

Summary
Burkitt lymphoma arising in paediatric post-solid-organ transplantation-Burkitt lymphoma (PSOT-BL) is a clinically aggressive malignancy and a rare form of post-transplant lymphoproliferative disorder (PTLD). We evaluated 35 patients diagnosed with PSOT-BL at 14 paediatric medical centres in the United States. Median age at organ transplantation was 2.0 years (range: 0.1–14) and age at PSOT-B L diagnosis was 8.0 years (range: 1–17). All but one patient had late onset of PSOT-BL (≥2 years post-transplant), with a median interval from transplant to PSOT-BL diagnosis of 4.0 years (range: 0.4–12). Heart (n = 18 [51.4%]) and liver (n = 13 [37.1%]) were the most frequently transplanted organs. No patients had loss of graft or treatment-related mortality. A variety of treatment regimens were used, led by intensive Burkitt lymphoma-specific French–American–British/Lymphomes Malins B (FAB/LMB), n = 13 (37.1%), and a low-intensity regimen consisting of cyclophosphamide, prednisone and rituximab (CPR) n = 12 (34.3%). Median follow-up was 6.7 years (range: 0.5–17). Three-year event-free and overall survival were 66.2% and 88.0%, respectively. Outcomes of PSOT-BL patients receiving BL-specific intensive regimens are comparable to reported BL outcomes in immunocompetent children. Multi-institutional collaboration is feasible and provides the basis of prospective data collection to determine the optimal treatment regimen for PSOT-BL.
Keywords: Burkitt lymphoma, lymphoproliferative disorder, post-solid-organ transplantation
INTRODUCTION
Most monomorphic post-transplant lymphoproliferative disorders (M-PTLD) are high-grade mature B-c ell non-Hodgkin lymphomas (B-NHL). Of these, diffuse large B-cell type (DLBCL) is the most common; Burkitt lymphoma (BL) type M-PTLD is reported much less frequently in the literature. Post-solid-organ transplant BL (PSOT-BL) is a late form of M-PTLD characterized by a more aggressive clinical presentation and distinct pathologic and biologic features.1–5 The incidence of BL is increased 23-fold in solid-organ transplant recipients compared to the general population.1
Intensive chemotherapy regimens, such as the specific French–American–British/Lymphomes Malins B (FAB/LMB) regimen, are required for immunocompetent paediatric patients with BL (IC-BL) to achieve excellent outcomes.6–11 The addition of rituximab has further improved outcomes based on the recent Inter-B-NHL Ritux 2010 (Children Oncology Group ANHL1131) clinical trial in paediatric patients with high-risk B-NHL that showed a three-year event-free survival (EFS) of 93.9% for patients in the rituximab plus chemotherapy group and 82.3% for patients in the chemotherapy-only group.11
In contrast, there is no defined standard treatment regimen for PSOT-BL. Reports on both adult and paediatric patients have used approaches ranging from low-dose chemotherapy regimens with or without the addition of rituximab to more intensive immunochemotherapeutic regimens similar to those used for IC-BL. The latter approach appears to be more effective though treatment-related morbidity and mortality remain major limitations, particularly in adult patients. Of particular concern are the risk of infection-related death, risk of rejection and higher risk of end-organ toxicities specific to the allograft.4,5,12 It is unclear whether treatment approaches should be extrapolated from the non-PSOT-BL experience or from the approaches taken with other forms of M-PTLD. For example, DLBCL-type M-P TLD may respond well to reduction of immunosuppression (RIS) and/or specific anti-B-cell immunotherapy, such as rituximab, and adequate treatment does not always require chemotherapy.13–17 However, published data about treatment and outcomes specifically for PSOT-BL are limited by the small number of patients with PSOT-BL compared to other types of M-PTLD.3,4,12,18–20 In the limited available data in children, patients with PSOT-BL were often grouped with other types of PTLD despite its more aggressive clinical phenotype.13,21 This study aimed to retrospectively identify paediatric patients with PSOT-BL in a multicentre collaborative to describe treatment approaches and clinical outcomes.
METHODS
Patients
This multicentre retrospective study included children with PSOT-BL diagnosed before they were 18 years of age and treated from 2000 to 2020 at 14 academic paediatric centres across the United States. The study was approved by the Institutional Review Boards at all institutions. Patients were identified based on institutional pathologic diagnosis employing World Health Organization (WHO) criteria and clinical data and were entered into a centralized, de-identified database. Notable pathology variables included positivity for CD19/CD20 and CD10, lack of BCL-2 expression, and a high Ki67 proliferation index (>95%). Central pathology review was not performed. Patients without MYC rearrangement were required to meet other criteria for classic BL (e.g., medium-sized CD20+ B cells of germinal centre origin with a high proliferation index [Ki67 > 95%] with intermixed tingible body macrophages).22 Specific treatments varied among respective institutions.
Statistical analysis and end points
Data from all sites were abstracted similarly using a standardized form with reliability among the sites confirmed. Abstracted data included Murphy/St Jude stage,23,24 risk group stratification based on FAB/LMB risk group,7,9 serum lactate dehydrogenase (LDH) values standardized relative to the institutional upper limit of normal (ULN), and use of computed tomography (CT) for response assessment. Events were defined as any of the following: progression, relapse, second PTLD, or death. Time to event was determined as the time from PSOT-BL diagnosis to the earliest time of any event or censorship at last follow-up. Overall survival was assessed as time from PSOT-BL diagnosis to death from any cause or censorship at last follow-up. The log-rank test was used to evaluate potential differences in survival outcomes. Statistical analyses were performed with SAS software, version 9.4 (SAS Institute Inc., Cary, NC, USA) and GraphPad Prism version 9.3.1 (GraphPad Software, San Diego, CA, USA).
RESULTS
Patients and disease characteristics at diagnosis of PSOT-BL
Baseline characteristics of the 35 study patients are presented in Table 1. Nine (25.7%) were female. Median year of PSOT-BL diagnosis was 2013 (range 2000–2020). More than half of the patients were diagnosed over eight years between 2013 and 2020. Median age at organ transplantation was 2.0 years (range, 0.1–14) and age at diagnosis of PSOT-BL was 8.0 years (range, 1–17). Thirty-four of 35 (97.1%) patients had late onset of PSOT-BL (≥2 years since transplant), with a median interval from transplant to PSOT-BL diagnosis of 4.0 years (range, 0.4–12). Heart (n = 18 [51.4%]) and liver (n = 13 [37.1%]) were the most frequently transplanted organs, followed by bowel (n = 2 [5.7%)], kidney (n = 1 [2.9%]) and lung (n = 1 [2.9%]).
TABLE 1.
Patient characteristics of paediatric BL-PSOT patients, N = 35
| Characteristic | N | % |
|---|---|---|
| Median age at organ transplant (range)—years | 2.0 (0.1–14.0) | |
| Median age at PTLD diagnosis (range)—years | 8.0 (1.0–17.0) | |
| Time from transplant to PTLD (range)—years | 4.0 (0.43–11.7) | |
| Median year of PTLD diagnosis (range) | 2013(2000–2020) | |
| Male:female ratio | 26:9 | |
| Organ transplanted | ||
| Heart | 18 | 51.4 |
| Liver | 13 | 37.1 |
| Bowel | 2 | 5.7 |
| Kidney | 1 | 2.9 |
| Lung | 1 | 2.9 |
| EBV viraemiaa at diagnosis of BL (N = 33) | ||
| Positive | 29 | 87.9 |
| Negative | 4 | 12.1 |
| EBV serologic statusb at diagnosis of BL (N = 17) | ||
| Positive | 14 | 82.4 |
| Negative | 3 | 17.6 |
| Immunosuppressive therapy at PSOT-BL diagnosis | ||
| Tacrolimus | 28 | |
| Alone | 12 | 34.3 |
| In combination with other immunosuppressive drugs | 16 | 45.7 |
| Ciclosporin combined with other immunosuppressive drugs | 6 | 17.1 |
| Sirolimus only | 1 | 2.9 |
Abbreviations: EBV, Epstein-Barr Virus; PTLD, post-transplant lymphoproliferative disorder; PSOT-BL, post-solid-organ transplant Burkitt lymphoma.
Viremia detected in whole blood or plasma.
Serologic testing of viral capsid antigen IgG.
Thirty-four patients were receiving a calcineurin inhibitor at the time of PSOT-BL diagnosis: tacrolimus alone (n = 12 [34%]), tacrolimus with an additional immunosuppressive agent (prednisone, azathioprine, and/or mycophenolate mofetil) (n = 16 [45.7%]), or ciclosporin with an additional immunosuppressive agent: mycophenolate (n = 4 [11.4%]), sirolimus (n = 1 [2.9%]), or azathioprine (n = 1 [2.9%]). One patient was on sirolimus alone.
Disease characteristics are shown in Table 2. Lactate dehydrogenase (LDH) was above the ULN in 28 of 35 patients (80%) at the time of BL diagnosis and two times ULN or higher in 16 (45.7%) patients. Most patients (n = 30 [85.7%]) had Murphy stage III or IV. Abdomen and mediastinum were the most frequently involved sites (n = 25 [71.4%]) and (n = 14 [40%]) respectively, followed by peripheral lymph nodes (n = 18 [51.4%]), bone marrow (n = 9 [25.7%]), central nervous system (CNS) (n = 2 [5.7%]), with cerebrospinal fluid (CSF) involvement (n = 1 [2.9%]), or intracranial mass (n = 1 [2.9%]). Baseline disease characteristics by treatment regimen are depicted in Table 3.
TABLE 2.
Tumour characteristics of paediatric BL-PSOT patients (N = 35)
| Characteristic | N | % |
|---|---|---|
| Tumour stage | ||
| I | 1 | 2.9 |
| II | 4 | 11.4 |
| III | 19 | 54.3 |
| IV | 11 | 31.4 |
| FAB/LMB risk stratification | ||
| A | 1 | 2.9 |
| B | 27 | 77.1 |
| C | 7 | 20.0 |
| Elevated LDH | 28 | 80.0 |
| LDH ≥2 x upper limit of normal | 16 | 45.7 |
| CNS positive | 2 | 5.7 |
| CSF (N = 1) | ||
| Intracranial mass (N = 1) | ||
| BM positive | 9 | 25.7 |
| <25% (N=4) | ||
| ≥25% (N=5) | ||
| Abdominal mass | 25 | 71.4 |
| Mediastinal mass | 14 | 40.0 |
| Peripheral lymphadenopathy | 18 | 51.4 |
| Tumour EBV positive (N=31) | 27 | 87.1 |
| MYC by FISH (N=29) | ||
| Positive | 28 | 96.6 |
| Negative | 1 | 3.4 |
Abbreviations: BM, bone marrow; CNS, central nervous system; CSF, cerebrospinal fluid; EBV, Epstein-Barr Virus; FAB/LMB, French-American-British/Lymphomes Malins B; FISH, fluorescent in-situ hybridization; LDH, lactate dehydrogenase; PSOT-BL, post-solid-organ transplant Burkitt lymphoma.
TABLE 3.
Comparison of disease characteristics by treatment regimen
| Regimen | CPR | FAB/LMB (gr B, C) | EPOCH-R | SC-EPOCH-R | R-only | R-COPAD | R-CHOP | Other | Total |
|---|---|---|---|---|---|---|---|---|---|
| Total | 13 | 12 | 1 | 2 | 2 | 2 | 1 | 2 | 35 |
| Stage | |||||||||
| I | 1 | 1 | |||||||
| II | 1 | 2 | 1 | 4 | |||||
| III | 9 | 5 | 1 | 1 | 1 | 1 | 1 | 19 | |
| IV | 3 | 5 | 1 | 1 | 1 | 11 | |||
| FAB/LMB risk stratification | |||||||||
| A | 1 | 1 | |||||||
| B | 11 | 9 | 1 | 2 | 1 | 1 | 1 | 1 | 27 |
| C | 2 | 3 | 1 | 1 | 7 | ||||
| Elevated LDH | |||||||||
| Yes | 12 | 11 | 1 | 1 | 1 | 1 | 1 | 1 | 28 |
| No | 1 | 1 | 1 | 1 | 1 | 1 | 7 | ||
| LDH ≥ 2xupper limit of normala | |||||||||
| Yes | 6 | 6 | 1 | 1 | 1 | 1 | 16 | ||
| No | 7 | 6 | 2 | 1 | 1 | 1 | 1 | 19 | |
In cases with elevated LDH; CPR, cyclophosphamide, prednisone, rituximab21; EPOCH-R, etoposide, prednisone, vincristine, doxorubicin, cyclophosphamide, and rituximab39,40; FAB/LMB, French-American-British/Lymphomes Malins B8,9,11; LDH, lactate dehydrogenase; R-COPAD, rituximab, cyclophosphamide, vincristine prednisolone and doxorubicin42; R-CHOP, rituximab, cyclophosphamide, doxorubicin, vincristine and prednisolone43; R-only, rituximab only; other: UKCCSG9001 (n = 1),29 POG9317 (n = 1)44; SC-EPOCH-R, short-course EPOCH.27,34
Pathology
Tumour morphology in all 35 patients supported the diagnosis of BL. All 35 tumours were CD20-positive. CD10 was tested in 30 tumours, and all were positive. MIB1/Ki67 proliferation index was >95% in all but one of 28 patients with available results.
Thirty-one tumours underwent Epstein–B arr virus (EBV)-encoded RNA (EBER) in-situ hybridization (ISH), among which 27 (87%) were EBER ISH-positive. Latent membrane protein type-1 (LMP-1) was available on only five tumours and was positive in all but one. Fluorescence in-situ hybridization (FISH) for MYC rearrangements was positive in 28 of 29 patients. Only one tumour was negative by MYC FISH and further cytogenetic testing of this tumour was not performed.
The diagnosis of PSOT-BL was made from bone marrow aspiration in three patients, all of which had classic Burkitt morphology and flow cytometric features. MYC testing by FISH was positive in the two bone marrow samples with MYC testing available.
Treatment regimens
Treatment regimens are depicted in Figure 1. All patients had RIS after diagnosis of PTLD. Rituximab-containing regimens were given in all but two patients. The most common regimens were cyclophosphamide, prednisone, and rituximab21,25 (CPR, n = 13) and conventional paediatric BL therapy (FAB/LMB therapy, n = 12: group B regimen in n = 9 and group C in n = 3).6–11 Five patients were previously reported.20,26,27 None of the patients in our study who received CPR were enrolled on the Children’s Oncology Group (COG) ANHL0221 trial investigating this regimen in PTLD.21 Duration of therapy varied based on the treatment regimen used. Patients who received FAB/LMB group B therapy received reduction phase followed by four cycles of chemotherapy over a duration of 13 weeks (n = 9) and followed by 6–8 cycles over a duration of 19–25 weeks in group C therapy (n = 3). Six cycles of CPR were administered over 18 weeks in eight of 13 patients. Lesser numbers of cycles were administered in the remaining five patients because of partial response (n = 2); acute rejection (n = 1); fear of rejection (n = 1); and subsequent confirmation of BL diagnosis (n = 1). Duration of therapy in the remaining treatment regimens was: six cycles of R-CHOP (18 weeks [n = 1]), six cycles of EPOCH-R (18 weeks [n = 1]), three cycles of SC-EPOCH-R (10 weeks [n = 2]), two cycles of COPAD (cyclophosphamide, vincristine, prednisolone and doxorubicin) (six weeks [n = 2]), four and six weekly doses on rituximab monotherapy (4–6 weeks [n = 2]).
FIGURE 1.
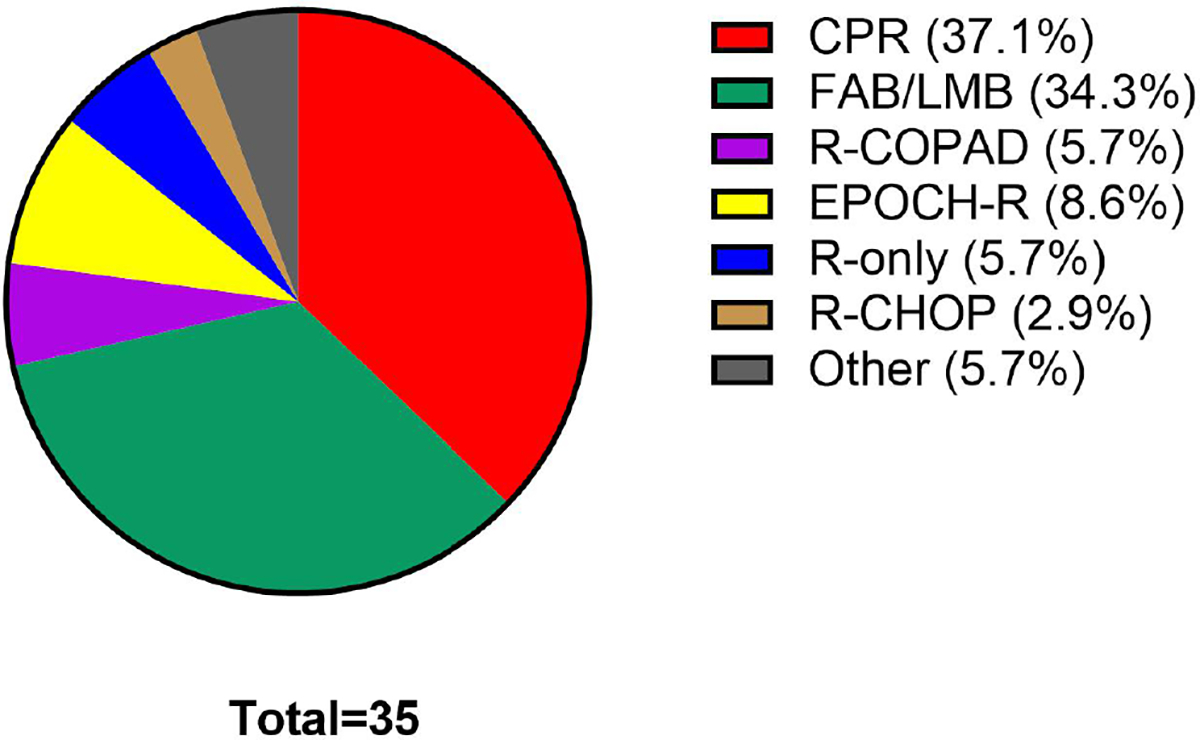
Treatment regimens used in paediatric PSOT-BL patients across the various institutions of this study. Shown is the frequency with which paediatric PSOT-BL patients were treated with various regimens. PSOT-BL, post-solid-organ transplant Burkitt lymphoma; CPR, cyclophosphamide, prednisone, rituximab21; FAB/LMB, French–American–British/Lymphomes Malins B8,9,11; R-COPAD, rituximab, cyclophosphamide, vincristine, prednisolone and doxorubicin38; EPOCH-R, etoposide, prednisone, vincristine, doxorubicin, cyclophosphamide, and rituximab39,40; R-CHOP, rituximab, cyclophosphamide, doxorubicin, vincristine and prednisolone41; R-only, rituximab only; Other, UKCCSG9001 (n = 1)29; POG9317 (n = 1).43 [Colour figure can be viewed at wileyonlinelibrary.com]
All patients who were treated with a FAB/LMB regimen received prophylactic intrathecal (IT) chemotherapy per group B or C regimen. No patients received CNS radiotherapy, consolidative haematopoietic stem cell transplantation, third-party cytotoxic T cells (CTLs) or chimaeric antigen receptor-T (CART) cells.
Outcome summary
The three-year EFS and overall survival (OS) for these 35 patients were 66.2% and 88.0% respectively (Figure 2). Median follow-up was 6.7 years (range, 0.5–17). Overall, 13 patients had secondary events, including progression, relapse, second PTLD or death (Table 4). Although limited by sample size and potential for outcome bias due to potential individual physician or institutional treatment selection preference, patients who were treated with FAB/LMB (n = 12) or CPR (n = 13) demonstrated a respective 3-year EFS of 76.4% and 52.7%, and OS of 100% and 84.6%. However, statistical comparison between those receiving CPR and those in the intensively treated FAB/LMB group was not feasible due to the small numbers in each subgroup and the inability to control for underlying unidentified factors in treatment choice. Similarly, due to the small number of events the evaluation of multivariate models was not feasible. Statistical comparison of outcomes for other treatment regimens was not possible due to the diverse regimens used, limitations of sample size and retrospective study design.
FIGURE 2.
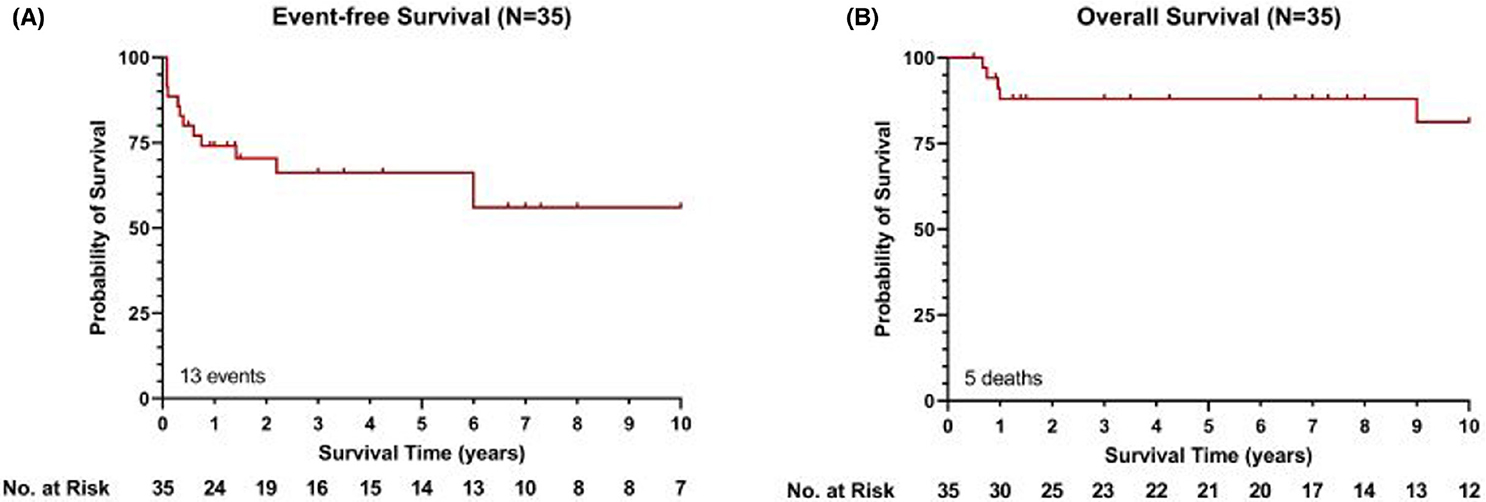
Event-free survival and overall survival (OS) in paediatric PSOT-BL patients. Kaplan–Meier plots for event-free survival (EFS) (A) and OS (B) in paediatric PSOT-BL patients. PSOT-BL, post-solid-organ transplant Burkitt lymphoma. Three-year EFS and OS 66.2% and 88.0%. [Colour figure can be viewed at wileyonlinelibrary.com]
TABLE 4.
Outcomes in patients with PSOT-BL by treatment regimen
| Regimen | CPR | FAB/LMB (Gr B, C) | EPOCH-R | SC-EPOCH-R | R-only | R-COPAD | R-CHOP | Other | Total |
|---|---|---|---|---|---|---|---|---|---|
| Total | 13 | 12 | 1 | 2 | 2 | 2 | 1 | 2 | 35 |
| Total adverse outcome events | 7 | 2 | 1 | 1 | 2 | 0 | 0 | 0 | 13 |
| Progression | 1 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 4 |
| Relapse | 5 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 6 |
| Second PTLD | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 3 |
| Survival | |||||||||
| Alive in CR Last FU | 10 | 12 | 0 | 2 | 1 | 2 | 1 | 2 | 30 |
| All deaths | 3 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 5 |
| Death from PSOT-BL | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 3 |
Abbreviations: CPR, cyclophosphamide, prednisone, rituximab21; CR, complete remission; EPOCH-R, etoposide, prednisone, vincristine, doxorubicin, cyclophosphamide, and rituximab35,36; FAB/LMB, French-American-British/Lymphomes Malins B8,9,11; FU, follow up; Other, UKCCSG9001 (n = 1),29 POG9317 (n = 1)44; PSOT-BL, postsolid-organ transplant Burkitt lymphoma; PTLD, post-transplant lymphoproliferative disorders; R-CHOP, rituximab, cyclophosphamide, doxorubicin, vincristine and prednisolone43; R-COPAD, rituximab, cyclophosphamide, vincristine prednisolone and doxorubicin42; R-only, rituximab only; SC-ECPOCH-R, short-course etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin.27,34
Progression
Four patients progressed on first-line therapy (rituximab monotherapy [n = 1]; EPOCH-R [n = 1]; CPR [n = 1]; and FAB/LMB [n = 1]), resulting in three deaths 0.7–1 year from BL diagnosis. Death from progression occurred in patients receiving the following regimens: rituximab monotherapy (n = 1); EPOCH-R (n = 1); CPR (n = 1). Of the 12 patients treated with FAB/LMB, one patient progressed on treatment and was successfully salvaged with an individualized intensive chemotherapy. All 12 patients are alive in complete remission (CR).
Relapse
There were six relapses. A higher relapse rate (41.6%) was observed in patients who received mild chemoimmunotherapy regimen CPR compared to BL-specific intensive chemotherapy or chemoimmunotherapy. Five of the six relapses occurred in patients who received low-dose chemoimmunotherapy regimen with CPR (five of 13 patients, 38.5%; Table 4). All five who relapsed after CPR were successfully salvaged by FAB/LMB regimen in four cases and an individualized regimen in one. Three of these relapses occurred shortly after completion of six cycles of CPR (5–7 months after start of CPR). The remaining two relapses after CPR were late relapses 2.2 and six years from initial BL diagnosis. These were the only two late relapses (over 1 year from BL diagnosis) in all 35 patients.
Treatment failure occurred in the two patients who received rituximab monotherapy (one progression and one relapse). One achieved CR but relapsed at the end of a 6-week rituximab course. He was subsequently treated with FAB/LMB regimen, achieved CR and is alive in CR 7.6-y ears following BL diagnosis. The other progressed, failed FAB/LMB salvage therapy and died 10 months from initial diagnosis. Outcomes in the remaining smaller subgroups are shown in Table 4.
Second PTLD
There were three subsequent non-BL episodes of PTLD (DLBCL/PTLD 0.3–6 years after initial BL diagnosis [n = 2], polymorphic PTLD [n = 1]). All were on immune suppression at the time of development of the second PTLD. One of these patients who developed DLBCL/PTLD died of the second PTLD. The other two were successfully treated and alive in CR 6 and 17 years after initial BL diagnosis.
Death
A total of five patients died (14.3%). Causes of death were progression of PSOT-BL on up-front regimen (n = 3), second PTLD (n = 1), and death from complications of a subsequent transplant, (n = 1). Early rejection of transplanted organ occurred in two (5.7%) patients within a year after PSOT-BL diagnosis. Neither of these two rejections resulted in graft loss.
Treatment modifications
Of patients who received FAB/LMB regimen, four had methotrexate dose modification and/or omission due to renal dysfunction secondary to tumour lysis syndrome (n = 2), renal dysfunction of unspecified cause in a heart transplant recipient (n = 1) and malignant ascites (n = 1). Doxorubicin was electively eliminated in one heart transplant recipient due to concern for cardiotoxicity. There were no treatment-related deaths among all 35 patients.
DISCUSSION
This multi-institutional retrospective work was undertaken to better characterize paediatric PSOT-BL. Despite the limitations inherent in a retrospective design, this study provides the following crucial observations: (1) the majority of patients received a heart or liver transplantation; (2) diverse regimens were used to treat BL, the most common being an intensive BL-specific regimen (FAB/LMB) or low-intensity regimen (CPR); (3) identification of comparable three-year OS rates (88.0%) in children with PSOT-BL compared to 87%–95% in paediatric IC-BL,7,9,11 but inferior three-year EFS rates (66.2%) compared to 82.3%–93.9% in paediatric IC-BL7,9,11; (4) a high relapse rate (41.6%) in patients who received mild chemoimmunotherapy regimen CPR compared to BL-specific intensive chemotherapy or chemoimmunotherapy; (5) late recurrence of PSOT-BL in two patients (5.7%); (6) treatment failure in the two patients who received rituximab monotherapy; (7) second occurrence of a different PTLD in three patients (8.6%); (8) absence of treatment-related mortality; and (9) low rejection rate (n = 2, 5.7%), with no graft loss.
Findings in our study support previous reports in PSOT-BL,1 and IC-BL,6 of predominance of male gender, and predominance of liver and heart transplant recipients.1 Another consistent finding is late onset of the PSOT-BL with a duration between organ transplant and PSOT-BL diagnosis of more than two years.1,3,4,28,29 It is unknown whether there is a change in incidence of PSOT-BL over the last few decades. In the US Transplant Cancer Match Study (1987–2009) there was no change in incidence over the study period.1 In our study it was not possible to determine an increase in the trend of occurrence of PSOT-BL because of potential selection bias or missed cases.
Chronic immune suppression remains the major factor predisposing solid-organ transplant recipients to this rare form of M-PTLD.1,12 The majority of tumours with available EBER testing in our study were EBV-positive (87.1% of 31 patients). This is similar to the reported incidence of EBV positivity of up to 90% in tumours from paediatric patients with PSOT-BL,1 and in endemic-BL tumours (nearly 95%). In contrast only 20%–30% of sporadic BL-IC tumours are EBV-positive.30 On the other hand, very limited data are available about potential differences in the biology of PSOT-BL compared to IC-BL, including limited MYC data.31 We propose that future prospective work should include MYC testing, with cytogenetic testing in all patients particularly when MYC is negative by FISH. Whether tumours share the mutational profile of that described in sporadic settings remains to be determined and is the subject of future studies, a next step within our collaborative. In a study by Ferreiro et al., 11q gain/loss aberration was reported to be more frequent in g PSOT-Bl (three of seven patients) compared to its rare occurrence IC-BL (3%). They proposed that immunosuppression may favour its formation and suggested that studies of PSOT-BL are needed to explore this potential association further.31
Barriers to prospectively studying optimal treatment regimens in PSOT-BL include the rarity of patients and the high rate of end-organ dysfunction because of their transplant. Given the unique pathobiology of paediatric PSOT-BL, in recent clinical trials these patients were either excluded from trials of other forms of M-PTLD,32 constituted a small fraction of the participants,13,21,33 or were excluded from clinical trials of IC-BL.7,9–11 This has limited the ability to gain information from these studies.
As in our study, prior data suggest that rituximab monotherapy is insufficient to treat PSOT-BL.4,13 On the other hand, the optimal chemotherapy regimen and intensity required for successful treatment of PSOT-BL has not been fully investigated. A previously published Children’s Oncology Group study reported on 55 paediatric patients with EBV-positive PTLD treated with CPR; five had Burkitt histology, four of whom were long-term disease-free survivors.21 However, the study did not report on partial response (PR) or subsequent relapse requiring treatment escalation or salvage therapy, rendering it impossible to conclude if CPR was sufficient frontline therapy for PSOT-BL. Interestingly, in our study, despite the high relapse rate in patients who received CPR regimen, salvage therapy was effective in all five who relapsed after such a regimen.
In two response-adapted clinical studies of PTLD, all patients with PSOT-BL required up-front lymphoma-specific chemotherapy (R-CHOP in adults and mCOMP in children), rather than RIS and rituximab alone.4,13 The superior efficacy of FAB/LMB regimen observed is supported by a report of paediatric patients with PSOT from the French Society of Pediatric Oncology (SFOP). In their study 14 of 16 children treated with the LMB regimen were alive in first CR with a median follow-up of 9 years,28 supporting the use of an intensive regimen. Whether intensive chemotherapy regimens such as LMB regimen are superior to moderate-intensity chemotherapy such as mCOMP13 in PSOT-BL remains to be determined.
Other regimens used in our study were short-course etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin (SC-EPOCH-R),27,34 and EPOCH-R.35,36 It is important to note that SC-EPOCH-R27,34 and EPOCH-R,35,36 while successful in adults with BL, have not been studied in paediatric BL.
Selection bias is likely to be an important factor in the choice of less intensive regimens such as CPR. The choice of CPR could be related to comorbidities, particularly concern for nephrotoxicity or existing renal dysfunction, and therefore avoiding high dose methotrexate.
In BL, relapses beyond 1 year are rare and should raise suspicion of an underlying predisposing condition such as XLP1 and other primary or acquired immunodeficiencies.37–40 On the other hand, there are no reports of two consecutive BLs in solid-organ transplant recipients. In the two patients who experienced late BL relapses in this study, clonality studies were not available to distinguish a relapse from a distinct second BL. We also found three PTLD occurrences. The risk of getting two distinct types of PTLD is unknown in solid-organ transplant recipients,41 and requires prospective evaluation. In our cases ongoing immune suppression is felt to be the main factor in these occurrences. To study this further, our collaborative is currently performing clonality testing on multiple occurrences of PTLD in individual patients.
The retrospective nature of this study posed several limitations, including potential selection bias, based on the retrospective review of records and choice of cases. Another is lack of central pathology review, absence of MYC data in some patients and lack of genomic studies. A diversity of treatment regimens was used, and it was also not possible to determine the rationale for treatment selection, as physician and institutional choice appear to play a role in the treatment regimen used.
In this paediatric PSOT-BL study, the three-year OS rates in children with PSOT-BL was comparable to paediatric IC-BL but inferior to the three-year EFS rates reported in paediatric IC-BL. Intensive BL regimens resulted in OS comparable to those observed in children with IC-BL, fewer treatment failures compared to low-intensity immunochemotherapy and acceptable treatment toxicities. This study also illustrates that multi-institutional collaboration is feasible and provides an excellent platform for establishing a paediatric post-solid-organ transplant PTLD registry. Linking such a registry to existing paediatric solid-organ transplant registries would capture their strength by expanding pathologic and oncologic components of the current organ transplant databases. This will be an important step in providing the needed foundation to prospectively study these challenging areas.
FUNDING INFORMATION
Clinton C. Mason acknowledges funding from the Paediatric Cancer Program, which is supported by the Intermountain Healthcare and Primary Children’s Hospital Foundations as well as the Department of Paediatrics and Division of Paediatric Haematology/Oncology at the University of Utah.
REFERENCES
- 1.Mbulaiteye SM, Clarke CA, Morton LM, Gibson TM, Pawlish K, Weisenburger DD, et al. Burkitt lymphoma risk in U.S. solid organ transplant recipients. Am J Hematol. 2013;88(4):245–50. 10.1002/ajh.23385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yanik EL, Shiels MS, Smith JM, Clarke CA, Lynch CF, Kahn AR, et al. Contribution of solid organ transplant recipients to the pediatric non-Hodgkin lymphoma burden in the United States. Cancer. 2017;123(23):4663–71. 10.1002/cncr.30923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bobillo S, Abrisqueta P, Sánchez-González B, Giné E, Romero S, Alcoceba M, et al. Posttransplant monomorphic Burkitt’s lymphoma: clinical characteristics and outcome of a multicenter series. Ann Hematol. 2018;97(12):2417–24. 10.1007/s00277-018-3473-8 [DOI] [PubMed] [Google Scholar]
- 4.Zimmermann H, Reinke P, Neuhaus R, Lehmkuhl H, Oertel S, Atta J, et al. Burkitt post-transplantation lymphoma in adult solid organ transplant recipients: sequential immunochemotherapy with rituximab (R) followed by cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) or R-CHOP is safe and effective in an analysis of 8 patients. Cancer. 2012;118(19):4715–24. 10.1002/cncr.27482 [DOI] [PubMed] [Google Scholar]
- 5.Gong JZ, Stenzel TT, Bennett ER, Lagoo AS, Dunphy CH, Moore JO, et al. Burkitt lymphoma arising in organ transplant recipients: a clinicopathologic study of five cases. Am J Surg Pathol. 2003;27(6):818–27. 10.1097/00000478-200306000-00014 [DOI] [PubMed] [Google Scholar]
- 6.Egan G, Weitzman S, Burkitt AS. Lymphoma and diffuse large B-cell lymphoma. In: Abla O, Attarbaschi A, editors. Non-Hodgkin’s lymphoma in childhood and adolescence. 1st ed. Switzerland: Springer Nature; 2019. p. 167–183l. 10.1007/978-3-030-11769-6 [DOI] [Google Scholar]
- 7.Cairo MS, Sposto R, Gerrard M, Auperin A, Goldman SC, Harrison L, et al. Advanced stage, increased lactate dehydrogenase, and primary site, but not adolescent age (≥15 years), are associated with an increased risk of treatment failure in children and adolescents with mature B-cell non-Hodgkin’s lymphoma: results of the FAB LMB 96 study. J Clin Oncol. 2012;30(4):387–93 10.1200/JCO.2010.33.3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cairo MS, Gerrard M, Sposto R, Auperin A, Pinkerton CR, Michon J, et al. Results of a randomized international study of high-r isk central nervous system B non-Hodgkin lymphoma and B acute lymphoblastic leukemia in children and adolescents. Blood. 2007;109(7):2736–43. 10.1182/blood-2006-07-036665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patte C, Auperin A, Gerrard M, Michon J, Pinkerton R, Sposto R, et al. Results of the randomized international FAB/LMB96 trial for intermediate risk B-cell non-H odgkin lymphoma in children and adolescents: it is possible to reduce treatment for the early responding patients. Blood. 2007;109(7):2773–80. 10.1182/blood-2006-07-036673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woessmann W, Seidemann K, Mann G, Zimmermann M, Burkhardt B, Oschlies I, et al. The impact of the methotrexate administration schedule and dose in the treatment of children and adolescents with B-cell neoplasms: a report of the BFM group study NHL-BFM95. Blood. 2005;105(3):948–58. 10.1182/blood-2004-03-0973 [DOI] [PubMed] [Google Scholar]
- 11.Minard-Colin V, Aupérin A, Pillon M, Burke GAA, Barkauskas DA, Wheatley K, et al. Rituximab for high-risk, mature B-cell non-Hodgkin’s lymphoma in children. N Engl J Med. 2020;382(23):2207–19. 10.1056/NEJMoa1915315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Afify Z, Punnett A. Post solid organ transplant Burkitt lymphoma. In: Berthelot DV, editor. Burkitt lymphoma: diagnosis, risk factors and treatment. New York: Nova; 2021. p. 143–78. 10.52305/VODC3429 [DOI] [Google Scholar]
- 13.Maecker-Kolhoff B, Beier R, Zimmermann M, Schlegelberger B, Baumann U, Mueller C, et al. Response-adapted sequential Immuno-chemotherapy of post-transplant lymphoproliferative disorders in pediatric solid organ transplant recipients: results from the prospective ped-PTLD 2005 trial. [abstract] Blood 2014;124:4468. Abstract 624. 10.1182/blood.V124.21.4468.4468 [DOI] [Google Scholar]
- 14.Trappe RU, Dierickx D, Zimmermann H, Morschhauser F, Mollee P, Zaucha JM, et al. Response to rituximab induction is a predictive marker in B-cell post-transplant lymphoproliferative disorder and allows successful stratification into rituximab or R-CHOP consolidation in an international, prospective, multicenter phase II trial. J Clin Oncol. 2017;35(5):536–43. 10.1200/JCO.2016.69.3564 [DOI] [PubMed] [Google Scholar]
- 15.Zimmermann H, Xu H, Barlev AA, Feng A, Li X, Navarro W, Trappe R, et al. Clinical outcomes of solid organ transplant patients with EBV+PTLD who fail first-line rituximab or rituximab plus chemotherapy: an analysis of German PTLD registry [Abstract]. Eur Hematol Assoc. 2019;3(S1):3148. Abstract PF719. https://library.ehaweb.org/eha/2019/24th/266518/heiner.zimmermann.clinical.outcomes.of.solid.organ.transplant.patients.with.html [Google Scholar]
- 16.Kanzelmeyer NK, Maecker-Kolhoff B, Zierhut H, Lerch C, Verboom M, Haffner D, et al. Graft outcomes following diagnosis of post-transplant lymphoproliferative disease in pediatric kidney recipients: a retrospective study. Transpl Int. 2018;31(4):367–76. 10.1111/tri.13071 [DOI] [PubMed] [Google Scholar]
- 17.Trappe R, Oertel S, Leblond V, Mollee P, Sender M, Reinke P, et al. Sequential treatment with rituximab followed by CHOP chemotherapy in adult B-cell post-transplant lymphoproliferative disorder (PTLD): the prospective international multicentre phase 2 PTLD-1 trial. Lancet Oncol. 2012;13(2):196–206. 10.1016/S1470-2045(11)70300-X [DOI] [PubMed] [Google Scholar]
- 18.Picarsic J, Jaffe R, Mazariegos G, Webber SA, Ellis D, Green MD, et al. Post-transplant Burkitt lymphoma is a more aggressive and distinct form of post-transplant lymphoproliferative disorder. Cancer. 2011;117(19):4540–50. 10.1002/cncr.26001 [DOI] [PubMed] [Google Scholar]
- 19.AbdelHameid D, Felice A, Cooper LB, Katugaha SB. Long-term remission in an adult heart transplant recipient with advanced Burkitt’s lymphoma post-transplant lymphoproliferative disorder after anthracycline-free chemotherapy: a case report and literature review. Transpl Infect Dis. 2020;22(4):e13265. 10.1111/tid.13265 [DOI] [PubMed] [Google Scholar]
- 20.El-Atoum B, Williams M, Molina K, Orjuela-Grimm MA, Prince J, Afify Z. Burkitt lymphoma after cardiac transplantation: therapeutic considerations. J Pediatr Hematol Oncol. 2022;44(3):100–2. 10.1097/MPH.0000000000002260 [DOI] [PubMed] [Google Scholar]
- 21.Gross TG, Orjuela MA, Perkins SL, Park JR, Lynch JC, Cairo MS, et al. Low-dose chemotherapy and rituximab for posttransplant lymphoproliferative disease (PTLD): a Children’s oncology group report. Am J Transplant. 2012;12(11):3069–75. 10.1111/j.1600-6143.2012.04206.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leoncini L, Campo E, Stein H, Harris NL, Jaffe ES, Kluin PM. Burkitt lymphoma. In: Swerdlow SH, Campo E, Harris NL, Jaffe HS, Pileri SA, Stein H, Theile J, editors. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon, France: International Agency for Research on Cancer; 2017. p. 330–4. [Google Scholar]
- 23.Murphy SB. Classification, staging and end results of treatment of childhood non-Hodgkin’s lymphomas: dissimilarities from lymphomas in adults. Semin Oncol. 1980;7(3):332–9. [PubMed] [Google Scholar]
- 24.Rosolen A, Perkins SL, Pinkerton CR, Guillerman RP, Sandlund JT, Patte C, et al. Revised international pediatric non-Hodgkin lymphoma staging system. J Clin Oncol. 2015;33(18):2112–8. 10.1200/JCO.2014.59.7203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Orjuela MA, Alobeid B, Liu X, Siebert AL, Kott ER, Addonizio LJ, et al. CD20 expression predicts survival in paediatric post-transplant lymphoproliferative disease (PTLD) following solid organ transplantation. Br J Haematol. 2011;152(6):733–42. 10.1111/j.1365-2141.2010.08448.x [DOI] [PubMed] [Google Scholar]
- 26.Orjuela M, Gross TG, Cheung YK, Alobeid B, Morris E, Cairo MS. A pilot study of chemoimmunotherapy (cyclophosphamide, prednisone, and rituximab) in patients with post-transplant lymphoproliferative disorder following solid organ transplantation. Clin Cancer Res. 2003;9(10 Pt 2):3945S–52S. [PubMed] [Google Scholar]
- 27.Rubinstein JD, Shah R, Breese EH, Burns KC, Mangino JL, Norris RE, et al. Treatment of posttransplant lymphoproliferative disorder with poor prognostic features in children and young adults: short-course EPOCH regimens are safe and effective. Pediatr Blood Cancer 2021;68(8):e29126. 10.1002/pbc.29126 [DOI] [PubMed] [Google Scholar]
- 28.Cohen-Gogo S, Landman-Parker GJ, Michel H, Pacquement H, Michon J, Bertrand Y, et al. LMB chemotherapy is effective for Burkitt lymphoma occurring after sold organ transplantation in children: a report from the societe francaise des cancers de L’enfante (SFCE). [abstract]. Pediatr Blood Cancer. 2019;66(Suppl):S260. Abstract 197.4. 10.1002/pbc.27989 [DOI] [Google Scholar]
- 29.Windebank K, Walwyn T, Kirk R, Parry G, Hasan A, Bown N, et al. Post cardiac transplantation lymphoproliferative disorder presenting as t(8;14) Burkitt leukaemia/lymphoma treated with low intensity chemotherapy and rituximab. Pediatr Blood Cancer. 2009;53(3): 392–6. 10.1002/pbc.22070 [DOI] [PubMed] [Google Scholar]
- 30.Thacker N, Abla O. Epidemiology of non-Hodgkin lymphomas in childhood and adolescence. In: Abla O, Attarbaschi A, editors. Non-Hodgkin’s lymphoma in childhood and adolescenc. Cham: Springer; 2019. p. 15–22. 10.1007/978-3-030-11769-6_2 [DOI] [Google Scholar]
- 31.Ferreiro JF, Morscio J, Dierickx D, Marcelis L, Verhoef G, Vandenberghe P, et al. Post-transplant molecularly defined Burkitt lymphomas are frequently MYC-negative and characterized by the 11q-gain/loss pattern. Haematologica. 2015;100(7):e275–9. 10.3324/haematol.2015.124305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rituximab and LMP-specific T-cells in treating pediatric solid organ recipients with EBV-p ositive, CD20-positive post-transplant lymph oproliferative disorder. https://clinicaltrials.gov/ct2/show/NCT02900976. Last updated April 30, 2021. Accessed May 4, 2022.
- 33.Schober T, Framke T, Kreipe H, Schulz TF, Grohennig A, Hussein K, et al. Characteristics of early and late PTLD development in pediatric solid organ transplant recipients. Transplantation. 2013;95(1):240–6. 10.1097/TP.0b013e318277e344 [DOI] [PubMed] [Google Scholar]
- 34.Dunleavy K, Little RF, Pittaluga S, Grant N, Wayne AS, Carrasquillo JA, et al. The role of tumor histogenesis, FDG-P ET, and short-course EPOCH with dose-d ense rituximab (SC-EPOCH-RR) in HIV-a ssociated diffuse large B-cell lymphoma.Blood 2010;115(15):3017–24.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dunleavy K, Pittaluga S, Shovlin M, Steinberg SM, Cole D, Grant C, et al. Low-intensity therapy in adults with Burkitt’s lymphoma. N Engl J Med. 2013;369(20):1915–25. 10.1056/NEJMoa1308392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roschewski M, Dunleavy K, Abramson JS, Powell BL, Link BK, Patel P, et al. Multicenter study of risk-adapted therapy with dose-adjusted EPOCH-R in adults with untreated Burkitt lymphoma. J Clin Oncol. 2020;38(22):2519–29. 10.1200/JCO.20.00303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iglesias Cardenas F, Agarwal AM, Vagher J, Maese L, Fluchel M, Afify Z. Two clonally distinct B-cell lymphomas reveal the diagnosis of XLP1 in a male child and his asymptomatic male relatives: case report and review of the literature. J Pediatr Hematol Oncol. 2021;43(8):e1210–3. 10.1097/MPH.0000000000002049 [DOI] [PubMed] [Google Scholar]
- 38.Zhou D, Paxton CN, Kelley TW, Afify Z, South ST, Miles RR. Two unrelated Burkitt lymphomas seven years apart in a patient with X-linked lymphoproliferative disease type 1 (XLP1). Am J Clin Pathol. 2016;146(2):248–53. 10.1093/ajcp/aqw036 [DOI] [PubMed] [Google Scholar]
- 39.Afify Z, Abla O. Reducing treatment burden matters in special populations with pediatric B-non-Hodgkin lymphoma. J Pediatr Hematol Oncol. 2021;43(8):e1270. 10.1097/MPH.0000000000002335 [DOI] [PubMed] [Google Scholar]
- 40.Afify Z, Abla O. Burkitt lymphoma in patients with primary immune deficiency disorders. In: Berthelot DV, editor. Burkitt lymphoma: diagnosis, risk factors and treatment. Hauppauge, New York: Nova Publishers; 2021. p. 89–142. 10.52305/VODC3429 [DOI] [Google Scholar]
- 41.Dalal M, Liu C, Drew K, Slayton W, Shih R, Collins W, et al. Concurrent subtypes of post-transplant lymphoproliferative disorders after solid organ transplant. [abstract] Pediatr Blood Cancer 2021;68(Suppl):S186. 10.1002/pbc.29060 [DOI] [Google Scholar]
- 42.Gerrard M, Cairo MS, Weston C, Auperin A, Pinkerton R, Lambilliote A, et al. Excellent survival following two courses of COPAD chemotherapy in children and adolescents with resected localized B-cell non-Hodgkin’s lymphoma: results of the FAB/LMB 96 international study. Br J Haematol. 2008;141(6):840–7. 10.1111/j.1365-2141.2008.07144.x [DOI] [PubMed] [Google Scholar]
- 43.Coiffier B, Lepage E, Briere J, Herbrecht R, Tilly H, Bouabdullah R, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med. 2002;346(4):235–42. 10.1056/NEJMoa011795 [DOI] [PubMed] [Google Scholar]
- 44.Laver JH, Kraveka JM, Hutchison RE, Chang M, Kepner J, Schwenn M, et al. Advanced-stage large-cell lymphoma in children and adolescents: results of a randomized trial incorporating intermediate-dose methotrexate and high-dose cytarabine in the maintenance phase of the APO regimen: a pediatric oncology group phase III trial. J Clin Oncol. 2005;23(3):541–7. 10.1200/JCO.2005.11.075 [DOI] [PubMed] [Google Scholar]