

Abstract
INTRODUCTION
Previous approaches pursuing in vivo staging of tau pathology in Alzheimer's disease (AD) have typically relied on neuropathologically defined criteria. In using predefined systems, these studies may miss spatial deposition patterns which are informative of disease progression.
METHODS
We selected discovery (n = 418) and replication (n = 132) cohorts with flortaucipir imaging. Non‐negative matrix factorization (NMF) was applied to learn tau covariance patterns and develop a tau staging system. Flortaucipir components were also validated by comparison with amyloid burden, gray matter loss, and the expression of AD‐related genes.
RESULTS
We found eight flortaucipir covariance patterns which were reproducible and overlapped with relevant gene expression maps. Tau stages were associated with AD severity as indexed by dementia status and neuropsychological performance. Comparisons of flortaucipir uptake with amyloid and atrophy also supported our model of tau progression.
DISCUSSION
Data‐driven decomposition of flortaucipir uptake provides a novel framework for tau staging which complements existing systems.
Highlights
NMF reveals patterns of tau deposition in AD.
Data‐driven staging of flortaucipir tracks AD severity.
Learned flortaucipir patterns overlap with AD‐related gene expression.
Keywords: Alzheimer's disease, AV‐1451, Braak, data‐driven, disease staging, flortaucipir, machine learning, NMF, non‐negative matrix factorization, tau, tau staging
1. BACKGROUND
The accumulation and spread of tau is a key pathological process underlying Alzheimer's disease (AD). Hyperphosphorylation of tau proteins leads to destabilization of microtubules and the formation of intracellular neurofibrillary tangles (NFTs). Presence of NFTs, along with amyloid‐β plaques, constitute the two proteinopathies which define AD. 1 Tau pathology is tightly linked to disease severity, displaying stronger associations with neurodegeneration and cognitive decline than measures of amyloid. 2 , 3 , 4
Tau pathology exhibits a characteristic pattern of spatial spread across the brain as AD progresses. The most commonly used system for histopathological assessment of tau severity is that of Braak and Braak, who assessed tau burden in post mortem brain specimens. 5 , 6 This system describes a progression of tau arising in transentorhinal cortex (stage I), spreading to broader entorhinal cortex and hippocampus (stage II), progressing outward to limbic cortex of the temporal lobe (stage III/IV), and finally infiltrating much of the neocortex, including primary sensory areas (stage V/VI). Recently, tau‐specific positron emission tomography (PET) tracers have enabled in vivo investigation of tau topography and staging. 7 Several studies have measured uptake of tau‐binding tracers in Braak‐like regions of interest (ROIs) and assigned corresponding disease stages. 8 , 9 , 10 , 11 , 12 , 13 These investigations have consistently shown associations between PET‐based Braak stages and disease severity, as indicated by cognitive assessments and other AD‐related biomarkers. However, the dependence on ROIs based on neuropathological studies might pose limitations, given that these studies typically examine a restricted set of preselected brain regions tractable to methods of gross post mortem pathology. Potentially, PET assessments of tau pathology contain different or additional spatial topographies which may be informative in a disease staging context.
Data‐driven methods offer a way to investigate tau‐PET signal across the whole brain without a priori assumptions of where it occurs. Several studies have applied unsupervised machine learning techniques to learn areas where tau‐PET signal covaries. 14 , 15 , 16 , 17 , 18 , 19 Prior work has recapitulated Braak‐like aggregation patterns (especially in the temporal lobe) but has also revealed distinct areas of tau accumulation in non‐Braak areas, such as the frontal and occipital cortex. However, several of these studies have relied on factorization techniques 14 , 15 , 17 , 20 that produce mixed‐sign spatial maps (i.e., components) and (subject‐specific) coefficients, which makes interpretation challenging considering the PET signal is inherently non‐negative. Other approaches have employed clustering techniques 16 , 18 , 19 which provide a hard assignment of regions to clusters, potentially masking the involvement of areas in multiple spatial patterns. Moreover, tau staging based on uptake in learned covarying regions remains largely uninvestigated. Such a system may be important for monitoring the spread of tau in AD while incorporating learned regions of tau accumulation which may lie outside the traditional Braak framework. Notably, recent work used machine learning to develop staging systems for subtypes of tau accumulation. 21 While this work has been crucial for revealing disease‐related heterogeneity, it may be more difficult to operationalize than a single staging system. Furthermore, it was trained on relatively coarse regions (i.e., the four major brain lobes and the medial temporal lobe [MTL]) and may be limited in capturing tau accumulation in smaller brain areas.
Here, we aimed to identify a simple staging model of AD‐related tau pathology that leverages data‐driven covariance patterns of tau‐PET signal. Accordingly, we first applied non‐negative matrix factorization on tau‐PET data from Alzheimer's Disease Neuroimaging Initiative (ADNI) participants spanning the AD spectrum (ADS) to discover interpretable and reproducible patterns of coordinated tau‐PET deposition. We then used the identified patterns to develop a tau pathology staging model, which was subsequently validated using longitudinal imaging and clinical data. Lastly, we examined the covariance patterns in association with other imaging markers in AD and AD‐related gene expression maps. The results were validated in an independent dataset, part of the Open Access Series of Imaging Studies (OASIS‐3). 22 Our results indicate a novel decomposition of tau progression which is informative of AD pathology in the majority of included individuals.
RESEARCH IN CONTEXT
Systematic review: PubMed and Google Scholar were used to identify papers which investigate spatial or temporal patterns in tau pathology using positron emission tomography (PET). Additional studies were found by investigating cited sources, where relevant.
Interpretation: Our results revealed a spatio‐temporal progression of flortaucipir which tracked the severity of cognitive impairment and biomarkers sensitive to Alzheimer's disease (AD). Despite being derived with hypothesis‐independent, data‐driven methods, the patterns we observed were highly reproducible and consistent with existing frameworks for tau staging. These results show predictive features in tau‐PET not previously described by related methods of staging and imaging‐based inference.
Future directions: We derived flortaucipir staging which was specific to sporadic AD. Future investigations would usefully address tau spatial patterns beyond those predictive for the most common presentations of AD, for example, dominantly inherited AD and atypical variants.
2. METHODS
2.1. Overview
We constructed a discovery dataset consisting of individuals from ADNI (Section 2.2) and a replication dataset consisting of individuals from OASIS‐3 22 (Section 2.3). From both cohorts, we selected individuals on the ADS who underwent flortaucipir (FTP) imaging. Note that inclusion in the ADS cohorts was only dependent on amyloid positivity, not clinical or cognitive status. Our analysis consisted of four major steps. First, we applied non‐negative matrix factorization (NMF) to FTP regional uptakes to learn regions of coordinated tau accumulation, which we refer to as Patterns of Tau Covariance (PTCs) (Section 2.4). PTCs were learned using solely the discovery cohort. Second, we derived a staging system for FTP by ordering PTCs according to their respective cross‐sectional frequency of increased tau deposition (Section 2.5‐2.6), similar to previous work. 5 , 23 , 24 , 25 , 26 , 27 , 28 The cross‐sectional ordering of PTCs was evaluated separately in discovery and replication cohorts, while our staging system was developed solely in the discovery cohort. Third, we applied the tau staging system and investigated associations between FTP staging and disease severity (Section 2.6‐2.7). The replication cohort was used to repeat and validate associations observed in the discovery cohort. Finally, we compared the spatial topography of PTCs with expression maps of AD‐related genes to identify genetic markers potentially implicated in tau spread (Section 2.8). Additional methodological details are presented in the Supplementary Methods (SM).
2.2. Discovery dataset (ADNI‐ADS)
2.2.1. Participants
Data used in the preparation of this article were obtained from the ADNI database (adni.loni.usc.edu). The ADNI was launched in 2003 as a public‐private partnership, led by Principal Investigator Michael W. Weiner, MD. The primary goal of ADNI has been to test whether serial magnetic resonance imaging (MRI), PET, other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and early AD.
Our analysis included individuals who underwent tau‐PET imaging, T1‐weighted (T1w) imaging, and amyloid‐PET imaging. Amyloid‐PET imaging was used for determination of amyloid positivity. Most amyloid scans were selected to be within 1 year of the tau acquisition; amyloid scans acquired greater than 1 year before tau were only used to assign amyloid status if they were positive (12 subjects). Two cohorts were selected based on amyloid status (Section 2.2.2 and SM1.1.2). The first cohort consisted of 418 amyloid positive participants on the ADS and was used for the primary investigation of AD‐related tau patterns. The second cohort consisted of 300 control (i.e., cognitively unimpaired [CU], amyloid negative) individuals who were included for normative comparison. We refer to these cohorts as ADNI‐ADS and ADNI‐CU, respectively.
ADNI data were accessed through the ADNIMERGE R package. 29 Data included in this manuscript were downloaded on November 23, 2022. A list of subjects used in this analysis will be made available at https://github.com/sotiraslab/earnest_nmf_tau_staging.
2.2.2. PET imaging
Detailed information on image acquisition and processing in ADNI is provided in the Supplementary Methods (SM1.1). Briefly, all participants underwent tau‐PET imaging with FTP and amyloid‐PET imaging with either florbetapir (AV45) or florbetaben (FBB). PET images were registered to the nearest T1w image and processed with FreeSurfer to derive average standardized uptake value ratios (SUVRs) in ROIs. FTP uptakes in 68 cortical gray matter ROIs were extracted for NMF analyses. Individual amyloid positivity was determined using established cutoffs for each tracer (see SM1.1.2). Amyloid Centiloid values were calculated using previously validated equations. 30
2.2.3. Clinical and cognitive assessments
Assessments were only included if they occurred within 1 year of FTP imaging. Participants were assessed for dementia with the Clinical Dementia Rating (CDR) Scale. 31 As a measure of dementia‐related neuropsychological impairment, we derived Preclinical Alzheimer Cognitive Composite (PACC) scores 32 (see SM1.3.1). We also computed neuropsychological composites for memory, executive functioning, and language (SM1.3.2).
2.3. Replication dataset: OASIS‐3
2.3.1. Participants
We included participants from OASIS‐3 22 who underwent tau‐PET, amyloid‐PET, and T1w imaging. Most amyloid scans were selected to be within 1 year of FTP acquisition; amyloid scans acquired greater than 1 year before FTP were only used to assign amyloid status if they were positive (seven subjects). Like the discovery dataset, two cohorts were selected based on amyloid status (Section 2.3.2 and SM1.2.2). The first cohort consisted of 132 amyloid positive participants on the ADS. The second cohort consisted of 268 control (i.e., CDR = 0, amyloid negative) individuals. We refer to these cohorts as OASIS3‐ADS and OASIS3‐CU, respectively.
2.3.2. PET imaging
Detailed information on image acquisition and processing in OASIS‐3 is provided in the Supplementary Methods (SM1.2). Briefly, all included participants underwent tau‐PET imaging with FTP and amyloid‐PET imaging with either AV45 or Pittsburgh Compound B (PIB). PET and T1w images from the same visit were processed with the PET Unified Pipeline (https://github.com/ysu001/PUP) 33 , 34 to derive average SUVRs in ROIs. FTP uptakes in 68 cortical gray matter ROIs (same regions as in ADNI) were extracted for replication analyses. Individual amyloid positivity was determined using established cutoffs for each tracer (see SM1.2.2). Amyloid Centiloid values were computed using previously validated equations. 35
2.3.3. Clinical and cognitive assessments
Assessments were only included if they occurred within 1 year of FTP imaging. Similar to ADNI, OASIS‐3 participants were assessed for dementia with the CDR Scale 31 and for neuropsychological impairment with the PACC (see SM1.3.1) as well as composites for memory, executive functioning, and language (see SM1.3.2).
2.4. Non‐negative matrix factorization
NMF is an unsupervised machine learning method, which is used for dimensionality reduction and matrix decomposition. 36 NMF has been applied in neuroimaging to identify interpretable covariance structures and reveal patterns in high‐dimensional imaging data. 37 , 38 , 39 , 40 , 41 A detailed description of the formulation of NMF is provided in the Supplementary Methods (SM1.4.1). Briefly, NMF approximates an input data matrix as the product of a component matrix and a weight matrix , such that . The matrix contains components, which capture covarying signal in , and the matrix contains subject‐specific weights for each component. With this decomposition, any individual observation is approximated as a linear combination of components from .
Here, NMF was applied to regional FTP SUVRs of the ADNI‐ADS cohort. Only cross‐sectional, baseline scans were used for NMF training. The input matrix for ADNI‐ADS had a shape of (ROIs by subjects). The learned PTCs (i.e., the components of indicate regions where FTP signal tends to covary across the population. For each subject and PTC, we calculated an SUVR by normalizing the PTC (to sum to 1) and taking its inner product with the vector of the subject's regional SUVRs. Similarly, regional FTP SUVRs were computed for all PTCs in OASIS3‐ADS for replication analyses.
NMF requires the user to specify the number of components to estimate. Consistent with prior studies, 38 , 41 , 42 we varied this parameter (from 2 to 20, step size 1) and estimated multiple NMF solutions. We then applied a model selection procedure which assesses data fit and reproducibility to identify the optimal number of components for describing FTP deposition (see SM1.4.2). For an assessment of replicability across datasets, we compared ADNI‐ADS components to components estimated in OASIS3‐ADS at the same dimensionality using indices of spatial similarity (see SM1.4.3).
2.5. Determination of FTP progression across PTCs
Like approaches previously employed in neuropathologic and neuroimaging staging studies, 5 , 23 , 24 , 25 , 26 , 27 , 28 we estimated a model of tau progression based on the frequency of elevated FTP uptake in each PTC. Specifically, we assumed that the proportion of participants who exhibit tau pathology in each PTC is an indicator of the PTC's involvement in spatially progressing tau pathology. Accordingly, the earliest areas of tau accumulation would be detected as elevated at a higher frequency than areas of later tau accumulation. This approach depends on identifying PTCs with elevated tau pathology. Accordingly, we used a modified W‐score approach 25 to assign a binary tau status (elevated/not elevated) to each PTC for each subject in the ADNI‐ADS cohort (SM1.5.1). Following prior work, W‐scores were thresholded at 2.5 to determine PTC‐wise tau positivity. 25 Importantly, we conducted a sensitivity analysis to evaluate the effect of W‐score thresholds on PTC ordering and subsequent staging (SM1.5.2). To assess the generalizability of the FTP progression pattern we determined, we repeated this process in the OASIS3‐ADS dataset.
2.6. FTP staging system
2.6.1. Staging system
We next developed a staging system for gauging an individual's position in our estimated progression of tau pathology. We first aimed to group our input regions (PTCs) into a smaller number of tau stages to enable a more concise staging system (fewer stages). Prior methods for developing novel PET staging systems have used heuristic strategies for determining the number of pathological stages such as placing a similar number of regions in each stage or applying quantiles to the frequencies of positivity. 24 , 27 These methods require the developer to either arbitrarily pick the number of stages or select from several tested systems based on some criterion. We instead aimed to apply a data‐driven method which does not require a priori selection of the number of stages. To this end, we developed a bootstrap method which groups regions (PTCs) into larger stages based on statistical assessment of the differences in tau‐positivity between regions (SM1.6.1). This method groups regions with similar frequencies into stages, and automatically determines the number of stages in the final model.
Our staging system was estimated using the ADNI‐ADS dataset and was subsequently applied to both ADNI‐ADS and OASIS3‐ADS. To assign a stage to an individual subject, we determined if their FTP scan exhibited elevated tau levels in any of the corresponding PTCs associated with each stage. Scans had to be positive for all prior stages to meet the criteria for a given stage; scans positive in a later stage but negative in an earlier stage were labeled as nonstageable (NS).
2.6.2. Relationship between tau staging and cross‐sectional markers of AD
We assessed differences in the distribution of tau stages with respect to CDR status, amyloid pathology (Centiloid values binned at < 40, 40–60, 60–80, 80–100, > 100), and apolipoprotein E (APOE) genotype (has at least one E4 allele vs. none) using Chi‐squared tests. We additionally used a one‐way analysis of variance (ANOVA) to compare PACC scores across FTP stages. Tukey's method was used for computing post hoc significant differences in PACC between stages. Associations were investigated separately in ADNI‐ADS and OASIS3‐ADS datasets.
2.6.3. Relationship between tau staging and longitudinal clinical progression
We examined associations between tau stages and clinical progression in both ADNI‐ADS and OASIS3‐ADS using survival analyses. We modeled progression to CDR >= 1 as the event of interest. Subjects were only included in this analysis if they were CDR < 1 at their baseline FTP scan and had available longitudinal CDR assessments. Log‐rank tests were used to assess the model fit and pairwise differences between progression risk for tau stages. Post hoc comparisons were false discovery rate (FDR) corrected. 43
2.6.4. Longitudinal validity of tau staging
We used longitudinal FTP scanning to test the longitudinal validity of the cross‐sectional tau staging scheme. Specifically, we examined whether longitudinal stage transitions adhered to the previously derived staging scheme of regional tau pathology progression. A permutation test was used to statistically evaluate the stability of staging (SM1.6.2). Additionally, we estimated regional rates of tau accumulation using linear mixed effect models to test if longitudinal tau change was related to staging (SM1.6.3).
2.6.5. Comparison with Braak staging
We also staged all ADNI‐ADS and OASIS3‐ADS participants with a Braak‐based approach in order to evaluate how our proposed system compared to an established alternative for assessing AD‐related tau. We adopted previous protocols for constructing Braak regions of interest from the Desikan–Killiany atlas 11 , 44 (see SM1.7 for more detail). Uptakes in Braak regions were converted to binary measures using the same W‐scoring procedure described in the supplement (SM1.5.1). Individuals were marked as NS if they were positive for a given Braak stage but not all prior stages.
We then conducted analyses which directly compared our proposed tau staging model and the Braak system. First, we replicated analyses evaluating the distributions of CDR, Centiloid, APOE status, and PACC, but we used Braak stages instead of the staging model presented in the main text. We then computed effect sizes for these associations under both staging models (Cramér's V for chi‐squared tests, eta‐squared for one‐way ANOVAs) and compared the strength of associations with each. Second, we ran a supervised machine learning experiment to test how well each staging model performed in classification of dementia status (SM1.7.2).
2.6.6. Investigations of NS individuals
Finally, we conducted analyses to characterize individuals found to be NS with our proposed system. First, we ran linear models to compare demographic (age), cognitive/clinical (PACC, Mini‐Mental State Examination [MMSE]), and biological (global amyloid uptake, global tau uptake, tau uptake in PTCs, tau laterality) measures between individuals who had stageable tau pathology (i.e., not stage 0 or NS) and those who were NS. For models including tau measures in PTCs, global tau uptake was included as a covariate to identify regional differences in tau deposition between stageable and NS individuals. These linear models were FDR‐corrected. 43 Second, we manually identified the tau presentations which resulted in individuals being NS (e.g., positive for stage 2 but not stage 1).
2.7. Association of regional tau with other AD‐related imaging markers
2.7.1. Relationship between FTP uptake in PTCs and global amyloid burden
We examined the relationship between regional estimates of tau deposition and global amyloid burden as quantified by Centiloid scale values. In ADNI‐ADS, linear regressions were used to model the association between Centiloid and tau uptake in each PTC (as measured by W‐scores) with age and sex as additional covariates. We compared the estimated marginal means of each pair of PTCs to test for differences in regional associations between tau and global amyloid burden. FDR correction was applied to correct for multiple comparisons. 43 This analysis was then repeated in the OASIS3‐ADS cohort.
2.7.2. Intra‐ and inter‐regional relationship between FTP and amyloid uptake
We further explored the relationship of tau and amyloid by comparing the uptake of each within and between PTCs. In each ADS dataset, we calculated amyloid uptake in each PTC by projecting the regional amyloid SUVRs onto the rescaled NMF component matrix. Partial correlations were used to measure the association between FTP and amyloid uptake in each pair PTCs, with age included as a covariate. p‐values for all comparisons were FDR corrected. 43 These analyses were run separately for each amyloid tracer.
2.7.3. Intra‐ and inter‐regional relationship between FTP and gray matter volume
In each ADS dataset, we performed a cross‐modality PTC‐based correlation analysis to assess the association of regional FTP accumulation with regional gray matter volume. Regional gray matter volumes estimated by FreeSurfer were provided by each dataset for the T1w image used in processing each FTP image. The average gray matter volume within each PTC was calculated by projecting the cortical ROI volumes onto the rescaled NMF component matrix. Partial correlations were used to measure the association between FTP and gray matter volume in each pair of PTCs, with age included as a covariate. p‐values for all comparisons were FDR corrected. 43
2.8. Spatial overlap with AD genes
Last, we sought to investigate whether PTCs overlapped with the expression of 42 genes linked to single nucleotide polymorphisms (SNPs) shown to be associated with AD in a recent genome‐wide association study (GWAS). 45 We used abagen (version 1.3) to access the respective gene expression cortical maps from the Allen Human Brain Atlas. 46 , 47 , 48 Of the 42 genes, 45 expression data were available for 35 of them. Parameters for sampling gene expression were set to the defaults provided by abagen. Notably, no mirroring of hemisphere data was performed, allowing us to investigate gene expression associations with asymmetric PTCs. We used neuromaps software (version 0.0.3) to statistically test the spatial association of PTCs with each gene. 49 For each PTC, a spatial null map 50 was created with 5000 permutations. The spatial overlap of each gene and the PTC was compared to this null distribution to derive a Pearson correlation and p‐value. p‐values were corrected for multiple comparisons using the FDR method. 43
2.9. Code availability
NMF was implemented in MATLAB (https://github.com/asotiras/brainparts). All other analyses were conducted in either R or Python. Code for this project will be made available at the following repository: https://github.com/sotiraslab/earnest_nmf_tau_staging.
3. RESULTS
3.1. Dataset characteristics
Descriptive statistics for the ADNI‐ADS and OASIS3‐ADS cohorts are provided in Table 1. The distribution of age and sex between cohorts was not equal, with relatively more females in OASIS3‐ADS (X2 = 3.96, df = 1, p = 0.047) and older individuals in ADNI‐ADS (t = 2.87, df = 267.73, p = 0.004). The ADNI‐ADS cohort had generally more advanced disease than OASIS3‐ADS, as indicated by lower MMSE scores (t = −2.66, df = 246.40, p = 0.008) and a higher proportion of individuals with CDR > 0 (X2 = 14.45, df = 2, p = 0.001). However, while amyloid burden in ADNI‐ADS was higher on average than OASIS3‐ADS, the difference was not statistically significant (t = 1.74, df = 235.07, p = 0.082). The difference in APOE genotype (based on presence of at least one E4 allele) was also not different across cohorts (X2 = 0.43, df = 1, p = 0.514).
TABLE 1.
Descriptive statistics for the ADNI‐ADS and OASIS3‐ADS datasets.
| Parameter | ADNI‐ADS | OASIS3‐ADS | p‐value |
|---|---|---|---|
| N | 418 | 132 | |
| Age | 75.47 (7.80) | 73.56 (6.32) | 0.004** |
| Sex (M / F) | 205 / 213 | 51 / 81 | 0.047* |
| CDR (0.0 / 0.5 / 1.0+) | 187 / 179 / 52 | 84 / 36 / 12 | < 0.001*** |
| MMSE | 27.00 (3.49) | 27.84 (3.07) | 0.008** |
| Centiloid | 71.98 (37.10) | 65.87 (34.66) | 0.082 |
| APOE (E4+ / E4‐ / NA) | 216 / 159 / 43 | 81 / 51 / 0 | 0.514 |
Note: All continuous variables (Age, MMSE, Centiloid) are represented as mean (standard deviation). p‐values for tests of statistical difference between the two datasets are shown in the last column. Continuous variables were tested with two‐sided t‐tests; categorical variables were tested with chi‐squared tests.
Abbreviations: APOE, apolipoprotein E; CDR, Clinical Dementia Rating; MMSE, Mini‐Mental State Exam; NA, missing.
Descriptive statistics for the ADNI‐CU and OASIS3‐CU cohorts are provided in Table S1. When comparing each CU cohort to their respective ADS counterpart (i.e., ADNI‐ADS vs. ADNI‐CU; OASIS3‐ADS vs. OASIS3‐CU), the CU cohorts differed in the expected direction, exhibiting lower age, higher MMSE scores, lower amyloid burden, and less APOE risk (all p < 0.001). Only the distribution of sex was similar across ADS/CU cohorts in ADNI (X2 = 2.90, df = 1, p = 0.089) and OASIS (X2 = 0.84, df = 1, p = 0.358). Between datasets, the ADNI‐CU and OASIS3‐CU only differed in terms of age, with higher age in ADNI‐CU (t = 5.11, df = 524.43, p < 0.001). There was a trend toward worse MMSE scores in ADNI‐CU relative to OASIS3‐CU, though this difference was not significant (t = −1.81, df = 562.84, p = 0.071).
3.2. NMF identifies eight regions of coordinated FTP deposition
NMF model selection analyses indicated eight components as the optimal dimensionality for describing FTP uptake in ADNI‐ADS. From repeated split‐half training of NMF in ADNI‐ADS, we observed sharp peaks in reproducibility metrics for the two‐ and eight‐component solutions (Figure S1A and B). Furthermore, the curve of reconstruction error against number of components displayed an elbow at eight PTCs, indicating that solutions beyond eight components offered relatively lower gains in approximation (Figure S1C and D).
We focused on the eight PTC solution for subsequent analyses of tau progression in the main text (Figure 1). We assigned shorthand names to refer to these PTCs, based on their approximate anatomical coverage. The frontal lobe was spanned by PTC6‐LateralFrontal and PTC8‐Oribitofrontal, the former capturing primarily lateral superior areas and the latter capturing more medial and insular cortex. The pre‐ and postcentral gyri were captured in a single component, PTC7‐Sensorimotor. PTC4‐Precuneus incorporated medial parietal cortices, particularly the precuneus and posterior cingulate. Lateral parietal cortices were instead grouped into components PTC2‐LeftParietalTemporal and PTC3‐RightParietalTemporal, which also included lateral aspects of the temporal lobe. PTC1‐MedialTemporal separately highlighted entorhinal and parahippocampal cortices. Finally, posterior cortices were captured in PTC5‐Occipital. Despite being trained on unilateral ROIs, most learned PTCs were highly symmetrical. The major exception to this was PTC2‐LeftParietalTemporal and PTC3‐RightParietalTemporal, which were constrained to left and right hemispheres (and symmetrical with each other). This pattern of symmetry indicates lateralized tau deposition in parietal and temporal cortices and bilateral deposition in other brain regions.
FIGURE 1.
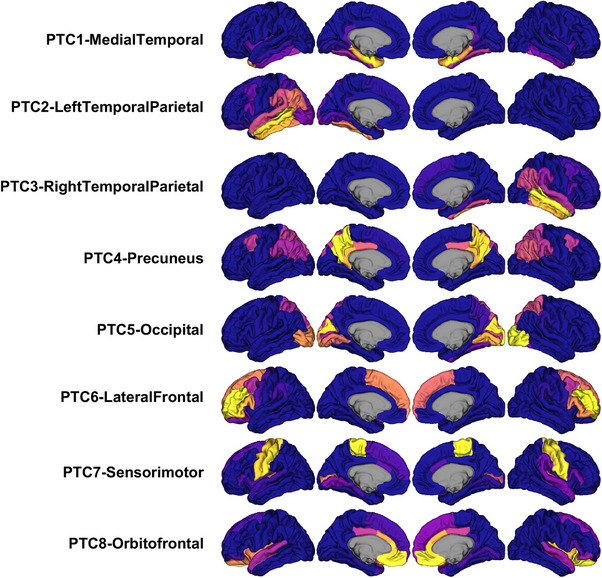
Representations of the eight PTCs derived with NMF. Each PTC (one per row) indicates regions where FTP signal covaries across the dataset. Within each PTC, hotter regions (yellow/orange) indicate regions of stronger correlated FTP change. PTCs were estimated in the discovery cohort (ADNI‐ADS). FTP, flortaucipir; NMF, non‐negative matrix factorization; PTC, Pattern of Tau Covariance.
We found reasonable similarity between the ADNI‐ADS PTCs and components estimated in OASIS3‐ADS (Figure S2; Adjusted Rand Index = 0.513, mean inner product = 0.710). Particularly, the medial temporal, temporal‐parietal, occipital, and sensorimotor PTCs had clear analogues in OASIS3‐ADS. A spatial permutation test indicated significant similarity of the NMF decompositions estimated separately in ADNI‐ADS and OASIS3‐ADS (p < 0.001).
3.3. Cross‐sectional staging identifies a reproducible model of tau progression
We next aimed to estimate the temporal ordering of tau spread by examining the frequency of elevated FTP signal across each PTC. In ADNI‐ADS, tau deposition was most frequently observed in medial temporal cortex (PTC1‐MedialTemporal: 45.7% of ADNI‐ADS), followed by broader temporal and parietal cortices (PTC3‐RIghtParietalTemporal: 38.3%, PTC2‐LeftParietalTemporal: 37.8%, PTC4‐Precuneus: 36.4%), occipital cortex (PTC5‐Occipital: 28.2%), and last frontal and sensorimotor areas (PTC6‐LateralFrontal: 26.8%, PTC7‐Sensorimotor: 21.1%, PTC8‐Orbitofrontal: 20.8%) (Figure 2A and C). We observed a very similar hierarchy of tau deposition in OASIS3‐ADS despite lower levels of tau pathology on average (Figure 2B and D). Like ADNI‐ADS, tau elevation was most frequently seen in medial temporal areas (PTC1‐MedialTemporal: 33.3% of OASIS3‐ADS) followed by temporal and parietal cortices (PTC3‐RightParietalTemporal: 28.8%, PTC2‐LeftParietalTemporal: 28.0%, PTC4‐Precuneus: 22.0%), occipital cortex (PTC5‐Occipital: 13.6%), and was least frequently observed in frontal and sensorimotor areas (PTC6‐LateralFrontal: 10.6%, PTC7‐Sensorimotor: 10.6%, PTC8‐Orbitofrontal: 9.8%).
FIGURE 2.
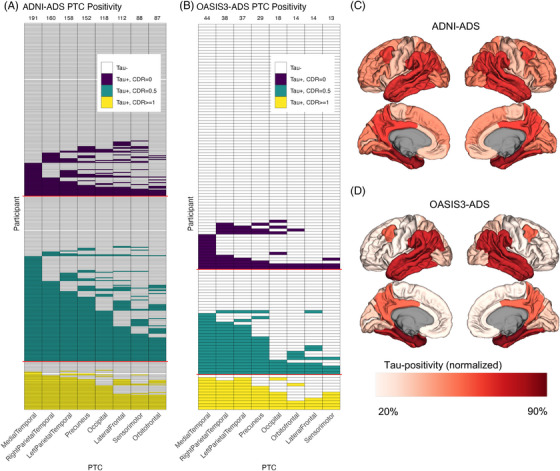
Determination of the spread of tau using a cross‐sectional modeling approach. (A) Plot showing the distribution of elevated FTP signal in each PTC for the ADNI‐ADS cohort. Each row indicates one participant, and each column represents one PTC. Filled cells indicate elevated FTP signal in a PTC for a given participant, while empty cells (white) indicate nonelevated FTP. A W‐score method was used to derive PTC positivity. Participants with elevated FTP are colored according to their CDR status (purple: CDR = 0, teal: CDR = 0.5, yellow: CDR = 1.0+). The frequency of elevated signal (i.e., the number of filled cells) in each PTC is used to order the columns from left to right (frequencies are shown as numbers above each column). (B) Same plot as (A), but in OASIS3‐ADS. New W‐score models were calculated for this cohort, and ordering of PTCs is based on the frequency of positivity in OASIS3‐ADS. (C) Brain map showing the rate of positivity in each PTC for ADNI‐ADS. (D) Same plot as (C), but in OASIS3‐ADS. For (C and D), each region was linked to a single PTC by normalizing the PTCs to sum to 1 and then using winner‐take‐all assignment. The fill represents the percent of individuals with elevated FTP in that region, relative to the total number of participants with elevated FTP in any region (ADNI‐ADS: 225, OASIS3‐ADS: 51). CDR, Clinical Dementia Rating; FTP, flortaucipir; PTC, Pattern of Tau Covariance.
3.4. PTC‐based tau staging tracks AD severity
3.4.1. Staging development and comparison with cross‐sectional markers of AD
Next, we defined a data‐driven staging model of regional tau progression by grouping PTCs into larger anatomical divisions (Figure 3A). Application of this process in ADNI‐ADS resulted in a four‐stage system of tau pathology, which captured the majority of participants in the dataset (90.0% stageable, 10.0% NS) (Figure 3B). Individuals with no tau elevation were assigned to stage 0 (45.5%). Stage 1 consisted of solely medial temporal tau (5.7% of ADNI‐ADS), stage 2 indicated tau spread throughout the temporal and parietal lobe (6.9%), stage 3 incorporated occipital and lateral frontal areas (8.4%), and stage 4 spanned the remaining orbitofrontal, insular, and sensorimotor areas (23.4%).
FIGURE 3.
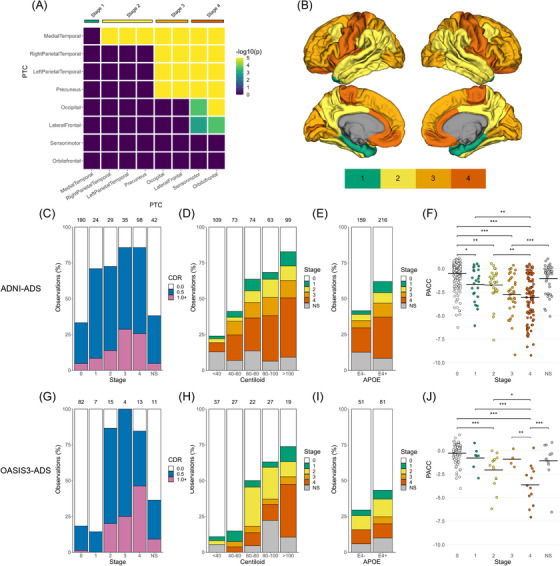
Development of an FTP staging system and comparison with cross‐sectional measures of AD severity. (A) PTCs were grouped into stages using a bootstrapped significance test for differences in rates of FTP positivity. (B) Maps showing the brain regions corresponding to each FTP stage. (C‐E) Bar plots showing the distributions of CDR status across FTP stages (C) and FTP stages across Centiloid (D) and APOE status (E) in the discovery cohort (ADNI‐ADS). (F) Swarm plots showing the distribution of PACC scores across FTP stages. An ANOVA was used to test the effect of staging on PACC scores, with FDR‐corrected post hoc Tukey comparisons used to assess pairwise stage differences (*p < 0.05, **p < 0.01, ***p < 0.001). (G‐J) Same as (C‐F), but with the replication cohort (OASIS3‐ADS). ANOVA, analysis of variance; APOE, apolipoprotein E; CDR , Clinical Dementia Rating; FDR, false discovery rate; FTP, flortaucipir; NS, nonstageable; PACC, Preclinical Alzheimer's Cognitive Composite; PTC, Pattern of Tau Covariance.
We investigated the stability of this staging system in a sensitivity analysis where the threshold for tau‐positivity was varied. We found that the ordering of PTCs (derived from tau‐positivity frequencies) was highly stable across the thresholds tested, with variations only occurring within regions grouped into the same stage (Figure S3A and B). Moreover, the estimated staging system was consistent, displaying either the model reported above or a similar model with an additional stage for precuneus deposition (after temporal and prior to frontal/occipital involvement) (Figure S3C). This alternative five‐stage model resulted in highly consistent groupings of individuals relative to the four‐stage system we report on herein (ARI for ADNI‐ADS: 0.95, ARI for OASIS3‐ADS: 0.98), even when omitting individuals who were stage 0 (ARI for ADNI‐ADS: 0.83, ARI for OASIS3‐ADS: 0.78).
We next validated the utility of our staging system by comparing tau stages with cross‐sectional variables related to AD. We found that our tau staging was associated with measures assessing clinical status, cognitive performance, amyloid burden, and genetic disease risk in ADNI‐ADS. CDR status was significantly associated with tau stage (X2 = 109.03, df = 10, p < 0.001), increasing in severity with advancing tau stage (Figure 3C). Most stage 0 individuals were CDR = 0, while in all other stages the majority of individuals were CDR > 0. Tau staging was also related to global amyloid burden (X2 = 109.6, df = 20, p < 0.001), increasing on average with Centiloid (Figure 3D and S4). Furthermore, APOE status was associated with staging (X2 = 23.8, df = 5, p < 0.001), with E4+ individuals showing more advanced tau than E4‐ individuals (Figure 3E). Finally, we investigated differences in PACC, which was significantly associated with tau staging (F = 32.31, df = 5, p < 0.001). Post hoc comparisons between stages revealed a progressive increase in cognitive impairment as the tau stage advanced (Figure 3F, Table S2). Further investigation of neuropsychological domains indicated that tau stages were associated with memory (F = 25.8, df = 5, p < 0.001), executive function (F = 11.85, df = 5, p < 0.001), and language (F = 10.87, df = 5, p < 0.001) scores (Figure S5A‐C). Specifically, memory deficits were observed as early as stage 1, while executive function impairments were not seen until stages 3 and 4. Furthermore, the pattern of deficits across stages corresponded with the gross functional role of implicated brain regions, that is, memory deficits were observed after tau involvement in the MTL and hippocampus, while impairments in executive function and language were only observed after tau deposition in the frontal lobe.
We next assessed the generalizability of our staging system by applying it in OASIS3‐ADS. As in the discovery cohort, most OASIS3‐ADS subjects were stageable (stage 0: 62.1%, stage 1: 5.3%, stage 2: 11.4%, stage 3: 3.0%, stage 4: 9.8%, NS: 8.3%). Additionally, we were able to reproduce most associations with tau staging when examining the same cross‐sectional markers of AD in OASIS3‐ADS (Figure 3G‐J). Like ADNI‐ADS, CDR status (X2 = 63.0, df = 10, p < 0.001), amyloid burden (X2 = 58.7, df = 20, p < 0.001), and PACC was associated with increased tau staging (Table S3, F = 16.11, df = 5, p < 0.001). APOE status was not significantly related to tau staging in OASIS3‐ADS (X2 = 4.6, df = 5, p = 0.473). While memory (F = 3.5, df = 5, p < 0.001), executive functioning (F = 17.7, df = 5, p < 0.001), and language (F = 7.6, df = 5, p < 0.001) scores were associated with staging in OASIS3‐ADS, statistically significant deficits (relative to stage 0) were only observable in stage 4 individuals (Figure S5D‐F).
3.4.2. Tau staging predicts longitudinal risk for dementia
We next used our tau staging to model risk for disease progression using survival analyses (Figure 4A and B). Longitudinal CDR data were available for 326 individuals in ADNI‐ADS who had none or very mild dementia at baseline (CDR = 0 or CDR = 0.5; 181 stage 0, 22 stage 1, 25 stage 2, 25 stage 3, 73 stage 4) and 110 similar individuals in OASIS3‐ADS (81 stage 0, 7 stage 1, 12 stage 2, 3 stage 3, and 7 stage 4). Data from 40 NS individuals in ADNI‐ADS and 10 NS individuals in OASIS3‐ADS were omitted (see Figure S6 for the same figures with their inclusion). In ADNI‐ADS, the baseline FTP stage had a significant effect on the conversion to mild or more severe dementia (CDR >= 1; p < 0.001; Figure 4A). Individuals in tau stages 2 (p < 0.001), 3 (p < 0.01), and 4 (p < 0.001) exhibited a higher progression risk compared to those in stage 0. Additionally, stage 4 individuals showed more severe progression than stage 1 individuals (p < 0.05) (Table S4). Repeating the survival analysis in OASIS3‐ADS reproduced this association between tau staging and dementia conversion (p < 0.001, Figure 4B). Similarly to ADNI‐ADS, individuals in tau stages 2 (p < 0.01), 3, (p < 0.05), and 4 (p < 0.001) exhibited a faster progression to dementia compared to those in stage 0 (Table S5). Furthermore, stage 4 demonstrated a quicker progression to dementia compared to stage 1 (p < 0.05) and stage 2 (p < 0.05).
FIGURE 4.
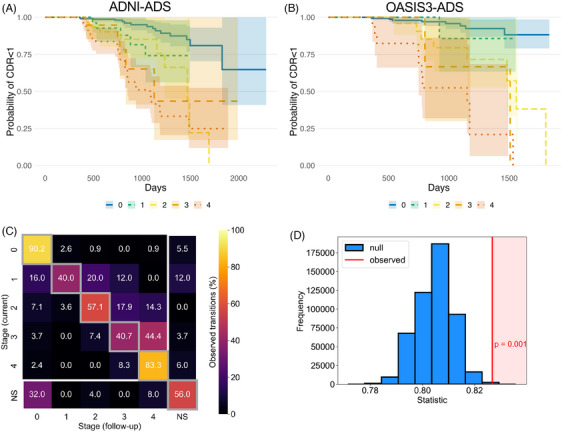
Assessment of FTP staging with longitudinal clinical and imaging data. (A) A survival analysis evaluated the risk of progressing to dementia (CDR >= 1) for each FTP stage in ADNI‐ADS. Nonstageable (NS) individuals are omitted for visual clarity (see Figure S6). (B) Same as (A), but with OASIS3‐ADS. (C) Heatmap showing the longitudinal progression of FTP staging in ADNI‐ADS. For all individuals with longitudinal FTP scanning, we tabulated the stage labels for all successive scan pairs. Rows indicate the stage of the current scan while columns indicate the stage of the follow‐up scan. The counts within each row are normalized to percentages. Boxes on the diagonal (gray squares) represent no change in stage between visits. Boxes above the diagonal represent increasing stage between visits, while boxes below the diagonal represent reverting stage (omitting the NS scans shown in the last row and column). (D) A permutation test was applied to the stage transition data (C) to evaluate longitudinal FTP staging. The statistic of interest was the proportion of scan pairs where the stage increases or stays the same, corresponding to the upper triangle of the heatmap matrix (including the diagonal but omitting the last column). The histogram shows the simulated null distribution of the statistic, while the red line shows the observed statistic and p‐value. Longitudinal FTP staging analysis was only conducted in ADNI‐ADS, due to a lack of data in OASIS3‐ADS. CDR, Clinical Dementia Rating; FTP, flortaucipir; NS, nonstageable.
3.4.3. Longitudinal FTP imaging follows tau staging
We compared our estimated progression of tau pathology with longitudinal FTP scanning available in ADNI. In ADNI‐ADS, 356 subjects (223 stage 0, 17 stage 1, 16 stage 2, 17 stage 3, 54 stage 4, 29 NS) had FTP imaging following baseline (minimum follow‐up scans = 1, maximum follow‐up scans = 5, mean = 1.57). In ADNI‐ADS, 74.8% of FTP scans had the same stage at the next visit, while 7.8% showed a model‐conforming transition to a higher stage and 3.3% showed a model‐violating decrease in stage (13.9% either were either NS initially or NS at follow‐up) (Figure 4C). The greatest proportion of reversions occurred at the earliest stages (16.0% of stage 1 individuals were stage 0 at follow‐up), while the greatest proportion of progressions occurred at later stages (44.4% of stage 3 individuals were stage 4 at follow‐up). A permutation method applied to ADNI‐ADS FTP follow‐up scans supported the notion that our tau stages progressed in the expected direction with longitudinal follow‐up (p = 0.001, Figure 4D). Similar results were found when using only the first and last tau scan instead of all consecutive tau visits (Figure S7).
Investigation of continuous rates of tau change provided further indication that the staging model captured the hierarchy of advancing tau accumulation. Most individuals with stageable (stage 1–4) and NS tau showed increased longitudinal tau accumulation across the brain relative to individuals without tau pathology (stage 0) (Figure S8, blue stars). However, NS individuals did not exhibit faster MTL‐tau uptake than stage 0 individuals (p > 0.05). Additionally, individuals assigned a given stage showed increased tau accumulation in the ROI corresponding to their stage, relative to individuals in the preceding stage (Figure S8, red stars). That is, stage 1 individuals showed faster rates of tau accumulation in the MTL relative to stage 0 individuals; differences were also observed in other brain regions but were largest in the MTL (Figure S8B). Stage 2 individuals accumulated tau more rapidly in temporal and parietal areas (relative to stage 1 individuals, Figure S8C), while stage 3 individuals accumulated tau more rapidly in frontal and occipital regions (relative to stage 2, Figure S8D). Stage 4 individuals were the only individuals to show increased longitudinal deposition in sensorimotor and orbitofrontal regions relative to the previous stage (Figure S8E).
3.4.4. Head‐to‐head comparison with Braak staging
We compared our proposed staging model to a PET‐based Braak staging protocol. We found similar amounts of NS individuals using each method in both ADNI‐ADS (our system: 10.0%, Braak stage: 13.4%) and OASIS3‐ADS (PTC stage: 8.3%, Braak stage: 6.1%). Like our staging model, Braak staging exhibited significant associations with CDR, Centiloid, APOE genotype, and PACC in ADNI‐ADS (Figure S9A‐D, all p < 0.001). These associations were replicated in OASIS3‐ADS (Figure S9E‐H), except for APOE genotype which was nonsignificant (p = 0.077). Comparison of effect sizes between the two staging systems did not provide a clear indication that one system exhibited stronger associations with AD‐related variables of interest (Table S6).
However, the proposed staging model generally surpassed Braak staging when testing the utility of each system to perform dementia classification in a set of supervised machine learning experiments (Table S7). Using only tau stages as features, our staging model exhibited significantly higher accuracy in predicting CDR status in both ADNI‐ADS (t = 2.087, p = 0.039) and OASIS3‐ADS (t = 6.961, p < 0.001). When including covariates along with tau stages, the two models performed equally in ADNI‐ADS (t = 0.227, p = 0.821). However, our model still outperformed Braak in OASIS3‐ADS with covariates included (t = 11.617, p < 0.001).
3.4.5. Non‐stageable individuals are characterized by MTL‐sparing tau deposition
Further characterization of NS individuals indicated that most of these tau presentations involved neocortical tau deposition without MTL involvement. In ADNI‐ADS, 37/42 NS individuals did not exhibit MTL (stage 1) positivity but showed tau positivity in other stages; of these cases, 28/37 showed temporal and parietal (stage 2) pathology while 9/37 did not. The remaining 5/42 cases showed tau positivity in all stages except stage 3, indicating a lack of tau involvement in frontal and occipital areas. In OASIS3‐ADS, we also found 6/11 NS individuals were negative for MTL (stage 1) pathology. Four of the remaining five NS individuals showed positivity for all stages except stage 3 (frontal/occipital); one individual was instead positive for stages 1 (MTL) and 3 (frontal/occipital), but not 2 (lateral temporal/parietal).
In ADNI‐ADS, direct comparison of individuals with stageable pathology and NS individuals (Table S8) indicated that the former group had worse cognitive impairment (PACC: p < 0.001) and more severe dementia (MMSE: p = 0.001), as well as more global amyloid (Centiloid: p = 0.001) and global tau deposition (total average cortical tau: p < 0.001). The two groups did not differ significantly in age (p = 0.130). Adjusting for total tau deposition, we found that NS individuals had significantly less tau uptake in PTC1‐MedialTemporal (p < 0.001), PTC6‐LateralFrontal (p = 0.032), and PTC7‐Sensorimotor (p = 0.002). Comparisons were repeated in OASIS3‐ADS, but no significant differences were found (all p > 0.05).
3.5. Regional differences in associations between tau, amyloid, and gray matter
We next investigated how continuous accumulation of FTP in each PTC related to other imaging markers of AD progression, namely amyloid‐PET and gray matter volume. In ADNI‐ADS, regression modelling indicated FTP uptake was positively correlated with Centiloid (R2 = 0.219, F = 54.83, p < 0.001; Figure 5A). Post hoc comparisons of estimated marginal means indicated significant differences in the strength of the amyloid‐FTP association across PTCs (Table S9, Figure S10A). The strength of association between amyloid and FTP uptake in each PTC was roughly inversely related to the cross‐sectional ordering of PTCs, that is, strongest in temporal and parietal areas, weakest in frontal and sensorimotor areas. These findings were reproducible in OASIS3‐ADS: tau deposition was associated with amyloid burden (R2 = 0.299, F = 26.07, p < 0.001; Figure 5B) and the strength of this association within PTCs was roughly inversely proportional to its position within our estimated tau progression model (Table S10, Figure S10B). The strength of the association between tau signal and gray matter loss was also related to the cross‐sectional ordering of PTCs (Figure 5B and D). While significant negative associations between FTP and volume were present for nearly all intra‐ and inter‐regional comparisons across both ADNI‐ADS and OASIS3‐ADS, relationships were strongest when considering the volume in areas estimated to be earlier in the tau progression (Table S11 and S12).
FIGURE 5.
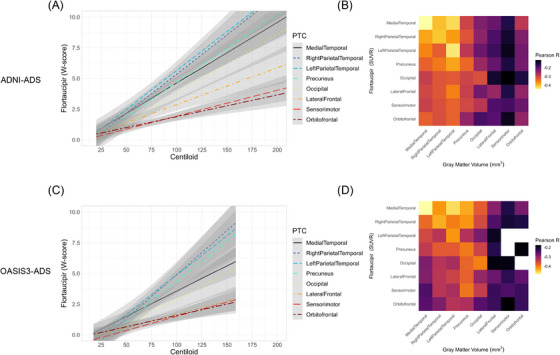
Comparison of FTP uptake in PTCs with amyloid burden and gray matter loss. (A) In ADNI‐ADS, linear regressions were used to model FTP accumulation with increasing Centiloid in each PTC. Tau uptakes are measured as W‐scores in each PTC. (B) Heatmap showing partial correlations between FTP uptake and gray matter volume in all pairs of PTCs. Cells on the diagonal represent correlations between FTP and gray matter volume in the same PTC, while off‐diagonal cells represent inter‐regional associations (rows = FTP, columns = gray matter). The fill is proportional to the partial correlation (adjusted for age); correlations which were not significant after correction for multiple comparisons are filled with white. (C‐D) Same as (A‐B), but with OASIS3‐ADS. FTP, flortaucipir; PTC, Pattern of Tau Covariance.
In contrast to volume, we observed statistically significant positive correlations between amyloid and tau in (and between) most PTCs across both ADNI‐ADS (Figure S11A and C) and OASIS3‐ADS (Figure S12A and C). While there was some indication of stronger associations when considering tau in PTCs modeled to have earlier tau accumulation, the gradient was not as clear as that seen in associations between tau and atrophy. These analyses also indicated an asymmetry in amyloid deposition, with stronger associations observed when considering amyloid in PTC3‐RightParietalTemporal than PTC2‐LeftParietalTemporal. Further investigation of the distribution of tau and amyloid in these PTCs indicated that this pattern was caused by lower deposition of amyloid in PTC2‐LeftParietalTemporal; this was observable across tracers and datasets (Figure S11B and D; Figure S12B and D).
3.6. Overlap of PTCs and AD‐related gene expression
We compared the spatial distribution of PTCs with the expression of genes linked to SNPs identified in a recent GWAS study. Of the 35 genes assessed, 14 had significant spatial correlation with at least one PTC (Figure 6). Most significant correlations were found with PTC1‐MedialTemporal (12/17), but there were also three genes associated with PTC5‐Occipital (CD33, HAVCR2, TREM2), one with PTC7‐Sensorimotor (TNIP1), and one with PTC8‐Orbitofrontal (TNIP1). The strongest association found was between APOE expression and PTC1‐MedialTemporal (R = 0.822, p = 0.014). Both positive and negative correlations between PTC intensity and gene expression were found, with most (9/12) associations with PTC1‐MedialTemporal being positive and most with other components being negative (4/5).
FIGURE 6.
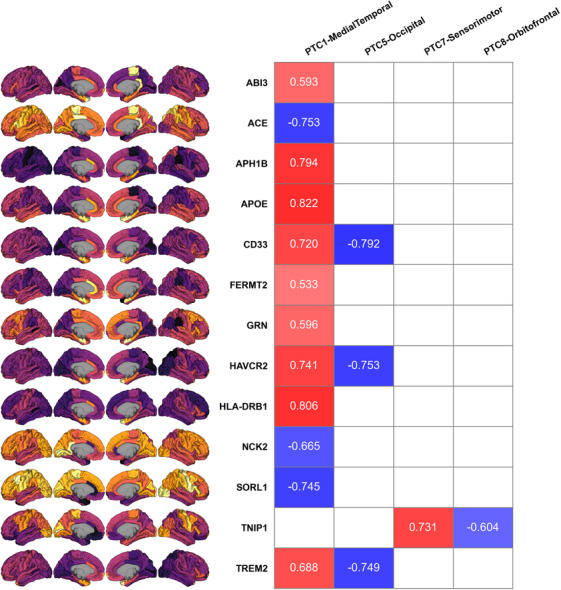
Comparison of PTCs with expression maps for AD‐related genes. Numbers in the table are Pearson R coefficients for significant associations between genes (rows) and PTCs (columns) (0.01 < p < 0.05 for all p‐values). Redder coloring indicates stronger positive correlation, while bluer coloring indicates stronger negative correlation. Nonsignificant associations are shown as empty cells. Brain panels on the left show Allen Brain Atlas gene expression for each gene. No significant associations were found for four of the eight PTCs (PTC2, PTC3, PTC4, and PTC6) and are not included in this figure. AD, Alzheimer's Disease; PTC, Pattern of Tau Covariance.
4. DISCUSSION
In this investigation, we applied data‐driven methods to estimate a novel progression model and staging system for tau pathology in AD. The major contributions of our work are as follows. First, we apply NMF to identify regions of coordinated tau aggregation (PTCs). Our results provide a low‐dimensional, interpretable, and reproducible framework for describing subject‐specific patterns of tau accumulation which is readily applicable to new data. Second, we define a novel system for staging tau pathology. This model was able to stage most AD‐related tau presentations in two independent datasets, showing consistent associations with multiple markers related to AD progression. Our work extends the sparse literature regarding data‐driven approaches of tau staging, 19 , 21 providing a simple model for categorizing tau presentations along a clear regional hierarchy (Figure 3B). Third and finally, we demonstrated an open scientific process by using publicly available data and providing a repository clearly documenting all our analyses (https://github.com/sotiraslab/earnest_nmf_tau_staging).
4.1. Spatial patterns of FTP deposition
Most PTCs corresponded to brain regions which are important nodes of tau pathology in AD. PTC1‐MedialTemporal covered regions which have been extensively investigated as the origin of pathological tau in typical AD. 5 , 25 , 51 , 52 , 53 PTC4‐Precuneus included regions of the posterior default mode network, which experiences dysregulation in AD that may be related to tau pathology. 15 , 17 , 54 A precuneus predominant variant of deposition has also been observed in a cohort of individuals with preclinical AD. 55 PTC5‐Occipital spans posterior regions of the brain, where accelerated tau deposition is observed in the posterior cortical atrophy variant of AD. 21 , 56 PTC7‐Sensorimotor covered regions which typically are last to exhibit FTP deposition. 26 , 51 Our reproducibility analyses further supported the relevance of these PTCs, as similar patterns were observed in independent data subsamples.
Correlated tau accumulation between hemispheres is typically observed in AD 7 ; correspondingly, most PTCs were symmetrical. The major exception was PTC2‐LeftParietalTemporal and PTC3‐RightParietalTemporal, which each covered unilateral regions of the temporal and parietal lobes. Previous research has shown tau lateralization in these areas: accumulation of tau in the left temporal lobe is implicated in AD with language impairment, specifically logopenic primary progressive aphasia. 56 , 57 Right‐dominant tau presentations have been identified in preclinical AD. 55 Other data‐driven investigations of tau‐PET have reported lateralized left and right tau deposition in temporal and parietal lobes. 15 , 17 , 20 , 21 The presence of asymmetrical temporal and parietal PTCs in our study supports the possibility of hemisphere‐specific tau spread from the MTL in AD. Notably, this lateralization was detectable in both discovery and replication datasets. While hemisphere‐specific tau covariance may stem from a subset of individuals with atypical disease, there is also some evidence of lateralized tau spread in typical AD. 51
Previous studies have applied data‐driven methods to reveal regions of correlated FTP deposition. Some of these studies have identified FTP regions with similar topology to the PTCs we report, namely in the precuneus and medial parietal areas, 15 , 17 , 20 bilateral 15 , 17 or lateralized 20 occipital regions, sensorimotor cortex, 16 , 18 and either bilateral 14 , 16 , 18 or unilateral 20 temporal and parietal areas. Few of these studies have identified an FTP factor specific to the entorhinal cortex, 19 sometimes instead reporting a wider component spanning the anterior and inferior temporal lobe. 15 , 16 , 18 , 20 This difference may stem from the fact that many previous studies applied their learning algorithms to voxel‐wise FTP images, for which PET statistical noise and biological variability are more influential. Along with this previous work, our study indicates that tau aggregates in neocortical regions which are not highlighted by Braak staging, particular the in frontal, occipital, and precuneus cortices.
Adding to the biological relevance of our learned FTP patterns, we found significant spatial overlap between PTCs and the expression of genes linked to AD. APOE had the strongest spatial association, indicating elevated expression of this gene in MTL structures. Previous studies have established a link between APOE genotype and tau pathophysiology in this area of the brain. 58 , 59 , 60 We also found more advanced tau deposition in those with APOE genetic risk (Figure 3). Beyond APOE, some other genes showing overlap with PTC1‐MedialTemporal have been implicated in early AD development and MTL pathology. 61 , 62 , 63 , 64 , 65 Our gene expression results should be interpreted with caution, as they are purely correlational, and assessment of the function of these genes is impossible based on this analysis alone. Still, they may be an indication of the genetic factors underpinning tau accumulation and progression.
4.2. Staging AD‐related tau progression
Our results indicated a progression of tau pathology starting in the MTL, spreading to broader temporal and parietal areas, further advancing to frontal and occipital cortices, and depositing ultimately in primary sensorimotor areas. Using a four‐stage system based on this model, we showed that higher FTP stages were associated with several clinical, cognitive, and biological indices related to AD severity. Furthermore, staging was associated with AD‐severity in independent datasets, longitudinally stable, and able to capture the tau deposition patterns of most individuals in our sample.
Previous research applying Braak staging to PET data has been important for confirming that histopathologically defined stages are useful for in vivo disease assessment. 8 , 9 , 10 , 11 , 66 We expand on this line of work by developing and testing a staging system which is informed by empirical FTP covariance. Our system exhibits some alignment with Braak stages, showing earliest tau accumulation in the MTL followed by spread to the broader temporal lobe. Diverging from Braak, we show tau development in the precuneus cortex cooccurring with temporal aggregation as well as a distinct stage for frontal and occipital binding. Interestingly, the staging system we learned bears resemblance to the tau stages estimated in a recent paper by Leuzy et al., 19 despite our use of a different dataset, model, and tau tracer. While these authors focused on prediction on longitudinal tau change in their learned regions, we show that similar regions can be used for categorical staging of AD and indexing disease progression.
We observed a small portion of individuals (∼ 10% of each cohort) who were incompatible with our estimated FTP progression pattern. While some of the NS presentations may be attributable to imaging noise or simplicity in our system, heterogeneity in AD and its underlying pathophysiology may also be a factor. We found that many of the NS individuals we observed were unstageable due to a lack of tau deposition in the MTL. This presentation is consistent with the hippocampal‐ or MTL‐sparing AD phenotype, which has been reported in previous literature. 21 , 67 Other examples of tau heterogeneity are observed in the visualization of tau positivity across PTCs (Figure 2), such as individuals with asymmetrical temporal positivity.
Our estimated model of tau progression was further supported by comparison of regional tau accumulation with amyloid burden and gray matter volume loss. While tau increased with amyloid burden in each PTC, tau‐elevation in early‐stage PTCs was observable at lower levels of amyloid, relative to later‐stage PTCs, which only showed tau elevation with extensive amyloid burden. Similarly, both inter‐ and intraregional associations between tau and gray matter were strongest when considering gray matter in regions estimated to be earlier in the tau progression. This result is consistent with the notion that pathological tau precedes neurodegeneration: FTP in “later” tau regions is still related to gray matter loss in “earlier” ones, where neurodegenerative processes should be already underway.
4.3. Strengths and limitations
Our study has strengths worth noting. We specifically only included amyloid‐positive subjects, regardless of cognitive status. Thus, the PTCs we learned are specifically relevant to the spatial spread of tau pathology in AD rather than other dementias or tau deposition unrelated to dementia. 1 Furthermore, most of our results were reproducible in independent cohorts, both of which are freely accessible to the scientific community (following formal data use agreements). This generalizability supports the relevance of the patterns we identify and their value in assessment of tau progression.
There are several limitations which should also be considered, however. Using the preprocessed data provided by ADNI and OASIS‐3, our analysis was limited to assessment of FTP uptake in FreeSurfer cortical gray matter ROIs. Because of this, our results do not assess tau accumulation in subcortical gray matter and are unable to detect patterns which occur at the sub‐ROI level. Furthermore, we were not able to include partial volume correction, which is an important PET processing step for recovering accurate measures of tracer uptake. Thus, spill‐in and spill‐out effects will contribute to the spatial patterns we report, but no worse than that arising from an 8 mm full‐width half‐maximum scanner. Our assessment of tau progression is also limited in assessment of the heterogeneity of tau deposition, as we learned one staging system which we applied to all individuals. Other data‐driven approaches have been able to identify variants of tau spread and corresponding staging systems. 21 Our staging system instead provides inference for the average progression of tau in AD. However, we found that few individuals were NS with our model, and most of these individuals appeared to have MTL‐sparing pathology. Importantly, the PTCs themselves may constitute areas of tau accumulation which are relevant in more diverse variants of the disease, as our learning was not constrained to any clinical presentation, only the presence of amyloid pathology.
5. CONCLUSIONS
In this paper, we identified reproducible spatial patterns of tau deposition using data‐driven approaches. We found a consistent pattern of tau accumulation and progression which was replicable in independent datasets. Our decomposition of tau uptake provides a low‐dimensional framework for assessing the extent of tau pathology in AD.
CONFLICT OF INTEREST STATEMENT
Author A.S. has equity in TheraPanacea and has received personal compensation for serving as grant reviewer for BrightFocus Foundation. J.J.L. and Washington University may receive royalty income based on a technology licensed by Washington University to Sora Neuroscience. The remaining authors have no conflicting interests to report. Author disclosures are available in the supporting information.
CONSENT STATEMENT
All participants included in this study provided informed consent to participate.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
The authors thank the staff for the Washington University Center for High Performance Computing who helped enable this work. This work was supported by the National Institutes of Health (NIH) (R01‐AG067103). Computations were performed using the facilities of the Washington University Research Computing and Informatics Facility, which were partially funded by NIH grants S10OD025200, 1S10RR022984‐01A1 and 1S10OD018091‐01. Additional support is provided The McDonnell Center for Systems Neuroscience. Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. Data were provided (in part) by OASIS (OASIS‐3: Longitudinal Multimodal Neuroimaging: Principal Investigators: T. Benzinger, D. Marcus, J. Morris; NIH P30 AG066444, P50 AG00561, P30 NS09857781, P01 AG026276, P01 AG003991, R01 AG043434, UL1 TR000448, R01 EB009352. AV‐45 doses were provided by Avid Radiopharmaceuticals, a wholly owned subsidiary of Eli Lilly.)
Earnest T, Bani A, Ha SM, et al.; for the Alzheimer's Disease Neuroimaging Initiative . Data‐driven decomposition and staging of flortaucipir uptake in Alzheimer's disease. Alzheimer's Dement. 2024;20:4002–4019. 10.1002/alz.13769
Collaborators from ADNI
Data used in preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp‐content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf
REFERENCES
- 1. Jack CR, Bennett DA, Blennow K, et al. NIA‐AA research framework: toward a biological definition of Alzheimer's disease. Alzheimer Dement. 2018;14:535‐562. doi: 10.1016/j.jalz.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gordon BA, McCullough A, Mishra S, et al. Cross‐sectional and longitudinal atrophy is preferentially associated with tau rather than amyloid β positron emission tomography pathology. Alzheimers Dement (Amst). 2018;10:245‐252. doi: 10.1016/j.dadm.2018.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. La Joie R, Visani AV, Baker SL, et al. Prospective longitudinal atrophy in Alzheimer's disease correlates with the intensity and topography of baseline tau‐PET. Sci Transl Med. 2020;12:eaau5732. doi: 10.1126/scitranslmed.aau5732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ossenkoppele R, Smith R, Mattsson‐Carlgren N, et al. Accuracy of tau positron emission tomography as a prognostic marker in preclinical and prodromal alzheimer disease: a head‐to‐head comparison against amyloid positron emission tomography and magnetic resonance imaging. JAMA Neurol. 2021;78:961‐971. doi: 10.1001/jamaneurol.2021.1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol. 1991;82:239‐259. doi: 10.1007/BF00308809 [DOI] [PubMed] [Google Scholar]
- 6. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389‐404. doi: 10.1007/s00401-006-0127-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hansson O. Biomarkers for neurodegenerative diseases. Nat Med. 2021;27:954‐963. doi: 10.1038/s41591-021-01382-x [DOI] [PubMed] [Google Scholar]
- 8. Schwarz AJ, Yu P, Miller BB, et al. Regional profiles of the candidate tau PET ligand 18 F‐AV‐1451 recapitulate key features of Braak histopathological stages. Brain. 2016;139:1539‐1550. doi: 10.1093/brain/aww023 [DOI] [PubMed] [Google Scholar]
- 9. Biel D, Brendel M, Rubinski A, et al. Tau‐PET and in vivo Braak‐staging as prognostic markers of future cognitive decline in cognitively normal to demented individuals. Alzheimer Res Ther. 2021;13:137. doi: 10.1186/s13195-021-00880-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schöll M, Lockhart SN, Schonhaut DR, et al. PET imaging of tau deposition in the aging human brain. Neuron. 2016;89:971‐982. doi: 10.1016/j.neuron.2016.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Therriault J, Pascoal TA, Lussier FZ, et al. Biomarker modeling of Alzheimer's disease using PET‐based Braak staging. Nat Aging. 2022;2:526‐535. doi: 10.1038/s43587-022-00204-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pascoal TA, Therriault J, Benedet AL, et al. [18F]MK‐6240 depicts early and late Braak stages of neurofibrillary tangles in preclinical and symptomatic subjects. Alzheimer Dement. 2020;16:e045584. doi: 10.1002/alz.045584 [DOI] [Google Scholar]
- 13. Pascoal TA, Benedet AL, Tudorascu DL, et al. Longitudinal 18F‐MK‐6240 tau tangles accumulation follows Braak stages. Brain. 2021;144:3517‐3528. doi: 10.1093/brain/awab248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brier MR, Gordon B, Friedrichsen K, et al. Tau and Aβ imaging, CSF measures, and cognition in Alzheimer's disease. Sci Transl Med. 2016;8:338ra66. doi: 10.1126/scitranslmed.aaf2362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jones DT, Graff‐Radford J, Lowe VJ, et al. Tau, amyloid, and cascading network failure across the Alzheimer's disease spectrum. Cortex. 2017;97:143‐159. doi: 10.1016/j.cortex.2017.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sepulcre J, Grothe MJ, Sabuncu M, et al. Hierarchical organization of tau and amyloid deposits in the cerebral cortex. JAMA Neurol. 2017;74:813. doi: 10.1001/jamaneurol.2017.0263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hoenig MC, Bischof GN, Seemiller J, et al. Networks of tau distribution in Alzheimer's disease. Brain. 2018;141:568‐581. doi: 10.1093/brain/awx353 [DOI] [PubMed] [Google Scholar]
- 18. Vogel JW, Mattsson N, Iturria‐Medina Y, et al. Data‐driven approaches for tau‐PET imaging biomarkers in Alzheimer's disease. Hum Brain Mapp. 2019;40:638‐651. doi: 10.1002/hbm.24401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Leuzy A, Smith R, Cullen NC, et al. Biomarker‐based prediction of longitudinal tau positron emission tomography in Alzheimer disease. JAMA Neurol. 2022;79:149. doi: 10.1001/jamaneurol.2021.4654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Franzmeier N, Dewenter A, Frontzkowski L, et al. Patient‐centered connectivity‐based prediction of tau pathology spread in Alzheimer's disease. Sci Adv. 2020;6:eabd1327. doi: 10.1126/sciadv.abd1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vogel JW, Young AL, Oxtoby NP, et al. Four distinct trajectories of tau deposition identified in Alzheimer's disease. Nat Med. 2021;27:871‐881. doi: 10.1038/s41591-021-01309-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. LaMontagne PJ, Benzinger TL, Morris JC, et al. OASIS‐3: Longitudinal Neuroimaging, Clinical, and Cognitive Dataset for Normal Aging and Alzheimer Disease 2019:2019.12.13.19014902. doi: 10.1101/2019.12.13.19014902 medRxiv [DOI]
- 23. Ferris SH, Aisen PS, Cummings J, et al. ADCS prevention instrument project: overview and initial results. Alzheimer Dis Assoc Disord. 2006;20:S109‐S123. doi: 10.1097/01.wad.0000213870.40300.21 [DOI] [PubMed] [Google Scholar]
- 24. Collij LE, Heeman F, Salvadó G, et al. Multitracer model for staging cortical amyloid deposition using PET imaging. Neurology. 2020;95:e1538‐e1553. doi: 10.1212/WNL.0000000000010256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee WJ, Brown JA, Kim HR, et al. Regional Aβ‐tau interactions promote onset and acceleration of Alzheimer's disease tau spreading. Neuron. 2022;110:1932‐1943.e5. doi: 10.1016/j.neuron.2022.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cho H, Choi JY, Hwang MS, et al. In vivo cortical spreading pattern of tau and amyloid in the Alzheimer disease spectrum. Ann Neurol. 2016;80:247‐258. doi: 10.1002/ana.24711 [DOI] [PubMed] [Google Scholar]
- 27. Grothe MJ, Barthel H, Sepulcre J, et al. In vivo staging of regional amyloid deposition. Neurology. 2017;89:2031‐2038. doi: 10.1212/WNL.0000000000004643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thal DR, Rüb U, Orantes M, Braak H. Phases of Aβ‐deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791‐1800. doi: 10.1212/WNL.58.12.1791 [DOI] [PubMed] [Google Scholar]
- 29. The ADNI Team . ADNIMERGE: Alzheimer's Disease Neuroimaging Initiative. 2022.
- 30. Klunk WE, Koeppe RA, Price JC, et al. The Centiloid Project: standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement. 2015;11:1‐15.e1‐4. doi: 10.1016/j.jalz.2014.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Morris JC. Clinical dementia rating: a reliable and valid diagnostic and staging measure for dementia of the Alzheimer type. Int Psychogeriatr. 1997;9:173‐176. doi: 10.1017/S1041610297004870 [DOI] [PubMed] [Google Scholar]
- 32. Donohue MC, Sperling RA, Salmon DP, et al. The preclinical alzheimer cognitive composite. JAMA Neurol. 2014;71:961‐970. doi: 10.1001/jamaneurol.2014.803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Su Y, Blazey TM, Snyder AZ, et al. Partial volume correction in quantitative amyloid imaging. Neuroimage. 2015;107:55‐64. doi: 10.1016/j.neuroimage.2014.11.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Su Y, D'Angelo GM, Vlassenko AG, et al. Quantitative analysis of PiB‐PET with FreeSurfer ROIs. PLoS One. 2013;8:e73377. doi: 10.1371/journal.pone.0073377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Su Y, Flores S, Hornbeck RC, et al. Utilizing the Centiloid scale in cross‐sectional and longitudinal PiB PET studies. NeuroImage: Clinical. 2018;19:406‐416. doi: 10.1016/j.nicl.2018.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee DD, Seung HS. Learning the parts of objects by non‐negative matrix factorization. Nature. 1999;401:788‐791. doi: 10.1038/44565 [DOI] [PubMed] [Google Scholar]
- 37. Sotiras A, Finding imaging patterns of structural covariance via Non‐Negative Matrix Factorization 2015:16. [DOI] [PMC free article] [PubMed]
- 38. Sotiras A, Toledo JB, Gur RE, Gur RC, Satterthwaite TD, Davatzikos C. Patterns of coordinated cortical remodeling during adolescence and their associations with functional specialization and evolutionary expansion. P Natl Acad Sci USA. 2017;114:3527‐3532. doi: 10.1073/pnas.1620928114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Patel R, Steele CJ, Chen AGX, et al. Investigating microstructural variation in the human hippocampus using non‐negative matrix factorization. Neuroimage. 2020;207:116348. doi: 10.1016/j.neuroimage.2019.116348 [DOI] [PubMed] [Google Scholar]
- 40. Patel R, Mackay CE, Jansen MG, et al. Inter‐ and intra‐individual variation in brain structural‐cognition relationships in aging. Neuroimage. 2022;257:119254. doi: 10.1016/j.neuroimage.2022.119254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nazeri A, Krsnik Ž, Kostović I, et al. Neurodevelopmental patterns of early postnatal white matter maturation represent distinct underlying microstructure and histology. Neuron. 2022;110:4015‐4030.e4. doi: 10.1016/j.neuron.2022.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sun D, Adduru VR, Phillips RD, et al. Adolescent alcohol use is linked to disruptions in age‐appropriate cortical thinning: an unsupervised machine learning approach. Neuropsychopharmacology. 2023;48:317‐326. doi: 10.1038/s41386-022-01457-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289‐300. doi: 10.1111/j.2517-6161.1995.tb02031.x [DOI] [Google Scholar]
- 44. Pascoal TA, Therriault J, Benedet AL, Savard M, Lussier FZ, Chamoun M, et al. 18F‐MK‐6240 PET for early and late detection of neurofibrillary tangles. Brain. 2020;143:2818‐2830. doi: 10.1093/brain/awaa180 [DOI] [PubMed] [Google Scholar]
- 45. Wightman DP, Jansen IE, Savage JE, et al. A genome‐wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer's disease. Nat Genet. 2021;53:1276‐1282. doi: 10.1038/s41588-021-00921-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Markello R, Golia S, Zheng Y‐Q, Mišić B, abagen: A toolbox for the Allen Brain Atlas genetics data. 2021.
- 47. Arnatkevic̆iūtė A, Fulcher BD, Fornito A. A practical guide to linking brain‐wide gene expression and neuroimaging data. Neuroimage. 2019;189:353‐367. doi: 10.1016/j.neuroimage.2019.01.011 [DOI] [PubMed] [Google Scholar]
- 48. Hawrylycz MJ, Lein ES, Guillozet‐Bongaarts AL, et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012;489:391‐399. doi: 10.1038/nature11405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Markello RD, Hansen JY, Liu Z‐Q, et al. neuromaps: structural and functional interpretation of brain maps. Nat Methods. 2022;19:1472‐1479. doi: 10.1038/s41592-022-01625-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Alexander‐Bloch AF, Shou H, Liu S, et al. On testing for spatial correspondence between maps of human brain structure and function. Neuroimage. 2018;178:540‐551. doi: 10.1016/j.neuroimage.2018.05.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vogel JW, Iturria‐Medina Y, Strandberg OT, et al. Spread of pathological tau proteins through communicating neurons in human Alzheimer's disease. Nat Commun. 2020;11:2612. doi: 10.1038/s41467-020-15701-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ridler C. Tau seeding starts early in the entorhinal cortex. Nat Rev Neurol. 2018;14:380‐380. doi: 10.1038/s41582-018-0016-9 [DOI] [PubMed] [Google Scholar]
- 53. Sanchez JS, Becker JA, Jacobs HIL, et al. The cortical origin and initial spread of medial temporal tauopathy in Alzheimer's disease assessed with positron emission tomography. Sci Transl Med. 2021;13:eabc0655. doi: 10.1126/scitranslmed.abc0655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Putcha D, Eckbo R, Katsumi Y, Dickerson BC, Touroutoglou A, Collins JA. Tau and the fractionated default mode network in atypical Alzheimer's disease. Brain Commun. 2022;4:fcac055. doi: 10.1093/braincomms/fcac055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Young CB, Winer JR, Younes K, et al. Divergent cortical tau positron emission tomography patterns among patients with preclinical Alzheimer disease. JAMA Neurol. 2022. doi: 10.1001/jamaneurol.2022.0676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tetzloff KA, Graff‐Radford J, Martin PR, et al. Regional distribution, asymmetry, and clinical correlates of tau uptake on [18F]AV‐1451 PET in atypical Alzheimer's disease. J Alzheimers Dis. 2018;62:1713‐1724. doi: 10.3233/JAD-170740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ossenkoppele R, Schonhaut DR, Schöll M, et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer's disease. Brain. 2016;139:1551‐1567. doi: 10.1093/brain/aww027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Singh NA, Tosakulwong N, Graff‐Radford J, et al. APOE ε4 influences medial temporal atrophy and tau deposition in atypical Alzheimer's disease. Alzheimer Dement. doi: 10.1002/alz.12711. n.d.;n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shi Y, Yamada K, Liddelow SA, et al. ApoE4 markedly exacerbates tau‐mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017;549:523‐527. doi: 10.1038/nature24016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Therriault J, Benedet AL, Pascoal TA, et al. Association of Apolipoprotein E ε4 with medial temporal tau independent of amyloid‐β. JAMA Neurol. 2020;77:470‐479. doi: 10.1001/jamaneurol.2019.4421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sleegers K, den Heijer T, van Dijk EJ, et al. ACE gene is associated with Alzheimer's disease and atrophy of hippocampus and amygdala. Neurobiol Aging. 2005;26:1153‐1159. doi: 10.1016/j.neurobiolaging.2004.09.011 [DOI] [PubMed] [Google Scholar]
- 62. Assareh AA, Piguet O, Lye TC, et al. Association of SORL1 gene variants with hippocampal and cerebral atrophy and Alzheimer's disease. Curr Alzheimer Res. 2014;11:558‐563. doi: 10.2174/1567205011666140618101408 [DOI] [PubMed] [Google Scholar]
- 63. Cao M, Liu J, Zhang X, et al. ABI3 is a novel early biomarker of Alzheimer's disease. J Alzheimers Dis. 2022;87:335‐344. doi: 10.3233/JAD-215635 [DOI] [PubMed] [Google Scholar]
- 64. Zhu B, Liu Y, Hwang S, et al. Trem2 deletion enhances tau dispersion and pathology through microglia exosomes. Mol Neurodegener. 2022;17:58. doi: 10.1186/s13024-022-00562-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Jain N, Lewis CA, Ulrich JD, Holtzman DM. Chronic TREM2 activation exacerbates Aβ‐associated tau seeding and spreading. J Exp Med. 2023;220:e20220654. doi: 10.1084/jem.20220654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Maass A, Landau S, Baker SL, et al. Comparison of multiple tau‐PET measures as biomarkers in aging and Alzheimer's disease. Neuroimage. 2017;157:448‐463. doi: 10.1016/j.neuroimage.2017.05.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ferreira D, Mohanty R, Murray ME, Nordberg A, Kantarci K, Westman E. The hippocampal sparing subtype of Alzheimer's disease assessed in neuropathology and in vivo tau positron emission tomography: a systematic review. Acta Neuropathologica Communications. 2022;10:166. doi: 10.1186/s40478-022-01471-z [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information