

Abstract
A 34-year-old Japanese man presented with blurred vision, headache, nausea, anemia, thrombocytopenia, and severe renal dysfunction. Thrombotic microangiopathy was initially suspected to have been caused by malignant hypertension. Antihypertensive medications did not improve his thrombocytopenia or renal dysfunction, and other diseases causing thrombotic microangiopathy were ruled out. Therefore, the patient was diagnosed with atypical hemolytic uremic syndrome. A renal biopsy revealed an overlap of thrombotic microangiopathy and C3 glomerulopathy. Genetic testing revealed c.848A>G (p.Asp283Gly), a missense heterozygous variant in the gene encoding complement factor I. Overlapping atypical hemolytic uremic syndrome and C3 glomerulopathy with complement factor I mutation is very rare, especially in Japan.
Keywords: aHUS, TMA, C3 glomerulopathy, CFI, case report
Introduction
Dysregulation of the complement pathway has been reported to be associated with aggravation of renal damage (1). Atypical hemolytic uremic syndrome (aHUS) and C3 glomerulopathy are representative complement-mediated kidney diseases. Although genetic and acquired factors related to the complement pathway reportedly play a role in the etiology and disease susceptibility of both disorders, aHUS and C3 glomerulopathy generally differ in terms of renal pathology and clinical findings.
aHUS clinically shows thrombotic microangiopathy (TMA), characterized by the triad of acute kidney injury, microangiopathic hemolytic anemia, and thrombocytopenia. The diagnosis of aHUS requires the exclusion of other diseases that cause TMA, such as thrombotic thrombocytopenic purpura (TTP) and Shiga toxin-producing Escherichia coli hemolytic uremic syndrome. In renal pathology, aHUS exhibits TMA findings, such as endothelial swelling, subendothelial accumulation of proteins, and thrombosis in glomerular capillaries on light microscopy with no deposition of immune complexes or complements on immunofluorescence staining.
aHUS is caused by overactivation of the alternative pathway of the complement system at the endothelial cell surface (2). It is triggered by congenital or acquired abnormalities in the complement activation regulators. Genetic abnormalities are found in approximately 46% of patients with aHUS (3). Genetic cases of aHUS have been caused by pathological mutations in genes encoding complement factor H (CFH), complement factor I (CFI), complement factor B (CFB), complement C3 (C3), CD46 (CD46), thrombomodulin (THBD), diacylglycerol kinase ε (DGKE), plasminogen (PLG), and inverted formin 2 (INF2), whereas reported acquired cases have included those with anti-factor H antibody positivity (4). There are extensive reports on anti-factor H antibodies and mutations in CFH or C3 in Japanese patients with aHUS (3). However, there have been no reported Japanese cases of aHUS due to mutations in CFI.
C3 glomerulopathy often presents with membranoproliferative glomerulonephritis patterns and glomerular accumulation of complement proteins, characterized by bright C3 staining on immunofluorescence microscopy with minimal or no staining for immunoglobulins. C3 glomerulopathy often progresses to end-stage kidney disease and recurs following renal transplantation. Pathological observations in patients with C3 glomerulopathy indicate selective alternative pathway overactivation and C3 consumption during the fluid phase (1). In addition, C3 glomerulopathy is subclassified according to electron microscopy findings, either as C3 glomerulonephritis or dense deposit disease. C3 glomerulonephritis is characterized by mesangial, subendothelial, intramembranous, and sometimes subepithelial capillary wall deposits, while in dense deposit disease, the deposits are dense, osmiophilic, sausage-shaped, intramembranous, and mesangial (5,6).
We herein report a rare case of a Japanese patient with overlapping aHUS and C3 glomerulopathy with the p.Asp283Gly mutation in CFI.
Case Report
A 34-year-old Japanese man with no remarkable medical history presented to our hospital with a 1-week history of blurred vision in both eyes, headaches, and nausea. On the day of the presentation, the patient was alert and oriented. There were no episodes of diarrhea or medication use in the previous few months. His body temperature was normal (36.2°C). His pulse rate was 90 beats/min with a regular rhythm; however, his blood pressure was 230/130 mmHg. He had a family history of hypertension, including his mother and brother; however, his personal blood pressure history was unknown because he had not undergone any medical examinations. None of his close relatives had a history of kidney disease or urinary abnormalities.
A physical examination revealed no noticeable neurological abnormalities, mucosal ulceration, lymphadenopathy, or skin rashes. A fundus examination revealed discoid edema, flaming hemorrhaging, hard exudate, and arteriolar narrowing, consistent with grade-4 hypertensive retinopathy (Fig. 1). The laboratory data at the time of the presentation are presented in Table. Blood tests revealed anemia (hemoglobin, 8.1 g/dL), thrombocytopenia (platelet count, 7.6×104/μL), renal dysfunction [serum creatinine, 8.16 mg/dL; estimated glomerular filtration rate (eGFR), 7.1 mL/min/1.73 m2], elevated lactate dehydrogenase levels (928 U/L), and decreased haptoglobin levels (3 mg/dL). The white blood cell count and C-reactive protein level were within normal ranges. Tests for antinuclear antibodies, myeloperoxidase-anti-neutrophil cytoplasmic antibody, proteinase3-anti-neutrophil cytoplasmic antibody, hepatitis B virus, and hepatitis C virus were negative. A urinalysis showed proteinuria with 3.5 g/gCr and microhematuria with 30-49 glomerular red blood cells/high-power field. Computed tomography (CT) findings of the brain, chest, and abdomen were all unremarkable, and an electrocardiogram showed a sinus rhythm.
Figure 1.
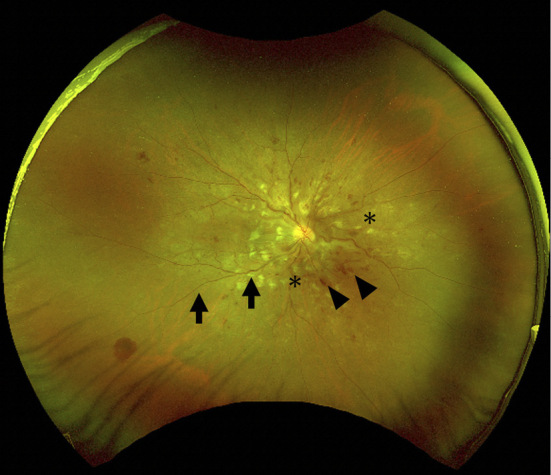
Fundus examination findings. Grade 4 hypertensive retinopathy is indicated by discoid edema, flaming hemorrhaging (arrowheads), hard exudate (asterisks), and arteriolar narrowing (arrows).
Table.
Laboratory Data.
| <Blood> | ||||||||
| WBC | 8,230 | /μL | Cr | 8.16 | mg/dL | Anti-DNA antibody | Negative | |
| RBC | 275 | ×104/μL | BUN | 45.6 | mg/dL | Anti-Scl70 antibody | Negative | |
| Hb | 8.1 | g/dL | Na | 131 | mEq/L | HBsAb | 18.6 | mIU/mL |
| Ht | 23.6 | % | K | 3.8 | mEq/L | HBsAg | Negative | |
| MCV | 85.8 | Fl | Cl | 98 | mEq/L | HCVAb | Negative | |
| Plt | 7.6 | ×104/μL | ||||||
| CRP | 0.34 | mg/dL | PT-INR | 1.05 | ||||
| AST | 37 | U/L | C3 | 97 | mg/dL | aPTT | 28.9 | s |
| ALT | 20 | U/L | C4 | 42 | mg/dL | FDP | 10.7 | μg/mL |
| LDH | 928 | U/L | CH50 | 39 | U/mL | Fibrinogen | 414 | mg/dL |
| T-bil | 1.2 | mg/dL | Haptoglobin | 3 | mg/dL | ADAMTS13 | ||
| D-Bil | 0.1 | mg/dL | Activity | 61 | % | |||
| ANA | <40 | Inhibitor | <0.5 | BU/mL | ||||
| Total Protein | 5.5 | g/dL | MPO-ANCA | <1.0 | U/mL | Direct coombs’ | Negative | |
| Albumin | 3.2 | g/dL | PR3-ANCA | <1.0 | U/mL | Indirect coombs’ | Negative | |
| <Urine> | ||||||||
| pH | 6.0 | Occult blood | 3+ | |||||
| Specific gravity | 1.009 | UPCR | 5.01 | g/gCr | ||||
| Protein | 3+ | RBC | 30-49 | /HPF | ||||
| <stool> | ||||||||
| Occult blood | Negative | STEC | Negative |
WBC: white blood cell, RBC: red blood cell, Hb: hemoglobin, Ht: hematocrit, Plt: platelet, AST: aspartate aminotransferase, ALT: alanine aminotransferase, LDH: lactate dehydrogenase, T-Bil: total bilirubin, D-Bil: direct bilirubin, Cr: creatinin, BUN: blood urea nitrogen, CRP: C-reactive protein, ANA: antinuclear antibody, MPO/PR3-ANCA: myeloperoxidase/proteinase 3-anti-neutrophil cytoplasmic antibody, HBsAg/Ab: anti-hepatitis B virus surface antigen/antibody, HCVAb: anti-hepatitis C virus antibody, PT-INR: prothrombin time-international normalized ratio, aPTT: activated partial thromboplastin time, FDP: fibrin degradation product, ADAMTS13: a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13, UPCR: urine protein/creatinine ratio, HPF: high-power field, STEC: Shiga toxin-producing Escherichia coli
TMA due to malignant hypertension was initially suspected based on these clinical findings. The patient was immediately admitted to our hospital and started on antihypertensive treatment with nicardipine (50 mg/day). Simultaneously, we carried out several tests to rule out other conditions known to cause TMA. The a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13 (ADAMTS13) activity was normal, and its inhibitor was negative, excluding the possibility of TTP. The stool culture was negative for STEC, which ruled out Shiga toxin-producing E. coli hemolytic uremic syndrome. No hypocomplementemia was observed. In addition, there was no evidence of collagen disease, cancer, drug reactions, or infectious disease, which could be causes of secondary TMA, as assessed by laboratory and imaging examinations.
After antihypertensive medication was started, the headache and nausea improved, and urinary protein excretion gradually decreased to approximately 2 g/gCr. The platelet counts increased to 160,000 /μL but soon decreased again to 50,000 /μL, and the improvement in the renal function was poor. Drug-induced thrombocytopenia, infection, and disseminated intravascular coagulation were excluded as causes of the decrease in the platelet count. In addition, there was no bleeding tendency during clinical observation at this time point. Therefore, we considered that pathological information from a renal biopsy was necessary for a definitive diagnosis, treatment decision, and prognostic estimation.
A percutaneous renal biopsy was performed on day 13 of hospitalization to determine the cause of the severe renal insufficiency. Renal biopsy specimens included eight glomeruli, four of which were globally sclerosed. Periodic acid-Schiff staining of the glomeruli revealed diffuse global endocapillary proliferative changes with endothelial swelling (Fig. 2a). Periodic acid-silver methenamine staining showed double contours of partial capillary walls (Fig. 2b), and Masson's trichrome staining showed fibrin thrombi (Fig. 2c). There were no adhesions or crescents in Bowman's space. Focal cellular infiltration, tubular atrophy, and interstitial fibrosis were observed in the tubulointerstitium. There was also intimal fibrosis with narrowed lumina and concentric lamination of intimal fibrosis, a so-called “onion skin” appearance (Fig. 2d). Immunofluorescence staining predominantly showed C3 deposition along the basement membrane and mesangial area (Fig. 2e), whereas IgA, IgG, IgM, C1q, and C4d staining were all negative. Electron microscopy revealed widespread foot process effacement and electron-dense deposits in the subendothelial spaces (Fig. 2f). Therefore, the diagnosis of the renal pathology was considered to be overlapping TMA and C3 glomerulopathy.
Figure 2.

Kidney biopsy findings. Periodic acid-Schiff staining revealed diffuse global endocapillary proliferative changes with endothelial swelling (a). Periodic acid-silver-methenamine staining showed double contours of the partial capillary walls (b). Masson’s trichrome staining revealed fibrin thrombi (c). There was intimal fibrosis with narrowed lumina and concentric lamination of intimal fibrosis, which causes an “onion skin” appearance (d). Immunofluorescence microscopy showed prominent C3 positivity (+) along the basement membrane and mesangium (e), whereas IgA, IgG, IgM, C1q, and C4d staining were all negative. Electron microscopy of one glomerulus showed widespread foot process effacement and electron-dense deposits in the subendothelial spaces (f) (Original magnification, a-e, 400×; f, 5,000×).
The clinical course of the patient is shown in Fig. 3. Thrombocytopenia and renal dysfunction did not improve with antihypertensive treatment; therefore, the differential diagnosis of TMA was re-assessed, and aHUS was diagnosed. Three sessions of plasma exchange were performed, followed by eculizumab with meningococcal vaccine administration. After plasma exchange and subsequent initiation of eculizumab, the platelet counts increased and remained stable. In addition, the serum creatinine levels gradually decreased. Hypertension was controlled at approximately 130/80 mmHg with oral antihypertensive medications, nifedipine, and olmesartan medoxomil. The patient was discharged from the hospital on day 40 and switched from eculizumab to ravulizumab after discharge, eventually continuing treatment with ravulizumab every eight weeks.
Figure 3.

Clinical course. PEx: plasma exchange, LDH: lactate dehydrogenase, UPCR: urine protein-to-creatinine ratio
To confirm activation of the complement cascade, we sent the patient's plasma to the Department of Nephrology at Nagoya University, Japan for a quantitative hemolytic assay using sheep blood cells and human citrated plasma (7). Only 11-15% hemolysis was observed in the patient's plasma compared to healthy human plasma supplemented with the monoclonal antibody which inhibits CFH activity. This result suggests that this patient may not have either any CFH mutations nor anti-factor H antibodies. Furthermore, no anti-factor H autoantibodies were detected in the patient's plasma by an enzyme-linked immunosorbent assay. In addition, genetic testing was performed for the significant genes in the complement pathway related to aHUS (CFH, CFI, CFB, C3, CD46, THBD, DGKE) at the Kazusa DNA Research Institute in Chiba, Japan. One missense variant was found in the compound heterozygous form, c.848A>G (p. Asp283Gly), in CFI. Franklin by Genoox (Palo Alto, CA, USA) (8) and VarSome (Saphetor, Lausanne, Switzerland) (9) were used to classify variants. These classifications are based on population, predictive, computational, functional, segregation, de novo, allelic, and other types of data. This gene candidate was classified as a likely pathogenic variant by Gennox and a pathogenic variant by VarSome.
Discussion
We encountered a rare Japanese case of overlapping aHUS and C3 glomerulopathy with the p.Asp283Gly mutation in CFI. The clinical diagnosis of aHUS is TMA with the triad of microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury, excluding Shiga toxin-producing E. coli hemolytic uremic syndrome, TTP, and secondary TMA such as metabolic diseases, autoimmune diseases, gestational hypertension, HELLP syndrome, malignancy, infection, drug-induced hypertension, and malignant hypertension. However, differentiating aHUS from other TMAs is often difficult, particularly in cases with malignant hypertension. The practical guidelines for aHUS in 2023 (Japan) also recommend reassessing the cause of TMA in cases of poor remission with secondary TMA (10). In our case, TMA due to malignant hypertension was initially suspected, but the TMA findings did not improve with antihypertensive drugs; therefore, the patient was re-evaluated and finally diagnosed with aHUS.
Interestingly, the renal pathology in this case showed an overlap between TMA and C3 glomerulopathy. Light microscopy revealed characteristic findings of TMA, such as glomerular endothelial swelling, double contours, and fibrin thrombi, as well as segmental lobulation and membranoproliferative glomerulonephritis-like findings. Immunofluorescence staining also revealed bright glomerular staining for C3 and no staining for other immunoglobulins or complement factors. Furthermore, electron microscopy revealed electron-dense deposits in subendothelial spaces. These findings are incompatible with those of TMA alone. Therefore, the diagnosis of renal pathology in our case was considered to be overlapping TMA and C3 glomerulopathy.
Both aHUS and C3 glomerulopathy are representative of complement-mediated kidney disease caused by genetic or acquired dysregulation of the complement pathway (11). In the present case, a heterozygous missense variant, c.848A>G (p.Asp283Gly), in CFI was identified by screening for mutations in genes related to the complement pathway. The variant in this patient (c.848A>G) resulted in the substitution of the Asp residue with Gly at position 283 in the low-density lipoprotein receptor domain class A of the CFI protein, which has been identified as a crucial calcium-binding site in this domain (12). Some variants in genes related to the complement pathway, which result in dysregulation of the complement pathway, have been implicated in the pathogenesis of both aHUS and C3 glomerulopathy (1). To date, various pathological mutations in CFH, CFI, CFB, and C3 have been reported as congenital causes of aHUS and C3 glomerulopathy. In addition, both diseases share the same genetic variations (12,13). Ravindran et al. recently reported a series of five patients with overlapping C3 glomerulopathy and TMA from 114 patients with C3 glomerulopathy in native kidney biopsies. Among them, three cases underwent complement evaluations, of which two cases were abnormal; one case showed a pathogenic mutation of CFH, and the other showed multiple variants of unknown significance along with anti-factor H autoantibody and C4 nephritic factor (14). Interestingly, a previously reported case of C3 glomerulopathy and TMA in a transplanted kidney after pulmonary infection in a young man had two CFI mutations, one of which was c.848A>G (p.Asp283Gly), similar to that in our patient (15). This report suggests that patients with the missense variant c.848A>G (p.Asp283Gly) in CFI may be genetically susceptible to C3 glomerulopathy and TMA.
Furthermore, although this missense variant in CFI was classified as a likely pathogenic variant by Gennox (8) and as a pathogenic variant by VarSome (9), its allele frequency was 0.1% according to the Japanese genome database, which is notably higher than the global database frequency of 0.0007% (16). The penetrance of this variant in Japanese populations, even if pathogenic, is therefore considered low.
To date, differences in the frequency of genetic variations in patients with aHUS have been reported between Japan and other countries, such as Europe and the USA. In a nationwide epidemiological survey of 118 Japanese aHUS patients enrolled between 1998 and 2016, C3 mutations were the most common (25.3%), and there have been no reports of CFI mutations (3). In Italy, France, and the USA, C3 mutations are rare, and CFH mutations account for more than 20%; CFI mutations were reported at rates of 3.7% in Italy, 8.4% in France, and 8.3% in the USA (17-19). Therefore, our case of a Japanese patient with a CFI in aHUS is considered rare.
In conclusion, we encountered a rare case of a Japanese individual with overlapping aHUS and C3 glomerulopathy and a heterozygous p.Asp283Gly mutation in CFI. Complement-mediated kidney diseases, such as sHUS and C3 glomerulopathy, are rare. Data on Japanese patients remain insufficient; therefore, further case series and findings from both experimental and clinical studies of complement-mediated kidney disease are needed to elucidate the pathology.
notes
The patient described in this case report provided his informed consent for publication of the details of his case.
Author's disclosure of potential Conflicts of Interest (COI).
Norimitsu Inoue: Research funding, Alexion Pharmaceuticals, Shoichi Maruyama: Honoraria, Alexion Pharmaceuticals.
Acknowledgments
We appreciate the support of the Kazusa DNA Research Institute and the Department of Nephrology at Nagoya University for the genetic and complement analyses. We also thank Benjamin Phillis of the Clinical Study Support Center of Wakayama Medical University for proofreading and editing the manuscript.
References
- 1.Goodship THJ, Cook HT, Fakhouri F, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int 91: 539-551, 2017. [DOI] [PubMed] [Google Scholar]
- 2.Richards A, Goodship JA, Goodship THJ. The genetics and pathogenesis of haemolytic uraemic syndrome and thrombotic thrombocytopenic purpura. Curr Opin Nephrol Hypertens 11: 431-435, 2002. [DOI] [PubMed] [Google Scholar]
- 3.Fujisawa M, Kato H, Yoshida Y, et al. Clinical characteristics and genetic backgrounds of Japanese patients with atypical hemolytic uremic syndrome. Clin Exp Nephrol 22: 1088-1099, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoshida Y, Kato H, Ikeda Y, Nangaku M. Pathogenesis of atypical hemolytic uremic syndrome. J Atheroscler Thromb 26: 99-110, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bomback AS, Appel GB. Pathogenesis of the C3 glomerulopathies and reclassification of MPGN. Nat Rev Nephrol 8: 634-642, 2012. [DOI] [PubMed] [Google Scholar]
- 6.Pickering MC, D'Agati VD, Nester CM, et al. C3 glomerulopathy: consensus report. Kidney Int 84: 1079-1089, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoshida Y, Miyata T, Matsumoto M, et al. A novel quantitative hemolytic assay coupled with restriction fragment length polymorphisms analysis enabled early diagnosis of atypical hemolytic uremic syndrome and identified unique predisposing mutations in Japan. PLoS One 10: e0124655, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Franklin by Genoox [Internet]. [cited 2023 May 25]. Available from: https://franklin.genoox.com/clinical-db/home
- 9.Varsome [internet]. [cited 2023 May 25]. Available From: https://varsome.com/
- 10.Committee for the Revision of the Clinical Guide for Atypical Hemolytic Uremic Syndrome. Clinical Guide for Atypical Hemolytic Uremic Syndrome 2023. Tokyo-Igakusha, Tokyo, 2023: (in Japanese). [Google Scholar]
- 11.Noris M, Remuzzi G. Genetics of immune-mediated glomerular diseases: focus on complement. Semin Nephrol 37: 447-463, 2017. [DOI] [PubMed] [Google Scholar]
- 12.Osborne AJ, Breno M, Borsa NG, et al. Statistical validation of rare complement variants provides insights into the molecular basis of atypical hemolytic uremic syndrome and C3 glomerulopathy. J Immunol 200: 2464-2478, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bu F, Borsa NG, Jones MB, et al. High-throughput genetic testing for thrombotic microangiopathies and C3 glomerulopathies. J Am Soc Nephrol 27: 1245-1253, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ravindran A, Pereira Palma LM, Fervenza FC, Sethi S. Overlap of C3 glomerulopathy and thrombotic microangiopathy: a case series. Kidney Int Rep 8: 619-627, 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wen J, Wang W, Xu F, Sun J, Chen J, Ni X. C3 glomerulonephritis and thrombotic microangiopathy of renal allograft after pulmonary infection in a male with concomitant two complement factor I gene variations: a case report. BMC Nephrol 19: 148, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.jMorp [Internet]. [cited 2023 May 24]. Available from: https://jmorp.megabank.tohoku.ac.jp/
- 17.Noris M, Caprioli J, Bresin E, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol 5: 1844-1859, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fremeaux-Bacchi V, Fakhouri F, Garnier A, et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol 8: 554-562, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maga TK, Nishimura CJ, Weaver AE, Frees KL, Smith RJH. Mutation in alternative pathway complement proteins in American patients with atypical hemolytic syndrome. Hum Mutat 31: E1445-E1460, 2010. [DOI] [PubMed] [Google Scholar]