

Abstract
Extracellular vesicles (EVs) derived from mesenchymal stem cells are promising nanotherapeutics in liver diseases due to their regenerative and immunomodulatory properties. Nevertheless, a concern has been raised regarding the rapid clearance of exogenous EVs by phagocytic cells. Here we explore the impact of protein corona on EVs derived from two culturing conditions in which specific proteins acquired from media were simultaneously adsorbed on the EV surface. Additionally, by incubating EVs with serum, simulating protein corona formation upon systemic delivery, further resolved protein corona–EV complex patterns were investigated. Our findings reveal the potential influences of corona composition on EVs under in vitro conditions and their in vivo kinetics. Our data suggest that bound albumin creates an EV signature that can retarget EVs from hepatic macrophages. This results in markedly improved cellular uptake by hepatocytes, liver sinusoidal endothelial cells and hepatic stellate cells. This phenomenon can be applied as a camouflage strategy by precoating EVs with albumin to fabricate the albumin-enriched protein corona–EV complex, enhancing non-phagocytic uptake in the liver. This work addresses a critical challenge facing intravenously administered EVs for liver therapy by tailoring the protein corona–EV complex for liver cell targeting and immune evasion.
Subject terms: Nanoparticles, Drug delivery
In regenerative medicine, stem-cell-derived extracellular vesicles are emerging as cell-free nanotherapeutics. Here, the authors show that coating these nanovesicles with blood proteins such as albumin improves their uptake by liver cells, offering a better treatment strategy for liver diseases.
Main
Although chronic liver diseases can be caused by a variety of different aetiologies, they are all characterized by a continuous process of inflammation, oxidative stress and scarring, which concur in promoting liver deterioration, cirrhosis and ultimately liver failure and death1. This pathophysiological progression is underpinned by a variety of different liver cell subsets, including hepatocytes, hepatic stellate cells, liver sinusoidal endothelial cells (LSECs), macrophages and lymphoid cells. Developing a broad-spectrum treatment that can be utilized to ameliorate a range of liver diseases requires therefore the potential to target different liver cell populations2,3.
There is a growing body of evidence suggesting that the immunomodulatory and rejuvenative properties of mesenchymal stem cells (MSCs) are mediated through the paracrine effects of the extracellular vesicles (EVs) they secrete4. In preclinical models of liver injury, studies have demonstrated that MSC-derived EVs (MSC EVs) can reduce collagen deposition and hepatic inflammation5, suppress hepatic stellate cell activation6, promote hepatocyte proliferation7 and inhibit hepatocyte apoptosis8. The use of EVs, as opposed to MSCs, as therapeutic agents brings many additional benefits including feasibility of large-scale production, the ease of storage and batch-to-batch quality control. Unlike cell-based approaches, EVs may also be engineered, for example, by surface modification, to improve pharmacokinetic and pharmacodynamic properties9.
We have previously shown that EVs from cancer and non-cancer cell origin can accumulate in the liver after intravenous administration although their cellular uptake profile remains unknown10,11. Recently, it has been shown that the so-called ‘protein corona’ can greatly affect the behaviour of synthesized particulates in vivo, including cellular targeting12,13. Nevertheless, the effects of protein corona on the in vivo fate of EVs have never been unravelled14.
The protein corona is formed when nanoparticles (NPs) come into contact with biological fluids, and proteins are adsorbed onto the NP surface. In contrast to synthetic NPs, culture-derived EVs encounter proteins at two time points: first, during the production in the cell culture condition; and second, on injection. Their corona can therfore be composed of both a ‘primary’ and ‘secondary’ corona formed at the production site and on contact with bodily fluids, respectively. The relative contribution of these two coronas in terms of particle properties and in vivo behaviour has yet to be described.
In this study, we aimed to determine how different good manufacturing practice (GMP)-compatible culturing methods affect EVs’ corona compositions and subsequently impact of liver cell targeting and accumulation, and to identify the key proteins mediating the in vivo behaviour of EVs. We hypothesize that the corona is composed, in part, of proteins derived from the cell culture media and the in vivo environment upon administration. MSC EVs were isolated from two-dimensional (2D) (EV2D) and 3D (EV3D) cultures15. Comparative proteomic analysis and measurement of in vivo organ biodistribution and uptake of EVs in liver cell subpopulations were undertaken. Principal component analysis (PCA) was applied to correlate corona compositions with biological functions.
Results
EVs from 2D and 3D cultures are physically comparable
Serum-free conditioned culture media (CCM) and KnockOut serum replacement (KO)-containing CCM derived from 2D and 3D MSC cultures, respectively, were subjected to EV isolation. Dot-blot analysis (Fig. 1a) shows that EV2D and EV3D were positive for canonical tetraspanins, that is, CD81, CD9 and CD63. The expression of an endosome-associated protein, TSG101, in both EVs confirms their endosomal origin15. The null microBCA read-outs of isolated unconditioned media for 2D and 3D culture confirmed that free proteins present in the media could not be co-isolated (Supplementary Fig. 1). The measurement of total EV protein content correlated with the particle concentration, as determined by micro bicinchoninic acid assay (microBCA) and nanoparticle tracking analysis (NTA), respectively (Fig. 1b). EV3D had a higher protein content than EV2D in agreement with the threefold increase in particle to protein ratio (P:P) achieved for EV2D (3.0 ± 0.5 × 1011) versus EV3D (9.7 ± 0.5 × 1010), respectively, both of which were within the acceptable purity range reported16. EV2D and EV3D were identical in surface charge as determined by zeta-potential values (Fig. 1c) and size (Fig. 1d), determined by mean, mode and particle size distribution (see Supplementary Fig. 2 for representative NTA and transmission electron microscopy results), indicating the acceptable EV charge/size range obtained17,18.
Fig. 1. Physicochemical and biochemical characterization of EV2D and EV3D.

a, Expression of EV surface markers (CD81, CD9, CD63) and internal marker (TSG101) analysed by chemiluminescence dot-blot. Equal numbers of EVs (5 × 1010 particles per ml) were spotted on the nitrocellulose membrane prior to staining. b, Protein concentration measured by microBCA assay correlated with particle concentration measured by NTA (n = 5). c, Zeta-potential of EVs in deionized water (n = 4, biologically independent samples). Data are presented as mean ± s.d. d, Size of EVs in PBS measured by NTA (n = 22, biologically independent samples). Percentiles (D10, D50 and D90) determine particle size distribution. EV2D and EV3D exhibited comparable physicochemical and biochemical characteristics except for protein content (EV3D > EV2D). Data are presented as mean and mode ± s.d. Statistical analysis was performed by two-tailed unpaired t-test: NS, not significant, P > 0.05.
Culturing conditions affects protein corona identity of EVs
To mimic the EV–serum protein interaction in vivo, EV2D and EV3D were exposed to 50% (v/v) EV-depleted fetal bovine serum (FBS) (EV-D FBS) and subjected to ultracentrifugation and protein corona desorption to obtain the EV with bound hard corona (HC-EV2D and HC-EV3D), hard corona (that is, protein associated with EV following stripping, HC2D and HC3D), and stripped EVs (strip-EV2D and strip-EV3D) (see Supplementary Fig. 3 for procedure). The protocol was optimized to efficiently desorb HC (Supplementary Tables 1 and 2 and Supplementary Fig. 4).
As shown in Fig. 2a, after the removal of unbound FBS proteins, the HC-EV2D and HC-EV3D were found to be significantly larger in size than non-incubated EV. Despite this, no significant change in zeta-potential could be observed. The protein bound per surface area measured by microBCA assay was also found to be increased, indicating the additional protein was acquired from EV-D FBS (Fig. 2b). Visualization of protein bands was carried out by loading digested EV, HC-EV, HC, stripped EV, EV-D FBS and KO on sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) gels and performing silver staining (Fig. 2c). KO and EV-D FBS were included to determine which proteins were acquired from FBS incubation compared to those intrinsically found in EVs. Distinct protein profiles between EV2D and EV3D can be observed, with EV3D showing similarities to KO medium proteins which were absent in EV2D. This finding was confirmed to be independent of the cells’ geometry (Supplementary Fig. 5). Upon exposure to EV-D FBS, an HC is formed on the EV surface, causing the protein adsorption on the EV with different preferential compositions when comparing HC-EV2D and HC-EV3D. Similar protein patterns were also observed in HC2D and HC3D bands. The differences in protein bands disappeared in the stripped EV2D and stripped EV3D samples. Altogether, the results suggest that culturing supplementation affects the protein identity of EVs and the protein corona formed upon exposure to exogenous proteins, that is, from serum. To distinguish the type of corona formed, we refer to the protein corona acquired from CCM and FBS incubation as primary and secondary corona, respectively.
Fig. 2. Protein corona formation on EVs derived from different culturing conditions evaluated by NTA, microBCA assay, SDS–PAGE and LC–MS.

a, Size changes due to protein corona formation (n = 3; 1*P = 0.014, 2*P = 0.019). b, Protein bound per surface area of EVs measured by microBCA assay (n = 3; 3*P = 0.012, and *4P = 0.031). Data are presented as mean ± s.d. Statistical analysis is performed using two-tailed paired t-test (*P < 0.05). c, Representative silver-stained SDS–PAGE of EVs (n ≥ 2), HC-EV, HC and stripped EVs (strip-EV). EV-D FBS and KO are FBS incubation controls and culture media, respectively. Similarities and differences are depicted as green and red arrows, respectively. The results confirmed HC formation of both EV types although the compositions are qualitatively different. d,e, Quantitative LC–MS analysis of protein corona formation. The most abundant proteins in the non-FBS-incubated (d) and FBS-incubated (e) EVs identified by LC–MS against human protein and bovine protein databases, respectively, are displayed in the heatmap (R1 and R2 represent two biological replicates). Both EV3D and HC-EV3D, unlike their 2D counterpart, are rich in albumin. Values are expressed as percentage abundance of total protein amounts identified (n = 2, biological replicates with n = 3, technical replicates per sample).
Proteomic analysis of EV2D and EV3D was performed by liquid chromatography–mass spectrometry (LC–MS) and analysed using the human protein database. The top 31 identified proteins expressed as a relative protein abundance in percentage (%RPA) are displayed in the heatmap as shown in Fig. 2d (see Supplementary Table 3 for full details). The differences in the protein enrichment of EV2D and EV3D indicate that although EVs are secreted from the same parental cells, differential culturing conditions significantly affect their protein profiles. As EV2D were isolated from serum-free media, it is expected that the proteins detected in EV2D will be free from cell-culture supplements. The most abundant proteins found in EV2D were actin cytoplasmic 1 and immunoglobulin heavy constant mu, which are reported as subcellular components and secretomes of MSCs, respectively19,20. In contrast, the EV3D media had to be supplemented with KO to maintain the spheroidal condition. Three protein components, that is, serum albumin, m7GpppN-mRNA hydrolase (DCP2), methylosome subunit pICln (CLNS1A) and peripherin (PRPH) were the most abundant in EV3D. DCP2, CLNS1A, and PRPH are identified as MSC cytoplasmic proteins, whereas only albumin does not originate from MSCs and is classified as secreted protein21–23. It was therefore suggested that albumin was derived from the KO supplement and formed as the first corona during culture. This is also confirmed by comparing the LC–MS data of EV3D and KO supplement (Supplementary Fig. 6). The protein profile of HC adsorbed on the EVs after exposure to FBS was analysed against a bovine protein database to exclude all human-derived proteins in the samples and focus on the protein layer obtained from FBS incubation only (that is, the second corona). Figure 2e shows the heatmap of the top 30 identified proteins as %RPA (Supplementary Table 4 for full details). The differences in HC profile reveal that differential culturing conditions also affect the protein compositions of the second corona. Bovine-derived albumin (Supplementary Fig. 7) was found to be prominently enriched on EV3D compared with EV2D. Altogether, the LC–MS data support the previous silver-stained SDS–PAGE results and suggest that the culturing conditions of 3D culture enabled the formation of the first and second coronas enriched with albumin on EV3D.
Protein corona modulates cellular uptake in vitro
Because the EVs in this study were prepared with the aim of increasing the accumulation in liver cells other than phagocytic cells, the uptake of EVs (HC-free EVs and HC-coated EVs) by macrophages and liver parenchymal cells were in vitro simulated in phagocytic cells, that is, J774 and human-monocyte-derived macrophages (Hu-Ø) and human hepatocellular carcinoma cell line, HepG2, respectively, for 1 h, 4 h and 24 h. Uptake was evaluated by the fold increase in the mean fluorescence intensity signal per cell (MFI) compared with non-treated cells (see Supplementary Figs. 8 and 9 for labelling rationale). The presence of HC on EV2D significantly increased the uptake by phagocytic cells during 1 h incubation in J774 and 1 and 4 h incubation in Hu-Ø (Fig. 3a,b), respectively, while the increase in uptake by HepG2 was observed only at 24 h incubation (Fig. 3c). In the case of HC-EV3D, a significant reduction of phagocytic uptake at 24 h incubation was observed compared with EV3D (Fig. 3d,e). In HepG2, a significant increase in EV3D uptake mediated by HC was observed at 4 h incubation. Remarkably, both EV3D and HC- EV3D could be preferentially taken up by HepG2 compared with their 2D counterparts. This preferential behaviour could also be observed when labelling EVs by lipophilic dye incorporation (Supplementary Fig. 10). Altogether, the results suggest that HC-EV3D shows favourable uptake profiles by the liver microenvironment indicated by reduced and increased uptake by phagocytic cells and hepatocytes, respectively.
Fig. 3. Cellular uptake of non-incubated EVs and HC-EVs in phagocytic and HepG2 cells.

a–f, Cellular uptake of EV2D ± HC (a–c) and EV3D ± HC (d–f) in phagocytic J774 cells (a,d), human-monocyte-derived macrophages (Hu-Ø) (b,e) and HepG2, representing non-phagocytic liver hepatocytes (c,f). AF488-labelled EVs or HC-EVs were incubated with cells at a dose of 2 × 109 particles per well (24-well plate) for 1 h, 4 h and 24 h. Cellular uptake was measured by flow cytometry, and uptake was expressed as fold increase of the mean AF488 signal per cell (MFI) compared with non-treated cells. Doping of EVs with albumin-rich HC, that is, the case of EV3D, significantly reduces uptake in phagocytic cells while keeping uptake in HepG2 cells unchanged (1**P = 0.00417, 2***P = 0.00021, 3**P = 0.00243, 4*P = 0.04176, 5**P = 0.00871, 6****P = 0.00003, 7**P = 0.00522). Data are presented as mean ± s.d. (n = 3) with two-tailed unpaired t-test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
Non-phagocytic liver cells show preference for EV3D in vivo
A biodistribution study was carried out in mice to determine if the differences found in vitro prevail in vivo. DiR-labelled EV2D and EV3D were intravenously administered for optical imaging (see Supplementary Fig. 8d for labelling and Supplementary Table 5 for dose information). At 24 h post-injection, whole-body imaging was performed as shown in Fig. 4a (see Supplementary Fig. 11 for 1 and 4 h biodistribution). The brightest signals could be detected in the upper abdominal area, corresponding to the location of the liver and spleen, with mice injected with EV3D illustrating the highest DiR signal. This is supported by the ex vivo imaging of major organs, which revealed that EV3D accumulated most in the liver and spleen over a 24 h period (Fig. 4b). Semiquantitative analysis of ex vivo organ images (normalized to organ weights) was carried out (Fig. 4c) and suggested that both EVs recorded the highest accumulation in the spleen and liver, followed by lungs and kidneys. When comparing the accumulations between two types of EVs in the liver, higher DiR signals were detected in the EV3D group (P < 0.05), agreeing with the in vivo imaging. Mice were also individually housed in a metabolic cage for urine correction to check for renal clearance (Fig. 4d). Interestingly, the highest DiR signals in the urine were detected in the EV2D group, confirming the shorter half-life of EV2D in the circulation system.
Fig. 4. In vivo organ biodistribution and cellular uptake by liver subpopulations of EVs.

a–d, Animals were intravenously injected with 2 × 1011 DiR-labelled EVs, PBS or the free dye (control): representative whole-body live (ventral) imaging (a), ex vivo images of whole major organs (b), semiquantitative analysis of the ex vivo images (n = 3 for EV samples, 1*P = 0.0464) (c) and urine clearance at 24 h post-injection (n = 3, 2*P = 0.0393) (d). Fluorescence intensity measured as total radiant efficiency per gramme of tissue was obtained by using an ROI tool and Living Image v.4.7.3 software. e,f, Cellular uptake of EVs by liver subpopulations (hepatocytes, Kupffer cells, endothelial cells and stellate cells) by flow cytometry shown as cell number positive for the signals (n ≥ 3, 3**P = 0.0032, 4**P = 0.0089 and 5*P = 0.0371) (e) or MFI (n ≥ 3, 6**P = 0.0014 and 7**P = 0.0033) (d). EV3D showed significantly higher uptake in hepatocytes, endothelial cells and stellate cells but not Kupffer cells. Data are presented as mean ± s.d. with two-tailed unpaired t-test (c,d) and one-way ANOVA with post hoc Tukey test (e,f) (*P < 0.05, **P < 0.01).
To investigate in vivo uptake by liver subpopulations, that is, hepatocytes, Kupffer cells, endothelial cells and stellate cells, DiD-labelled EV2D and EV3D were intravenously administered (see Supplementary Table 5 for dose information). At 24 h post-injection, mice were anaesthetized, followed by liver perfusion (see Supplementary Fig. 12 for liver perfusion procedure). Digested livers were subjected to differential centrifugation to isolate hepatocytes and non-parenchymal cell (NPC) population (that is, Kupffer cells, endothelial cells and stellate cells) (see Supplementary Fig. 13 for isolation procedure)5. Cell fractions were then stained with fluorescently conjugated antibodies for the markers reportedly expressed by these subpopulations to enable cell type identification co-localized with DiD signal of EVs by flow cytometry (see Supplementary Fig. 14 for antibody panel and gating strategy). Liver subpopulations could be isolated with consistency in numbers (Supplementary Fig. 15). DiD signal was also co-localized with the signal of anti-human anti-CD9 antibody in hepatocytes, confirming the labelling stability of EVs in vivo (Supplementary Fig. 16). As shown in Fig. 4e,f, the majority of Kupffer cells (∼90%) took up EVs with no preference between EV2D and EV3D, while differences could be observed in the other cell subpopulations: 48.38% of hepatocytes, 68.15% of endothelial cells and 62.38% of stellate cells were positive for EV3D with mean fluorescence intensity (MFI) values of 13,338.5, 5,219.75 and 4,478, respectively. Values were significantly reduced for EV2D samples with percentage of positive cells and MFI values of 24.25%, 33.88%, 23.15% and 1,868, 1,538, 1,237, obtained for hepatocytes, endothelial cells and stellate cells, respectively. In summary, macrophages are indifferent to EV type for uptake, whereas hepatocytes and endothelial cells, followed by stellate cells appeared to favour uptake of EV3D over EV2D which could explain the overall higher liver signal for EV3D.
EV opsonization is predictably protein corona dependent
To understand if there is a correlation between EVs’ in vitro and in vivo behaviours, biological processes and protein corona compositions, EV and HC proteins identified by LC–MS were subjected to Gene Ontology (GO) analysis. To focus on EV proteins potentially playing a role in cellular uptake, only identified proteins coded for extracellular space with highly significant enrichment (P < 0.001, Supplementary Fig. 17) were selected for further analysis (see Supplementary Table 6 for protein accession codes).
Subsequent enrichment analysis was then performed by biological processes for both EV and HC sets against the human and bovine databases (UniProt), respectively. Proteins related to significantly enriched processes (Fig. 5a,b) were quantitatively analysed by PCA to obtain a set of new variables called principal component (PC) corresponding to a linear combination of the originals. The score plot as shown in Fig. 5c,d displayed a difference between EVs and HCs by considering only PC1, respectively (see Supplementary Fig. 18 for scree plot). When examining the loading plots to identify variables with the highest impact of each component, the immunoglobulin heavy constant mu strongly influenced the separation between EV2D and EV3D on PC1 (Fig. 5e). The higher abundance of this protein on EV2D (see Supplementary Fig. 19a for Bi-plot) made them favoured for engulfment by phagocytes and to induce complement activation (classical pathway) as interpreted from associated protein functions (UniProt database). The loading plots of HC samples (Fig. 5f) suggest the separation of HC2D and HC3D by α-2-HS-glycopotein is more abundant in HC3D. The second most influential protein was complement C3 which shows collinearity with various proteins that play a major role in the EV clearance via several processes. Supplementary Tables 3 and 4 display protein contents and Supplementary Tables 7 and 8 show the contribution to the biological processes. The higher abundance of these highly related proteins in HC2D (see Supplementary Fig. 19b for Bi-plot) suggests that upon systemic exposure EV2D clearance is promoted via opsonization, complement activation and phagocytosis24–26.
Fig. 5. GO analysis for classification of the protein identified by LC–MS.

a,b, Proteins detected in both EV3D/2D and HC2D/3D were decoded to obtain a gene list involved in biological processes contributing to the clearance of non-incubated EVs (against in-built UniProt human database, GO analysis with Bonferroni correction (1*P = 0.01 and 2**P = 0.007) (a) and hard protein corona of incubated EVs (against in-built UniProt non-human mammal (bovine) database, GO analysis with Bonferroni correction, 3****P = 1.6 × 10−6, 4****P = 4.97 × 10−11 and 5****P = 4.89 × 10−35) (b). c,d, GO analysis was performed using FunRich software v.3.1.3. Significantly enriched proteins (hypergeometric and Bonferroni analysis *P < 0.05, **P < 0.01 and ****P < 0.0001) by quantity underwent PCA for separation of EV2D and EV3D (c) and HC2D and HC3D (d), shown as score plots. e,f, Loading plots were used to illustrate how each protein influences the computed PCs in c,d, respectively. Overall, the intrinsic properties of EV protein corona constituents are fundamentally different. HC2D proteins are associated with higher extents of complement activation. The PCA was performed using mean of the LC–MS data (two biological and three technical replicates), and EVs were pooled from three batches.
Albumin contributes to uptake of EVs in liver cells in vivo
We hypothesized that the presence of albumin in the first corona acts as an attraction site for additional albumin molecules to adsorb onto the EV surface to form the second corona layer. We further hypothesized that these albumin molecules in the HC potentially increase the uptake in liver cells due to the abundance of albumin receptors and their subtype (SPARC) (Supplementary Fig. 20).
To evaluate the first hypothesis, we simulated EV3D culturing supplementation (formation of the first corona) through exposure to Alexa Fluor 488 (AF488)-labelled albumin (ALB-AF488) during the 2D culture, and subsequently incubated the isolated EV2D with Cy5-labelled albumin (ALB-Cy5) to simulate the formation of the second corona. A similar phenomenon to the behaviour of EV3D was observed, that is, isolated EV2D with an albumin-enriched first corona attracted more albumin molecules which formed a second corona layer, thereby proving our hypothesis (Extended Data Fig. 1a). The lack of spillover between two fluorophores was confirmed to ensure the measurement accuracy of EVs with two co-localized fluorescent signals (Supplementary Fig. 21). We visually confirmed that the primary corona is associated with EVs by dual-tracking fluorescence microscopy (Supplementary Fig. 22). A negatively charged liposome (LIP) was included as a synthetic NP control. When implementing dual corona formation, the albumin-enriched corona formed on the LIP was disrupted upon ultracentrifugation, suggesting a weaker association with LIP than observed with EVs (Extended Data Fig. 1b).
Extended Data Fig. 1. In vitro study simulating formation of primary protein corona (1st corona) and secondary protein corona (2nd corona) on EVs and liposomes.

(a) EV2D and (b) Negatively charged liposomes (LIP, as representative of synthetic particles with lipid bilayers) were incubated firstly with albumin labelled with Alexa fluor 488 (ALB-AF488) (1st corona) then with albumin labelled with Cyanide 5 (ALB-Cy5) (2nd corona). Fluorescence was evaluated by a plate reader to determine the degree of albumin adsorption. Results confirmed that EV precoating with ALB (1st corona) induces deposition of more albumin (2nd corona) on EVs (**p = 0.0015). This is mimicking EVs interaction with blood when injected intravenously. In case of LIP, quenching was observed. Data are presented as mean ± SD (n = 3, biologically independent samples) with two-tailed unpaired t-Test analysis. The figure was created with BioRender.com.
To test the second hypothesis, we attempted to block albumin receptors (reviewed in Supplementary Table 9 and Supplementary Fig. 23). Confirming our hypothesis, the uptake of EV3D was reduced in vitro (Supplementary Fig. 24). The in vivo biodistribution and liver uptake was performed by intravenous injection of bovine serum albumin (BSA) followed by DiR or DiD-labelled EV3D, respectively. A reduction of EV3D accumulation in the liver at 1 h, 4 h and 24 h and uptake by all liver subpopulations at 24 h could be seen when BSA was coadministered (Supplementary Fig. 25 and Fig. 6a, respectively). In another study, we compared the uptake of DiD-labelled EV2D with and without albumin coating. Albumin-coated EV2D showed the highest uptake by all cell types (Fig. 6b). Surprisingly, preincubation of isolated EV2D with KO media, however, did not lead to cell uptake enhancement (Supplementary Fig. 26), implying that the formation of the first corona at the site of secretion is essential (see Supplementary Fig. 27 for a summary of the in vivo results). Collectively, these studies confirmed the role of albumin receptors in the internalization of albumin-enriched EVs.
Fig. 6. In vivo proof-of-concept studies confirming the effect of albumin receptor saturation or EV coating on liver cell internalization.
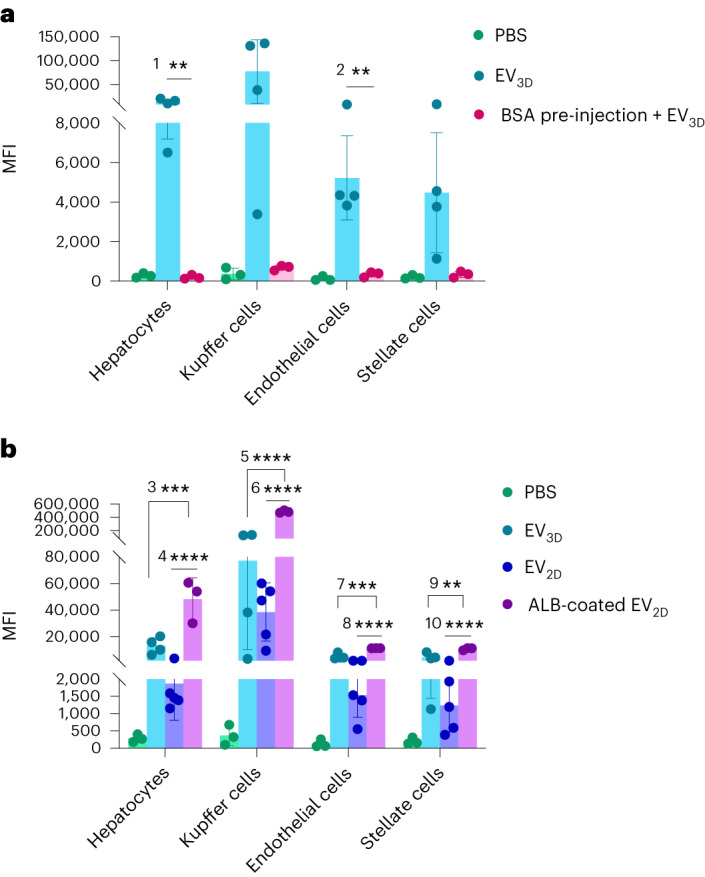
a, In vivo uptake of EV3D in liver cells is reduced/blocked when mice were injected with BSA (5 min pre-EV injection, 10 mg ml−1, 100 µl) followed by intravenous injection of DiD-labelled EV3D (2 × 1011 particles per mouse, n = 3, 1**P = 0.0091 and 2**P = 0.0063). b, In vivo uptake of albumin-coated DiD-labelled EV2D in liver cells after intravenous administration (2 × 1011 particles per mouse, n = 3, 3***P = 0.0004, 4****P < 0.0001, 5****P < 0.0001, 6****P < 0.0001, 7***P = 0.0001, 8****P < 0.0001, 9**P = 0.0018, 10****P < 0.0001). This confirms the involvement of albumin and albumin receptor in the cellular internalization of EVs. Data are presented as mean ± s.d. with one-way ANOVA with post hoc Tukey (*P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001).
Discussion
It is well established that protein corona formation around synthetic NPs, upon systemic exposure, can be a determining factor for in vivo fate. For EVs, only a few studies have reported the adsorption of proteins acquired from CCM and/or after exposure to serum. Protein corona on THP-1-derived EVs was shown to mediate proinflammatory responses by human-monocyte-derived dendritic cells in vitro27. A corona layer of protein secretome was also found on human-platelet-lysate-derived EVs after EV isolation using tangential-flow filtration. This corona exerted regenerative and immunomodulatory properties on the skin-organoid model and T cells in vitro28. In line with this study, the presence of corona containing proangiogenic factors secreted from the parent cells, human placental expanded stromal cells, on placental expanded stromal-cell-derived EVs could mediate vascularized skin regeneration in vivo29. Our study also confirms that protein corona can form on EVs during the culture process and could contribute to biological changes.
Albumin coating of a range of synthetic nanocarriers has shown beneficial effects, including enhanced lipid membrane interactions, reduced toxicity, augmented receptor-mediated (gp60, gp30, gp18 and SPARC receptors) uptake in tumour cells30–36, and prolonged blood circulation37. Albumin corona on PHBHHx biopolymer inhibited plasma protein adsorption (that is, free immunoglobulin-G and complement activation fragments, considered as ‘opsonins’), resulting in a lower degree of opsonization37. Similarly, albumin-rich corona adsorbed on polymeric molecularly imprinted nanogels reduced non-specific protein binding and increased NP half-life38. In our study, we also observed that EVs with albumin-enriched corona adsorbed less immunoglobulin and complement fragments. Albumin has also been employed as drug–NP conjugates for liver non-phagocytic-cell-directed targeting. Naproxen, a conventional anti-inflammatory drug, conjugated with human serum albumin (HSA) was efficiently targeted to LSECs due to the recognition of HSA by scavenger receptors39. Correspondingly, liposomes coupled with HSA derivatized with cis-aconitic anhydride were mostly taken up by LSECs due to the action of the albumin-scavenger receptor40. The uptake of albumin-bound oleate by hepatocytes was also shown to be facilitated by albumin receptors41. In line with this, the albumin-dominated corona formed on lipid-like NPs was shown to preferentially bind to gp18 and gp30 expressed on primary hepatocytes, leading to enhanced cellular uptake via macropinocytosis and endocytosis42. The hepatic-albumin receptors, therefore, evidently contribute to the uptake of exogenous albumin-bound compounds. In terms of EVs, it was reported recently that EVs decorated with albumin-binding domains could capture more albumin in the circulation, resulting in prolonged half-life43. Our results broadly showed hepatic persistence of EV3D, whereas EV2D showed susceptibility to renal clearance. The ability of EV3D to evade renal excretion might be explained by the binding affinity of albumin to the receptors responsible for mediating the reabsorption of filtered proteins in a renal proximal tubule, cubilin and megalin, resulting in the albumin–EV complex being rescued from renal clearance12,13. Nevertheless, the ability to prolong the blood-circulation time of albumin-enriched corona on EVs should be further investigated in depth.
Although we have confirmed here the contributory effects of corona enriched with albumin, the main component in culture media and systemic environment, the depletion of the other subcomponents, for example, complement proteins, in heat-inactivated serum, which was used to simulate hard corona formation in this study, might affect the profile and amount of adsorbed protein44,45. Therefore, the formation of protein corona on EVs in non-heat-activated serum requires additional scrutiny.
Conclusions
Our delivery complex is proposed as liver cell targeting EVs enabled by albumin preloading during culture processes. Although our study focused on the effect of albumin, the most enriched protein in the corona, on modulation of uptake, a whole series of other studies can follow looking at the function of the individual proteins. Our prototypic model might be applied to develop efficient carrier for EVs from other cell sources. Future work will focus on exploring if these changes can result in improved therapeutic responses in a liver disease model.
Methods
Two-dimensional culture of umbilical-cord-derived MSCs
MSCs from umbilical cord (ucMSCs) were obtained from the Anthony Nolan Cord Bank, cut in small pieces and plated in basal medium (that is, αMEM + 1% v/v penicillin–streptomycin) containing 5% (v/v) human platelet lysate at 37 °C, 5% CO2. After removal of non-adherent cells and washing with PBS, the media was replenished and the cells were cultured until reaching 70–90% confluency. Trypsinization was then performed for further characterization and cell passaging. ucMSCs were characterized for positivity of CD90, 105, CD106 and CD73, human leukocyte antigen class I, and the lack of expression of CD14, CD31 and CD45. Early passages of 1 × 106 ucMSCs (passages 2–5) were continuously cultured in a filtered 175 cm2 flask (Corning) in basal media + 5% (v/v) human platelet lysate. When the cells reached 70%–80% confluency, the media were replaced with basal medium prior to harvesting CCM on the following day for EV isolation and subculturing by a conventional trypsinization protocol.
Three-dimensional culture of ucMSCs
Aggrewell400 microwell culture plate was used to generate ucMSC spheroids following a previously established protocol15. Briefly, 500 µl anti-adherence rinsing solution was added to each well prior to centrifugation at 2,000g for 2 min to remove bubbles and incubation for 30 min–2 h at room temperature. The Aggrewell was then washed with 500 µl PBS per well, followed by addition of 500 µl basal medium and centrifugation at 2,000g for 2 min to remove bubbles. The basal medium was then replaced with the cell suspension obtained from 2D trypsinization (prepared at a density of 1.2 × 105 cells per 500 µl per well in the basal medium supplemented with 20% v/v KO serum replacement according to the manufacturer’s instructions). The plate was then centrifuged at 200g for 5 min for cell aggregation at the bottom of each microwell and kept undisturbed in the incubator. CCM was harvested on day 3 and every 2–3 days for EV isolation. Fresh medium was replenished after medium harvesting to maintain spherical ucMSC culture for 12 days.
Isolation of EVs
EV isolation was performed as described in detail previously46. Ultraclear polycarbonate ultracentrifuge tubes (catalogue number 355631, Beckman Coulter) were filled with 22.5 ml filtered CCM (prepared by filtering CCM using a 0.22 µm syringe filter). Then, 3 ml 25% w/w sucrose solution (prepared in D2O) was layered slowly below the CCM using a glass Pasteur pipette. Centrifugation using a swing-out rotor (SW32 Ti, Beckman Coulter) was performed at 100,000g for 1.5 h at 4 °C. The sucrose solution was then collected (2 ml per tube) and subjected to a washing step for EV purification by adding to prefilled polycarbonate ultracentrifuge tubes (catalogue number 355618; Beckman Coulter) with 20 ml filtered PBS prior to ultracentrifugation at 100,000g for 1.5 h at 4 °C using a fixed-angle rotor (70 Ti, Beckman Coulter). Supernatant was discarded and the pellet of EVs obtained was resuspended in filtered PBS. EVs were kept at 4 °C for 1 week storage and −80 °C for long-term storage.
Detection of EV markers by dot-blot
The analysis was performed following a previously published protocol15. First, 50 µl EVs at a concentration of 5 × 1010 particles per ml were spotted on a nitrocellulose membrane (one membrane per marker) (Bio-Rad). The membrane was dried by nitrogen gas prior to the blocking step using blocking buffer (that is, 3% skim milk prepared in Tris-buffered saline with 0.1% Tween 20 (TBS-T)) for 1 h at room temperature. Primary antibodies (CD9, CD63, CD81 and TSG101) were individually added in fresh blocking buffer followed by incubation overnight at 4 °C (1:1,000 dilution). The membrane was then washed three times in TBS-T, 5 min each wash. Horseradish-peroxidase-conjugated secondary antibody was then added to the fresh blocking buffer prior to further incubation for 1 h at room temperature (1:20,000 for anti-mouse and 1:1,000 for anti-rabbit). The membrane was washed as previously mentioned, and the signals were developed by substrate addition (SuperSignal West Femto Maximum Sensitivity Substrate), followed by imaging using the Gel Doc system (Bio-Rad) and analysis by Image Lab software (Bio-Rad).
Protein assay for determining the protein concentration of isolated EVs
Protein concentrations were determined by microBCA assay in a 96-well plate following the manufacturer’s instructions adapted for EVs. Briefly, EV samples (minimum concentration, 5 × 1010 particles per ml) were diluted 1:1 in PBS. MicroBCA reagent mix (prepared according to the manufacturer’s instructions) was added to duplicate 40 µl diluted samples (50 µl per well), followed by incubating at 37 °C for 1 h. The measurement was compared against serially diluted BSA as standard (prepared in duplicates). The absorbance was read at 562 nm using a FLUOStar Omega plate reader (BMG LabTech). MARS v.2.40 software (BMG LabTech) was used for analysis by extrapolating the values from the standard curve using a third-order polynomial equation, with r2 > 0.999 for each assay.
NTA
The size and concentration of EVs was measured by NTA using a Nanosight LM10 (Malvern Instruments) equipped with a 488 nm laser. The camera level was automatically adjusted, and the analysis detection threshold was set at 3–4. EV samples were diluted in filtered PBS to obtain optimal concentrations (20–80 particles per frame). Four video recordings with a duration of 40 s were carried out for each EV preparation. Nanosight NTA 3.2 software (Malvern Instruments) was used to analyse the recorded video.
Zeta-potential
The dynamic electrophoretic mobility of EVs and HC-EVs was measured with a Malvern Zetasizer Nano ZS and Zetasizer v.7.12 software (Malvern Instruments). Prior to the measurements, EV samples (minimum concentration, 1 × 1011 particles per ml) were diluted 1:50 in deionized water. Measurements were carried out at 25 °C for each experimental triplicate.
Protein corona preparation
Protein corona coating was performed following a published protocol47 with modifications. To remove EVs and aggregated protein serum, FBS was subjected to ultracentrifugation at 100,000g for 18 h at 4 °C. Supernatant was collected and filtered through 0.22 µm filters (EV-D FBS). EVs (∼7 × 1011 particles, equivalent to 0.05 m2) were prepared in a total volume of 300 µl sterile PBS supplemented with 1% penicillin–streptomycin solution (sPBS-PS) in 1.5 ml Eppendorf tubes prior to incubating with 300 µl EV-D FBS for 1 h at 37 °C, 300 r.p.m. Upon incubation, the mixture was transferred to an ultracentrifuge tube (catalogue number 343778, Beckman Coulter). The samples were ultracentrifuged at 100,000g for for 1 h at 4 °C. Supernatant was carefully removed without disturbing the pellet. The pellet was further washed with 1 ml sPBS-PS twice, following the above-mentioned condition. In corona desorption studies, upon the final wash, the pellet (HC-EVs) was dispersed in freshly prepared 100 µl 2% w/v SDS, 62.5 mM Tris–HCl solution, followed by incubation at 95 °C for 5 min to desorb the proteins constituting the HC. The HC was then separated from EVs by ultracentrifugation as above. The supernatant containing HC was collected and subjected to protein determination by microBCA assay. HC solutions were stored at −80 °C before further analysis.
SDS–PAGE
EVs, HC-EVs, EV-D FBS, KO and HC (15 µg) samples were mixed with LDS sample buffer and RIPA buffer (mixed with 1:100 protease inhibitor cocktail) and incubated for 10 min at 70 °C. The samples were then applied on a NuPAGE 4–12% Bis–Tris protein gel and run for 30 min at 100 mV and a further 1 h at 150 mV. For silver staining, the gel was fixed in 100 ml fixing solution (50% methanol, 10% acetic acid, 50 µl formaldehyde) overnight, followed by washing with 50% ethanol three times. The gel was then sensitized by hypo-solution (0.02% w/v sodium thiosulfate) for 1 min, followed by impregnating the gel with 0.2% w/v silver nitrate for 30 min. The gel was then washed with deionized water three times and subjected to band development using 100 ml developing solution (6 g sodium carbonate, 2 ml hypo-solution and 50 µl formaldehyde). Once developed, the reaction was stopped by 5% v/v acetic acid. A digital photograph of the gel was taken against a white background.
Digestion of the protein corona for mass spectrometry analysis
Proteomic analysis of FBS proteins, EVs and their HC by LC–MS was conducted using the method described by Schottler et al.47. Briefly, SDS was removed from the samples by Pierce detergent removal columns, and 25 µg of each protein sample was precipitated by ProteoExtract protein precipitation kit following the manufacturer’s manual. The resulting protein pellets were resuspended in 0.1% RapiGest SF in 50 mM ammonium bicarbonate and incubated for 15 min at 80 °C. Dithiothreitol (final concentration, 5 mM) was added to reduce the proteins, and the mixture was incubated at 56 °C for 45 min. Iodoacetamide (final concentration, 15 mM) was then added, and the mixture further incubated in the dark for 1 h at room temperature. The protein samples were digested for 18 h at 37 °C by trypsin with an enzyme:protein ratio of 1:50 (w/w). The reaction was stopped by the addition of 2 μl hydrochloric acid. The digested peptides were diluted with 0.1% v/v formic acid in UPLC-MS grade water and spiked with 50 fmol μl−1 Hi3 Escherichia coli standard for absolute quantification.
In vitro cellular uptake of EVs and HC-EVs
EV2D and EV3D were labelled as per our published protocol48. Concentration in particles per ml were obtained by NTA as mentioned above. The fluorescence intensity of freshly prepared EVs (100 µl) was measured using a FLUOStar Optima plate reader (BMG Labtech), with excitation and emission wavelengths of 485 nm and 520 nm, respectively. When there is a difference in labelling efficiencies between the batches or EV types, samples were mixed with non-labelled EVs so that fluorescence intensity and particles numbers are comparable for the different samples. Phagocytic cells, that is, J774 and human-monocyte-derived macrophages, and non-phagocytic cells, that is, HepG2, were seeded at a density of 1.5 × 105 cells per well in 24-well plates (Corning). After 24 h, the media were changed to serum-free media, and the cells were treated with labelled EV2D, HC-EV2D, EV3D and HC-EV3D of comparable fluorescence unit values and a dose of 2 × 109 particles and incubated for 1 h, 4 h and 24 h. To evaluate the contribution of albumin-binding receptor in mediating the EV internalization, HepG2 cells were treated with BSA (12.5 mg ml−1), followed by the addition of DiD-labelled EV3D, for 24 h. Flow cytometry was performed on a BD FACSCalibur using BD FACStation v.6.0 software (BD Biosciences) on the detached cells by trypsin–EDTA 0.05%, washed and resuspended in 200 µl PBS. All samples were analysed using FlowJo v.10.7.2 (TreeStar/BD Bioscience). Cells were gated by their forward and side scatter. The uptake of EVs was evaluated by the MFI of Alexa Fluor 488. Fold change in MFI was calculated with respect to the relative MFI increase over untreated controls.
Animals
All animal experiments were performed in compliance with the UK Animals (Scientific Procedures) Act 1986 and the UK Home Office Code of Practice for the Housing and Care of Animals Used in Scientific Procedures (Home Office 1989). In vivo experimentation adhered to the project license approved by the King’s College London animal welfare and ethical review body (AWERB) and the UK Home Office (PBE6EB195 and PP8950634). Animal research and veterinary care was performed at Franklin-Wilkins Building, King’s College London under the protocol approved for this study by a Named Training and Competency Officer (Julie Keeble) and a Named Animal Care and Welfare Officer (Jayne Morgan). Female CD-1 mice (~25–35 g, 5 weeks old) were obtained from Charles River for in vivo biodistribution study. Male and female C57BL/6 mice (18–25 g, 6–8 weeks old) were obtained from Charles River for in vivo cellular uptake study. All mice were housed in a 12 h light/12 h dark cycle with the temperature maintained between 65 and 75 °F (∼18–23 °C) and ∼50% humidity. Both sexes were used in the study, complying with the recently published recommendations by the UK Medical Research Council for conducting research on animals.
In vivo biodistribution of EVs by fluorescence optical imaging
Filtered CCM was incubated with 1 µM fluorescent lipophilic tracer DiR (1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyanine iodide) at room temperature for 1 h with agitation prior to EV isolation by sucrose cushion plus ultracentrifugation as described above to remove unbound dye. The experiment was performed in CD1 mice, randomly divided into groups. Freshly purified DiR-labelled EV2D and EV3D (2 × 1011 particles in 200 µl) were injected intravenously via the tail vein. Control mice were injected with PBS only or PBS containing DiR. Isoflurane-sedated live mice were imaged using the IVIS Lumina III system (excitation, 740 nm; emission, 840 nm) at 1, 4 and 24 h following intravenous injection prior to killing the animals. Major organs (brain, heart, lung, liver, stomach, spleen, kidney and intestine) were harvested for ex vivo fluorescence imaging. The fluorescence intensity in each organ (total radiant efficiency) was obtained using Living Image v.4.7.3 Software (PerkinElmer) to determine the organ biodistribution of DiR-labelled EV2D and EV3D by drawing the regions of interest (ROIs) of each organ. The values were then normalized to organ weights (total radiant efficiency per g). For the renal clearance study, upon injection with DiR-labelled EVs, mice were individually housed in a standard circular metabolic cage (Nalgene Nunc) for 24 h, and urine was collected into a Nalgene tube at the bottom of a funnel system for further fluorescence intensity determination using an IVIS Lumina III system and Living Image v.4.7.3 Software (PerkinElmer) by drawing ROIs covering a urine container to obtain total radiant efficiency per urine sample.
In vivo cellular uptake of EVs in liver subpopulations
EV2D and EV3D were labelled with DiD (DiIC18(5); 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine, 4-chloro-benzenesulfonate salt) using the same procedure as for DiR labelling in the in vivo studies. The experiment was performed in C57BL/6 mice, randomly divided into groups. Freshly purified DiD-labelled EV2D and EV3D (2 × 1011 particles in 200 µl) were intravenously injected via the tail vein. Control mice were injected with PBS only or PBS containing DiD. For the albumin receptor blocking study, 100 µl BSA diluted in PBS (10 mg ml−1, dosage selected based on the literature49–52) was injected 5 min prior to EV3D injection (2 × 1011 particles in 100 µl). For the study of albumin-coated EV internalization, to prepare albumin-coated, DiD-labelled EV2D, 2D ucMSCs (70–80% confluency) were cultured in basal medium supplemented with 2.5 mg ml−1 BSA, followed by medium collection after 24 h of culture, DiD labelling and EV isolation as previously mentioned. Albumin-coated DiD-labelled EV2D were then intravenously injected (2 × 1011 particles in 200 µl). Twenty-four hours later, mice were anaesthetized with phenobarbital and subjected to skin dissection on the ventral midline to open the peritoneal cavity for liver perfusion following the previously published protocol with modifications53. Mice were perfused with 30 ml HBSS–EGTA warmed at 41 °C through the inferior vena cava using a peristaltic pump (SciQ 300, Watson Marlow) and a 27G × 0.38″ × 12″ winged infusion set (BD Valu-Set) at a speed of 15 r.p.m. The hepatic portal vein was cut after the liver became swollen and discoloured, after which the speed was adjusted to 20 r.p.m. Mice were then perfused with 25 ml collagenase-containing HBSS–CaCl2 (concentration, 1 mg ml−1) warmed at 41 °C, at a speed of 15 r.p.m. The liver was transferred to a Petri dish containing cold HBSS–CaCl2 buffer. Hepatic cells were released by gentle breaking of Glisson capsule and shaking the liver. Upon obtaining homogeneous cell suspension, cells were filtered through a 70 µm strainer (Corning). Hepatocytes were separated by centrifugation for 3 min at 50g at 4 °C with low brake, three spins. After each spin, the pellet was resuspended in 30 ml cold HBSS–CaCl2, and the supernatant was collected for further NPC fractionation. The NPC fraction was pelleted at 650g at 4 °C and subjected to incubation with RBC lysis buffer for 5 min on ice, followed by addition of 20 ml PBS to stop the reaction and further centrifugation at 650g at 4 °C to pellet the purified NPC fraction. The identification of each subpopulation was performed based on the marker expression using fluorescently labelled antibodies for analysis by flow cytometry (BD FACSCelesta, operated by BD FACSDiva v.9.2 software, BD Biosciences) and FlowJo v.10.7.2 software (TreeStar/BD Bioscience). Hepatocytes were stained with anti-mouse ASGPR1 antibody (1:200)54–56 and anti-human CD9 via intracellular staining using 0.1% Triton-X 100 in PBS. Kupffer cells were stained with anti-mouse CD45, F4-80 and CD11b (1:200, each)57–59. Endothelial cells were stained with anti-mouse CD45 (1:200), CD31 (1:200) and CD146 (1:100)60–62. Stellate cells were stained with anti-mouse CD45 (1:200), GFAP (1:50) and detected using a 450/50 filter and a 405 nm violet laser63–65. All fractions were stained with Zombie Aqua to determine the viability of the cells. Staining was performed for 30 min at room temperature. The frequency of parent cells taking up EVs was defined by gating the DiD-positive cell population against the control (PBS) group. The MFIs of the DiD signal expressed by each cell type were used to evaluate the uptake amount of EVs.
Statistical analysis
Statistical analyses of the data were performed using Prism 9.4.1 (GraphPad Software) by using one-way analysis of variance (ANOVA) with Tukey post hoc test for all P values (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; P > 0.05 was non-significant). All results are expressed as mean ± s.d. Data distribution was assumed to be normal but this was not formally tested. No statistical methods were used to predetermine sample sizes, but the sample sizes were chosen based on previous experiments similarly performed by our group with proven statistically significant effects10. All graphs were made in Prism 9.4.1. MATLAB 9.11 was used to generate heatmaps of data. Data collection and analysis were not performed blind to the conditions of the experiments. All data points were included for analyses.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41565-023-01585-y.
Supplementary information
Supplementary Materials, Methods, Results, Figs. 1–27, Tables 1–9 and References.
Source data for Supplementary Figs. 4, 9, 10, 15, 17, 18, 20, 24, 26.
Source data
Numerical source data and uncropped dot-blots, respectively.
Numerical source data and uncropped gels, respectively.
Numerical source data.
Numerical source data.
Numerical source data.
Numerical source data.
Numerical source data.
Acknowledgements
This work is supported by a King’s PGR International Scholarship granted to R.L., and by the Biotechnology and Biological Sciences Research Council (BB/J008656/1 to K.A.J.), the Wellcome Trust (WT103913 to K.A.J.) and the Brain Tumour Charity (GN-000398 to K.T.A.-J.). Images were drawn on BioRender.com.
Extended data
Author contributions
R.L.-O. devised the concept, performed and designed the experiments, and wrote the manuscript. A.W., J.T.-W.W., F.N.F., L.X., A.S.F., G.L. and F.D. provided experimental support and supervision. S.H. performed experiments. J.O. and V.M. designed and performed the LC–MS experiments. K.T.A.-J. developed the concept, designed the experiments, supervised the work and wrote the manuscript. All authors reviewed and edited the manuscript before submission.
Peer review
Peer review information
Nature Nanotechnology thanks Bhagyashree Joshi and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Data availability
UniProtKB/SwissProt Homo sapiens and Bos taurus fasta databases (Proteome ID: UP000005640 and UP000009136, respectively) were used in this study. The authors declare that all data supporting the findings of this study are available in the provided Source Data and the Supplementary Information. Additional data are also available from the corresponding author upon reasonable request. Source data are provided with this paper.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
is available for this paper at 10.1038/s41565-023-01585-y.
Supplementary information
The online version contains supplementary material available at 10.1038/s41565-023-01585-y.
References
- 1.Asrani SK, et al. Burden of liver diseases in the world. J. Hepatol. 2019;70:151–171. doi: 10.1016/j.jhep.2018.09.014. [DOI] [PubMed] [Google Scholar]
- 2.Kwak KA, et al. Current perspectives regarding stem cell-based therapy for liver cirrhosis. Can. J. Gastroenterol. Hepatol. 2018;2018:4197857. doi: 10.1155/2018/4197857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guo Y, et al. Current status and future prospects of mesenchymal stem cell therapy for liver fibrosis. J. Zhejiang Univ. Sci. B. 2016;17:831–841. doi: 10.1631/jzus.B1600101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zagoura DS, et al. Therapeutic potential of a distinct population of human amniotic fluid mesenchymal stem cells and their secreted molecules in mice with acute hepatic failure. Gut. 2012;61:894–906. doi: 10.1136/gutjnl-2011-300908. [DOI] [PubMed] [Google Scholar]
- 5.Li T, et al. Exosomes derived from human umbilical cord mesenchymal stem cells alleviate liver fibrosis. Stem Cells Dev. 2013;22:845–854. doi: 10.1089/scd.2012.0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hyun J, et al. MicroRNA125b-mediated Hedgehog signaling influences liver regeneration by chorionic plate-derived mesenchymal stem cells. Sci. Rep. 2015;5:14135. doi: 10.1038/srep14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tan CY, et al. Mesenchymal stem cell-derived exosomes promote hepatic regeneration in drug-induced liver injury models. Stem Cell Res. Ther. 2014;5:76. doi: 10.1186/scrt465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen L, et al. Exosomes derived from human menstrual blood-derived stem cells alleviate fulminant hepatic failure. Stem Cell Res. Ther. 2017;8:9. doi: 10.1186/s13287-016-0453-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bottger R, et al. Lipid-based nanoparticle technologies for liver targeting. Adv. Drug Deliv. Rev. 2020;154-155:79–101. doi: 10.1016/j.addr.2020.06.017. [DOI] [PubMed] [Google Scholar]
- 10.Lázaro-Ibáñez E, et al. Selection of fluorescent, bioluminescent, and radioactive tracers to accurately reflect extracellular vesicle biodistribution in vivo. ACS Nano. 2021;15:3212–3227. doi: 10.1021/acsnano.0c09873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Faruqu FN, et al. Membrane radiolabelling of exosomes for comparative biodistribution analysis in immunocompetent and immunodeficient mice—a novel and universal approach. Theranostics. 2019;9:1666–1682. doi: 10.7150/thno.27891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiao Q, et al. The effects of protein corona on in vivo fate of nanocarriers. Adv. Drug Deliv. Rev. 2022;186:114356. doi: 10.1016/j.addr.2022.114356. [DOI] [PubMed] [Google Scholar]
- 13.Bai X, et al. In vivo protein corona formation: characterizations, effects on engineered nanoparticles’ biobehaviors, and applications. Front. Bioeng. Biotechnol. 2021;9:646708. doi: 10.3389/fbioe.2021.646708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heidarzadeh M, et al. Protein corona and exosomes: new challenges and prospects. Cell Commun. Signal. 2023;21:64. doi: 10.1186/s12964-023-01089-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Faruqu FN, et al. Defined serum-free three-dimensional culture of umbilical cord-derived mesenchymal stem cells yields exosomes that promote fibroblast proliferation and migration in vitro. FASEB J. 2021;35:e21206. doi: 10.1096/fj.202001768RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Webber J, Clayton A. How pure are your vesicles? J. Extracell. Vesicles10.3402/jev.v2i0.19861 (2013). [DOI] [PMC free article] [PubMed]
- 17.Ramirez MI, et al. Technical challenges of working with extracellular vesicles. Nanoscale. 2018;10:881–906. doi: 10.1039/C7NR08360B. [DOI] [PubMed] [Google Scholar]
- 18.Tkach M, Théry C. Communication by extracellular vesicles: where we are and where we need to go. Cell. 2016;164:1226–1232. doi: 10.1016/j.cell.2016.01.043. [DOI] [PubMed] [Google Scholar]
- 19.Jønson L, et al. Molecular composition of IMP1 ribonucleoprotein granules. Mol. Cell Proteom. 2007;6:798–811. doi: 10.1074/mcp.M600346-MCP200. [DOI] [PubMed] [Google Scholar]
- 20.Schroeder HW, Jr., Cavacini L. Structure and function of immunoglobulins. J. Allergy Clin. Immunol. 2010;125:S41–S52. doi: 10.1016/j.jaci.2009.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cougot N, Babajko S, Séraphin B. Cytoplasmic foci are sites of mRNA decay in human cells. J. Cell Biol. 2004;165:31–40. doi: 10.1083/jcb.200309008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chari A, et al. An assembly chaperone collaborates with the SMN complex to generate spliceosomal SnRNPs. Cell. 2008;135:497–509. doi: 10.1016/j.cell.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 23.Sunesson L, Hellman U, Larsson C. Protein kinase cepsilon binds peripherin and induces its aggregation, which is accompanied by apoptosis of neuroblastoma cells. J. Biol. Chem. 2008;283:16653–16664. doi: 10.1074/jbc.M710436200. [DOI] [PubMed] [Google Scholar]
- 24.van Asbeck EC, et al. Mannose binding lectin plays a crucial role in innate immunity against yeast by enhanced complement activation and enhanced uptake of polymorphonuclear cells. BMC Microbiol. 2008;8:229. doi: 10.1186/1471-2180-8-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takahashi K, et al. Relative roles of complement factor 3 and mannose-binding lectin in host defense against infection. Infect. Immun. 2005;73:8188–8193. doi: 10.1128/IAI.73.12.8188-8193.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalia N, Singh J, Kaur M. The ambiguous role of mannose-binding lectin (MBL) in human immunity. Open Med. 2021;16:299–310. doi: 10.1515/med-2021-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tóth E, et al. Formation of a protein corona on the surface of extracellular vesicles in blood plasma. J. Extracell. Vesicles. 2021;10:e12140. doi: 10.1002/jev2.12140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gomes FG, et al. Synergy of human platelet-derived extracellular vesicles with secretome proteins promotes regenerative functions. Biomedicines. 2022;10:238. doi: 10.3390/biomedicines10020238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolf M, et al. A functional corona around extracellular vesicles enhances angiogenesis, skin regeneration and immunomodulation. J. Extracell. Vesicles. 2022;11:e12207. doi: 10.1002/jev2.12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Churchman AH, et al. Serum albumin enhances the membrane activity of ZnO nanoparticles. Chem. Commun. (Camb.) 2013;49:4172–4174. doi: 10.1039/c3cc37871c. [DOI] [PubMed] [Google Scholar]
- 31.Rastogi L, Kora AJ, J A. Highly stable, protein capped gold nanoparticles as effective drug delivery vehicles for amino-glycosidic antibiotics. Mater. Sci. Eng. C. 2012;32:1571–1577. doi: 10.1016/j.msec.2012.04.044. [DOI] [PubMed] [Google Scholar]
- 32.Zaloga J, et al. Development of a lauric acid/albumin hybrid iron oxide nanoparticle system with improved biocompatibility. Int J. Nanomed. 2014;9:4847–4866. doi: 10.2147/IJN.S68539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vauthier C, Lindner P, Cabane B. Configuration of bovine serum albumin adsorbed on polymer particles with grafted dextran corona. Colloids Surf. B. 2009;69:207–215. doi: 10.1016/j.colsurfb.2008.11.017. [DOI] [PubMed] [Google Scholar]
- 34.Qi J, et al. Nanoparticles with dextran/chitosan shell and BSA/chitosan core–doxorubicin loading and delivery. Int. J. Pharm. 2010;393:176–184. doi: 10.1016/j.ijpharm.2010.03.063. [DOI] [PubMed] [Google Scholar]
- 35.Li Z, et al. Insight into the preformed albumin corona on in vitro and in vivo performances of albumin-selective nanoparticles. Asian J. Pharm. Sci. 2019;14:52–62. doi: 10.1016/j.ajps.2018.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nigam P, et al. Graphene quantum dots conjugated albumin nanoparticles for targeted drug delivery and imaging of pancreatic cancer. J. Mater. Chem. B. 2014;2:3190–3195. doi: 10.1039/C4TB00015C. [DOI] [PubMed] [Google Scholar]
- 37.Peng Q, et al. Preformed albumin corona, a protective coating for nanoparticles based drug delivery system. Biomaterials. 2013;34:8521–8530. doi: 10.1016/j.biomaterials.2013.07.102. [DOI] [PubMed] [Google Scholar]
- 38.Takeuchi T, et al. Molecularly imprinted nanogels acquire stealth in situ by cloaking themselves with native dysopsonic proteins. Angew. Chem. Int. Ed. 2017;56:7088–7092. doi: 10.1002/anie.201700647. [DOI] [PubMed] [Google Scholar]
- 39.Albrecht C, et al. Effect of chronic bile duct obstruction and LPS upon targeting of naproxen to the liver using naproxen–albumin conjugate. J. Drug Target. 1998;6:105–117. doi: 10.3109/10611869808997886. [DOI] [PubMed] [Google Scholar]
- 40.Kamps JAAM, et al. Massive targeting of liposomes, surface-modified with anionized albumins, to hepatic endothelial cells. Proc. Natl Acad. Sci. USA. 1997;94:11681–11685. doi: 10.1073/pnas.94.21.11681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weisiger R, Gollan J, Ockner R. Receptor for albumin on the liver cell surface may mediate uptake of fatty acids and other albumin-bound substances. Science. 1981;211:1048–1051. doi: 10.1126/science.6258226. [DOI] [PubMed] [Google Scholar]
- 42.Miao L, et al. Synergistic lipid compositions for albumin receptor mediated delivery of mRNA to the liver. Nat. Commun. 2020;11:2424. doi: 10.1038/s41467-020-16248-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liang X, et al. Extracellular vesicles engineered to bind albumin demonstrate extended circulation time and lymph node accumulation in mouse models. J. Extracell. Vesicles. 2022;11:e12248. doi: 10.1002/jev2.12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lesniak A, et al. Serum heat inactivation affects protein corona composition and nanoparticle uptake. Biomaterials. 2010;31:9511–9518. doi: 10.1016/j.biomaterials.2010.09.049. [DOI] [PubMed] [Google Scholar]
- 45.Simon J, et al. Protein denaturation caused by heat inactivation detrimentally affects biomolecular corona formation and cellular uptake. Nanoscale. 2018;10:21096–21105. doi: 10.1039/C8NR07424K. [DOI] [PubMed] [Google Scholar]
- 46.Faruqu FN, Xu L, Al-Jamal KT. Preparation of exosomes for siRNA delivery to cancer cells. J. Vis. Exp. 2018;5:10.3791/58814. doi: 10.3791/58814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schottler S, et al. Protein adsorption is required for stealth effect of poly(ethylene glycol)- and poly(phosphoester)-coated nanocarriers. Nat. Nanotechnol. 2016;11:372–377. doi: 10.1038/nnano.2015.330. [DOI] [PubMed] [Google Scholar]
- 48.Xu L, et al. Design of experiment (DoE)-driven in vitro and in vivo uptake studies of exosomes for pancreatic cancer delivery enabled by copper-free click chemistry-based labelling. J. Extracell. Vesicles. 2020;9:1779458. doi: 10.1080/20013078.2020.1779458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boysen L, et al. Formation and glomerular deposition of immune complexes in mice administered bovine serum albumin: evaluation of dose, frequency, and biomarkers. J. Immunotoxicol. 2019;16:191–200. doi: 10.1080/1547691X.2019.1680776. [DOI] [PubMed] [Google Scholar]
- 50.Yoshikawa N, et al. Interaction of lipoplex with albumin enhances gene expression in hepatitis mice. Pharmaceutics. 2020;12:341. doi: 10.3390/pharmaceutics12040341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eddy AA, et al. Interstitial fibrosis in mice with overload proteinuria: deficiency of TIMP-1 is not protective. Kidney Int. 2000;58:618–628. doi: 10.1046/j.1523-1755.2000.00208.x. [DOI] [PubMed] [Google Scholar]
- 52.Wheeler DS, et al. The immunomodulatory effects of albumin in vitro and in vivo. Adv. Pharmacol. Sci. 2011;2011:691928. doi: 10.1155/2011/691928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aparicio-Vergara M, et al. Isolation of Kupffer cells and hepatocytes from a single mouse liver. Methods Mol. Biol. 2017;1639:161–171. doi: 10.1007/978-1-4939-7163-3_16. [DOI] [PubMed] [Google Scholar]
- 54.Shi B, Abrams M, Sepp-Lorenzino L. Expression of asialoglycoprotein receptor 1 in human hepatocellular carcinoma. J. Histochem. Cytochem. 2013;61:901–909. doi: 10.1369/0022155413503662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Braun JR, et al. The major subunit of the asialoglycoprotein receptor is expressed on the hepatocellular surface in mice lacking the minor receptor subunit. J. Biol. Chem. 1996;271:21160–21166. doi: 10.1074/jbc.271.35.21160. [DOI] [PubMed] [Google Scholar]
- 56.Bon C, et al. Capacity limits of asialoglycoprotein receptor-mediated liver targeting. MAbs. 2017;9:1360–1369. doi: 10.1080/19420862.2017.1373924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Movita D, et al. Inflammatory monocytes recruited to the liver within 24 hours after virus-induced inflammation resemble Kupffer cells but are functionally distinct. J. Virol. 2015;89:4809–4817. doi: 10.1128/JVI.03733-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.De Simone G, et al. Identification of a Kupffer cell subset capable of reverting the T cell dysfunction induced by hepatocellular priming. Immunity. 2021;54:2089–2100.e8. doi: 10.1016/j.immuni.2021.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kinoshita M, et al. Characterization of two F4/80-positive Kupffer cell subsets by their function and phenotype in mice. J. Hepatol. 2010;53:903–910. doi: 10.1016/j.jhep.2010.04.037. [DOI] [PubMed] [Google Scholar]
- 60.Connolly MK, et al. In hepatic fibrosis, liver sinusoidal endothelial cells acquire enhanced immunogenicity. J. Immunol. 2010;185:2200. doi: 10.4049/jimmunol.1000332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Strauss O, et al. Immunofluorescence identifies distinct subsets of endothelial cells in the human liver. Sci. Rep. 2017;7:44356. doi: 10.1038/srep44356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hutchins NA, et al. Kupffer cells protect liver sinusoidal endothelial cells from Fas-dependent apoptosis in sepsis by down-regulating gp130. Am. J. Pathol. 2013;182:742–gp154. doi: 10.1016/j.ajpath.2012.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mederacke I, et al. High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat. Protoc. 2015;10:305–315. doi: 10.1038/nprot.2015.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hwang I, et al. Retinol from hepatic stellate cells via STRA6 induces lipogenesis on hepatocytes during fibrosis. Cell Biosci. 2021;11:3. doi: 10.1186/s13578-020-00509-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ichikawa S, et al. Hepatic stellate cells function as regulatory bystanders. J. Immunol. 2011;186:5549–5555. doi: 10.4049/jimmunol.1003917. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials, Methods, Results, Figs. 1–27, Tables 1–9 and References.
Source data for Supplementary Figs. 4, 9, 10, 15, 17, 18, 20, 24, 26.
Numerical source data and uncropped dot-blots, respectively.
Numerical source data and uncropped gels, respectively.
Numerical source data.
Numerical source data.
Numerical source data.
Numerical source data.
Numerical source data.
Data Availability Statement
UniProtKB/SwissProt Homo sapiens and Bos taurus fasta databases (Proteome ID: UP000005640 and UP000009136, respectively) were used in this study. The authors declare that all data supporting the findings of this study are available in the provided Source Data and the Supplementary Information. Additional data are also available from the corresponding author upon reasonable request. Source data are provided with this paper.