targets and biological efficacy in inhibition of multiple hallmark pathways in cancer cells (Fig. 6).
Figure 6. Proposed model for compound 17 targeting of PRMT5:MEP50 and functional consequence.
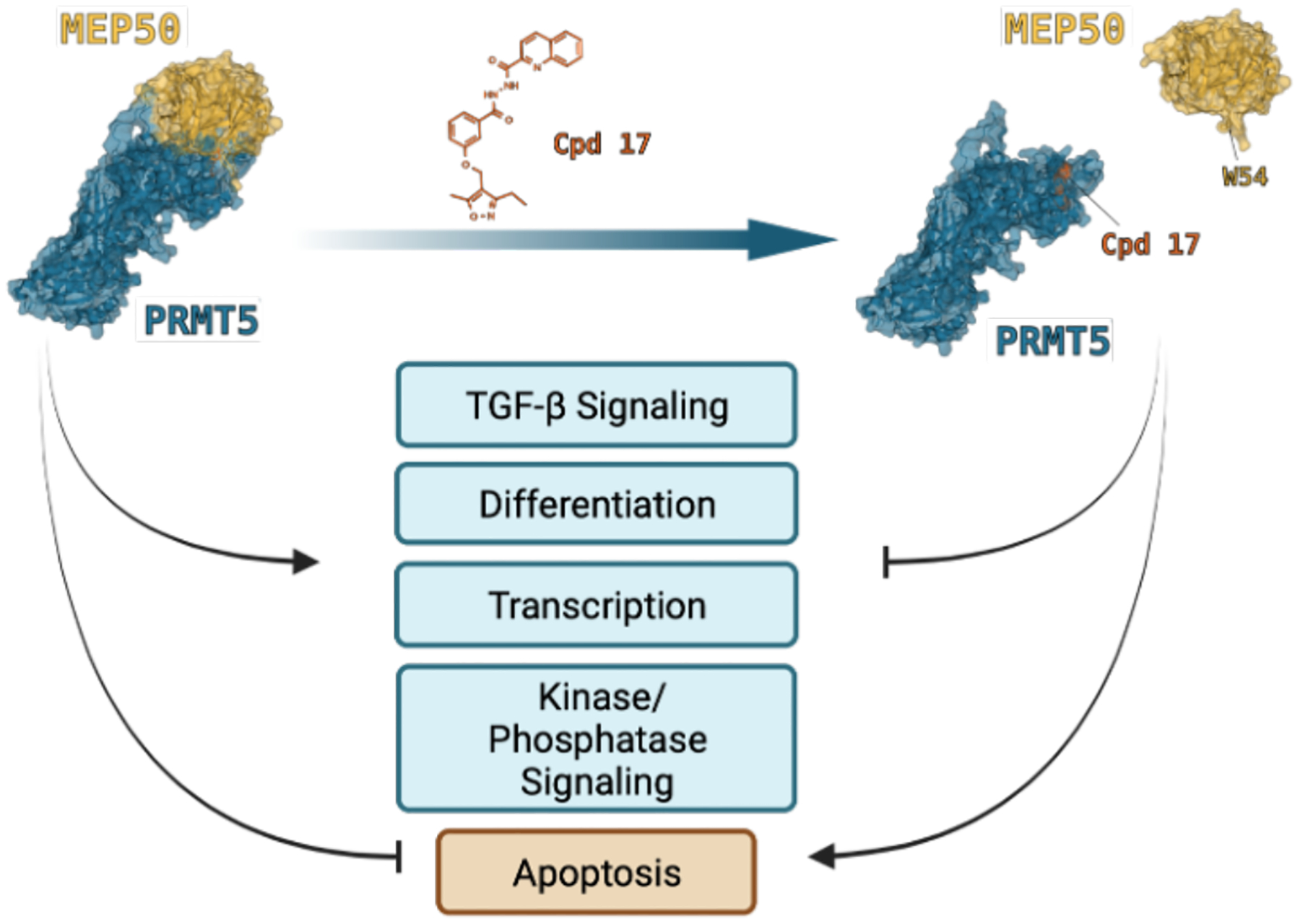
Compound 17 prevents cancer cell proliferation and reduces viability by suppressing differentiation/development, TGF-β signaling, dysregulation of kinase/phosphatase-mediated signaling, transcription, and resulting activation of T53 induced apoptosis. PRMT5:MEP50 PPI interface with PRMT5 (blue) and MEP50 (gold) from PDB 4GQB. MEP50 W54 residue binding to PRMT5 pocket is occluded by presence of compound 17.
Discussion
PRMT5 has been validated as a therapeutic target in multiple cancers with ten active clinical trials at the time of writing (clinicaltrials.gov).2,52 All compounds undergoing active trials are either SAM- or Substrate-competitive inhibitors. Given the multiple roles of PRMT5 in virtually all developing normal cells, the clinical applicability of these PRMT5 inhibitors remains unknown until such clinical trials are complete and adverse effect data becomes available. One recent approach was the development of a proteolysis targeting chimera (PROTAC) molecule targeting PRMT5 via the SAM binding site to the VHL E3 ligase.53 While our manuscript is preparation, Mulvaney et. al., have recently identified a conserved PRMT5 binding motif (PBM) that mediates interaction with PRMT5 cofactors COPR5, RioK1, and pICln and reported the discovery of an inhibitor targeting the interaction of PBM with RioK1.21,22 This inhibitor also appears to be effective in suppressing the growth of MTAP-deleted cancer cells.21 Given the unique cofactor-dependency of PRMT5 among the PRMT family of proteins, we proposed targeting the PPI interface directly between PRMT5 and MEP50 by occluding the MEP50 W54 binding pocket in the PRMT5 TIM barrel. Our virtual screen and BiFC screens led to the identification of compound 8 as an initial hit. Further synthesis and screening of additional analogs resulted in the identification of compound 17 with almost 4-fold improvement in potency based on IC50 in LNCaP cells. Significantly, we provided several pieces of evidence supporting that compound 17 is specific and on-target. Firstly, compound 17 decreased global histone H4R3me2s, an epigenetic mark mediated by PRMT5:MEP50. Second, treatment with compound 17 resulted in decreased repression of IVL gene normally repressed by PRMT5:MEP50, especially in non-keratinocyte cell types40, without affecting the expression of AR, which is regulated by PRMT5:pICln instead20. Lastly, we utilized Co-IP to demonstrated decreased binding of endogenous MEP50 to endogenous PRMT5 in LNCaP cell lysate. Thus, compound 17 represents a novel class of PRMT5:MEP50 inhibitors that merits further development based on the high level of target specificity.
PRMT5 has been extensively investigated in multiple human cancers.4–6,10,54 Overexpression of PRMT5 correlates with disease progression, therapeutic resistance, and poor survival55. However, few studies have evaluated the role of PRMT5 cofactors or adaptors including MEP50. Our findings that PRMT5 cooperates with pICln, but not MEP50, to activate transcription of AR and DDR genes in prostate cancer cells provide evidence that transcriptional regulation of PRMT5 target gene expression is likely dependent on the cofactors involved and potentially context-dependent19,20. Indeed, we have also demonstrated that during the course of fractionated ionizing radiation (FIR)-induced NED, PRMT5:MEP50 mediates FIR-induced neuroendocrine differentiation (NED) and that knockdown of PRMT5 significantly increased the sensitivity of LNCaP xenograft tumors to FIR, reduced tumor recurrence, and improved overall survival.19 Thus, identification of compound 17 as a lead compound for future development may offer several opportunities for clinical implications. As NED is associated with therapeutic resistance and contributes to the development of neuroendocrine prostate cancer (NEPC)56, targeting PRMT5:MEP50 could be used to prevent treatment-induced neuroendocrine NED or even NEPC. Future development of the lead compound 17 will offer a unique opportunity for further evaluation of the inhibitor in FIR-induced NED in vitro and in vivo and potentially NEPC treatment.
Because PRMT5 can repress transcription of PTEN and RBL2 in leukemia/lymphoma cell lines7,8, it would be interesting to validate the role of PRMT5:MEP50 PPI in regulation of PTEN and RB1 (or RB family members) in these human cancers. If confirmed, targeting PRMT5:MEP50 PPI with compound 17 or future analogs could be utilized for leukemia/lymphoma treatment or as a sensitizer for other therapies by activating PTEN/RB-family in conjunction with other disease-specific targeted therapy. As PTEN is deficient in multiple cancers and PTEN negatively regulates the PI3K-AKT-mTOR pathway, targeting PRMT5:MEP50 under specific contexts may allow indirect re-activation of PTEN and deactivation of mTOR signaling as an indirect alternative to targeting PTEN/mTOR signaling, known to be therapeutically challenging.57–59
PRMT5 also plays a critical role in RNA splicing by forming a complex with MEP50 and pICln to catalyze the methylation of Sm proteins and to facilitate the assembly of spliceosome for both normal and cancer cells.60–63 PRMT5 regulates splicing in both hematopoietic and neuronal stem/progenitor cells; recently, in a panel of patient-derived glioblastoma cell lines, inhibition of PRMT5-mediated alternative splicing was found to impair proliferation, induce senescence, and trigger apoptosis.60,61,63 It is plausible that compound 17 or its future analogs could be used to treat multiple alternative-splicing driven diseases or progression stages (AR reactivation via AR-V7 in prostate cancer, TAK1/CD44 alternative splicing in EMT, or PTPMT1-mediated radioresistance in lung cancer).64–67 As the field continues to evolve, distinct cellular roles of PRMT5:MEP50 will continue to be uncovered, providing specific disease/context dependencies and mechanisms for patient stratification. We anticipate that targeting the PRMT5/MEP50 interaction with future analogs of compound 17 with improved pharmacokinetics and potency may be explored for treatment of various human diseases at different stages or processes that are dependent on the formation of the PRMT5/MEP50 complex.
RNA-seq identified significant dysregulation of TP53 signaling pathway, cellular proliferation/differentiation, and MAP Kinase signaling, each of which is a core function of PRMT5 activity in normal and cancer cells. Interestingly, only a small subset of genes was identified when compared to RNA-seq data from PRMT5 or MEP50 knockdown, suggesting a narrow scope of mechanism of action for therapeutic compounds targeting PRMT5:MEP50 PPI. Such a narrow scope may in fact provide an added layer of specificity and selectivity for future therapeutic approaches. Further, unbiased approaches such as ChIP-seq targeting PRMT5 and MEP50 with and without compound 17 or future analogs would help to identify PRMT5:MEP50-specific target genes, which may facilitate patient selection in the clinical setting.
Methylation of histone and non-histone substrates is a critical mediator of normal cell development and fate determination in differentiation as well as cancer cell proliferation and therapy resistance, necessitating clear delineation of therapeutic window and context-specific targeting strategies. PRMT5-mediated epigenetic activation/repression, alternative splicing, and PTEN/TP53 methylation, and growth factor (e.g., TGFβ / FGFR / EGFR) coactivation are all cancer cell dependencies that may be exploited via PRMT5-targeting therapies. In prostate cancer specifically, we have demonstrated PRMT5:MEP50 has separate and distinct roles compared to PRMT5:pICln19,39, and it is a logical progression that more research will uncover additional cofactor-specific roles. We realize that compound 17 is not an ideal lead compound for in vivo study or clinical development, due mostly to low solubility (predicted LogP 3.06, Molinspiration Software68) potentially hindering bioavailability or distribution as well as the presence of a hydrazide linker, which may present challenges for toxicity in vivo. Additional work is needed to improve potency and pharmacokinetic/pharmacodynamic properties, but we feel that we have demonstrated successful development of a chemical probe targeting the PRMT5:MEP50 PPI, and that this class of compounds could provide the foundation for potent and selective therapeutic compounds in the future.
EXPERIMENTAL SECTION
Computational Modeling, Docking, and Binding Energy Prediction.
Virtual screening35 was conducted with ZINCPharmer35. Refinement was performed (list of compounds was minimized) using SMINA36. Binding energies were predicted via web server for FastContact33.
Cell Lines and Cell Culture.
LNCaP, COS-1, and A549 cell lines were purchased from ATCC. Routine mycoplasma screening was performed as described previously by Owens et. al. using the LookOut PCR Mycoplasma Detection Kit (Sigma)19. Cells were stored as frozen stock in vapor phase of LN2 and thawed prior to use. Cell lines were cultured 3 passages after thawing prior to experimentation and maintained for no longer than 30 total passages. LNCaP cells were cultured in RPMI 1640 (Corning), and COS-1 cells were cultured in DMEM (Corning) medium. A549 cells were cultured in Hink’s F12K Medium (Corning). All media were supplemented with 10% FBS (Atlanta Biologicals), 1 mM sodium pyruvate (Corning), penicillin (100 units/mL) and streptomycin (100 μg/mL) combination (Gibco), and 2 mM/L L-glutamine (Corning). Knockdown cell lines were generated using the pLKO-Tet-On system. The pLKO-Tet-On plasmid for shRNA expression was obtained from Addgene69, and shRNA sequences that target PRMT5 (5′- CCCATCCTCTTCCCTATTAAG-3′: referring to #1832), SC (5′- CAACAAGATGAAGAGCACCAA-3′), MEP50 (5′- CCTCACAAGGACTCTGTGTTT-3′) were utilized as described previously for stable cell line generation41.
For dox-induced PRMT5, MEP50, or scrambled control knockdown cell lines, doxycycline was applied at the final concentration of 1 μg/mL every 48 h to establish and maintain PRMT5 knockdown (shPRMT5), MEP50 knockdown (shMEP50), or express scramble control shRNA (shSC). Cells were harvested in Trizol and RNA was purified for RNAseq (Ambion) following methodology described previously39.
BiFC Assay and Screening.
BiFC Mutation Assay:
COS-1 cells were grown in DMEM and seeded to 100,000 cells / well of a 12-well plate and allowed to attach for 24 h. Cells were transfected with 400 ng/well of pMyc-VN-PRMT5 (WT or mutant) BiFC plasmid, 400 ng/well pHA-VC-MEP50 (WT or mutant) BiFC plasmid, and 200 ng/well pHA-Cerulean transfection control. Following 18 h after transfection, the cells were imaged on a Nikon TE-2000U microscope and images for CFP, YFP, and phase contrast were acquired using MetaMorph software (Nikon) with 10X objective. Images were analyzed with ImageJ70. Regions of Interest (ROI) were selected around each cell, and mean intensity was measured for each selection. A YFP:CFP ratio was calculated for DMSO as well as control treatment cells. The YFP:CFP ratio was then normalized to that of DMSO to generate the BiFC Efficiency score. All BiFC mutant experiments are performed as three biological replicates. To ensure comparable expression of BiFC plasmids, cells were subsequently washed with PBS and harvested in 100 μL of 2X SDS sample buffer and analyzed via western blot. Anti-HA antibody was used to detect MEP50 fusions and Cerulean expression. Anti-Myc antibody was used to detect PRMT5 fusions.
BiFC Drug Screens:
COS-1 cells were grown in DMEM and seeded to 50,000 cells/well of a 12-well plate and allowed to attach for 24 h. Cells were then transfected with three plasmids pMyc-VN155-PRMT5, pHA-VC-MEP50, and pFLAG-NLS-CFP in order to visualize the interaction between PRMT5:MEP50. For BiFC screens, the COS-1 cells were treated with compound or DMSO to final concentration (10 μM for compound 1 – 12 screen and 0.25, 0.50, 0.75, 1.0, and 5.0 μM or subsequent compound 8 screen) six hours after transfection and returned to the incubator. Following 24 hours after transfection, cells were imaged on a Nikon TE-2000U microscope and images for CFP, YFP, and phase contrast were acquired using MetaMorph software (Nikon) with 20X objective. Images were analyzed with ImageJ70. Regions of Interest (ROI) were selected around each cell, and mean intensity was measured for each selection. A YFP:CFP ratio was calculated for DMSO as well as control treatment cells. The YFP:CFP ratio was then normalized to that of DMSO to generate the BiFC Efficiency score. The lower score indicated less PRMT5:MEP50 PPI detected in a given cell or treatment group. Inhibition (% Inhibition) is calculated as a percent of 100% - the BiFC Efficiency. For the BiFC screen of compounds 13 – 22 (including 8a and 8b), COS-1 cells were transfected for 24 h, treated with 0.5 μM of compounds for 18 h, and then imaged as described above. For the BiFC screen of compounds 23 – 27 (including 8a and 8b), COS-1 cells were transfected for 24 h, treated with 0.25 μM of compounds for 18 h, and then imaged as described above. BiFC drug screens were performed in single biological replicate as a high throughput screen to produce ranked order of compounds, although multiple biological replicate data was used whenever available.
MTT Assay.
LNCaP cells were seeded at 7,000 cells per well of a 96-well cell culture plate and incubated 24 h to allow for attachment. Test compounds were diluted in RPMI-1640/25% DMSO pre-dilutions and added to respective wells of the assay plate to maintain constant concentration of 0.25% DMSO. The treatment concentration range for each compound was designed as a DMSO control plus an 11-point curve such that the top two concentrations killed all cells following the 72 hour treatment window to ensure regression curve had acceptable asymptote readings. After addition of compounds, cells were returned to incubator (37 °C, 5% CO2) for 72 h. Following incubation, assay plates were removed from incubator and media aspirated. 30 μL complete RPMI supplemented with 0.5 mg/mL MTT (Sigma) was added to the plate, and plate returned to incubator for 4 h. Plates were removed, and 88 μL DMSO was added. Plates were shaken at 700 rpm for 1 minute and read on spectrophotometer at 570 nm.
RT-qPCR Assay.
LNCaP cells were seeded to either 6 cm or 10 cm dishes at 800,000 or 2,200,000 cells/dish respectively. Cells were allowed to attach for 24 h and then subsequently treated with either compound 17 (500 nM) or DMSO for 72 h. Cells were then harvested with Trizol reagent (Ambion) and RNA integrity was verified via agarose gel electrophoresis. Promega High Capacity cDNA Reverse Transcription Kit (Promega) was utilized following manufacturer instructions and as described previously41,71,72. RT-qPCR was performed with FastStart Universal SYBR Green Master Mix (Thermo Fisher Scientific) and detected on a QuantStudio 6 Flex with QuantStudio Real-Time PCR control software (Thermo Fisher Scientific). QuantStudio Design and Analysis software (Thermo Fisher Scientific) was used for data analysis. Technical triplicates were run for all samples, samples without detectable amplification were deemed undetected. Primer sets were validated via melt curve and agarose gel analysis of RT-qPCR product. AR primers were used as described previousy41 and IVL primers were used as described previously.40 All primer sequences utilized are described in Supplementary Information.
Co-Immunoprecipitation.
LNCaP cells were treated with either DMSO or compound 17 (500 nM, 24hrs) and harvested for Co-IP and WB in lysis buffer composed of 50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 10 mM EDTA, 0.1% Triton X-100, 1 mM DTT, 1 mM PMSF, and 5 μg each of chymostatin, leupeptin, pepstatin A, and antipan. Cells were lysed over 1 hour rotating in 4 °C and supernatant cleared. Total lysate (1 mg/mL) was used for Co-IP with 4 μg rabbit anti-PRMT5 pAb (Millipore Sigma 07–405), rabbit anti-MEP50 pAb (Cell Signaling Technologies S2823S) or normal rabbit IgG (Millipore Sigma N01–100UG) overnight. Antibody-bound proteins were precipitated with Pierce Protein A agarose beads (Thermo Scientific 20333). Antibodies and immunoprecipitated proteins were prepared for western blot by adding 50 μL 2X SDS buffer, boiling at 95 °C for 5 min, and storing at −80 °C or proceeding to western blot.
Western Blot Assay.
Co-IP product, input sample, or ladder were loaded into a 10% acrylamide/bisacrylamide gel (20 μL Co-IP, 20 μL input (0.4% total), 5 μL ladder per lane). Gel was run 90 min @ 125 V and transferred onto a nitrocellulose membrane for 75 min at 100 V. The membrane was washed and incubated with either anti-PRMT5 rabbit pAb (1:1000 in phosphate buffered saline, pH 7.4, supplemented with Tween-20 (PBST), Millipore 07–405) or anti-MEP50 mouse mAb (1:1000 in PBST, Invitrogen MA5–32970). Secondary anti-rabbit IgG-HRP conjugate (1:1000 in PBST, GE Healthcare) or anti-mouse IgG-HRP conjugate (1:1000 in PBST, GE Healthcare) was used to provide signal for the blot which was subsequently imaged on a Bio-Rad ChemiDoc Touch Imaging System (Bio-Rad). Band Intensity was determined with ImageLab software and ImageJ.
For global histone H4R3 and H4R3me2s western blots, cell lysate was prepared in RIPA buffer (10 mM Tris-HCl pH 8.0, 5 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 150 mM sodium chloride, and 5 μg/mL each chymostatin, leupeptin, pepstatin A, and antipan in DMSO, with 1 mM PMSF and total soluble protein was quantified using Bradford assay. 100 μL lysate was combined with 100 μL 2X SDS buffer, and 20 μg total lysate was loaded into the wells of a 15% acrylamide/bisacrylamide gel. The gel was run for 60 min at 125 V and transferred onto nitrocellulose membrane for 45 min at 100 V. Anti-H4R3 rabbit pAb (Abcam, ab10158) or anti-H4R3me2s rabbit pAb (Abcam, ab5823) was diluted 1:1000 in PBST and incubated overnight at 4 °C. Secondary IRDye® 800CW Donkey anti-Rabbit IgG Secondary Antibody (Li-Cor, 926–32213) was diluted 1:20,000 in PBST and incubated with the membrane, which was then imaged via LiCor Odyssey CLx imager and analyzed with ImageStudioLite software (Li-Cor). Integrated intensity of H4R3me2s band was normalized to H4R3 to determine relative abundance of H4R3me2s across DMSO and Cpd 17-treated samples.
For confirmation of BiFC plasmid expression, 100 μL 2X SDS buffer was loaded into the wells of the 24-well plated used for the BiFC screen and harvested. To the lanes of a 10% SDS-PAGE gel, 20 μL lysate was loaded. The gel was run for 60 min at 125 V and transferred onto nitrocellulose membrane for 75 min at 100 V. The membrane was incubated with either anti-HA tag antibody (Sigma-Aldrich, H3663) for detection of HA-fusion Cerulean protein and HA-fused MEP50 (wild type or mutants) or anti-Myc tag antibody (Abcam, Clone 9E10, ab32) for detection of Myc-fusion PRMT5 (wild type or mutants) at 1:1000 dilution in PBST. Secondary antibody IRDye® 800CW Donkey anti-Mouse IgG (LI-COR, 926–32212) was used for detection, and membranes were read on LI-COR Odyssey imager.
RNA Sequencing Analysis
The reads were mapped to the human genome hg38 using STAR (v2.7.2a)73. RNA-seq aligner with the following parameter: “--outSAMmapqUnique 60”. Uniquely mapped sequencing reads were assigned to GENCODE 31 gene using featureCounts (v2.0.1)74 with the following parameters: “–p –Q 10 -O”. The data was filtered using read count > 10 in at least 3 of the samples, normalized using TMM (trimmed mean of M values) method and subjected to differential expression analysis using edgeR (v3.34.1)75,76. Gene ontology and KEGG pathway functional analysis was performed on differential expression gene with p value cut-off of 0.05 using DAVID.77,78
Chemical Synthesis
General Methods.
NMR spectra were recorded on Bruker spectrometers (1H at 400 MHz, 500 MHz, 800 MHz and 13C at 100 MHz, 125 MHz, 200 MHz). Chemical shifts (δ) were given in ppm with reference to solvent signals [1H NMR: CHCl3 (7.26); 13C NMR: CDCl3 (77.2), C6D6 (128.02), CD3OD (49.0)]. Column chromatography was performed on silica gel. All reactions sensitive to air or moisture were conducted under argon atmosphere in dry and freshly distilled solvents under anhydrous conditions, unless otherwise noted. Anhydrous THF and toluene were distilled over sodium benzophenone ketyl under Argon. Anhydrous CH2Cl2 was distilled over calcium hydride under Argon. All other solvents and reagents were used as obtained from commercial sources without further purification. All compounds tested in the biological assays are >95% purity based on NMR analysis (Supplemental Information) or HPLC analysis (Figure S5).
(3,5-dimethylisoxazol-4-yl)methanol (28): To a 0 °C solution of 3,5-dimethyl-1,2-oxazole-4-carbaldehyde (1.0 g, 8.0 mmol) in anhydrous methanol (60 mL) was added sodium borohydride (450 mg, 12.0 mmol). The reaction mixture was stirred at room temperature overnight. Methanol was evaporated and water (50 mL) was added. The resultant mixture was extracted with EtOAc (3 × 50 mL). The organic extracts were combined, dried over Na2SO4, filtered, evaporated, and subjected to the flash column chromatography to afford 28 (817 mg, 80% yield) as white solid. 1H NMR (500 MHz, CDCl3) δ = 4.38 (s, 2H), 2.32 (s, 3H), 2.21 (s, 3H); 13C NMR (125 MHz, CDCl3) δ = 166.7, 159.8, 113.8, 53.4, 10.9, 9.9. MS (ESI): m/z 128.1 calc. for C6H10NO2+ [M+H]+, found 128.2.
Methyl 3-((3,5-dimethylisoxazol-4-yl)methoxy)benzoate (31): To a solution of 28 (540 mg, 4.2 mmol) in anhydrous CH2Cl2 (42 mL) was added dropwise phosphorus tribromide (1.3 mL, 12.7 mmol). The mixture was stirred at room temperature for 3 h. Water (50 mL) was added. The resultant mixture was extracted with CH2Cl2 (3 × 50 mL). The organic extracts were combined, dried over MgSO4, filtered, evaporated, and the residue was dried in vacuo, affording crude 29 for the next step without further purification.
To a stirred solution of 3-phenolic methyl ester 30 (571 mg, 3.8 mmol) in DMF (26 mL) at room temperature, was added potassium carbonate (1 g, 7.5 mmol) followed by 29 (710 mg, 3.8 mmol). The reaction mixture was stirred overnight. The mixture was filtered over a celite pad and washed with EtOAc (5 × 60 mL). The organic extracts were combined, dried over MgSO4, filtered, and evaporated. The crude residue was purified by flash column chromatography to afford 31 (900 mg, 93% yield). 1H NMR (500 MHz, CDCl3) δ 7.68 (dt, J = 7.8, 1.2 Hz, 1H), 7.61 (dd, J = 2.7, 1.5 Hz, 1H), 7.36 (t, J = 7.9 Hz, 1H), 7.12 (ddd, J = 8.2, 2.7, 1.0 Hz, 1H), 4.84 (s, 2H), 3.92 (s, 3H), 2.42 (s, 3H), 2.30 (s, 3H); 13C NMR (125 MHz, CDCl3) δ = 167.7, 166.8, 159.8, 158.2, 131.6, 129.6, 122.7, 120.5, 114.6, 110.0, 59.7, 52.3, 11.2, 10.2. MS (ESI): m/z 262.1 calc. for C14H16NO4+ [M+H]+, found 262.4.
3-((3,5-dimethylisoxazol-4-yl)methoxy)benzohydrazide (32): A solution of hydrazine hydrate (80%, 2.09 mL, 35 mmol) was added dropwise to a solution of 31 (2.3 mmol) in EtOH (12 mL). The reaction mixture was refluxed for 12 h until completion. After cooling, water (10 mL) was added, and the precipitate was filtered and washed with a small amount of ethanol and water. The crude product was subjected to the next step without further purification.
A general procedure for the synthesis of 8b, 13–20: Acyl chloride (0.12 mmol) in CH2Cl2 (1.0 mL) was added dropwise to a dried round flask containing the corresponding benzohydrazide (0.11 mmol), pyridine (44 μL, 0.55 mmol), and DMAP (1.5 mg, 0.01 mmol) in CH2Cl2 (1.0 mL) at 0 °C. The mixture was stirred at room temperature for 12 h and then washed with dilute aqueous HCl and water and dried over Na2SO4. After removal of the solvent at reduced pressure, the crude product was purified by flash column chromatography to obtain the desired product.
N′-(3-((3,5-dimethylisoxazol-4-yl)methoxy)benzoyl)quinoline-2-carbohydrazide (8b, 46%): 1H NMR (500 MHz, CDCl3) δ 10.70 (s, 1H), 10.07 (s, 1H), 8.31 – 8.25 (m, 1H), 8.19 (d, J = 8.4 Hz, 1H), 8.12 (dd, J = 8.4, 1.2 Hz, 1H), 7.86 (dd, J = 8.2, 1.4 Hz, 1H), 7.78 (ddd, J = 8.4, 6.9, 1.4 Hz, 1H), 7.64 (ddd, J = 8.1, 6.8, 1.2 Hz, 1H), 7.59 – 7.48 (m, 2H), 7.31 (t, J = 7.9 Hz, 1H), 7.05 (ddd, J = 8.3, 2.6, 0.9 Hz, 1H), 4.77 (s, 2H), 2.36 (s, 3H), 2.25 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 167.7, 164.5, 162.0, 159.8, 158.6, 147.8, 146.6, 137.6, 132.9, 130.5, 130.0, 129.9, 129.6, 128.5, 127.7, 120.2, 119.9, 118.6, 112.6, 110.0, 59.6, 11.1, 10.1. HRMS (ESI): m/z 417.1557 calc. for C23H21N4O4+ [M+H]+, found 417.1561.
N′-(3-((3,5-dimethylisoxazol-4-yl)methoxy)benzoyl)quinoline-8-sulfonohydrazide (13, 77%): 1H NMR (500 MHz, CDCl3) δ 9.39 (s, 1H), 9.18 (dd, J = 4.3, 1.7 Hz, 1H), 8.39 (dd, J = 7.3, 1.4 Hz, 1H), 8.30 (dd, J = 8.3, 1.8 Hz, 2H), 8.09 (dd, J = 8.2, 1.4 Hz, 1H), 7.65 – 7.58 (m, 2H), 7.30 (t, J = 7.9 Hz, 1H), 7.20 (d, J = 7.7 Hz, 1H), 7.15 (s, 1H), 7.04 (ddd, J = 8.3, 2.7, 1.0 Hz, 1H), 4.72 (s, 2H), 2.35 (s, 3H), 2.23 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 167.6, 165.0, 159.6, 158.6, 151.3, 143.8, 136.9, 136.0, 134.2, 132.6, 131.2, 130.0, 128.8, 125.3, 122.6, 119.7, 119.6, 113.0, 109.8, 59.6, 11.1, 10.1. HRMS (ESI): m/z 453.1227 calc. for C22H21N4O5S+ [M+H]+, found 453.1222.
N′-(3-((3,5-dimethylisoxazol-4-yl)methoxy)benzoyl)-2-naphthohydrazide (14, 47%): 1H NMR (500 MHz, CDCl3) δ 9.64 (q, J = 6.1 Hz, 2H), 8.42 (d, J = 1.4 Hz, 1H), 7.93 – 7.86 (m, 4H), 7.58 (m, 2H), 7.52 – 7.47 (m, 2H), 7.37 (t, J = 7.9 Hz, 1H), 7.13 – 7.09 (m, 1H), 4.80 (s, 2H), 2.40 (s, 3H), 2.28 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 167.7, 164.4, 163.9, 159.7, 158.7, 135.2, 132.8, 132.5, 130.1, 129.1, 128.8, 128.3, 127.8, 127.1, 123.2, 119.8, 119.8, 112.9, 109.9, 59.7, 11.2, 10.2. HRMS (ESI): m/z 416.1605 calc. for C24H22N3O4+ [M+H]+, found 416.1610.
N′-(3-((3,5-dimethylisoxazol-4-yl)methoxy)benzoyl)-5-(trifluoromethyl)picolinohydrazide (15, 96%): 1H NMR (500 MHz, CDCl3) δ 10.54 (d, J = 5.2 Hz, 1H), 9.72 (d, J = 5.2 Hz, 1H), 8.85 (dd, J = 1.5, 0.8 Hz, 1H), 8.24 (dt, J = 8.2, 0.8 Hz, 1H), 8.13 – 8.07 (m, 1H), 7.53 – 7.47 (m, 2H), 7.33 (t, J = 7.9 Hz, 1H), 7.08 (ddd, J = 8.3, 2.6, 0.9 Hz, 1H), 4.80 (s, 2H), 2.40 (s, 3H), 2.27 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 167.7, 164.4, 160.1, 159.7, 158.6, 151.0, 145.7, 145.7, 134.9, 132.7, 130.0, 129.5 (q, J= 33.75 Hz), 122.9 (q, J = 271.25 Hz), 122.3, 119.9 (d, J = 11.25 Hz), 112.9, 109.9, 59.6, 11.1, 10.1. 19F NMR (470 MHz, CDCl3) δ -63.8. HRMS (ESI): m/z 457.1094 calc. for C20H17F3N4NaO4+ [M+Na]+, found 457.1101.
N′-(3-((3,5-dimethylisoxazol-4-yl)methoxy)benzoyl)-6-(trifluoromethyl)picolinohydrazide (16, 99%): 1H NMR (500 MHz, CDCl3) δ 10.26 (d, J = 4.9 Hz, 1H), 9.10 (d, J = 4.9 Hz, 1H), 8.40 – 8.35 (m, 1H), 8.11 (td, J = 7.9, 0.7 Hz, 1H), 7.89 (dd, J = 7.9, 1.0 Hz, 1H), 7.53 – 7.46 (m, 2H), 7.39 (t, J = 7.9 Hz, 1H), 7.12 (ddd, J = 8.2, 2.6, 1.0 Hz, 1H), 4.84 (s, 2H), 2.43 (s, 3H), 2.30 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 167.8, 164.2, 159.8, 159.7, 158.7, 148.4, 147.5 (d, J = 36.25 Hz), 139.4, 132.9, 130.1, 125.3, 123.6, 119.9, 119.7, 113.1, 109.9, 59.7, 11.2, 10.2; 19F NMR (470 MHz, CDCl3) δ −69.0. HRMS (ESI): m/z 435.1275 calc. for C20H18F3N4O4+ [M+H]+, found 435.1281.
N′-(3-((3-ethyl-5-methylisoxazol-4-yl)methoxy)benzoyl)quinoline-2-carbohydrazide (17, 39%): 1H NMR (500 MHz, CDCl3) δ 10.51 (s, br, 1H), 9.64 (s, br, 1H), 8.33 (d, J = 8.5 Hz, 1H), 8.24 (d, J = 8.4 Hz, 1H), 8.17 (dd, J = 8.5, 1.1 Hz, 1H), 7.92 – 7.88 (m, 1H), 7.81 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.66 (ddd, J = 8.1, 6.8, 1.2 Hz, 1H), 7.57 – 7.54 (m, 1H), 7.51 (dt, J = 7.7, 1.2 Hz, 1H), 7.38 (t, J = 7.9 Hz, 1H), 7.11 (ddd, J = 8.3, 2.6, 0.9 Hz, 1H), 4.83 (s, 2H), 2.69 (q, J = 7.6 Hz, 2H), 2.41 (s, 3H), 1.29 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 167.8, 164.4, 164.0, 161.3, 158.7, 147.7, 146.7, 137.7, 133.1, 130.6, 130.0, 130.0, 129.6, 128.5, 127.8, 119.9, 119.8, 118.7, 112.8, 109.3, 59.6, 18.7, 12.2, 11.2. HRMS (ESI): m/z 453.1533 calc. for C24H22N4NaO4+ [M+Na]+, found 453.1539.
N′-(3-((5-ethyl-3-methylisoxazol-4-yl)methoxy)benzoyl)quinoline-2-carbohydrazide (18, 71%): 1H NMR (500 MHz, CDCl3) δ 10.70 (s, 1H), 9.53 (s, 1H), 8.33 (d, J = 8.4 Hz, 1H), 8.23 (d, J = 8.4 Hz, 1H), 8.16 (dd, J = 8.5, 1.2 Hz, 1H), 7.89 (dd, J = 8.3, 1.4 Hz, 1H), 7.80 (ddd, J = 8.4, 6.9, 1.4 Hz, 1H), 7.66 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 7.56 (dd, J = 2.6, 1.5 Hz, 1H), 7.52 (dt, J = 7.7, 1.2 Hz, 1H), 7.38 (t, J = 7.9 Hz, 1H), 7.13 – 7.08 (m, 1H), 4.83 (s, 2H), 2.78 (q, J = 7.6 Hz, 2H), 2.29 (s, 3H), 1.27 (t, J = 7.6 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 172.4, 164.0, 161.3, 159.7, 158.7, 147.7, 146.7, 137.7, 133.1, 130.5, 130.0, 130.0, 129.6, 128.5, 127.8, 119.9, 119.8, 118.7, 112.9, 109.0, 59.6, 19.3, 12.2, 10.2. LRMS (ESI): m/z 431.2 calc. for C24H23N4O4+ [M+H]+, found 431.7.
N′-(3-((3-methyl-5-propylisoxazol-4-yl)methoxy)benzoyl)quinoline-2-carbohydrazide (19, 78%): 1H NMR (500 MHz, CDCl3) δ 10.70 (s, 1H), 9.22 (s, 1H), 8.35 (d, J = 8.5 Hz, 1H), 8.26 (d, J = 8.4 Hz, 1H), 8.18 (d, J = 8.5 Hz, 1H), 7.91 (dd, J = 8.2, 1.4 Hz, 1H), 7.82 (ddd, J = 8.4, 6.8, 1.4 Hz, 1H), 7.67 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 7.55 (dd, J = 2.7, 1.5 Hz, 1H), 7.53 – 7.49 (m, 1H), 7.41 (t, J = 7.9 Hz, 1H), 7.14 (dd, J = 8.0, 2.6 Hz, 1H), 4.85 (s, 2H), 2.74 (t, J = 7.5 Hz, 2H), 2.31 (s, 3H), 1.73 (h, J = 7.4 Hz, 2H), 0.96 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 171.4, 163.9, 161.1, 159.7, 158.8, 147.7, 146.7, 137.7, 133.1, 130.5, 130.1, 130.0, 129.6, 128.5, 127.8, 119.8, 118.7, 113.0, 109.7, 59.6, 27.6, 21.2, 13.7, 10.2. MS (ESI): m/z 445.2 calc. for C25H25N4O4+ [M+H]+, found 445.7.
N′-(3-((3-methyl-5-phenylisoxazol-4-yl)methoxy)benzoyl)quinoline-2-carbohydrazide (20, 60%): 1H NMR (500 MHz, CDCl3) δ 10.67 (s, 1H), 9.43 (s, 1H), 8.31 (d, J = 8.4 Hz, 1H), 8.22 (d, J = 8.4 Hz, 1H), 8.16 (d, J = 8.5 Hz, 1H), 7.89 (dd, J = 8.2, 1.4 Hz, 1H), 7.80 (ddd, J = 8.5, 6.9, 1.4 Hz, 1H), 7.72 – 7.63 (m, 3H), 7.58 (dd, J = 2.6, 1.4 Hz, 1H), 7.53 (dt, J = 7.7, 1.2 Hz, 1H), 7.46 (qd, J = 4.8, 1.6 Hz, 3H), 7.40 (t, J = 7.9 Hz, 1H), 7.15 (dd, J = 8.2, 2.5 Hz, 1H), 4.99 (s, 2H), 2.38 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 168.4, 164.0, 161.3, 160.8, 158.6, 147.7, 146.6, 137.7, 133.2, 130.5, 130.4, 130.1, 130.0, 129.6, 129.1, 128.5, 127.8, 127.5, 127.4, 120.1, 119.7, 118.6, 113.1, 109.5, 59.9, 10.2. MS (ESI): m/z 479.2 calc. for C28H23N4O4+ [M+H]+, found 479.8.
N′-(3-((5-ethyl-3-methylisoxazol-4-yl)methoxy)benzoyl)quinoline-2-carbohydrazide (21): To a solution of 31 (200 mg, 0.77 mmol) in THF/H2O (7.0 mL, 1:1 in volume) was added LiOH (36.7 mg, 1.53 mmol) at room temperature. The resulting mixture was stirred for 12 h. Upon completion, the resultant mixture was acidified with aq. HCl then extracted with EtOAc. The organic extracts were combined, dried over Na2SO4, filtered, evaporated, and the residue was subjected to a quick flash column chromatography to afford the acid for the next step.
To a solution of the above acid (50 mg, 0.2 mmol) and catalytic amount of DMF in anhydrous CH2Cl2 (1 mL) at 0 °C was added (COCl)2 (34 μL, 0.4 mmol) dropwise and the resulting mixture was stirred for 1 h. The resulting mixture was concentrated under reduced pressure to afford the acid chloride 45 which was subjected to the next step without further purification.
To a solution of the acid chloride from the previous step in anhydrous CH2Cl2 at 0 °C was added DIPEA (42 μL, 0.25 mmol) dropwise followed by N-Boc-ethylenediamine (32 μL, 0.2 mmol). The resulting mixture was stirred at room temperature for 12 h. The resulting mixture was concentrated under reduced pressure to afford the amide, which was subjected to the next step without further purification.
To a solution of the amide from the previous step in CH2Cl2 (0.6 mL) at 0 °C was added TFA (0.2 mL) dropwise. After 10 min, the resulting mixture was concentrated under reduced pressure to afford the primary amine 48, which was subjected to the next step without further purification.
Quinaldoyl chloride 37 (26 mg, 0.1 mmol) in CH2Cl2 (1.0 mL) was added dropwise to a dried round flask containing the primary amine 48 from the previous step, Et3N (38 μL, 0.27 mmol), in CH2Cl2 (1.0 mL) at 0 °C. The mixture was stirred at room temperature for 12 h. The resulting mixture was concentrated under reduced pressure. The crude product was purified by flash column chromatography to obtain 21 (16.7 mg, 12% over 5 steps). 1H NMR (500 MHz, CDCl3) δ 8.73 (t, J = 6.3 Hz, 1H), 8.32 – 8.25 (m, 2H), 8.10 (dq, J = 8.6, 0.9 Hz, 1H), 7.90 – 7.87 (m, 1H), 7.77 (ddd, J = 8.4, 6.9, 1.4 Hz, 1H), 7.63 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 7.58 (d, J = 4.9 Hz, 1H), 7.52 (dd, J = 2.6, 1.6 Hz, 1H), 7.44 (ddd, J = 7.6, 1.6, 1.0 Hz, 1H), 7.35 (t, J = 7.9 Hz, 1H), 7.04 (ddd, J = 8.2, 2.6, 1.0 Hz, 1H), 4.83 (s, 2H), 3.84 (td, J = 6.9, 6.2, 4.3 Hz, 2H), 3.77 (ddd, J = 7.2, 4.8, 3.5 Hz, 2H), 2.41 (s, 3H), 2.28 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 167.6, 167.4, 166.6, 159.8, 158.5, 149.1, 146.5, 137.7, 135.8, 130.4, 129.8, 129.7, 129.4, 128.2, 127.8, 119.6, 118.9, 118.6, 112.8, 110.1, 59.6, 42.2, 39.3, 11.2, 10.2. HRMS (ESI): m/z 445.1870 calc. for C25H25N4O4+ [M+H]+, found 445.1876.
N′-(3-((3-methyl-5-propylisoxazol-4-yl)methoxy)benzoyl)quinoline-2-carbohydrazide (22): To a solution of acid chloride 45 (0.33 mmol) in anhydrous CH2Cl2 at 0 °C was added DIPEA (70 μL, 0.40 mmol) dropwise followed by N-Boc-1,3-propanediamine (58 μL, 0.33 mmol). The resulting mixture was stirred at room temperature for 12 h. The resulting mixture was concentrated under reduced pressure to afford the amide, which was subjected to the next step without further purification.
To a solution of the amide from the previous step in CH2Cl2 (1.5 mL) at 0 °C was added TFA (0.5 mL) dropwise. After 10 min, the resulting mixture was concentrated under reduced pressure to afford the primary amine 49, which was subjected to the next step without further purification.
Quinaldoyl chloride 37 (63 mg, 0.33 mmol) in CH2Cl2 (1.5 mL) was added dropwise to a dried round flask containing the primary amine 49 from previous step, pyridine (0.13 mL, 1.66 mmol), and DMAP (4.0 mg, 0.03 mmol) in CH2Cl2 (1.5 mL) at 0 °C. The mixture was stirred at room temperature for 12 h. The mixture was then washed with dilute aqueous HCl and water and dried over Na2SO4. After removal of the solvent at reduced pressure, the crude product was purified by flash column chromatography to obtain 22 (50 mg, 23% over 5 steps) as product. 1H NMR (500 MHz, CDCl3) δ 8.56 (t, J = 7.3 Hz, 1H), 8.37 – 8.27 (m, 2H), 8.12 (t, J = 8.8 Hz, 1H), 7.91 (t, J = 8.7 Hz, 1H), 7.78 (h, J = 6.4, 4.1 Hz, 2H), 7.68 – 7.61 (m, 2H), 7.57 (t, J = 8.4 Hz, 1H), 7.39 (q, J = 8.4, 7.9 Hz, 1H), 7.28 (d, J = 1.6 Hz, 1H), 7.10 – 7.05 (m, 1H), 4.89 (d, J = 9.1 Hz, 2H), 3.71 (q, J = 6.2 Hz, 2H), 3.56 (q, J = 5.9 Hz, 2H), 2.44 (d, J = 9.3 Hz, 3H), 2.31 (dd, J = 8.1, 1.9 Hz, 3H), 1.95 (q, J = 6.0 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 167.6, 166.9, 165.8, 159.8, 158.6, 149.2, 146.5, 137.7, 136.2, 130.3, 129.8, 129.7, 129.4, 128.2, 127.8, 119.6, 118.8, 118.7, 112.9, 110.2, 59.6, 36.4, 36.1, 29.8, 11.2, 10.2. HRMS (ESI): m/z 481.1846 calc. for C26H26N4NaO4+ [M+Na]+, found 481.1851.
N′-(3-((3-methyl-5-phenylisoxazol-4-yl)methoxy)benzoyl)quinoline-2-carbohydrazide (23): 2-Quinolineethanamine 50 (70 mg, 0.41 mmol) in CH2Cl2 (2.0 mL) was added dropwise to a dried round flask containing the acid chloride 45 (0.41 mmol), pyridine (0.16 mL, 2.02 mmol), and DMAP (5.0 mg, 0.04 mmol) in CH2Cl2 (2.0 mL) at 0 °C. The mixture was stirred at room temperature for 12 h and then washed with dilute aqueous HCl and water and dried over Na2SO4. After removal of the solvent at reduced pressure, the crude product was purified by flash column chromatography to obtain 23 (0.8 mg; ~5%). 1H NMR (500 MHz, CDCl3) δ 8.13 (dd, J = 8.4, 0.9 Hz, 1H), 8.06 – 7.99 (m, 2H), 7.82 (dd, J = 8.1, 1.4 Hz, 1H), 7.69 (ddd, J = 8.4, 6.9, 1.4 Hz, 1H), 7.53 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 7.49 (dd, J = 2.6, 1.5 Hz, 1H), 7.40 – 7.32 (m, 3H), 7.04 (ddd, J = 7.8, 2.7, 1.3 Hz, 1H), 4.82 (s, 2H), 4.03 – 3.95 (m, 2H), 3.31 – 3.25 (m, 2H), 2.41 (s, 3H), 2.28 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 167.6, 166.7, 160.5, 159.8, 158.6, 147.6, 136.9, 136.5, 129.8, 129.7, 128.6, 127.8, 126.9, 126.3, 122.0, 119.3, 118.5, 113.0, 110.1, 59.6, 38.6, 37.0, 11.2, 10.2. HRMS (ESI): m/z 402.1812 calc. for C24H24N3O3+ [M+H]+, found 402.1817.
N-(3-((3,5-dimethylisoxazol-4-yl)methoxy)benzoyl)quinoline-2-carboxamide (24): Concentrated ammonia (4.0 mL) was added dropwise to a solution of 31 (300 mg, 1.14 mmol) in MeOH (2.0 mL). The reaction mixture was heated to 60 °C and stirred for 12 h until completion, as determined by TLC. After cooling, water was added, and the precipitate was filtered and washed with a small amount of methanol and water. The crude product was put under vacuum to afford amide (150 mg, 53%) as product, which was subjected to the next step without further purification.
To a solution of the above amide (20 mg, 0.08 mmol) in anhydrous THF was added NaH (6.4 mg, 60% in mineral oil) at 0 °C. The resulting mixture was stirred at 0 °C for 30 min before adding 37 (15.3 mg, 0.08 mmol). The mixture was stirred at room temperature for 12 h and then diluted with EtOAc. The organic phase was washed with dilute aqueous HCl and water and dried over Na2SO4. After removal of the solvent at reduced pressure, the crude product was purified by flash column chromatography to obtain 24 (15 mg, 47%). 1H NMR (500 MHz, CDCl3) δ 11.77 (s, 1H), 8.42 (d, J = 1.3 Hz, 2H), 8.19 (dd, J = 8.6, 1.1 Hz, 1H), 7.96 (dd, J = 8.2, 1.4 Hz, 1H), 7.85 (ddd, J = 8.5, 6.9, 1.4 Hz, 1H), 7.74 – 7.63 (m, 3H), 7.52 (t, J = 7.9 Hz, 1H), 7.22 (ddd, J = 8.2, 2.7, 0.9 Hz, 1H), 4.92 (s, 2H), 2.47 (s, 3H), 2.33 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 167.8, 164.6, 162.2, 159.8, 158.9, 148.3, 146.2, 138.4, 135.1, 130.8, 130.2, 130.0, 129.9, 129.0, 128.0, 120.6, 119.9, 119.0, 113.8, 109.9, 59.8, 11.3, 10.2. MS (ESI): m/z 402.1 calc. for C23H20N3O4+ [M+H]+, found 402.6.
N-(3-((3,5-dimethylisoxazol-4-yl)methoxy)benzyl)quinoline-2-carboxamide (25): To a solution of 31 (390 mg, 1.49 mmol) in anhydrous THF (2 mL) at 0 °C was slowly added LiAlH4 (34 mg, 0.90 mmol) in anhydrous THF (2 mL) dropwise. The resulting mixture was stirred at room temperature for 3 h. The reaction was quenched by subsequent addition of water and EtOAc. The suspension was filtered, and the residue was extracted with EtOAc, then dried over Na2SO4. After removal of the solvent at reduced pressure, the crude residue was purified by flash column chromatography to afford the primary alcohol (340 mg, 98% yield). 1H NMR (500 MHz, CDCl3) δ 7.31 – 7.25 (m, 1H), 7.00 – 6.96 (m, 2H), 6.86 (ddd, J = 8.2, 2.6, 1.0 Hz, 1H), 4.80 (s, 2H), 4.69 (s, 2H), 2.40 (s, 3H), 2.28 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 167.5, 159.8, 158.6, 142.8, 129.8, 119.8, 114.1, 113.0, 110.3, 65.0, 59.5, 11.2, 10.2.
To a solution of the above primary alcohol (340 mg, 1.46 mmol) in anhydrous CH2Cl2 (14.6 mL) at 0 °C was added Dess-Martin periodinane (680 mg, 1.60 mmol). The mixture was stirred at room temperature for 3 h before water was added. The resultant mixture was extracted with CH2Cl2, washed with aq. Na2S2O3 and aq. NaHCO3. The organic extracts were combined, dried over Na2SO4, filtered, and evaporated. The crude residue was purified by flash column chromatography to afford aldehyde 51 (297 mg, 88% yield). 1H NMR (500 MHz, CDCl3) δ 9.96 (s, 1H), 7.53 – 7.39 (m, 3H), 7.19 (ddd, J = 7.6, 2.7, 1.7 Hz, 1H), 4.85 (s, 2H), 2.40 (s, 3H), 2.27 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 191.9, 167.7, 159.7, 158.9, 137.9, 130.3, 124.4, 122.3, 112.4, 109.9, 59.7, 11.2, 10.2.
To a solution of 51 in MeOH/H2O (2 mL, 1:1 in volume) was added H2NOH-HCl (36 mg, 0.52 mmol) and NaOH (21 mg, 0.52 mmol). The mixture was refluxed for 6 h. The resultant mixture was extracted with EtOAc. The organic extracts were combined, dried over Na2SO4, filtered, and evaporated. The crude was subjected to the next step without further purification.
To a solution of the oxime from the previous step in anhydrous THF (1 mL) at 0 °C was slowly added LiAlH4 (36 mg, 0.95 mmol) in anhydrous THF (1 mL) dropwise. The resulting mixture was raised to room temperature and then stirred at reflux for 3 h. The reaction was quenched by subsequent addition of water and EtOAc. The suspension was filtered, and the residue was extracted with EtOAc, then dried over Na2SO4. After removal of the solvent at reduced pressure, the crude was subjected to the next step without further purification.
Quinaldoyl chloride 37 (16.5 mg, 0.086 mmol) in CH2Cl2 (1.0 mL) was added dropwise to a dried round flask containing the primary amine from the previous step, DIPEA (18 μL, 0.103 mmol in CH2Cl2 (1.0 mL) at 0 °C. The mixture was stirred at room temperature for 12 h. After removal of the solvent at reduced pressure, the crude product was purified by flash column chromatography to obtain 25 (10 mg, 6% yield, over 3 steps) as product. 1H NMR (500 MHz, CDCl3) δ 8.63 (s, 1H), 8.37 – 8.31 (m, 2H), 8.08 (dd, J = 8.6, 1.1 Hz, 1H), 7.89 (dt, J = 8.1, 1.0 Hz, 1H), 7.76 (ddd, J = 8.4, 6.8, 1.5 Hz, 1H), 7.63 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 7.31 (t, J = 7.9 Hz, 1H), 7.08 – 7.04 (m, 1H), 7.01 (t, J = 2.1 Hz, 1H), 6.90 – 6.86 (m, 1H), 4.79 (s, 2H), 4.74 (d, J = 6.2 Hz, 2H), 2.38 (s, 3H), 2.27 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 167.5, 164.5, 159.8, 158.7, 149.6, 146.5, 140.2, 137.6, 130.2, 129.9, 129.7, 129.4, 128.0, 127.8, 120.9, 119.0, 114.1, 113.9, 110.2, 59.4, 43.5, 11.2, 10.2. HRMS (ESI): m/z 410.1475 calc. for C23H21N3NaO3+ [M+Na]+, found 410.1482.
N-(3-((3,5-dimethylisoxazol-4-yl)methoxy)phenethyl)quinoline-2-carboxamide (26): To a solution of 53 (428 mg, 1.58 mmol) in anhydrous THF (2.5 mL) at 0 °C was slowly added LiAlH4 (36 mg, 0.95 mmol) in anhydrous THF (2.5 mL) dropwise. The resulting mixture was stirred at room temperature for 3 h. The reaction was quenched by subsequent addition of water and EtOAc. The suspension was filtered, and the residue was extracted with EtOAc, then dried over Na2SO4. After removal of the solvent at reduced pressure, the crude residue was purified by flash column chromatography to afford the primary alcohol (300 mg, 78% yield). 1H NMR (500 MHz, CDCl3) δ 7.30 – 7.19 (m, 1H), 6.90 – 6.85 (m, 1H), 6.82 (dd, J = 7.5, 1.1 Hz, 2H), 4.78 (s, 2H), 3.85 (t, J = 6.6 Hz, 2H), 2.85 (t, J = 6.6 Hz, 2H), 2.39 (s, 3H), 2.28 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 167.6, 159.8, 158.6, 140.5, 129.7, 122.1, 115.7, 112.6, 110.4, 63.5, 59.4, 39.2, 11.2, 10.2.
To a solution of the above primary alcohol (300 mg, 1.21 mmol) in anhydrous CH2Cl2 (7 mL) at 0 °C was added Dess-Martin periodinane (618 mg, 1.46 mmol). The mixture was stirred at room temperature for 3 h. Water was added. The resultant mixture was extracted with CH2Cl2, washed with aq. Na2S2O3 and aq. NaHCO3. The organic extracts were combined, dried over Na2SO4, filtered, evaporated, and the crude residue was purified by flash column chromatography to afford the aldehyde (230 mg, 78% yield). 1H NMR (500 MHz, CDCl3) δ 9.73 (s, 1H), 7.29 (dd, J = 8.3, 7.5 Hz, 1H), 6.91 – 6.82 (m, 2H), 6.79 (dd, J = 2.6, 1.6 Hz, 1H), 4.78 (s, 2H), 3.66 (d, J = 2.4 Hz, 2H), 2.38 (s, 3H), 2.27 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 199.1, 167.6, 159.8, 158.8, 133.6, 130.2, 122.7, 116.2, 113.7, 110.2, 59.5, 50.5, 11.1, 10.1.
To a solution of the above aldehyde (125 mg, 0.51 mmol) in H2O (1 mL) was added H2NOH-HCl (46.7 mg, 0.66 mmol) and Na2CO3 (32.4 mg, 0.31 mmol). The mixture was refluxed for 6 h. The resultant mixture was extracted with EtOAc. The organic extracts were combined, dried over Na2SO4, filtered, and evaporated. The crude was subjected to the next step without further purification.
To a solution of the oxime from the previous step in anhydrous THF (1.5 mL) at 0 °C was slowly added LiAlH4 (28 mg, 0.75 mmol) in anhydrous THF (1.5 mL) dropwise. The resulting mixture was raised to room temperature for 3 h. The reaction was quenched by subsequent addition of water and EtOAc. The suspension was filtered and the residue was extracted with EtOAc, then dried over Na2SO4. After removal of the solvent at reduced pressure, the crude was subjected to the next step without further purification.
Quinaldoyl chloride 37 (27 mg, 0.14 mmol) in CH2Cl2 (1.4 mL) was added dropwise to a dried round flask containing the primary amine 54 from the previous step (35 mg), DIPEA (28 μL, 0.16 mmol in CH2Cl2 (1.4 mL) at 0 °C. The mixture was stirred at room temperature for 12 h. After removal of the solvent at reduced pressure, the crude product was purified by flash column chromatography to obtain 22 (7.4 mg, 10% yield, over 3 steps) as product. 1H NMR (500 MHz, CDCl3) δ 8.40 (s, 1H), 8.31 (s, 2H), 8.07 – 8.03 (m, 1H), 7.88 (dd, J = 8.3, 1.5 Hz, 1H), 7.74 (ddd, J = 8.4, 6.8, 1.4 Hz, 1H), 7.62 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 7.28 (t, J = 7.9 Hz, 1H), 6.95 (dt, J = 7.6, 1.2 Hz, 1H), 6.90 (t, J = 2.1 Hz, 1H), 6.84 (ddd, J = 8.2, 2.7, 1.0 Hz, 1H), 4.76 (s, 2H), 3.80 (dt, J = 7.5, 6.5 Hz, 2H), 3.00 (t, J = 7.2 Hz, 2H), 2.35 (s, 3H), 2.24 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 167.5, 164.5, 159.8, 158.7, 149.8, 146.5, 140.9, 137.5, 130.1, 129.8, 129.6, 129.3, 128.0, 127.8, 122.0, 118.8, 115.3, 113.0, 110.3, 59.5, 40.8, 36.1, 11.1, 10.1. HRMS (ESI): m/z 424.1632 calc. for C24H23N3NaO3+ [M+Na]+, found 424.1642.
Statistical Analysis
Quantified data are presented graphically via Prism 9.0.0 (GraphPad Software). For biological experiments, data were collected in triplicate and statistical significance was calculated via student’s t-test for normally distributed data. Statistical significance calculations and cutoff values for RNAseq-based data are described above. For all qPCR experiments, statistical analysis was performed on ΔCT values (CT value of gene normalized to CT value of GAPDH control). Statistical significance of western blotting was performed on normalized raw intensity values based on loading control (Either H4R3 for global histone methylation or corresponding band in 0.4% Input sample for Co-IP). All relevant statistics are reported in the corresponding legends.
Supplementary Material
ACKNOWLEDGEMENTS
Andrew Asberry and Hunter Sims were supported by drug discovery trainee fellowship under NIH T32 Fellowship Grant NIH T32GM125620. We thank all members of the Chang-Deng Hu lab, Mingji Dai lab, and Wen Jiang lab for constructive suggestions. We also thank Gyeon Oh and Yihang Wu for technical support.
FUNDING SOURCES
This work was partially supported by the following grants: U.S. Army Medical Research Acquisition Activity (W81XWH-16-10394), NCI RO1CA212403, NIH R35 GM128570, Purdue University Center for Cancer Research Small Grants, Purdue University Institute for Drug Discovery, Purdue Research Foundation Research Grant, Indiana University Simon Comprehensive Cancer Center (P30CA082709), Purdue University Center for Cancer Research (P30CA023168), Walther Cancer Foundation, and Indiana University Precision Health Initiative (PHI).
Footnotes
SUPPORTING INFORMATION
The data underlying this study are available in the published article and its online supplementary material. HPLC trace, NMR Spectra, biological reagents (shRNA sequences, antibodies, oligonucleotide primers), and SMILES structure strings are included in supporting information (SI) spreadsheets.
Associated Content
RNA-seq data for treatment of LNCaP cells with Cpd 17, or shRNA-mediated knockdown of PRMT5 or MEP50 in LCNaP cells, openly available in Gene Expression Omnibus at GSE206460 (Cpd 17 treatment), GSE206820 (MEP50 knockdown) and GSE206111 (PRMT5 knockdown).
RNA-seq data for knockdown of PRMT5 or MEP50 in A549 cells are openly available in Gene Expression Omnibus at GSE80182.
REFERENCES
- (1).Karkhanis V; Hu Y-J; Baiocchi RA; Imbalzano AN; Sif S Versatility of PRMT5-Induced Methylation in Growth Control and Development. Trends Biochem. Sci 2011, 36 (12), 633–641. 10.1016/j.tibs.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Hwang JW; Cho Y; Bae G-U; Kim S-N; Kim YK Protein Arginine Methyltransferases: Promising Targets for Cancer Therapy. Exp. Mol. Med 2021, 53 (5), 788–808. 10.1038/s12276-021-00613-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Tewary SK; Zheng YG; Ho M-C Protein Arginine Methyltransferases: Insights into the Enzyme Structure and Mechanism at the Atomic Level. Cell. Mol. Life Sci 2019, 76 (15), 2917–2932. 10.1007/s00018-019-03145-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Stopa N; Krebs JE; Shechter D The PRMT5 Arginine Methyltransferase: Many Roles in Development, Cancer and Beyond. Cell. Mol. Life Sci 2015, 72 (11), 2041–2059. 10.1007/s00018-015-1847-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Beketova E; Owens JL; Asberry AM; Hu C-D PRMT5: A Putative Oncogene and Therapeutic Target in Prostate Cancer. Cancer Gene Ther 2021. 10.1038/s41417-021-00327-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Shailesh H; Zakaria ZZ; Baiocchi R; Sif S Protein Arginine Methyltransferase 5 (PRMT5) Dysregulation in Cancer. Oncotarget 2018, 9 (94), 36705–36718. 10.18632/oncotarget.26404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Tae S; Karkhanis V; Velasco K; Yaneva M; Erdjument-Bromage H; Tempst P; Sif S Bromodomain Protein 7 Interacts with PRMT5 and PRC2, and Is Involved in Transcriptional Repression of Their Target Genes. Nucleic Acids Res 2011, 39 (13), 5424–5438. 10.1093/nar/gkr170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Chung J; Karkhanis V; Tae S; Yan F; Smith P; Ayers LW; Agostinelli C; Pileri S; Denis GV; Baiocchi RA; Sif S Protein Arginine Methyltransferase 5 (PRMT5) Inhibition Induces Lymphoma Cell Death through Reactivation of the Retinoblastoma Tumor Suppressor Pathway and Polycomb Repressor Complex 2 (PRC2) Silencing. J. Biol. Chem 2013, 288 (49), 35534–35547. 10.1074/jbc.M113.510669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Banasavadi-Siddegowda YK; Russell L; Frair E; Karkhanis VA; Relation T; Yoo JY; Zhang J; Sif S; Imitola J; Baiocchi R; Kaur B PRMT5-PTEN Molecular Pathway Regulates Senescence and Self-Renewal of Primary Glioblastoma Neurosphere Cells. Oncogene 2017, 36 (2), 263–274. 10.1038/onc.2016.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zhu F; Rui L PRMT5 in Gene Regulation and Hematologic Malignancies. Genes Dis 2019, 6 (3), 247–257. 10.1016/j.gendis.2019.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Antonysamy S The Structure and Function of the PRMT5:MEP50 Complex. Subcell. Biochem 2017, 83, 185–194. 10.1007/978-3-319-46503-6_7. [DOI] [PubMed] [Google Scholar]
- (12).Timm DE; Bowman V; Madsen R; Rauch C Cryo-Electron Microscopy Structure of a Human PRMT5:MEP50 Complex. PLOS ONE 2018, 13 (3), e0193205. 10.1371/journal.pone.0193205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Antonysamy S; Bonday Z; Campbell RM; Doyle B; Druzina Z; Gheyi T; Han B; Jungheim LN; Qian Y; Rauch C; Russell M; Sauder JM; Wasserman SR; Weichert K; Willard FS; Zhang A; Emtage S Crystal Structure of the Human PRMT5:MEP50 Complex. Proc. Natl. Acad. Sci 2012, 109 (44), 17960–17965. 10.1073/pnas.1209814109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ho M-C; Wilczek C; Bonanno JB; Xing L; Seznec J; Matsui T; Carter LG; Onikubo T; Kumar PR; Chan MK; Brenowitz M; Cheng RH; Reimer U; Almo SC; Shechter D Structure of the Arginine Methyltransferase PRMT5-MEP50 Reveals a Mechanism for Substrate Specificity. PLoS ONE 2013, 8 (2), e57008. 10.1371/journal.pone.0057008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Burgos ES; Wilczek C; Onikubo T; Bonanno JB; Jansong J; Reimer U; Shechter D Histone H2A and H4 N-Terminal Tails Are Positioned by the MEP50 WD Repeat Protein for Efficient Methylation by the PRMT5 Arginine Methyltransferase. J. Biol. Chem 2015, 290 (15), 9674–9689. 10.1074/jbc.M115.636894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Guderian G; Peter C; Wiesner J; Sickmann A; Schulze-Osthoff K; Fischer U; Grimmler M RioK1, a New Interactor of Protein Arginine Methyltransferase 5 (PRMT5), Competes with PICln for Binding and Modulates PRMT5 Complex Composition and Substrate Specificity. J. Biol. Chem 2011, 286 (3), 1976–1986. 10.1074/jbc.M110.148486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lacroix M; Messaoudi SE; Rodier G; Le Cam A; Sardet C; Fabbrizio E The Histone-Binding Protein COPR5 Is Required for Nuclear Functions of the Protein Arginine Methyltransferase PRMT5. EMBO Rep 2008, 9 (5), 452–458. 10.1038/embor.2008.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Krapivinsky G; Pu W; Wickman K; Krapivinsky L; Clapham DE PICln Binds to a Mammalian Homolog of a Yeast Protein Involved in Regulation of Cell Morphology. J. Biol. Chem 1998, 273 (18), 10811–10814. 10.1074/jbc.273.18.10811. [DOI] [PubMed] [Google Scholar]
- (19).Owens JL; Beketova E; Liu S; Tinsley SL; Asberry AM; Deng X; Huang J; Li C; Wan J; Hu C-D PRMT5 Cooperates with PICln to Function as a Master Epigenetic Activator of DNA Double-Strand Break Repair Genes. iScience 2020, 23 (1), 100750. 10.1016/j.isci.2019.100750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Beketova E; Fang S; Owens JL; Liu S; Chen X; Zhang Q; Asberry AM; Deng X; Malola J; Huang J; Li C; Pili R; Elzey BD; Ratliff TL; Wan J; Hu C-D Protein Arginine Methyltransferase 5 Promotes PICln-Dependent Androgen Receptor Transcription in Castration-Resistant Prostate Cancer. Cancer Res 2020, 80 (22), 4904–4917. 10.1158/0008-5472.CAN-20-1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).McKinney DC; McMillan BJ; Ranaghan MJ; Moroco JA; Brousseau M; Mullin-Bernstein Z; O’Keefe M; McCarren P; Mesleh MF; Mulvaney KM; Robinson F; Singh R; Bajrami B; Wagner FF; Hilgraf R; Drysdale MJ; Campbell AJ; Skepner A; Timm DE; Porter D; Kaushik VK; Sellers WR; Ianari A Discovery of a First-in-Class Inhibitor of the PRMT5–Substrate Adaptor Interaction. J. Med. Chem 2021, 64 (15), 11148–11168. 10.1021/acs.jmedchem.1c00507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Mulvaney KM; Blomquist C; Acharya N; Li R; Ranaghan MJ; O’Keefe M; Rodriguez DJ; Young MJ; Kesar D; Pal D; Stokes M; Nelson AJ; Jain SS; Yang A; Mullin-Bernstein Z; Columbus J; Bozal FK; Skepner A; Raymond D; LaRussa S; McKinney DC; Freyzon Y; Baidi Y; Porter D; Aguirre AJ; Ianari A; McMillan B; Sellers WR Molecular Basis for Substrate Recruitment to the PRMT5 Methylosome. Mol. Cell 2021, S1097276521005888. 10.1016/j.molcel.2021.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Waldmann H; Krzyzanowski A; Gasper R; Adihou H; ‘t Hart P Biochemical Investigation of the Interaction of PICln, RioK1 and COPR5 with the PRMT5-MEP50 Complex. ChemBioChem 2021, cbic.202100079. 10.1002/cbic.202100079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Greenblatt SM; Liu F; Nimer SD Arginine Methyltransferases in Normal and Malignant Hematopoiesis. Exp. Hematol 2016, 44 (6), 435–441. 10.1016/j.exphem.2016.03.009. [DOI] [PubMed] [Google Scholar]
- (25).Kaniskan HÜ; Martini ML; Jin J Inhibitors of Protein Methyltransferases and Demethylases. Chem. Rev 2018, 118 (3), 989–1068. 10.1021/acs.chemrev.6b00801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Li X; Wang C; Jiang H; Luo C A Patent Review of Arginine Methyltransferase Inhibitors (2010–2018). Expert Opin. Ther. Pat 2019, 29 (2), 97–114. 10.1080/13543776.2019.1567711. [DOI] [PubMed] [Google Scholar]
- (27).Hu C-D; Chinenov Y; Kerppola TK Visualization of Interactions among BZIP and Rel Family Proteins in Living Cells Using Bimolecular Fluorescence Complementation. Mol. Cell 2002, 9 (4), 789–798. 10.1016/S1097-2765(02)00496-3. [DOI] [PubMed] [Google Scholar]
- (28).Kodama Y; Hu C-D Bimolecular Fluorescence Complementation (BiFC): A 5-Year Update and Future Perspectives. BioTechniques 2012, 53 (5), 285–298. 10.2144/000113943. [DOI] [PubMed] [Google Scholar]
- (29).Dempster JM; Rossen J; Kazachkova M; Pan J; Kugener G; Root DE; Tsherniak A Extracting Biological Insights from the Project Achilles Genome-Scale CRISPR Screens in Cancer Cell Lines; preprint; Cancer Biology, 2019. 10.1101/720243. [DOI] [Google Scholar]
- (30).Dempster JM; Boyle I; Vazquez F; Root D; Boehm JS; Hahn WC; Tsherniak A; McFarland JM Chronos: A CRISPR Cell Population Dynamics Model; preprint; Bioinformatics, 2021. 10.1101/2021.02.25.432728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Pacini C; Dempster JM; Boyle I; Gonçalves E; Najgebauer H; Karakoc E; van der Meer D; Barthorpe A; Lightfoot H; Jaaks P; McFarland JM; Garnett MJ; Tsherniak A; Iorio F Integrated Cross-Study Datasets of Genetic Dependencies in Cancer. Nat. Commun 2021, 12 (1), 1661. 10.1038/s41467-021-21898-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).DepMap, Broad. DepMap 21Q3 Public, 2021, 14529771955 Bytes. 10.6084/M9.FIGSHARE.15160110.V2. [DOI]
- (33).Champ PC; Camacho CJ FastContact: A Free Energy Scoring Tool for Protein-Protein Complex Structures. Nucleic Acids Res 2007, 35 (Web Server), W556–W560. 10.1093/nar/gkm326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Kodama Y; Hu C-D Bimolecular Fluorescence Complementation (BiFC) Analysis of Protein–Protein Interaction. In Methods in Cell Biology; Elsevier, 2013; Vol. 113, pp 107–121. 10.1016/B978-0-12-407239-8.00006-9. [DOI] [PubMed] [Google Scholar]
- (35).Koes DR; Camacho CJ ZINCPharmer: Pharmacophore Search of the ZINC Database. Nucleic Acids Res 2012, 40 (W1), W409–W414. 10.1093/nar/gks378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Koes DR; Baumgartner MP; Camacho CJ Lessons Learned in Empirical Scoring with Smina from the CSAR 2011 Benchmarking Exercise. J. Chem. Inf. Model 2013, 53 (8), 1893–1904. 10.1021/ci300604z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hu C-D; Kerppola TK Simultaneous Visualization of Multiple Protein Interactions in Living Cells Using Multicolor Fluorescence Complementation Analysis. Nat. Biotechnol 2003, 21 (5), 539–545. 10.1038/nbt816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kerppola TK Design and Implementation of Bimolecular Fluorescence Complementation (BiFC) Assays for the Visualization of Protein Interactions in Living Cells. Nat. Protoc 2006, 1 (3), 1278–1286. 10.1038/nprot.2006.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Owens JL; Beketova E; Liu S; Shen Q; Pawar JS; Asberry AM; Yang J; Deng X; Elzey BD; Ratliff TL; Cheng L; Choo CR; Citrin DE; Polascik TJ; Wang B; Huang J; Li C; Wan J; Hu C-D Targeting Protein Arginine Methyltransferase 5 (PRMT5) Suppresses Radiation-Induced Neuroendocrine Differentiation and Sensitizes Prostate Cancer Cells to Radiation. Mol. Cancer Ther 2022, molcanther.MCT-21–0103-A.2021. 10.1158/1535-7163.MCT-21-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Saha K; Adhikary G; Eckert RL MEP50/PRMT5 Reduces Gene Expression by Histone Arginine Methylation and This Is Reversed by PKCδ/P38δ Signaling. J. Invest. Dermatol 2016, 136 (1), 214–224. 10.1038/JID.2015.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Deng X; Shao G; Zhang H-T; Li C; Zhang D; Cheng L; Elzey BD; Pili R; Ratliff TL; Huang J; Hu C-D Protein Arginine Methyltransferase 5 Functions as an Epigenetic Activator of the Androgen Receptor to Promote Prostate Cancer Cell Growth. Oncogene 2017, 36 (9), 1223–1231. 10.1038/onc.2016.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Chen H; Lorton B; Gupta V; Shechter D A TGFβ-PRMT5-MEP50 Axis Regulates Cancer Cell Invasion through Histone H3 and H4 Arginine Methylation Coupled Transcriptional Activation and Repression. Oncogene 2017, 36 (3), 373–386. 10.1038/onc.2016.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Hanahan D Hallmarks of Cancer: New Dimensions. Cancer Discov 2022, 12 (1), 31–46. 10.1158/2159-8290.CD-21-1059. [DOI] [PubMed] [Google Scholar]
- (44).Tarighat SS; Santhanam R; Frankhouser D; Radomska HS; Lai H; Anghelina M; Wang H; Huang X; Alinari L; Walker A; Caligiuri MA; Croce CM; Li L; Garzon R; Li C; Baiocchi RA; Marcucci G The Dual Epigenetic Role of PRMT5 in Acute Myeloid Leukemia: Gene Activation and Repression via Histone Arginine Methylation. Leukemia 2016, 30 (4), 789–799. 10.1038/leu.2015.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Alinari L; Mahasenan KV; Yan F; Karkhanis V; Chung J-H; Smith EM; Quinion C; Smith PL; Kim L; Patton JT; Lapalombella R; Yu B; Wu Y; Roy S; De Leo A; Pileri S; Agostinelli C; Ayers L; Bradner JE; Chen-Kiang S; Elemento O; Motiwala T; Majumder S; Byrd JC; Jacob S; Sif S; Li C; Baiocchi RA Selective Inhibition of Protein Arginine Methyltransferase 5 Blocks Initiation and Maintenance of B-Cell Transformation. Blood 2015, 125 (16), 2530–2543. 10.1182/blood-2014-12-619783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Jiang H; Zhu Y; Zhou Z; Xu J; Jin S; Xu K; Zhang H; Sun Q; Wang J; Xu J PRMT5 Promotes Cell Proliferation by Inhibiting BTG2 Expression via the ERK Signaling Pathway in Hepatocellular Carcinoma. Cancer Med 2018, 7 (3), 869–882. 10.1002/cam4.1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Mei M; Zhang R; Zhou Z-W; Ying Z; Wang J; Zhang H; Zheng H; Bao S PRMT5-Mediated H4R3sme2 Confers Cell Differentiation in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res 2019. 10.1158/1078-0432.CCR-18-2342. [DOI] [PubMed] [Google Scholar]
- (48).Paul C; Sardet C; Fabbrizio E The Histone- and PRMT5-Associated Protein COPR5 Is Required for Myogenic Differentiation. Cell Death Differ 2012, 19 (5), 900–908. 10.1038/cdd.2011.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Wrighton KH Cell Signalling: PRMT5 Restricts ERK Activity. Nat. Rev. Mol. Cell Biol 2011, 12 (11), 689. 10.1038/nrm3213. [DOI] [PubMed] [Google Scholar]
- (50).Yin S; Liu L; Brobbey C; Palanisamy V; Ball LE; Olsen SK; Ostrowski MC; Gan W PRMT5-Mediated Arginine Methylation Activates AKT Kinase to Govern Tumorigenesis. Nat. Commun 2021, 12 (1), 3444. 10.1038/s41467-021-23833-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Scoumanne A; Zhang J; Chen X PRMT5 Is Required for Cell-Cycle Progression and P53 Tumor Suppressor Function. Nucleic Acids Res 2009, 37 (15), 4965–4976. 10.1093/nar/gkp516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Rugo HS; Jacobs I; Sharma S; Scappaticci F; Paul TA; Jensen-Pergakes K; Malouf GG The Promise for Histone Methyltransferase Inhibitors for Epigenetic Therapy in Clinical Oncology: A Narrative Review. Adv. Ther 2020, 37 (7), 3059–3082. 10.1007/s12325-020-01379-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Shen Y; Gao G; Yu X; Kim H; Wang L; Xie L; Schwarz M; Chen X; Guccione E; Liu J; Bedford MT; Jin J Discovery of First-in-Class Protein Arginine Methyltransferase 5 (PRMT5) Degraders. J. Med. Chem 2020, 63 (17), 9977–9989. 10.1021/acs.jmedchem.0c01111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Wang Y; Hu W; Yuan Y Protein Arginine Methyltransferase 5 (PRMT5) as an Anticancer Target and Its Inhibitor Discovery. J Med Chem 2018, 13. [DOI] [PubMed] [Google Scholar]
- (55).Xiao W; Chen X; Liu L; Shu Y; Zhang M; Zhong Y Role of Protein Arginine Methyltransferase 5 in Human Cancers. Biomed. Pharmacother. Biomedecine Pharmacother 2019, 114, 108790. 10.1016/j.biopha.2019.108790. [DOI] [PubMed] [Google Scholar]
- (56).Zhang Y; Zheng D; Zhou T; Song H; Hulsurkar M; Su N; Liu Y; Wang Z; Shao L; Ittmann M; Gleave M; Han H; Xu F; Liao W; Wang H; Li W Androgen Deprivation Promotes Neuroendocrine Differentiation and Angiogenesis through CREB-EZH2-TSP1 Pathway in Prostate Cancers. Nat. Commun 2018, 9 (1), 4080. 10.1038/s41467-018-06177-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Hua H; Kong Q; Zhang H; Wang J; Luo T; Jiang Y Targeting MTOR for Cancer Therapy. J. Hematol. Oncol.J Hematol Oncol 2019, 12 (1), 71. 10.1186/s13045-019-0754-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).McLoughlin NM; Mueller C; Grossmann TN The Therapeutic Potential of PTEN Modulation: Targeting Strategies from Gene to Protein. Cell Chem. Biol 2018, 25 (1), 19–29. 10.1016/j.chembiol.2017.10.009. [DOI] [PubMed] [Google Scholar]
- (59).Stanford SM; Bottini N Targeting Tyrosine Phosphatases: Time to End the Stigma. Trends Pharmacol. Sci 2017, 38 (6), 524–540. 10.1016/j.tips.2017.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Bezzi M; Teo SX; Muller J; Mok WC; Sahu SK; Vardy LA; Bonday ZQ; Guccione E Regulation of Constitutive and Alternative Splicing by PRMT5 Reveals a Role for Mdm4 Pre-MRNA in Sensing Defects in the Spliceosomal Machinery. Genes Dev 2013, 27 (17), 1903–1916. 10.1101/gad.219899.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Tan DQ; Li Y; Yang C; Li J; Tan SH; Chin DWL; Nakamura-Ishizu A; Yang H; Suda T PRMT5 Modulates Splicing for Genome Integrity and Preserves Proteostasis of Hematopoietic Stem Cells. Cell Rep 2019, 26 (9), 2316–2328.e6. 10.1016/j.celrep.2019.02.001. [DOI] [PubMed] [Google Scholar]
- (62).Pesiridis GS; Diamond E; Van Duyne GD Role of PICLn in Methylation of Sm Proteins by PRMT5. J. Biol. Chem 2009, 284 (32), 21347–21359. 10.1074/jbc.M109.015578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Sachamitr P; Ho JC; Ciamponi FE; Ba-Alawi W; Coutinho FJ; Guilhamon P; Kushida MM; Cavalli FMG; Lee L; Rastegar N; Vu V; Sánchez-Osuna M; Coulombe-Huntington J; Kanshin E; Whetstone H; Durand M; Thibault P; Hart K; Mangos M; Veyhl J; Chen W; Tran N; Duong B-C; Aman AM; Che X; Lan X; Whitley O; Zaslaver O; Barsyte-Lovejoy D; Richards LM; Restall I; Caudy A; Röst HL; Bonday ZQ; Bernstein M; Das S; Cusimano MD; Spears J; Bader GD; Pugh TJ; Tyers M; Lupien M; Haibe-Kains B; Artee Luchman H; Weiss S; Massirer KB; Prinos P; Arrowsmith CH; Dirks PB PRMT5 Inhibition Disrupts Splicing and Stemness in Glioblastoma. Nat. Commun 2021, 12 (1), 979. 10.1038/s41467-021-21204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Zhang Y; Qian J; Gu C; Yang Y Alternative Splicing and Cancer: A Systematic Review. Signal Transduct. Target. Ther 2021, 6 (1), 78. 10.1038/s41392-021-00486-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Sheng J; Zhao Q; Zhao J; Zhang W; Sun Y; Qin P; Lv Y; Bai L; Yang Q; Chen L; Qi Y; Zhang G; Zhang L; Gu C; Deng X; Liu H; Meng S; Gu H; Liu Q; Coulson JM; Li X; Sun B; Wang Y SRSF1 Modulates PTPMT1 Alternative Splicing to Regulate Lung Cancer Cell Radioresistance. EBioMedicine 2018, 38, 113–126. 10.1016/j.ebiom.2018.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Tripathi V; Shin J-H; Stuelten CH; Zhang YE TGF-β-Induced Alternative Splicing of TAK1 Promotes EMT and Drug Resistance. Oncogene 2019, 38 (17), 3185–3200. 10.1038/s41388-018-0655-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Brown RL; Reinke LM; Damerow MS; Perez D; Chodosh LA; Yang J; Cheng C CD44 Splice Isoform Switching in Human and Mouse Epithelium Is Essential for Epithelial-Mesenchymal Transition and Breast Cancer Progression. J. Clin. Invest 2011, 121 (3), 1064–1074. 10.1172/JCI44540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Molinspiration Cheminformatics Free Web Services, Https://Www.Molinspiration.Com, Slovensky Grob, Slovakia. [Google Scholar]
- (69).Wiederschain D; Susan W; Chen L; Loo A; Yang G; Huang A; Chen Y; Caponigro G; Yao Y; Lengauer C; Sellers WR; Benson JD Single-Vector Inducible Lentiviral RNAi System for Oncology Target Validation. Cell Cycle 2009, 8 (3), 498–504. 10.4161/cc.8.3.7701. [DOI] [PubMed] [Google Scholar]
- (70).Schneider CA; Rasband WS; Eliceiri KW NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9 (7), 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Hsu C-C; Hu C-D Transcriptional Activity of C-Jun Is Critical for the Suppression of AR Function. Mol. Cell. Endocrinol 2013, 372 (1–2), 12–22. 10.1016/j.mce.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Zhang H-T; Zhang D; Zha Z-G; Hu C-D Transcriptional Activation of PRMT5 by NF-Y Is Required for Cell Growth and Negatively Regulated by the PKC/c-Fos Signaling in Prostate Cancer Cells. Biochim. Biophys. Acta 2014, 1839 (11), 1330–1340. 10.1016/j.bbagrm.2014.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Dobin A; Davis CA; Schlesinger F; Drenkow J; Zaleski C; Jha S; Batut P; Chaisson M; Gingeras TR STAR: Ultrafast Universal RNA-Seq Aligner. Bioinforma. Oxf. Engl 2013, 29 (1), 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Liao Y; Smyth GK; Shi W FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinforma. Oxf. Engl 2014, 30 (7), 923–930. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- (75).Robinson MD; McCarthy DJ; Smyth GK EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinforma. Oxf. Engl 2010, 26 (1), 139–140. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).McCarthy DJ; Chen Y; Smyth GK Differential Expression Analysis of Multifactor RNA-Seq Experiments with Respect to Biological Variation. Nucleic Acids Res 2012, 40 (10), 4288–4297. 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Dennis G; Sherman BT; Hosack DA; Yang J; Gao W; Lane HC; Lempicki RA DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 2003, 4 (5), P3. [PubMed] [Google Scholar]
- (78).Huang DW; Sherman BT; Lempicki RA Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc 2009, 4 (1), 44–57. 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.