

SUMMARY
We treated a 27-year-old patient with Duchenne’s muscular dystrophy (DMD) with recombinant adeno-associated virus (rAAV) serotype 9 containing dSaCas9 (i.e., “dead” Staphylococcus aureus Cas9, in which the Cas9 nuclease activity has been inactivated) fused to VP64; this transgene was designed to up-regulate cortical dystrophin as a custom CRISPR–transactivator therapy. The dose of rAAV used was 1×1014 vector genomes per kilogram of body weight. Mild cardiac dysfunction and pericardial effusion developed, followed by acute respiratory distress syndrome (ARDS) and cardiac arrest 6 days after transgene treatment; the patient died 2 days later. A postmortem examination showed severe diffuse alveolar damage. Expression of transgene in the liver was minimal, and there was no evidence of AAV serotype 9 antibodies or effector T-cell reactivity in the organs. These findings indicate that an innate immune reaction caused ARDS in a patient with advanced DMD treated with high-dose rAAV gene therapy. (Funded by Cure Rare Disease.)
Duchenne’s muscular dystrophy (DMD) is a fatal, x-linked myopathy caused by mutations in DMD, the large structural gene encoding dystrophin (2.4 Mb of genomic DNA with 79 exons).1,2 Dystrophin plays a major role in the function of cardiac myocytes and skeletal myofibers as part of the dystrophin–glycoprotein complex that anchors myofilaments to the extracellular matrix and prevents stress-induced damage to the sarcolemma membrane.3,4
Several recombinant adeno-associated virus (rAAV)–based approaches to gene therapy for this disorder have been developed.5–7 The large size of the dystrophin gene has presented a challenge for rAAV-based gene replacement, given the 5-kb packaging limit of the vector capsid and the need for cis-acting elements, including the AAV inverted terminal repeats (ITRs), a transcriptional promoter, and a polyadenylation signal.8 This issue has led to several innovative approaches, such as the development of mini-dystrophin9 and micro-dystrophin10,11 transgenes that include fewer internal rod domain repeats than full-length dystrophin. Another approach to dystrophin correction is to use in vivo gene-editing methods based on CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats and associated Cas9 endonuclease).12,13 RNA-sequence–guided DNA binding can be used to introduce double-strand breaks, as is accomplished with the original Cas9 nuclease.14 Alternatively, RNA-guided DNA binding can be used to direct new, engineered Cas9 fusion proteins in which the nuclease activity has been inactivated (“dead” Cas9 or dCas9) and transcriptional transactivating domains have been introduced.15–17
Another hurdle for rAAV gene therapy for DMD is the high dose of rAAV that is required to transduce the extensive mass of tissue that makes up the cardiac and skeletal musculature. The doses of rAAV used in clinical trials for DMD have ranged from 5×1013 to 2×1014 vector genomes (vg) per kilogram of body weight.18,19 After treatment with doses within this range, a number of toxic syndromes have been observed, including hepatotoxic effects, often linked to an effector T-cell response to capsid or transgene product; thrombocytopenia and thrombotic microangiopathy, sometimes associated with renal toxic effects in an atypical hemolytic uremic syndrome; and cardiac toxic effects.20 In this study involving a 27-year-old patient, we used an rAAV containing a Staphylococcus aureus dCas9 (dSaCas9) transgene with the aim of directing the binding of a VP64 transcriptional activation domain to up-regulate a nonmuscle, full-length isoform of dystrophin (Dp427c).
CAS9 FUSION PROTEIN AND VECTOR
A skeletal-muscle biopsy specimen that had been obtained when the patient was 23 years of age showed patchy dystrophin immunostaining (Fig. S1 in the Supplementary Appendix, available with the full text of this article at NEJM.org) that was approximately 3% of the control muscle value, as assessed by Western blot (Fig. S2). Whole-genome sequencing showed a hemizygous deletion of approximately 30 kb encompassing the promoter and exon 1 of the muscle transcript variant of dystrophin (Dp427m) inherited from the maternal genome (Fig. S3). The deletion left the promoter and exon 1 of the cortical (Dp427c) and Purkinje (Dp427p1) transcript variants of dystrophin intact.
We hypothesized that the cortical isoform of dystrophin may be compensating for the absence of the muscle isoform on the basis of reports of exon 1 deletions in Dp427m observed in patients with X-linked dilated cardiomyopathy that resulted in no overt skeletal-muscle phenotype.21,22 RNA sequencing of a sample of the patient’s muscle indeed showed low transcript expression of dystrophin derived from the cortical promoter (Fig. 1A). His unique deletion mutation, encompassing the promoter and exon 1 of the muscle transcript variant of dystrophin (Dp427m), prompted the design of a therapeutic agent based on CRISPR–transactivator technology to up-regulate an alternate full-length dystrophin variant (Dp427c), which was shown to be partially expressed in his muscle and differs from Dp427m in its promoter and exon 1 (Fig. 1A). Using a CRISPR activation approach, we identified the optimal single guide RNA (sgRNA) to target up-regulation of Dp427c, first in in vitro models and subsequently in the hDMD/D2-mdx mouse model with a humanized DMD locus. Figure 1B shows the design of the gene-therapy construct, which includes the muscle-specific promoter CK8e and transcription activator VP64 fused to dSaCas9. The size of the transgene insert between the ITRs was 4558 bp, and it was packaged into AAV serotype 9 (AAV9). The certificate of analysis for the final Good Manufacturing Practice–grade product is shown in Table S1.
Figure 1. Design of the CRISPR–Transactivator Therapeutic.

Panel A shows RNA sequencing reads, including exons 1 through 7 (right to left) of the full-length DMD transcripts Dp427c (cortical), Dp427m (muscle), and Dp427p1 (Purkinje). A novel exon 1 (GRCh38 chrX: 33,726,502–33,726,715) that has expression similar to or higher than that of Dp427c was also detected by RNA sequencing. Arcs indicate the numbers of reads that span exons. Panel B shows the therapeutic construct that was cloned into a plasmid backbone with adeno-associated virus (AAV) serotype 2 inverted terminal repeats (ITRs) (Addgene plasmid 99680). The single guide RNA (sgRNA) expression is regulated by a human U6 (hU6) promoter, and the expression of the “dead” Staphylococcus aureus Cas9 (dSaCas9)–VP64 fusion protein is regulated by a CK8e promoter (Hauschka Laboratory, University of Washington), which was engineered from the regulatory elements from mouse muscle-type creatine kinase. The abbreviation bGH poly(A) denotes bovine growth hormone polyadenylation signal, and NLS nuclear localization sequence.
PATIENT AND CLINICAL COURSE
The patient had DMD diagnosed when he was 5.5 years of age and had been treated over the course of 21 years with daily deflazacort (1.1 mg per kilogram per day). He lost the ability to ambulate independently when he was 18 years of age. He had a progressive decline in arm function and increasing cardiopulmonary dysfunction, including a forced expiratory volume in 1 second (FEV1) and forced vital capacity (FVC) that declined progressively to 36% of the predicted value (for each) and a left ventricular ejection fraction (LVEF) of 55 to 60% as measured by both cardiac magnetic resonance imaging (MRI) and echocardiography (lower limit of the normal range, 55%).
On October 4, 2022, the patient received 1×1014 vg per kilogram of intravenous AAV9 vector containing the CRISPR–transactivator therapeutic agent (CK8e.dSaCas9.VP64.U6.sgRNA). At that time, the patient had severe generalized muscle weakness with a low lean muscle mass of 45% (as assessed by dual-energy x-ray absorptiometry [DEXA] scan), a restrictive pulmonary defect (FEV1 and FVC, 36% of the predicted value), and mild left ventricular systolic dysfunction (LVEF, 55 to 60% [confirmed by both echocardiography and MRI]). His cardiac function had been stabilized with lisinopril (10 mg daily), eplerenone (12.5 mg daily), and carvedilol (6.25 mg twice daily). Carvedilol and lisinopril were held before administration of the gene therapy because of the potential for these drugs to worsen myocarditis and cardiogenic shock associated with the delivery of gene therapy. His LVEF was unchanged as measured by echocardiography after discontinuation of these drugs. Although his pulmonary function had been declining over time, the FEV1 and FVC remained above the cutoff (30% of the predicted value) for patients with neuromuscular conditions meeting criteria for major scoliosis surgeries with potential for postoperative pulmonary compromise.23 No such criteria exist for the AAV–CRISPR treatment, but scoliosis surgery represents a typical example of prolonged physiological stress due to anesthesia and immobilization. The patient had been using non-invasive mask ventilation intermittently before admission, and it was instituted continuously beginning on day 5 after treatment. He had been taking deflazacort at a dose of 39 mg daily (equivalent to 32.5 mg of prednisone daily at 1 mg per kilogram per day) for 21.5 years. Given the anticipated worsening of muscular symptoms of DMD without treatment and the lack of alternatives, the patient was deemed by his primary neurologist and the pulmonary, cardiology, and critical care consultants to be a candidate for the gene-therapy procedures. The patient was fully informed of the potential risks of the treatment and provided written informed consent to proceed.
Immunologic screening at baseline showed that the AAV9 total antibody level was below the limit of detection (titer <1:25 by enzyme-linked immunosorbent assay, Athena Diagnostics) (Table S2) and negative enzyme-linked immunosorbent spot (ELISPOT) responses to AAV9 and dSaCas9–VP64 (Fig. S4). Prophylactic immune-suppression therapy included rituximab (beginning 13 days before administration of gene therapy), glucocorticoids (beginning 1 day before administration), and sirolimus (beginning 1 day before administration) (Fig. 2A and Fig. S5). Before participation, the patient underwent an evaluation for infectious diseases similar to that used for solid-organ transplant recipients, including documentation of seronegative status for Epstein–Barr virus (EBV) capsid antigen (IgM and IgG), EBV nuclear antigen, and cytomegalovirus (IgM and IgG), as well as recommended immunizations and boosting with a Haemophilus influenzae type b conjugate vaccine. There was no evidence of active infection. Blood counts, a serum chemical profile, and levels of B-type natriuretic peptide (BNP) and troponin I were normal at baseline and were tested after treatment (Table S3).
Figure 2. Data from the Clinical Study.

Panel A shows the timeline of treatment administered. IVIG denotes intravenous immune globulin. Days on the timeline indicate days before or after vector administration (which occurred on day 0). Panel B shows cardiac, complement, and liver measures as assessed during the clinical study (beginning 30 days before vector administration). Days on the x axis in each graph in Panel B are based on the individual protocol convention, in which the day of treatment is day 1 rather than day 0. In the graph of C3 and C4, the SC5b-9 level shown is 453 ng per milliliter (normal value, ≤244) and the CH50 level shown is less than 10 U per milliliter (normal range, 31 to 60). Additional laboratory results from the periods before and after gene therapy are shown in Table S3. ALT denotes alanine aminotransferase, AST aspartate aminotransferase, BNP B-type natriuretic peptide, and NT-proBNP N-terminal pro-BNP.
One day after vector delivery (Fig. 2A), the patient had premature ventricular contractions followed by a downward trend in platelet counts (from a baseline of 437,000 per cubic millimeter on day −1 to 311,000 per cubic millimeter at 2 days after treatment, which triggered a first dose of eculizumab to be given at 3 days after treatment to avoid complement-driven platelet consumption) and increasing levels of BNP and N-terminal proBNP (from baseline levels of 11 pg per milliliter and 63 pg per milliliter, respectively, to levels of 125 pg per milliliter and 556 pg per milliliter at 3 days after treatment) (Fig. 2B]). Increases in aspartate aminotransferase and alanine aminotransferase were proportional to increases in creatine kinase, as is seen in patients with DMD (Fig. 2B), and levels of γ-glutamyltransferase remained normal. Asymptomatic hypercarbia (partial pressure of carbon dioxide, 59 mm Hg) with respiratory acidosis occurred 3 to 4 days after the administration of gene therapy and resolved when bilevel positive airway pressures were increased from 10/4 cm of water to 12/5 cm of water. Five days after gene therapy, worsening cardiac function developed, with a decline in LVEF to between 45% and 50%; the patient was presumed to have myopericarditis, because the troponin I level was elevated (0.59 ng per milliliter; normal range, 0.01 to 0.04), and pericardial effusion with physiological characteristics of tamponade was seen on echocardiography. Acute respiratory distress developed 6 days after gene therapy, with chest radiographic findings of acute respiratory distress syndrome (ARDS) (Fig. S6) and left ventricular systolic dysfunction (LVEF, 45 to 50%). Mitigating therapies that were attempted during this time included increased doses of glucocorticoids, eculizumab (anti-C5), tocilizumab (anti–interleukin-6R), and anakinra (interleukin-1R blocker) (Fig. S5). The patient had a cardiopulmonary arrest and was given extracorporeal membrane oxygenation, and 8 days after administration of the vector he died from multi-organ failure and severe hypoxic ischemic neurologic injury, as evidenced by coma and changes on brain imaging.
LABORATORY AND POSTMORTEM STUDIES
An elevation in interleukin-6 levels (to 2.8 pg per milliliter [normal value, <2]) occurred 5 days after gene therapy, along with a mixed picture of complement components, with a mild elevation in SC5b-9 to a level of 453 ng per milliliter (normal value, ≤244) at 3 days after therapy. Multiplex cytokine bead-based assays revealed elevations in interleukin-8 in the serum (Fig. S9) and high levels of interleukin-6 and monocyte chemoattractant protein 1, which would typically not be detectable, in a sample of pericardial fluid obtained 3 days after therapy (Fig. S10). Interleukin-6 levels in pericardial fluid were noted to be 100 times as high as the interleukin-6 levels in serum.
The consent for limited autopsy obtained from the patient’s family allowed for gross and microscopic examinations of the heart, lungs, brain, triceps, and liver. Muscle mass in the heart and skeletal muscle was markedly decreased, and severe cardiomyopathy was present, characterized by substantial gross and histologic fibrofatty replacement of biventricular myocardium (Fig. 3A), a finding consistent with what has been described in patients with dystrophin-deficient cardiomyopathy,24 and without overt features of active inflammation or myocarditis, thrombotic microangiopathy, or complement deposition (confirmed by immunohistochemical analysis) (Fig. S11). The lungs were heavy and edematous (combined weight, 600 g [expected weight, 475 g]) with diffuse alveolar damage characterized by hyaline membrane formation, along with interstitial and intraalveolar edema (Fig. 3B). The findings were in keeping with the clinical impression of ARDS. There was no evidence of thrombotic microangiopathy or substantial inflammation. Gross and microscopic examination of the brain showed infarctions approximating a “watershed” distribution in the cerebral cortex and cerebellum, as well as widespread neuronal injury (Fig. S12).
Figure 3. Postmortem Analysis of Tissue.
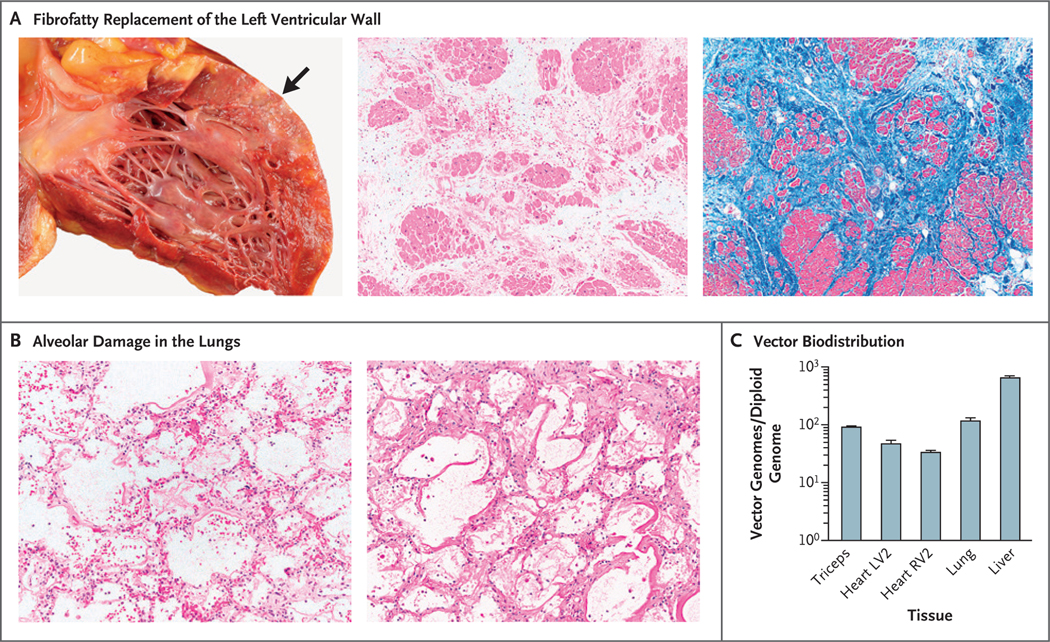
Panel A shows fibrofatty replacement of the left ventricular wall (arrow) in the patient’s heart. Histologic evaluation of this area shows marked interstitial fibrosis and fatty replacement with residual cardiac myocytes; there is no histologic evidence of myocarditis or thrombotic microangiopathy (hematoxylin–eosin and Masson’s trichrome staining). The findings are consistent with severe cardiomyopathy. Panel B shows a microscopic image of diffuse alveolar damage in the patient’s lungs, characterized by hyaline membrane deposition with interstitial and intraalveolar edema (hematoxylin–eosin and periodic acid–Schiff staining). The images in Panel B are shown at 2.5 times the magnification of the middle and right images in Panel A. Panel C shows vector biodistribution in patient tissues. Vector genomes were quantified by quantitative polymerase chain reaction and calculated to indicate the number of vector genomes per diploid genome for each tissue. Analyses were performed in technical triplicates; data are reported as means, and T bars indicate the standard deviation. LV2 and RV2 are specific sites within the left and right ventricles, respectively.
VECTOR BIODISTRIBUTION
Vector genomes were detected with the use of quantitative real-time polymerase chain reaction (PCR) in lung tissue and were present at a level of 119 vg per diploid genome; vector genomes were also detected in the myocardium (34 vg per diploid genome in the sample from the left ventricle and 48 vg per diploid genome in the sample from the right ventricle) (Fig. 3C). Relative to previous experience with AAV9,25 biodistribution of the vector in this patient was unexpectedly high in lung tissue as compared with cardiac and skeletal muscle tissue. Vector was present in the patient’s liver at a level of 680 vg per diploid genome. Droplet digital PCR was used to confirm the results of the vector genome analysis, but genome abundance in the liver exceeded the upper limit for accurate quantification (Fig. S13).
EXPRESSION OF TRANSGENE PRODUCTS
We assessed dSaCas9 transcript and protein expression, and no detectable levels were found, with the exception of trace amounts in the liver (Fig. S7). The absence of the transgene in skeletal and cardiac tissues did not warrant measurement of Dp427c up-regulation. Expression levels of sgRNA showed similar tissue trends (Fig. S8). No AAV9 capsid or Cas9 transgene–specific T-cell responses were detected by interferon-γ ELISPOT in the patient’s peripheral-blood mononuclear cells at day 4 or 7 after therapy (Fig. S4). Tests for anti-AAV9 neutralizing antibodies remained negative, which indicated that the time course of the toxic effects was not associated with humoral immune responses (Table S2).
DISCUSSION
This 27-year-old man with advanced DMD had severe cardiopulmonary toxic effects within 6 days after intravenous administration of rAAV9–dSaCas9–VP64 at a dose of 1×1014 vg per kilogram. We suggest that a cytokine-mediated capillary leak syndrome developed, which manifested as pericardial effusion on day 5 and ARDS on day 6 and led to worsening of preexisting right ventricle heart failure. Unlike some other patients with DMD in rAAV trials, this patient did not have evidence of thrombotic microangiopathy or of adaptive humoral or cell-mediated immune responses to AAV capsid or transgene products.26 Although thrombocytopenia may indicate that complement activation occurred in this patient, the absence of complement deposition in the lung and heart suggests that complement-mediated thrombotic microangiopathy was not the cause of his death. ARDS is not commonly described in association with AAV gene therapy, and other persons treated with the same dose of the recombinant AAV9 vector have not had this toxic effect, which suggests that both host factors and inherent properties of the vector led to unexpectedly high levels of vector genome in the lung and may have contributed to the outcome.
The acute toxic effects and shortened clinical course in this patient after transgene treatment precluded assessment of the safety and efficacy of the CRISPR–transactivator approach. Although trace amounts of the transgene product were present in the liver, none was detectable in cardiac or skeletal muscle, no effector T-cell responses to dSaCas9 or AAV9 were observed, and no anti-AAV9 neutralizing antibodies were detected. One additional factor may have been that the patient’s lower lean muscle mass of 45% resulted in his receiving a relatively higher vector dose per muscle nucleus than patients in other studies in which the same nominal dose of vector genomes per kilogram was used.
Another fatal case involved a nonambulatory 16-year-old patient with DMD who died 6 days after receiving gene therapy.20 The patient received fordadistrogene movaparvovec at a dose of 2×1014 vg per kilogram. It was speculated that the patient’s death was linked to an innate immune response against the viral capsid in the myocardium, which led to cardiogenic shock and heart failure. We have described a death that occurred after customized CRISPR gene-edited transgene therapy and that was caused by an innate immune reaction to a high load of rAAV. The late stage of DMD in our patient may have limited his physiological reserve and decreased his ability to survive the cardiopulmonary stress associated with acute toxic effects from rAAV gene therapy.
Supplementary Material
Acknowledgments
Supported by Cure Rare Disease.
Footnotes
References
- 1.Gao QQ, McNally EM. The dystrophin complex: structure, function, and implications for therapy. In: Comprehensive physiology. 2015;1223–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duan D, Goemans N, Takeda S, Mercuri E, Aartsma-Rus A. Duchenne muscular dystrophy. Nat Rev Dis Primers 2021;7: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lapidos KA, Kakkar R, McNally EM. The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ Res 2004;94:1023–31. [DOI] [PubMed] [Google Scholar]
- 4.Wilson DGS, Tinker A, Iskratsch T. The role of the dystrophin glycoprotein complex in muscle cell mechanotransduction. Commun Biol 2022;5:1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li C, Samulski RJ. Engineering adeno-associated virus vectors for gene therapy. Nat Rev Genet 2020;21:255–72. [DOI] [PubMed] [Google Scholar]
- 6.Kwon JB, Ettyreddy AR, Vankara A, et al. In vivo gene editing of muscle stem cells with adeno-associated viral vectors in a mouse model of Duchenne muscular dystrophy. Mol Ther Methods Clin Dev 2020;19:320–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manini A, Abati E, Nuredini A, Corti S, Comi GP. Adeno-associated virus (AAV)-mediated gene therapy for Duchenne muscular dystrophy: the issue of transgene persistence. Front Neurol 2022;12:814174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu Z, Yang H, Colosi P. Effect of genome size on AAV vector packaging. Mol Ther 2010;18:80–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y, Duan D. Novel mini-dystrophin gene dual adeno-associated virus vectors restore neuronal nitric oxide synthase expression at the sarcolemma. Hum Gene Ther 2012;23:98–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davies KE, Guiraud S. Micro-dystrophin genes bring hope of an effective therapy for Duchenne muscular dystrophy. Mol Ther 2019;27:486–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duan D. Micro-dystrophin gene therapy goes systemic in Duchenne muscular dystrophy patients. Hum Gene Ther 2018; 29:733–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Happi Mbakam C, Rousseau J, Tremblay G, Yameogo P, Tremblay JP. Prime editing permits the introduction of specific mutations in the gene responsible for Duchenne muscular dystrophy. Int J Mol Sci 2022;23:6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chemello F, Chai AC, Li H, et al. Precise correction of Duchenne muscular dystrophy exon deletion mutations by base and prime editing. Sci Adv 2021;7(18):eabg4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012;337:816–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qi LS, Larson MH, Gilbert LA, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013;152:1173–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mali P, Aach J, Stranges PB, et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol 2013;31:833–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lek A, Ma K, Woodman KG, Lek M. Nuclease-deficient clustered regularly interspaced short palindromic repeat-based approaches for in vitro and in vivo gene activation. Hum Gene Ther 2021;32:260–74. [DOI] [PubMed] [Google Scholar]
- 18.Duan D. Systemic AAV micro-dystrophin gene therapy for Duchenne muscular dystrophy. Mol Ther 2018;26:2337–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elangkovan N, Dickson G. Gene therapy for Duchenne muscular dystrophy. J Neuromuscul Dis 2021;8:Suppl 2:S303–S316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lek A, Atas E, Hesterlee SE, Byrne BJ, Bönnemann CG. Meeting report: 2022 muscular dystrophy association summit on ‘safety and challenges in gene transfer therapy’. J Neuromuscul Dis 2023;10:327–36. [DOI] [PubMed] [Google Scholar]
- 21.Ferlini A, Sewry C, Melis MA, Mateddu A, Muntoni F. X-linked dilated cardiomyopathy and the dystrophin gene. Neuromuscul Disord 1999;9:339–46. [DOI] [PubMed] [Google Scholar]
- 22.Cohen N, Muntoni F. Multiple pathogenetic mechanisms in X linked dilated cardiomyopathy. Heart 2004;90:835–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kurz LT, Mubarak SJ, Schultz P, Park SM, Leach J. Correlation of scoliosis and pulmonary function in Duchenne muscular dystrophy. J Pediatr Orthop 1983;3:347–53. [DOI] [PubMed] [Google Scholar]
- 24.Kamdar F, Garry DJ. Dystrophin-deficient cardiomyopathy. J Am Coll Cardiol 2016;67:2533–46. [DOI] [PubMed] [Google Scholar]
- 25.Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol Ther 2008;16:1073–80. [DOI] [PubMed] [Google Scholar]
- 26.Kumar SRP, Duan D, Herzog RW. Immune responses to muscle-directed adeno-associated viral gene transfer in clinical studies. Hum Gene Ther 2023;34:365–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.