

Abstract
Dasatinib, a second‐generation tyrosine kinase inhibitor, is approved for treating chronic myeloid and acute lymphoblastic leukemia. As a sensitive cytochrome P450 (CYP) 3A4 substrate and weak base with strong pH‐sensitive solubility, dasatinib is susceptible to enzyme‐mediated drug–drug interactions (DDIs) with CYP3A4 perpetrators and pH‐dependent DDIs with acid‐reducing agents. This work aimed to develop a whole‐body physiologically‐based pharmacokinetic (PBPK) model of dasatinib to describe and predict enzyme‐mediated and pH‐dependent DDIs, to evaluate the impact of strong and moderate CYP3A4 inhibitors and inducers on dasatinib exposure and to support optimized dasatinib dosing. Overall, 63 plasma profiles from perorally administered dasatinib in healthy volunteers and cancer patients were used for model development. The model accurately described and predicted plasma profiles with geometric mean fold errors (GMFEs) for area under the concentration–time curve from the first to the last timepoint of measurement (AUClast) and maximum plasma concentration (C max) of 1.27 and 1.29, respectively. Regarding the DDI studies used for model development, all (8/8) predicted AUClast and C max ratios were within twofold of observed ratios. Application of the PBPK model for dose adaptations within various DDIs revealed dasatinib dose reductions of 50%–80% for strong and 0%–70% for moderate CYP3A4 inhibitors and a 2.3–3.1‐fold increase of the daily dasatinib dose for CYP3A4 inducers to match the exposure of dasatinib administered alone. The developed model can be further employed to personalize dasatinib therapy, thereby help coping with clinical challenges resulting from DDIs and patient‐related factors, such as elevated gastric pH.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
As a sensitive cytochrome P450 (CYP) 3A4 substrate and weak base with strong pH‐sensitive solubility, dasatinib is susceptible to enzyme‐mediated and pH‐dependent drug–drug interactions (DDIs) with CYP3A4 perpetrator drugs and acid‐reducing agents, respectively.
WHAT QUESTION DID THIS STUDY ADDRESS?
This work aimed to develop a whole‐body physiologically‐based pharmacokinetic (PBPK) model of dasatinib to describe and predict enzyme‐mediated and pH‐dependent DDIs as well as to investigate a selection of clinically relevant DDIs, supporting optimized dasatinib precision dosing.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
A PBPK model for the CYP3A4 substrate dasatinib was developed and coupled with various strong and moderate CYP3A4 perpetrator models to provide model‐based dasatinib dosing recommendations within single as well as complex multiple DDIs.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
The presented dasatinib model may serve as a tool to further personalize dasatinib therapy, providing strategies to navigate clinical challenges that result from single and multiple DDIs and/or patient‐related factors or to perform DDI simulations with drugs under development involving dasatinib as sensitive CYP3A4 substrate.
INTRODUCTION
The introduction of tyrosine kinase inhibitors (TKIs) transformed the treatment and prognosis of chronic myeloid leukemia (CML), a myeloproliferative disorder accounting for approximately 15% of all newly diagnosed leukemias in adults. 1 Dasatinib (Sprycel®), an oral second‐generation and multi‐targeted TKI, is utilized in the treatment of Philadelphia chromosome positive (Ph+) CML in all phases and is also approved for Ph+ acute lymphoblastic leukemia (ALL). 2 Here, a comprehensive understanding of its pharmacokinetics (PK), encompassing absorption, distribution, metabolism and excretion (ADME) is vital for maximizing therapeutic efficacy and managing side effects.
Dasatinib, a weak base and a Biopharmaceutical Classification System (BCS) class II compound, demonstrates low solubility and high permeability, making it prone to drug–drug interactions (DDIs) with acid‐reducing agents (ARAs) because of its pH‐dependent solubility. 3 Given its strong pH‐dependent solubility with improved solubility in acidic conditions, co‐administration with proton pump inhibitors (PPIs), H2‐antagonists, and antacids can modify its absorption and consequently, therapeutic efficacy. 4 , 5
Dasatinib is primarily metabolized by cytochrome P450 (CYP) 3A4 via hydroxylation and N‐dealkylation to three major metabolites. 6 , 7 Hence, systemic exposure of dasatinib can be significantly impacted by DDIs with both CYP3A4 inhibitors and inducers. The United States Food and Drug Administration has listed dasatinib as a sensitive CYP3A4 substrate for use in clinical DDI studies. 8 For instance, the strong CYP3A4 inhibitor ketoconazole increased dasatinib's area under the concentration–time curve (AUC) over one dosing interval nearly five‐fold. 9 Conversely, pretreatment with rifampicin, a strong CYP3A4 inducer, can decrease the AUC by approximately 80%. 3 Moreover, dasatinib can affect the PK of other drugs, acting as a weak mechanism‐based inhibitor of CYP3A4 and a competitive inhibitor of CYP2C8 and several transporters, such as organic anion transporting polypeptide (OATP) 1B1 and OATP1B3. 10 , 11 , 12
Given dasatinib's susceptibility to both enzyme‐mediated and pH‐dependent DDIs, managing its therapeutic application presents clinical challenges. Here, physiologically‐based pharmacokinetic (PBPK) modeling emerges as a valuable tool for exploring DDI scenarios and supporting precision dosing of dasatinib. 13 Specifically, the PBPK approach can provide insights into how various drugs might influence dasatinib PK by modeling their complex biological ADME and interaction processes. Furthermore, these models can be utilized to simulate and predict the PK, guiding optimization of treatment regimens under complex conditions, such as multiple DDIs, thereby facilitating personalized and safer treatment strategies. 14
Considering dasatinib's complex PK profile and its susceptibility to DDIs, this study aimed to develop a whole‐body PBPK model with the objective to describe and predict the impact of various clinically studied DDIs including enzyme‐mediated and pH‐dependent DDIs on the exposure of dasatinib. As the package insert does not provide explicit dose adaptations for dasatinib when a specific perpetrator drug is co‐administered, 15 our PBPK model was applied to simulate a selection of clinically relevant DDI scenarios involving various CYP3A4 perpetrator drugs, not previously explored in clinical DDI trials. Among others, simulations with the potent antifungal agents fluconazole, itraconazole, ketoconazole, and voriconazole as well as macrolide antibiotics such as clarithromycin were performed as patients with blood malignancies are more susceptible to opportunistic infections. 16 , 17 Notably, since around a third of CML patients suffer on anxiety and depression particularly during TKI therapy, 18 co‐administration of selective serotonin reuptake inhibitors as effective antidepressants (e.g., fluvoxamine) frequently occurs. As a result, model‐based dose adaptations for dasatinib, under co‐treatment with these CYP3A4 inhibitors and additionally, with different CYP3A4 inducers, were performed to enhance support for precision dosing in patients. The model files will be made publicly available in the Clinical Pharmacy Saarland University GitHub repository (http://models.clinicalpharmacy.me/).
METHODS
Software
The dasatinib PBPK model was developed using PK‐Sim® and MoBi® (Open Systems Pharmacology Suite version 11.0, http://www.open‐systems‐pharmacology.org). 19 Digitization of published concentration–time profiles was performed with GetData Graph Digitizer version 2.26.0.20 (© S. Fedorov) according to Wojtyniak and coworkers. 20 Model parameter estimation via the Levenberg–Marquardt algorithm and local sensitivity analysis were conducted within PK‐Sim®. Calculation of PK parameters, model performance metrics as well as generation of graphics and dose adaptations were employed using R 4.2.1 (R Foundation for Statistical Computing, Vienna, Austria). 21
PBPK model building
PBPK model development was initiated with a comprehensive literature search to gather information about the physicochemical properties and ADME processes of dasatinib. Plasma concentration–time profiles following oral administration of dasatinib (as tablet, solution, or suspension) at fasted and fed state were extracted from 19 clinical trials including single‐ and multiple‐dose studies with healthy volunteers and cancer patients. An overview of all clinical studies, including administration protocols and demographics of participants, is presented in Tables S1, S7 and S9.
For the development of the dasatinib PBPK model, a middle‐out approach was employed. The concentration–time profiles were digitized and divided into a training and test dataset for model building (5 profiles) and evaluation (58 profiles), respectively. The middle‐out strategy merges prior information on drug‐ and system‐specific parameters with a parameter estimation step based on clinical trial data. 22 Initial model input parameters were informed through a combination of in vitro, in silico, and clinical data. If model parameter values could not be reliably sourced via literature or were pivotal for critical quantitative structure–activity relationship estimations, parameter estimation was performed by fitting model simulations to the plasma profiles of the training dataset.
Dissolution kinetics of dasatinib tablets, suspensions and solutions were described using a mechanistic Noyes–Whitney type model, selected for its applicability to particle dissolution processes. Model parameterization utilized either particle size distributions derived from the literature (for suspensions and tablets) 23 or immediately dissolved particle radii of <0.01 μm (for solutions). 24 Dasatinib supersaturation in the intestine was considered, as indicated by prior research, 25 but redissolution processes of precipitated drug were discarded because of dasatinib's low intestinal solubility. 23
Virtual “mean individuals” were created for each study as outlined in Section S1.2. In addition, population simulations were employed to predict the drug's PK across a virtual population, accounting for variability in physiological factors (see Section S1.3). Here, we created virtual populations of 100 individuals for each study, mirroring the demographics of the actual study populations. The ethnicity and demographic characteristics for these simulations were selected based on the specific study participant profiles, utilizing distributions from relevant databases. These included the third National Health and Nutrition Examination Survey (NHANES) for White Americans, 26 the International Commission on Radiological Protection (ICRP) database for Europeans 27 and the integrated database for the Japanese population. 28 Population variability in CYP3A4 expression was considered by varying the corresponding reference concentration within the virtual population according to Table S2.
PBPK model evaluation
PBPK model performance was evaluated using graphical and quantitative approaches. The predicted plasma concentration–time profiles were compared with corresponding observed profiles. Goodness of fit plots were generated to compare predicted and observed AUC from the first to the last timepoint of measurement (AUClast), maximum plasma concentration (C max) values and plasma concentrations, respectively. As quantitative measures, the mean relative deviation (MRD) of predicted plasma concentrations and the geometric mean fold error (GMFE) of predicted AUClast and C max values were calculated as previously described. 29 Additionally, a local sensitivity analysis was performed as described in Section S2.4.1.
PBPK drug–drug interaction modeling
Modeling of clinically studied DDI scenarios was performed with five perpetrator drugs (ketoconazole, rifampicin, rabeprazole, famotidine and Maalox®, an over‐the‐counter antacid containing aluminum hydroxide and magnesium hydroxide as active ingredients) and one victim drug (simvastatin). 3 , 4 , 5 , 9 The investigated DDI scenarios were categorized into two primary types: enzyme‐mediated DDIs and pH‐dependent DDIs. For the enzyme‐mediated DDIs, the dasatinib PBPK model was coupled with previously published PBPK models of ketoconazole, 29 rifampicin 30 and simvastatin. 31 Here, inhibition and induction processes were implemented as described in the Open Systems Pharmacology Suite manual, 32 using interaction parameters sourced from published PBPK models for each perpetrator drug. 29 , 30 For the pH‐dependent DDIs, the reduced gastric solubility due to intake of the ARAs rabeprazole, 4 famotidine 5 and Maalox® 5 was captured by increasing the gastric pH as previously performed 33 and adjusting the gastric emptying time for rabeprazole and Maalox®. Gastric pH values were only measured in the DDI study with rabeprazole. 4 Here, the mean gastric pH was measured to be 4.1 (2.8–5.2) following the administration of 20 mg rabeprazole and 0.7 (0.5–3.6) after intake of 20 mg rabeprazole plus 1500 mg betaine hydrochloride (BHCl), respectively, while the median gastric pH in the control group was 0.6 (0.5–1.8). 4 For administration of 40 mg famotidine (10 h prior to a 50 mg dasatinib intake), the gastric pH was adjusted to 2.8, as reported in the literature. 34 In a separate instance involving the concomitant administration of 30 mL Maalox®, the gastric pH was adjusted to 3.0, as documented in a different study. 35 For the control setting, the PK‐Sim® default gastric pH of 2.0 in the fasted state was utilized.
PBPK drug–drug interaction model evaluation
Performance of the DDI model was evaluated by graphical comparison of predicted and observed plasma concentration–time profiles with and without concomitant use of the perpetrator drugs. DDI effects were evaluated by calculating predicted AUClast and C max effect ratios according to Equations 1 and 2 comparing predicted ratios to the respective observed values. Here, the prediction success limits proposed by Guest et al. were applied to assess predictive accuracy for DDI ratios, representing a stricter criterium for DDI predictions than the traditional twofold range, especially when the relative AUC and C max change is small.
| (1) |
| (2) |
For DDI model performance evaluation, AUClast, effect and C max, effect represent the AUClast and C max values of the victim drug when administered with the perpetrator drug. Conversely, AUClast, control and C max, control represent the AUClast and C max values of the victim drug when administered alone.
Exposure simulations for model‐informed precision dosing
The developed PBPK model was applied to simulate dasatinib exposure in untested DDI scenarios with moderate and strong inhibitors as well as inducers of CYP3A4: The model was coupled with previously published PBPK perpetrator models of the inhibitors clarithromycin, 30 erythromycin, 36 fluconazole, 37 fluvoxamine, 38 grapefruit juice, 39 itraconazole, 30 and voriconazole 40 as well as the inducers carbamazepine 41 and efavirenz 41 , 42 to evaluate their impact on the exposure of dasatinib. Additionally, co‐administration of dasatinib with two perpetrator drugs simultaneously was investigated. Inhibition and induction processes were implemented using interaction parameters from the respective published PBPK models. The selected dosing regimens for each perpetrator drug and further information on the exposure simulations are given in Section S5.1. The magnitude of dose adjustments in the simulated DDI scenarios matching the exposure of dasatinib monotherapy was investigated for the two recommended daily dasatinib dosing regimens of 100 mg and 140 mg. Here, the simulated dasatinib doses were adapted in increments of 10 mg (in a range of 20–420 mg) until the steady‐state AUC (AUCss) matched the AUCss (80%–125%) of the monotherapy setting.
RESULTS
PBPK model building and evaluation
The training dataset for model building included five mean plasma concentration–time profiles from three single‐dose and one multiple‐dose study in healthy volunteers as well as a DDI study with ketoconazole in cancer patients, covering a dasatinib dose range from 20 to 140 mg. The test dataset consisted of 58 mean plasma profiles from healthy volunteers and cancer patients, who received single and multiple doses of dasatinib ranging from 15 to 200 mg.
Modeled elimination processes included metabolism via CYP3A4, implemented as Michaelis–Menten kinetics, an unspecific hepatic clearance to cover CYP3A4‐independent metabolism and renal excretion through passive glomerular filtration. The Michaelis–Menten constant (Km) for metabolism via CYP3A4, inhibition constants for CYP2C8, OATP1B1, and OATP1B3 as well as the CYP3A4 mechanism‐based inhibition parameters of dasatinib were derived from the literature. 7 , 11 , 12 Catalytic rate constant for CYP3A4‐mediated metabolism (kcat,CYP3A4) and the unspecific hepatic clearance were fitted. Here, kcat,CYP3A4 was informed by including the ketoconazole DDI study for model training. Based on the estimated kcat,CYP3A4 and the corresponding Km obtained from in vitro studies, an overall fraction metabolized via CYP3A4 resulting from metabolism in all tissues expressing this enzyme (see Table S3) was predicted to be ~92% of the absorbed drug. An overview of all integrated metabolic pathways and investigated DDIs is illustrated in Figure 1a–c, respectively. Drug‐dependent model input parameters of dasatinib are listed in Table S4. The PBPK model file of dasatinib can be found in Appendix S2.
FIGURE 1.
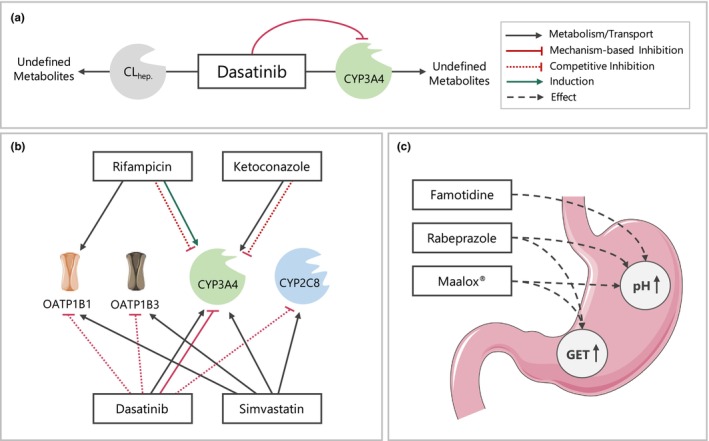
Schematic overview of implemented metabolic processes in the dasatinib PBPK model (a) as well as investigated DDIs including enzyme‐mediated (b) and pH‐dependent DDIs (c). Drawings by Servier, licensed under CC BY 3.0. 55 CLhep., unspecific hepatic clearance; CYP, cytochrome P450; DDIs, drug–drug interactions; GET, gastric emptying time; OATP, organic anion transporting polypeptide.
Plasma concentration–time profiles of the training dataset, a selection of plasma profiles of the test dataset and the corresponding goodness of fit plots showing predicted compared with observed AUClast, C max, and plasma concentrations are depicted in Figure 2.
FIGURE 2.
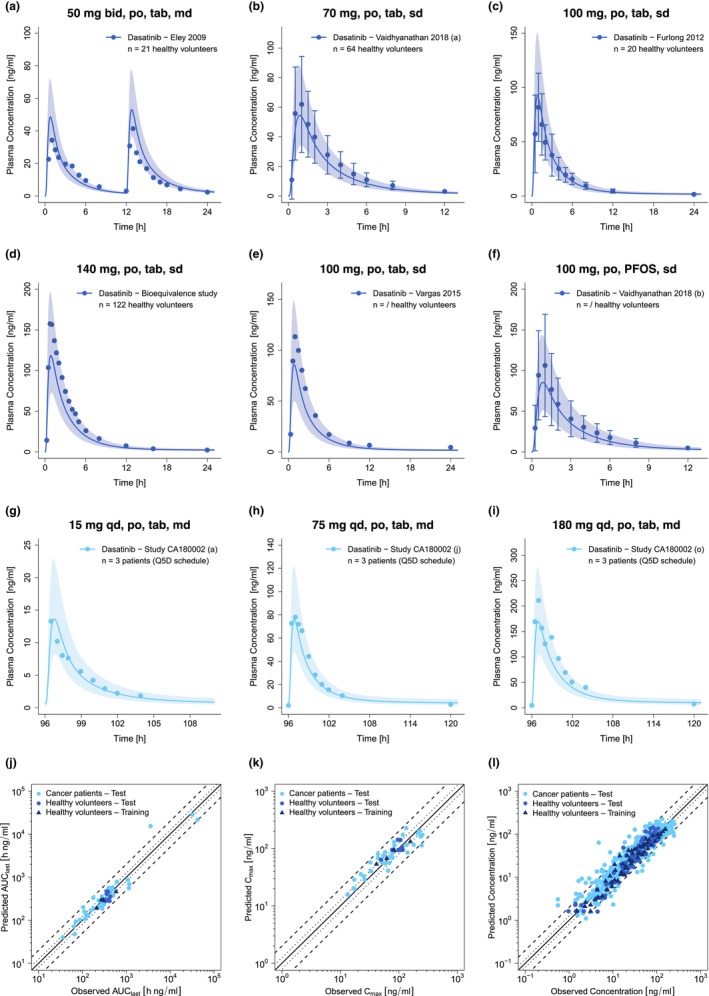
Selection of predicted and observed dasatinib plasma concentration–time profiles of the training (a–d) and the test dataset (e–i) on a linear scale as well as goodness of fit plots of predicted versus observed AUClast (j), C max (k) and plasma concentrations (l). Blue and light blue solid lines show predicted geometric mean concentration–time profiles in healthy volunteers and cancer patients, respectively, with colored ribbons illustrating the corresponding geometric standard deviation of the population simulations (n = 100). Points demonstrate the mean observed data of dasatinib with the corresponding standard deviation (if depicted in the respective publication). Linear and semilogarithmic predicted and observed plasma concentration–time profiles of all studies are shown in Sections S2.1 and S2.2. In the goodness of fit plots, solid lines mark the lines of identity, dotted lines indicate 1.25‐fold and dashed lines twofold deviation. /, no information available; AUClast, areas under the plasma concentration–time curves from the first to the last timepoint of measurement; bid, twice a day; C max, maximum plasma concentration; md, multiple dose; n, number of participants; PFOS, powder for oral suspension; po, peroral; Q5D, five consecutive days once daily dosing followed by two nontreatment days; qd, once a day; sd, single dose; tab, tablet.
Overall, the developed whole‐body PBPK model of dasatinib successfully described and predicted plasma concentration–time profiles of the training and test dataset including their shapes in both healthy volunteers and cancer patients. In addition, the model was able to capture minor effects of meal intake on dasatinib PK as demonstrated in Section S4. For the entire dasatinib dataset, 96% of predicted AUClast, 98% of predicted C max values, and 90% of predicted plasma concentrations were within the twofold acceptance criterion of their observed values. GMFEs for predicted AUClast and C max values were 1.27 (training dataset: 1.12 and test dataset: 1.28) and 1.29 (training dataset: 1.18 and test dataset: 1.30), respectively, while the overall MRD value for predicted plasma concentrations was 1.54. Detailed values are presented in Tables S5 and S6. Considering the training and test dataset separately, MRD values were calculated to be 1.35 and 1.56, respectively. Moreover, 49 out of 53 plasma concentration–times profiles show MRD values ≤2, supporting the adequate model predictions of longitudinal plasma concentration–time profile trajectories. As shown in Figure S6, residuals are randomly dispersed over time with no apparent trend, suggesting that the model does not exhibit systematic bias across the range of fits and predictions.
Of note, about half of the observed plasma profiles of cancer patients showed consistently lower plasma concentrations compared with the concentrations in healthy volunteers receiving similar dasatinib doses. As mentioned before, gastric pH plays a pivotal role in modulating dasatinib absorption. Moreover, co‐morbidities and administration of ARAs could negatively affect the absorption of dasatinib. To mitigate these factors and refine model predictions, we adjusted the gastric pH for a subset of cancer patients showing consistently lower dasatinib exposure compared with healthy volunteers to the upper end of the physiological range in the fasted state (1.5–2.5). 43 As a result, the model performance could be improved, reducing the MRD for the corresponding plasma profiles from 1.92 to 1.60. Local sensitivity analysis, conducted for a 100 mg once daily dosing regimen, identified the two pKa values, gastric pH and lipophilicity as the parameters most sensitive to dasatinib exposure at steady‐state (details are presented in Section S2.4.2).
PBPK drug–drug interaction modeling and evaluation
Eight dasatinib plasma profiles from five clinical DDI studies 3 , 4 , 5 , 9 were employed to prepare and evaluate the model predicting DDI scenarios. In enzyme‐mediated DDI studies, dasatinib (acting as victim drug) was administered with and without the perpetrator drugs ketoconazole (CYP3A4 competitive inhibition 29 ) and rifampicin (CYP3A4 induction and CYP3A4 competitive inhibition 30 ), respectively. Furthermore, plasma profiles of the victim drug simvastatin lactone and its metabolite simvastatin acid administered with and without dasatinib were included for model evaluation. Here, dasatinib acts as a CYP3A4 mechanism‐based inhibitor and CYP2C8, OATP1B1 and OATP1B3 competitive inhibitor. Simulated and observed plasma concentration–time profiles of all modeled enzyme‐mediated DDIs are depicted in Figure 3. The DDI model files are included in Appendix S2.
FIGURE 3.
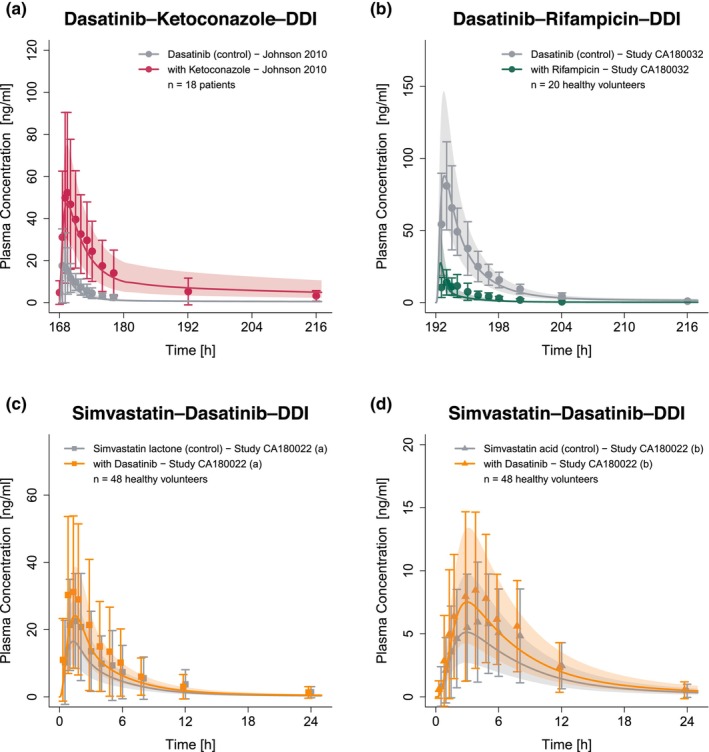
Predicted and observed plasma concentration–time profiles for enzyme‐mediated DDIs with dasatinib acting as victim (a, b) and perpetrator drug (c, d). The solid lines show predicted geometric mean concentration–time profiles with (colored) and without (gray) intake of the perpetrator drug and ribbons show the corresponding geometric standard deviation of the population simulations (n = 100). Points depict mean observed data with corresponding standard deviation of dasatinib, while squares and triangles depict the observed data with corresponding standard deviation of simvastatin lactone and simvastatin acid, respectively. Predicted and observed plasma concentration–time profiles of all studies on a semilogarithmic scale are shown in Section S3.2.1. DDIs, drug–drug interactions; n, number of participants.
For the investigation of pH‐dependent DDIs, plasma profiles of dasatinib with and without administration of the ARAs rabeprazole, 4 rabeprazole plus BHCl, 4 famotidine and Maalox® 5 were available for model evaluation. The corresponding plasma profiles of the different pH‐dependent DDI scenarios are depicted in Figure 4. Additional information on the DDI studies is provided in Table S7.
FIGURE 4.
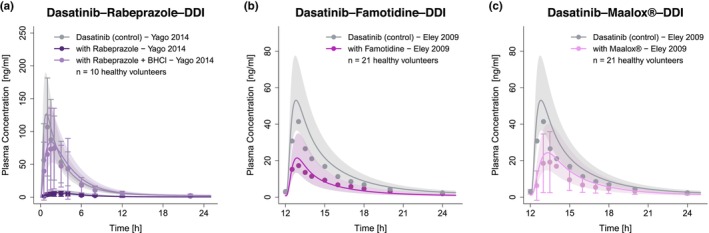
Predicted and observed plasma concentration–time profiles of dasatinib for the pH‐dependent DDIs. The solid lines show predicted geometric mean concentration–time profiles with (colored) and without (gray) the intake of the perpetrator drug and ribbons show the corresponding geometric standard deviation of the population simulations (n = 100). Points depict mean observed data with corresponding standard deviation of dasatinib (if depicted in the respective publication). Predicted and observed plasma concentration–time profiles of all studies on a semilogarithmic scale are shown in Section S3.2.2. BHCl, betaine hydrochloride; DDIs, drug–drug interactions; n, number of participants.
Figure 5 depicts the goodness of fit plots, comparing predicted to observed AUClast and C max ratios for dasatinib — modulated by intake of perpetrators ketoconazole, rifampicin, the antacid Maalox®, PPI rabeprazole, and H2‐blocker famotidine — as well as simvastatin lactone and its metabolite simvastatin acid, impacted by dasatinib administration. GMFEs for the predicted AUClast and C max ratios were 1.19 and 1.08 for pH‐dependent DDIs, 1.37 and 1.57 for enzyme‐mediated DDIs with dasatinib acting as victim drug as well as 1.21 and 1.07 for DDIs with dasatinib acting as perpetrator drug. Moreover, all AUClast and C max ratios lie within the limits proposed by Guest et al. (see Figure 5). Additionally, 7 out of 8 AUClast and C max ratios were within 1.5‐fold of observed ratios. Only the DDI with rifampicin, a strong CYP3A4 inducer and weak CYP3A4 inhibitor, was outside the stricter 1.5‐fold range with AUClast and C max ratios of 0.64 and 1.79, respectively. All predicted and observed values for AUClast and C max ratios are listed in Table S8.
FIGURE 5.
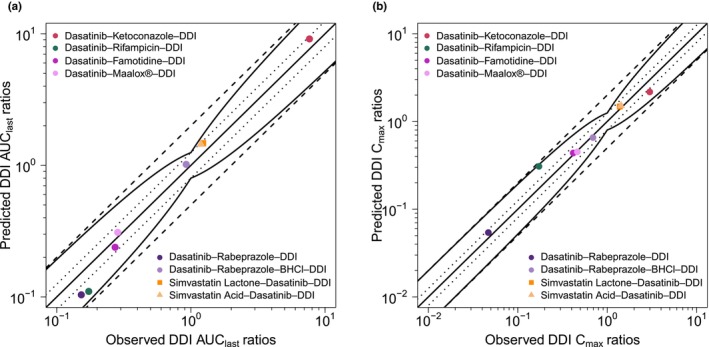
Predicted versus observed DDI AUClast ratios (a) and DDI C max ratios (b) of dasatinib (circles), simvastatin lactone (squares) and simvastatin acid (triangles). The straight solid lines mark the lines of identity, the curved lines show the limits of the predictive measure proposed by Guest et al. with 1.25‐fold variability. 56 Dotted lines indicate 1.25‐fold and dashed lines twofold deviation. AUClast, area under the plasma concentration–time curve from the first to the last timepoint of measurement; BHCl, betaine hydrochloride; C max, maximum plasma concentration; DDI, drug–drug interaction.
Exposure simulations for model‐informed precision dosing
The developed PBPK model was applied to simulate DDI scenarios of dasatinib with various strong and moderate CYP3A4 inhibitors and inducers. Subsequently, model‐based dasatinib dose adaptations were simulated based on the exposure matching principle. A selection of the corresponding plasma concentration–time profiles with and without adapted dasatinib doses is provided in Figure 6 and profiles for all investigated DDI settings in Figures S13 and S14.
FIGURE 6.
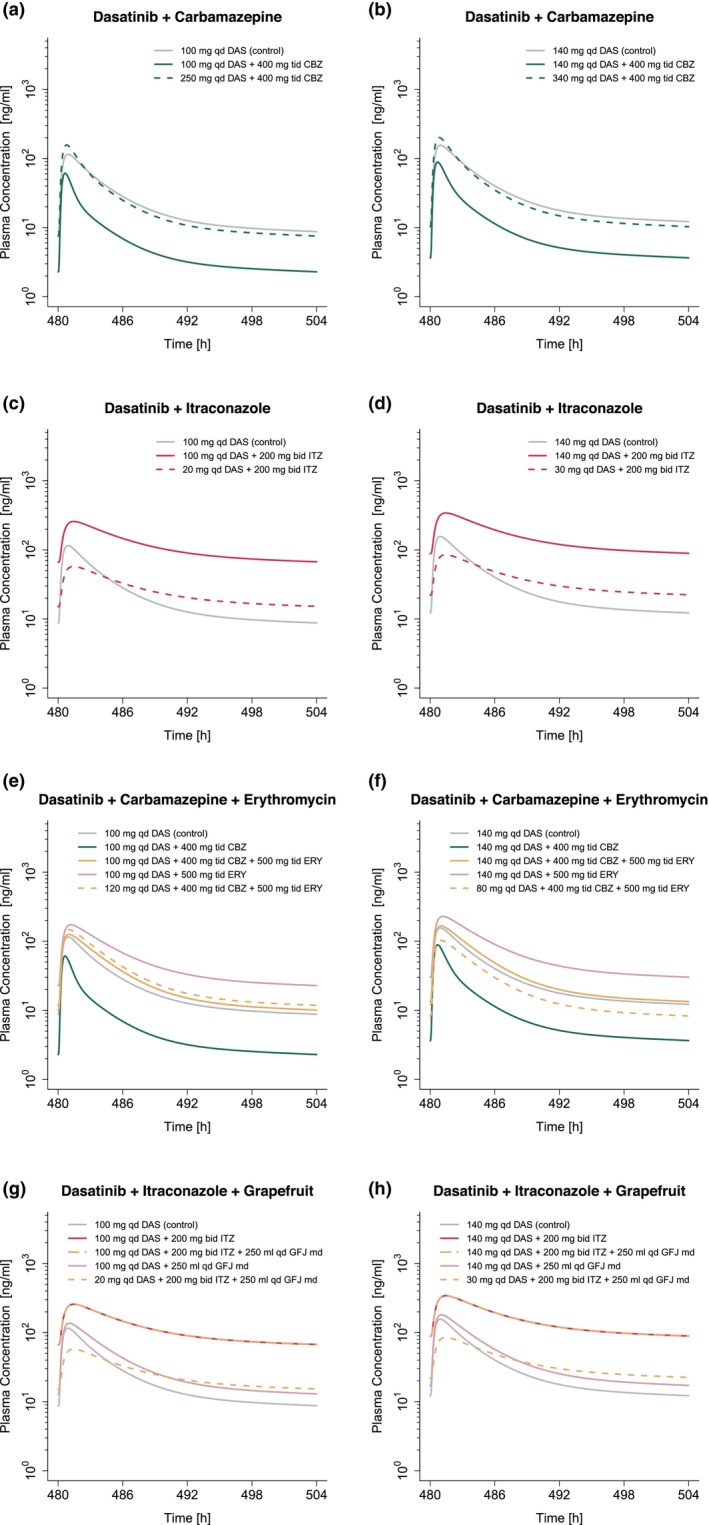
Model‐based dose adaptations for dasatinib within single (a–d) and multiple DDI scenarios (e–h) including moderate and strong CYP3A4 inhibitors and inducers. The first and second column represent the simulation results after administration of 100 mg and 140 mg dasatinib daily, respectively. Solid lines show the model predictions with (colored) and without (gray) intake of perpetrator drug. Colored dashed lines represent model predictions in the presence of perpetrator drug(s), using an adapted dasatinib dose. For the dosing simulations a virtual European male individual, 64 years of age and default values for body weight and height according to the International Commission on Radiological Protection (ICRP) database, was used. Bid, twice a day; CBZ, carbamazepine; DAS, dasatinib; DDI, drug–drug interaction; ERY, erythromycin; GFJ, grapefruit juice; ITZ, itraconazole; md, multiple dose; qd, once a day; tid, three times a day.
Model exposure simulations revealed that co‐administration of the perpetrator drugs may result in mean dasatinib AUCss increases of up to 4.6‐fold and reductions of up to 70% (see Table S12). Based on exposure matching simulations, this translated into a dasatinib dose range of 20–310 (30–360) mg to match the PBPK simulated monotherapy AUCss from 100 (140) mg dasatinib (see Figure 7). Model simulations revealed dose reductions of 50%–80% for strong and 0%–70% for moderate inhibitors. In contrast, during co‐administration of inducers, a 2.3–3.1‐fold increase of dasatinib dose was required to match the exposure of dasatinib monotherapy.
FIGURE 7.
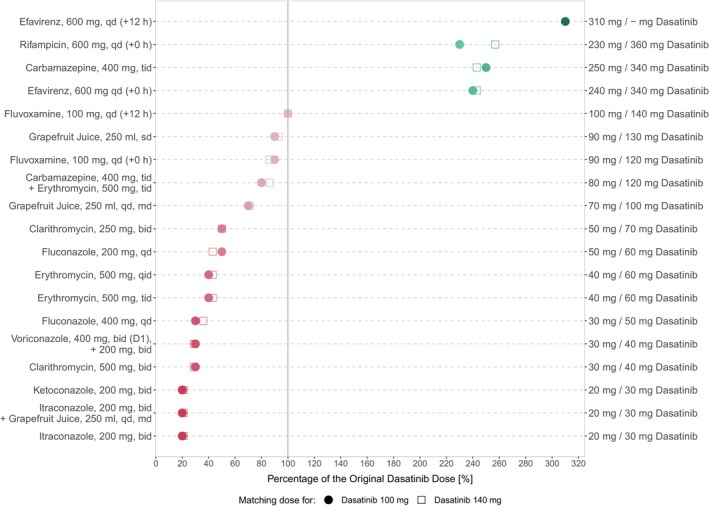
Overview of model‐based dose adaptations for dasatinib within single and multiple DDI scenarios based on the exposure matching principle, where points and squares show the percentage of the original dasatinib dose that match the PBPK simulated monotherapy AUCss at 100 mg and 140 mg, respectively. A virtual European male individual, 64 years of age and default values for body weight and height according to the International Commission on Radiological Protection (ICRP) database, was used for the dosing simulations. Red symbols depict dasatinib dose reductions and green symbols depict dasatinib dose elevations. The darker the color, the higher the magnitude of dasatinib dose adaptation. –, dose adaptations outside the simulated dose range; AUCss, steady‐state area under the concentration–time curve; bid, twice a day; D, day; md, multiple dose; qd, once daily; qid, four times a day; sd, single dose; tab, tablet; tid, three times a day.
Additionally, co‐administration with drugs like carbamazepine and erythromycin or itraconazole and grapefruit juice resulted in dose reductions of up to 20% and 80%, respectively. Table S12 provides an overview of all DDI scenarios, including AUCss effect ratios and dose adjustments to match monotherapy exposure for 100 mg and 140 mg daily dasatinib.
DISCUSSION
In this study, a whole‐body PBPK model of dasatinib was successfully developed integrating data from a total of 19 clinical trials. The model was able to describe and predict dasatinib plasma concentration–time profiles in healthy volunteers and cancer patients, covering a dasatinib dose range of 15–200 mg. Several dasatinib PBPK models have been published in the literature, including three models that investigated either the DDI potential of dasatinib mediated by enzymes and/or transporters 11 , 12 or the effects of varying formulations on dasatinib PK. 23 In contrast to previous work, the presented model was used to investigate a comprehensive range of clinically relevant enzyme‐mediated and pH‐dependent DDI scenarios in healthy volunteers and cancer patients within a single framework. For model development, a larger number of clinical studies involving a broader dose range (15–200 mg) of dasatinib was used compared with published PBPK models. Moreover, the model was utilized to simulate various DDI scenarios that have not yet been investigated in clinical trials and to provide model‐based dose adaptations, supporting precision dosing approaches for dasatinib. Furthermore, our established PBPK model extends the openly accessible PBPK model library with an additional sensitive CYP3A4 substrate and can be applied by the research community for various applications such as additional population‐specific DDI simulations and dose adaptations, DDI simulations with drugs under development involving dasatinib as a sensitive CYP3A4 substrate, or to develop and evaluate new PBPK models.
In our PBPK model, fraction absorbed after administration of dasatinib tablets across the dose range of 20–420 mg varied between 80% and 41% (Table S14). Additionally, the respective modeled absolute bioavailability ranged from 20% to 28% for the dose range of 20–420 mg (Table S14). Due to lack of intravenous data the modeled bioavailability values could not be compared with clinically observed data. 44 Dasatinib is primarily metabolized via CYP3A4, accounting for ~92% of the absorbed drug, consistent with literature reports. 7 Moreover, only 0.1% of absorbed dasatinib was excreted unchanged in urine, which is in line with results from a human ADME study. 6 In vitro studies suggested that dasatinib may be a substrate of the efflux transporters breast cancer resistance protein (BCRP) and P‐glycoprotein (P‐gp). 45 However, recent in vivo studies by Kamath et al. found no significant involvement of P‐gp in modulating the rate and extent of intestinal absorption. 46 Consequently, and due to the absence of in vitro Km values, P‐gp and BCRP were not incorporated into the PBPK model.
A good descriptive and predictive performance for DDIs was demonstrated by GMFEs of 1.24 and 1.18 for predicted AUClast and C max effect ratios, respectively. The DDIs emphasize the significant role of CYP3A4 in dasatinib metabolism and the importance to adapt dasatinib therapy in DDI settings to increase both patient safety and treatment efficacy. Due to the potential risk of adverse events resulting from increased dasatinib exposure during CYP3A4 inhibition, the package insert advises against the concomitant use of dasatinib and strong CYP3A4 inhibitors. 15 , 47 However, if such combination is unavoidable, the package insert recommends considering dose adaptations to 20 (40) mg dasatinib daily instead of 100 (140) mg for all strong inhibitors, while dosing recommendations for moderate CYP3A4 inhibitors are not provided. 15 Consequently, our developed PBPK model was applied in tandem with models of various strong and moderate CYP3A4 perpetrators to conduct DDI simulations to support dasatinib precision dosing.
The performed model simulations suggest that a “one‐dose‐fits‐all” approach during co‐treatment with strong or moderate inhibitors may lead to suboptimal dasatinib exposures: If combination of dasatinib with strong CYP3A4 inhibitors is unavoidable, dasatinib dose reductions of 50%–80% should be considered, depending on the inhibitor and selected dosing regimen, to match dasatinib AUCss during monotherapy. This translates into a dasatinib dose range of 20–50 mg for the 100 mg dosing regimen and 30–70 mg for the 140 mg dosing regimen. Moreover, simulations suggest dose reductions of 0%–70% for co‐treatment with moderate CYP3A4 inhibitors, depending on individual factors such as perpetrator drug and timing of drug administration (detailed numeric dasatinib dose adaptations are listed in Table S12). Furthermore, while simulations with a single dose of 250 mL grapefruit juice showed only a marginal increase in dasatinib exposure (~15%), a daily intake led to an AUCss increase of up to 40%, supporting the package insert guidance to avoid concomitant intake of dasatinib with grapefruit juice.
The PBPK model was additionally used to simulate the impact of CYP3A4 induction on dasatinib exposure, a situation for which only qualitative recommendations are available in the package insert: Concomitant strong inducers should be avoided, and if co‐administration is inevitable, a dose increase considered. 15 Our model simulations suggest that a 2.3–3.1‐fold increase in dasatinib dose would match the exposure of dasatinib monotherapy. However, if adapting the dasatinib dose to higher values that were neither part of the model training/test dataset nor clinically studied, it is crucial to carefully monitor patients for potential toxicities (e.g., pleural effusion or hematological toxicities). 15 , 47
Moreover, the effect of dose staggering was exemplarily investigated for fluvoxamine (moderate competitive CYP3A4 inhibitor) and efavirenz (moderate CYP3A4 inducer) as both drugs should be preferably taken in the evening according to the package insert. While no dose adaptations were needed for fluvoxamine with a 12 h staggered intake, the effect on dasatinib's AUCss increased when efavirenz was given 12 h staggered to dasatinib (see Figure 7 and Table S12).
Dasatinib's pH‐dependent DDIs represent another type of clinical interaction. In the investigated pH‐dependent DDIs, rabeprazole, famotidine and Maalox® co‐treatment showed strong impact on dasatinib PK with simulated AUClast effect ratios of 0.10, 0.24 and 0.31, respectively. Successful predictions of the DDI scenarios were attained by elevating the gastric pH constantly over time to the literature values of 4.1, 2.8, and 3.0, respectively, and estimating the elevated gastric emptying times to be 60.1 min for rabeprazole and 31.3 min for Maalox®. The impact of Maalox® on gastric emptying time has been reported in the literature, 48 while the effect of PPIs is volunteers of ongoing debate and necessitates further investigation. 49 , 50 Given the dynamic changes in gastric pH following the administration of ARAs, offering precise dosing recommendations for such DDI scenarios becomes challenging. However, the effect of antacids, which directly neutralize gastric acid, diminishes approximately 2 h post‐dose. 51 Consequently, antacids taken 2 h before dasatinib do not significantly alter dasatinib exposure. 5 In contrast, the reduction in gastric acid production through H2‐antagonists can persist for up to 12 h and the suppression induced by PPIs can continue for several days after stopping the PPI treatment. 51 Therefore, ARAs that have a shorter duration of pH elevation, such as antacids, should be preferred.
Besides single DDIs, two exemplary multiple DDIs were investigated. While the single DDI with carbamazepine (strong CYP3A4 inducer) required a 2.4‐fold increase in dasatinib dose to match the exposure of dasatinib monotherapy, the additional administration of erythromycin (moderate, irreversible CYP3A4 inhibitor) compensated this effect, overall resulting in a 20% dose reduction. In contrast, the additional intake of grapefruit juice to the strong CYP3A4 inhibitor itraconazole did not impact the simulated dose adaptation for the single dasatinib–itraconazole–DDI (80% dose reduction).
The dasatinib model was evaluated in a comprehensive PBPK DDI network, offering predictions for various clinical situations. It can explore single and multiple DDIs with dasatinib as the victim drug. The dasatinib perpetrator model, tested with the dasatinib–simvastatin DDI, holds potential for future research, especially concerning CYP3A4, CYP2C8, OATP1B1, and OATP1B3 substrates with a narrow therapeutic index. This underscores the importance of DDIs in long‐term dasatinib treatment.
There are limitations to this study, which merit consideration and will be explored in the forthcoming paragraphs. Metabolites of dasatinib were not incorporated into the PBPK model, as they are not considered clinically relevant. 6 However, during strong CYP3A4 induction, plasma concentrations of active metabolites could potentially increase to clinically relevant exposures, which was not addressed in our dosing recommendations. Contrary to CYP3A4 genetic variants, CYP3A5 polymorphisms are known to significantly affect the PK of many CYP3A substrates. 52 Consequently, due to the minor contribution of CYP3A5 to the metabolism of dasatinib, 7 CYP3A genetic variants were not considered in this work. While our PBPK model accounts for differences in patient demographics, such as age, which affects various physiological parameters including glomerular filtration rate (GFR), blood flow rates and tissue volumes, pathophysiologic changes for cancer patients (e.g., enzyme expression or α1‐acid glycoprotein levels [AGP]) were not integrated because of lack of specific information from clinical study reports. However, population variability in CYP3A4 enzyme expression or AGP levels was considered in the population simulations.
A less precise prediction performance compared with other explored DDIs could be observed for the DDI with rifampicin applying a stricter criterium of 1.5‐fold range. Here, predicted AUClast and C max ratios deviated more than 1.5‐fold from the observed ratios. Of note, rifampicin is not only an inhibitor and inducer of several enzymes, but also of several transporters like the efflux transporter P‐gp. Incorporation of P‐gp in the dasatinib PBPK model was investigated during model building; however, no significant improvement of DDI predictions involving rifampicin could be observed that has further encouraged to not include P‐gp in the final model. Similar limitations regarding DDI predictions with rifampicin have also been reported in previous work because of the complex inhibition and induction mechanisms for several enzymes and transporters. 53 , 54
The dosing simulations and subsequent dose adaptations were based on the exposure matching principle, utilizing dasatinib AUCss as exposure metric as described in the package insert. 15 However, it should be noted that despite achieving similar AUCss values for the control and DDI scenarios through dose adaptations, differences in plasma profile trajectories and thus C max and trough concentrations (C min) can lead to differences in drug efficacy and safety. Dose‐adapted plasma profiles during CYP3A4 inducer co‐treatment showed higher C max values while plasma profiles during inhibitor co‐treatment showed higher C min values compared with the simulated plasma profiles during monotherapy. An exposure–response analysis identified elevation in C min as the most significant predictor for pleural effusion, a key adverse event during dasatinib therapy. 47 Hence, close monitoring of patients for toxicities is inevitable particularly during CYP3A4 inhibitor co‐treatment and concomitant use with strong inhibitors should be avoided in clinical practice.
Finally, the recommended dasatinib dose for each DDI scenario represents an estimated average dose that can be affected by different sources of variability and uncertainties (e.g., enzyme abundance). For dosing simulations, a virtual European male individual, 64 years of age and default values for body weight and height according to the ICRP database was used. While also complex scenarios like multiple DDIs were simulated, the applicability of the provided dosing recommendations to patients with diverse clinical characteristics (e.g., hepatic impairment) is not warranted and was beyond the scope of this study. However, our PBPK model offers a foundation for future applications to personalize dasatinib therapy by using individual demographics, physiology, and enzyme activity, creating a “virtual twin” of the patient. Clinical studies are yet required to confirm the advantage of such a precision dosing approach for dasatinib therapy including efficacy and safety over the broad dose range that was required in the various DDI simulations to match the dasatinib exposure during monotherapy.
To conclude, a comprehensive whole‐body PBPK model was successfully developed for dasatinib, a sensitive CYP3A4 substrate. The established model was leveraged to simulate several previously unexplored DDI scenarios, resulting in model‐based dosing recommendations for dasatinib. Moreover, the model could serve as a tool to further optimize and personalize dasatinib therapy, providing strategies to navigate clinical challenges that result from single and multiple DDIs and/or patient‐related factors, such as elevated gastric pH. Finally, it extends the openly accessible PBPK model library with an additional sensitive CYP3A4 substrate and can be applied by the research community to investigate future single and multiple DDI scenarios involving dasatinib.
AUTHOR CONTRIBUTIONS
C.K., H.L.H.L., S.R., D.S., M.S. and T.L. wrote the manuscript; C.K., H.L.H.L., L.M.F., F.Z.M., S.R. and T.L. designed the research; C.K. performed the research; C.K. and H.L.H.L. analyzed the data.
FUNDING INFORMATION
Matthias Schwab was supported by the Robert Bosch Stiftung (Stuttgart, Germany), a grant from the German Federal Ministry of Education and Research (BMBF, 031L0188D, “GUIDE‐IBD”) and the DFG im Rahmen der Exzellenzstrategie des Bundes und der Länder‐EXC 2180–390900677. Thorsten Lehr was supported by the German Federal Ministry of Education and Research (BMBF, Horizon 2020 INSPIRATION grant 643271), under the frame of ERACoSysMed and the European Union Horizon 2021 SafePolyMed (grant 101057639).
CONFLICT OF INTEREST STATEMENT
The authors declared no competing interests for this work.
Supporting information
Appendix S1.
Appendix S2.
ACKNOWLEDGMENTS
We thank Elisabeth Emmerich for supporting this work during her internship at Saarland University. Open Access funding enabled and organized by Projekt DEAL.
Kovar C, Loer HLH, Rüdesheim S, et al. A physiologically‐based pharmacokinetic precision dosing approach to manage dasatinib drug–drug interactions. CPT Pharmacometrics Syst Pharmacol. 2024;13:1144‐1159. doi: 10.1002/psp4.13146
REFERENCES
- 1. Amercian Cancer Society . Cancer facts and figures. Amercian Cancer Society website. https://www.cancer.org/cancer/chronic‐myeloid‐leukemia/about/statistics.html. Accessed February 1, 2023.
- 2. European Medicines Agency (EMA) . Annex I: summary of product characteristics (Sprycel). European Medicines Agency website. https://www.ema.europa.eu/en/documents/product‐information/sprycel‐epar‐product‐information_en.pdf. Accessed January 18, 2023.
- 3. Center for Drug Evaluation . Clinical pharmacology and biopharmaceutics review(s): NDA Review – Dasatinib. U.S. Food and Drug Administration website. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2006/021986s000_Sprycel__ClinPharmR.pdf. Accessed August 04, 2022.
- 4. Yago MR, Frymoyer A, Benet LZ, et al. The use of betaine HCl to enhance dasatinib absorption in healthy volunteers with rabeprazole‐induced hypochlorhydria. AAPS J. 2014;16:1358‐1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eley T, Luo FR, Agrawal S, et al. Phase I study of the effect of gastric acid pH modulators on the bioavailability of oral dasatinib in healthy subjects. J Clin Pharmacol. 2009;49:700‐709. [DOI] [PubMed] [Google Scholar]
- 6. Christopher LJ, Cui D, Wu C, et al. Metabolism and disposition of dasatinib after oral administration to humans. Drug Metab Dispos. 2008;36:1357‐1364. [DOI] [PubMed] [Google Scholar]
- 7. Wang L, Christopher LJ, Cui D, et al. Identification of the human enzymes involved in the oxidative metabolism of dasatinib: an effective approach for determining metabolite formation kinetics. Drug Metab Dispos. 2008;36:1828‐1839. [DOI] [PubMed] [Google Scholar]
- 8. U.S. Food and Drug Administration (FDA) . Drug development and drug interactions | table of substrates, inhibitors and inducers. https://www.fda.gov/drugs/drug‐interactions‐labeling/drug‐development‐and‐drug‐interactions‐table‐substrates‐inhibitors‐and‐inducers. Accessed February 01, 2023.
- 9. Johnson FM, Agrawal S, Burris H, et al. Phase 1 pharmacokinetic and drug‐interaction study of dasatinib in patients with advanced solid tumors. Cancer. 2010;116:1582‐1591. [DOI] [PubMed] [Google Scholar]
- 10. Li X, He Y, Ruiz CH, Koenig M, Cameron MD, Vojkovsky T. Characterization of dasatinib and its structural analogs as CYP3A4 mechanism‐based inactivators and the proposed bioactivation pathways. Drug Metab Dispos. 2009;37:1242‐1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pahwa S, Alam K, Crowe A, et al. Pretreatment with rifampicin and tyrosine kinase inhibitor Dasatinib potentiates the inhibitory effects toward OATP1B1‐ and OATP1B3‐mediated transport. J Pharm Sci. 2017;106:2123‐2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chang M, Bathena S, Christopher LJ, Shen H, Roy A. Prediction of drug‐drug interaction potential mediated by transporters between dasatinib and metformin, pravastatin, and rosuvastatin using physiologically based pharmacokinetic modeling. Cancer Chemother Pharmacol. 2022;89:383‐392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gonzalez D, Rao GG, Bailey SC, et al. Precision dosing: public health need, proposed framework, and anticipated impact. Clin Transl Sci. 2017;10:443‐454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Grimstein M, Yang Y, Zhang X, et al. Physiologically based pharmacokinetic modeling in regulatory science: an update from the U.S. Food and Drug Administration's Office of Clinical Pharmacology. J Pharm Sci. 2019;108:21‐25. [DOI] [PubMed] [Google Scholar]
- 15. Bristol‐Myers Squibb Company . Sprycel U.S. prescribing information website. https://packageinserts.bms.com/pi/pi_sprycel.pdf. Accessed 01 March 2023.
- 16. Rodriguez GH, Ahmed SI, Al‐Akhrass F, Rallapalli V, Safdar A. Characteristics of, and risk factors for, infections in patients with cancer treated with dasatinib and a brief review of other complications. Leuk Lymphoma. 2012;53:1530‐1535. [DOI] [PubMed] [Google Scholar]
- 17. Shariati A, Moradabadi A, Chegini Z, Khoshbayan A, Didehdar M. An overview of the Management of the Most Important Invasive Fungal Infections in patients with blood malignancies. Infect Drug Resist. 2020;13:2329‐2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shi D, Li Z, Li Y, Jiang Q. Variables associated with self‐reported anxiety and depression symptoms in patients with chronic myeloid leukemia receiving tyrosine kinase inhibitor therapy. Leuk Lymphoma. 2021;62:640‐648. [DOI] [PubMed] [Google Scholar]
- 19. Lippert J, Burghaus R, Edginton A, et al. Open systems pharmacology community‐an open access, open source, Open Science approach to modeling and simulation in pharmaceutical sciences. CPT Pharmacometrics Syst Pharmacol. 2019;8:878‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wojtyniak J‐G, Britz H, Selzer D, Schwab M, Lehr T. Data digitizing: accurate and precise data extraction for quantitative systems pharmacology and physiologically‐based pharmacokinetic modeling. CPT Pharmacometrics Syst Pharmacol Pharmacol. 2020;9:322‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. R Core Team . R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2021. [Google Scholar]
- 22. Tsamandouras N, Rostami‐Hodjegan A, Aarons L. Combining the “bottom up” and “top down” approaches in pharmacokinetic modelling: fitting PBPK models to observed clinical data. Br J Clin Pharmacol. 2015;79:48‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vaidhyanathan S, Wang X, Crison J, et al. Bioequivalence comparison of pediatric Dasatinib formulations and elucidation of absorption mechanisms through integrated PBPK modeling. J Pharm Sci. 2019;108:741‐749. [DOI] [PubMed] [Google Scholar]
- 24. Willmann S, Thelen K, Becker C, Dressman JB, Lippert J. Mechanism‐based prediction of particle size‐dependent dissolution and absorption: cilostazol pharmacokinetics in dogs. Eur J Pharm Biopharm. 2010;76:83‐94. [DOI] [PubMed] [Google Scholar]
- 25. Tsume Y, Takeuchi S, Matsui K, Amidon GE, Amidon GL. In vitro dissolution methodology, mini‐gastrointestinal simulator (mGIS), predicts better in vivo dissolution of a weak base drug, dasatinib. Eur J Pharm Sci. 2015;76:203‐212. [DOI] [PubMed] [Google Scholar]
- 26. National Center for Health Statistics . Third National Health and Nutrition Examination Survey (NHANES III). Tech. rep., Hyattsville, MD, 20782. 1997.
- 27. Valentin J. Basic anatomical and physiological data for use in radiological protection: reference values. Ann ICRP. 2002;32:1‐277. [PubMed] [Google Scholar]
- 28. Schlender J. A report including the description of the physiology base of the Japanese population implemented in PK‐Sim. Github website. https://github.com/Open‐Systems‐Pharmacology/OSPSuite.Documentation/blob/master/Japanese_Population/Report.md. Accessed August 4, 2022.
- 29. Marok FZ, Wojtyniak JG, Fuhr LM, et al. A physiologically based pharmacokinetic model of ketoconazole and its metabolites as drug–drug interaction perpetrators. Pharmaceutics. 2023;15:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hanke N, Frechen S, Moj D, et al. PBPK models for CYP3A4 and P‐gp DDI prediction: a modeling network of rifampicin, Itraconazole, clarithromycin, midazolam, Alfentanil, and digoxin. CPT Pharmacometrics Syst Pharmacol. 2018;7:647‐659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wojtyniak J‐G, Selzer D, Schwab M, Lehr T. Physiologically based precision dosing approach for drug‐drug‐gene interactions: a simvastatin network analysis. Clin Pharmacol Ther. 2021;109:201‐211. [DOI] [PubMed] [Google Scholar]
- 32. Open Systems Pharmacology Suite Community . Open systems pharmacology suite manual. Github website. https://raw.githubusercontent.com/Open‐Systems‐Pharmacology/OSPSuite.Documentation/master/Open‐Systems‐Pharmacology‐Suite.pdf. Accessed November 23, 2021.
- 33. Dong Z, Li J, Wu F, et al. Application of physiologically‐based pharmacokinetic modeling to predict gastric pH‐dependent drug‐drug interactions for Weak Base drugs. CPT Pharmacometrics Syst Pharmacol. 2020;9:456‐465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Okada M, Yao T, Sakurai T, et al. A comparative study of once‐a‐day morning and once‐a‐day bedtime administration of 40 mg famotidine in treating gastric ulcers. Am J Gastroenterol. 1992;87:1009‐1013. [PubMed] [Google Scholar]
- 35. Decktor DL, Malcolm Robinson SG. Comparative effects of liquid antacids on esophageal and gastric pH in patients with heartburn. Am J Ther. 1995;2:481‐486. [DOI] [PubMed] [Google Scholar]
- 36. Dallmann A, Solodenko J, Wendl T, Frechen S. Building and evaluation of a PBPK model for erythromycin in healthy adults. Github website. https://github.com/Open‐Systems‐Pharmacology/OSP‐PBPK‐Model‐Library/blob/master/Erythromycin/Erythromycin_evaluation_report.pdf. Accessed October 18, 2022.
- 37. Eriksson J, Solodenko J. Building and evaluation of a PBPK model for fluconazole in healthy adults. Github website. https://github.com/Open‐Systems‐Pharmacology/OSP‐PBPK‐Model‐Library/blob/master/Fluconazole/fluconazole_evaluation_report.pdf. Accessed August 17, 2023.
- 38. Britz H, Hanke N, Volz AK, et al. Physiologically‐based pharmacokinetic models for CYP1A2 drug–drug interaction prediction: a modeling network of fluvoxamine, theophylline, caffeine, rifampicin, and midazolam. CPT Pharmacometrics Syst Pharmacol. 2019;8:296‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fuhr LM, Marok FZ, Fuhr U, Selzer D, Lehr T. Physiologically based pharmacokinetic modeling of bergamottin and 6,7‐dihydroxybergamottin to describe CYP3A4 mediated grapefruit‐drug interactions. Clin Pharmacol Ther. 2023;114:470‐482. [DOI] [PubMed] [Google Scholar]
- 40. Li X, Frechen S, Moj D, et al. A physiologically based pharmacokinetic model of Voriconazole integrating time‐dependent inhibition of CYP3A4, genetic polymorphisms of CYP2C19 and predictions of drug–drug interactions. Clin Pharmacokinet. 2020;59:781‐808. [DOI] [PubMed] [Google Scholar]
- 41. Fuhr LM, Marok FZ, Hanke N, Selzer D, Lehr T. Pharmacokinetics of the CYP3A4 and CYP2B6 inducer carbamazepine and its drug‐drug interaction potential: a physiologically based pharmacokinetic modeling approach. Pharmaceutics. 2021;13:1‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wendl T, Frechen S, Solodenko J, Dallmann A. Building and evaluation of a PBPK model for Efavirenz in healthy adults. Github website. https://github.com/Open‐Systems‐Pharmacology/OSP‐PBPK‐Model‐Library/blob/master/Efavirenz/efavirenz_evaluation_report.pdf. Accessed 18 October 2022.
- 43. Willmann S, Schmitt W, Keldenich J, Lippert J, Dressman JB. A physiological model for the estimation of the fraction dose absorbed in humans. J Med Chem. 2004;47:4022‐4031. [DOI] [PubMed] [Google Scholar]
- 44. Levêque D, Becker G, Bilger K, Natarajan‐Amé S. Clinical pharmacokinetics and pharmacodynamics of Dasatinib. Clin Pharmacokinet. 2020;59:849‐856. [DOI] [PubMed] [Google Scholar]
- 45. Hiwase DK, Saunders V, Hewett D, et al. Dasatinib cellular uptake and efflux in chronic myeloid leukemia cells: therapeutic implications. Clin Cancer Res. 2008;14:3881‐3888. [DOI] [PubMed] [Google Scholar]
- 46. Kamath AV, Wang J, Lee FY, Marathe PH. Preclinical pharmacokinetics and in vitro metabolism of dasatinib (BMS‐354825): a potent oral multi‐targeted kinase inhibitor against SRC and BCR‐ABL. Cancer Chemother Pharmacol. 2008;61:365‐376. [DOI] [PubMed] [Google Scholar]
- 47. Wang X, Roy A, Hochhaus A, Kantarjian HM, Chen T‐T, Shah NP. Differential effects of dosing regimen on the safety and efficacy of dasatinib: retrospective exposure‐response analysis of a phase III study. Clin Pharm. 2013;5:85‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Monés J, Carrio I, Sainz S, et al. Gastric emptying of two radiolabelled antacids with simultaneous monitoring of gastric pH. Eur J Nucl Med. 1995;22:1123‐1128. [DOI] [PubMed] [Google Scholar]
- 49. Jones MP, Shah D, Ebert CC. Effects of rabeprazole sodium on gastric emptying, electrogastrography, and fullness. Dig Dis Sci. 2003;48:69‐73. [DOI] [PubMed] [Google Scholar]
- 50. Anjiki H, Sanaka M, Kuyama Y. Dual effects of rabeprazole on solid‐phase gastric emptying assessed by the 13C‐octanoate breath test. Digestion. 2005;72:189‐194. [DOI] [PubMed] [Google Scholar]
- 51. Patel D, Bertz R, Ren S, Boulton DW, Någård M. A systematic review of gastric acid‐reducing agent‐mediated drug–drug interactions with orally administered medications. Clin Pharmacokinet. 2020;59:447‐462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wojnowski L, Kamdem LK. Clinical implications of CYP3A polymorphisms. Expert Opin Drug Metab Toxicol. 2006;2:171‐182. [DOI] [PubMed] [Google Scholar]
- 53. Jia G, Ren C, Wang H, Fan C. Prediction of drug–drug interactions between roflumilast and CYP3A4/1A2 perpetrators using a physiologically‐based pharmacokinetic (PBPK) approach. BMC Pharmacol Toxicol. 2024;25:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Loer HLH, Feick D, Rüdesheim S, et al. Physiologically based pharmacokinetic modeling of tacrolimus for food‐drug and CYP3A drug–drug‐gene interaction predictions. CPT Pharmacometrics Syst Pharmacol. 2023;12:724‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Les Laboratoires Servier . Servier Medical Art. https://smart.servier.com. Accessed July 10, 2023.
- 56. Guest EJ, Aarons L, Houston JB, Rostami‐Hodjegan A, Galetin A. Critique of the two‐fold measure of prediction success for ratios: application for the assessment of drug‐drug interactions. Drug Metab Dispos. 2011;39:170‐173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1.
Appendix S2.