

Abstract
Mucopolysaccharidosis IVA (MPS IVA) is a rare disease caused by mutations in the gene encoding the lysosomal enzyme N-acetylgalactosamine-6-sulfate sulfatase (GALNS). We report here two GALNS pharmacological chaperones, ezetimibe and pranlukast, identified by molecular docking-based virtual screening. These compounds bound to the active cavity of GALNS and increased its thermal stability as well as the production of recombinant GALNS in bacteria, yeast, and HEK293 cells. MPS IVA fibroblasts treated with these chaperones exhibited increases in GALNS protein and enzyme activity and reduced the size of enlarged lysosomes. Abnormalities in autophagy markers p62 and LC3B-II were alleviated by ezetimibe and pranlukast. Combined treatment of recombinant GALNS with ezetimibe or pranlukast produced an additive effect. Altogether, the results demonstrate that ezetimibe and pranlukast can increase the yield of recombinant GALNS and be used as a monotherapy or combination therapy to improve the therapeutic efficacy of MPS IVA ERT.
Keywords: Morquio A syndrome, MPS IVA, ezetimibe, pranlukast, GALNS, pharmacological chaperone, combination therapy, lysosomal storage disease
Graphical Abstract
1. INTRODUCTION.
Mucopolysaccharidosis type IVA (MPS IVA, OMIM 253000), also called Morquio A syndrome, is a lysosomal storage disease (LSD) caused by mutations in the gene encoding for the enzyme N-acetylgalactosamine-6-sulfate sulfatase (GALNS, EC 3.1.6.4)1. Worldwide incidence rates of MPS IVA range from 0.14 to 0.009 (per 10,000 at birth)2. GALNS deficiency leads to the lysosomal accumulation of the glycosaminoglycans (GAGs) keratan sulfate (KS) and chondroitin-6-sulfate (C6S)1, 3. Treatment options for MPS IVA patients currently include non-steroidal anti-inflammatory drugs, antibiotics, oxygen supplementation, surgical procedures to correct orthopedic deformities and tracheal obstructions, enzyme replacement therapy (ERT), and hematopoietic stem cell transplantation (HSCT)1, 3, 4. Patients under ERT with elosulfase alfa have shown some improvements in clinical symptoms of the disease5. A weekly intravenous administration of 2.0 mg/kg for 24 weeks produced a slight improvement in the 6-minute walk test and the reduction of urinary KS6 in the phase III studies, as well as an improvement in the maximal voluntary ventilation, performance of daily life activities, and height/growth rate7. Unfortunately, ERT has limited effects in correcting the skeletal, corneal, and cardiac abnormalities, due to tissue avascularity and short half-life of the enzyme5. A recent study showed that MPS IVA ERT-treated patients continue to exhibit poor growth despite early ERT intervention before 5 years of age. These findings indicate that current intravenous ERT is ineffective at correcting abnormal growth in MPS IVA8. In addition, all MPS IVA treated patients developed anti-drug antibodies at week 120, although antibodies titers did not correlate with clinical efficacy or adverse effects9. Treating MPS IVA patients with HSCT is relatively new, and recent studies have shown long-term and normal enzyme activities, increase in lumbar bone mineral density, improvement in ambulatory movement and remission of the narrow airway10–12. Nevertheless, HSCT is a high-risk procedure with many possible complications and high mortality. Limitations of current therapies indicate an unmet need for new therapeutic strategies to improve and expand the treatment options for MPS IVA patients.
GALNS protein is a 120 kDa homodimer with two 60 kDa monomers comprised of 40 and 15 kDa polypeptides connected by disulfide bonds and modified by two N-glycosylations13, 14. Currently, over 217 deleterious mutations have been reported in the GALNS gene including missense/nonsense (73%), minor mutations of splicing (9%), small deletions (10%), small insertions (2%), small indels (1%), gross deletions (2%), gross insertions/duplications (1%), and complex rearrangements (1%)15. GALNS genotype-phenotype correlations have predicted mutations that may affect the hydrophobic core, salt bridges, ligand affinity, solvent-accessible surface, and N-glycosylation sites14, 16–20. Based on these findings, pharmacological chaperones have been proposed as treatment alternative for MPS IVA1. Pharmacological chaperones are small molecules that bind to the target protein at the endoplasmic reticulum. These molecules usually serve as molecular templates that stabilize the native conformation of a protein, or promote the correct folding and trafficking of a mutant protein21, 22. Pharmacological chaperones usually bind to the active site of a protein at a concentration below 10 μM23–25 and increase its the thermal stability24. Compared to ERT, pharmacological chaperones have advantages of oral administration, wide tissue distribution profile, and fewer issues of immunogenicity24, 25. In LSD, pharmacological chaperones has been experimentally tested on Fabry disease with 1-deoxygalactonojirimycin (migalastat)26, as well as Gaucher disease, Pompe disease, and gangliosidosis (GM1 and GM2)27, 28. For MPS, pharmacological chaperones have been studied for MPS II29, MPS IIIC28, and MPS IVB30, but not yet for MPS IVA. Overall, it has been observed that these molecules increase the enzyme activity of the mutant proteins in a mutation-dependent manner, which may be sufficient to ameliorate most disease symptoms23, 27, suggesting that chaperones could be employed as a monotherapy. In addition, it has been observed that these drugs can enhance the stability of recombinant lysosomal enzymes, suggesting that co-adminsitration of a pharmacological chaperone and the enzyme may increase the efficacy of the ERT21, 22.
Previously, we reported computational docking for natural (KS and C6S) and artificial (4-methylumbelliferyl-β-D-galactopyranoside-6-sulfate, 4MUGPS) ligands of human GALNS20. Here, we describe the characterization of two pharmacological chaperones for GALNS identified from a molecular docking-based virtual screening against a library of experimental and approved drugs. Our data indicated that these compounds bind to the active cavity of GALNS, increasing the activity and thermostability of human recombinant GALNS (hrGALNS). These molecules reduced the lysosomal mass and normalized the abnormal autophagic processes in MPS IVA patient-derived fibroblasts. Therefore, these novel pharmacological chaperones have potential for further drug development to improve the treatment and outcomes for patients suffering from MPS IVA. The benefit of using the chaperones can be realized in three aspects: an increase in the production of proteins for ERT, as a monotherapy for MPS IVA patients, and combined with ERT to improve the efficacy of therapy.
2. RESULTS.
Virtual screening identified small molecule binders of human GALNS
We have previously reported that docking of human GALNS with different substrates suggested the importance of Asp39, Asp40, formylglycine (FGly) 79, Arg83, Tyr108, His142, His236, Asp288, Asn289, and Lys310 in protein-ligand interactions20. The modeled GALNS has high similarity to the reported crystal structure of human GALNS (RMSD of 1.2 Å) with the main differences observed in regions further from the active site cavity20. These results, in addition to other structure and docking results31, suggest that the modeled GALNS structure can be used to identify small molecules interacting with the active site cavity of the enzyme. In this study, we used the modeled structure of GALNS and performed a virtual screening that identified a set of compounds.
Among the top 20 interacting compounds (Supporting Information, Table S1), the antibiotic, antineoplastic, antiestrogen, antiviral, and epigenetic regulator compounds were not considered for further investigation due to the intended use for the treatment of pediatric patients and the potential side effects of long-term administration. Ezetimibe (ZINC3810860) and pranlukast (ZINC22001688), ranked third and fourth, respectively, among the 11,421 compounds present in ZINC In Man subset from ZINC (a free public resource for ligand discovery in silico), and were selected for further analysis (Figure 1A). In-silico simulations showed that ezetimibe and pranlukast docked in the active cavity of GALNS in a similar position to that predicted for natural and artificial substrates of the enzyme (Figure 1B).
Figure 1.
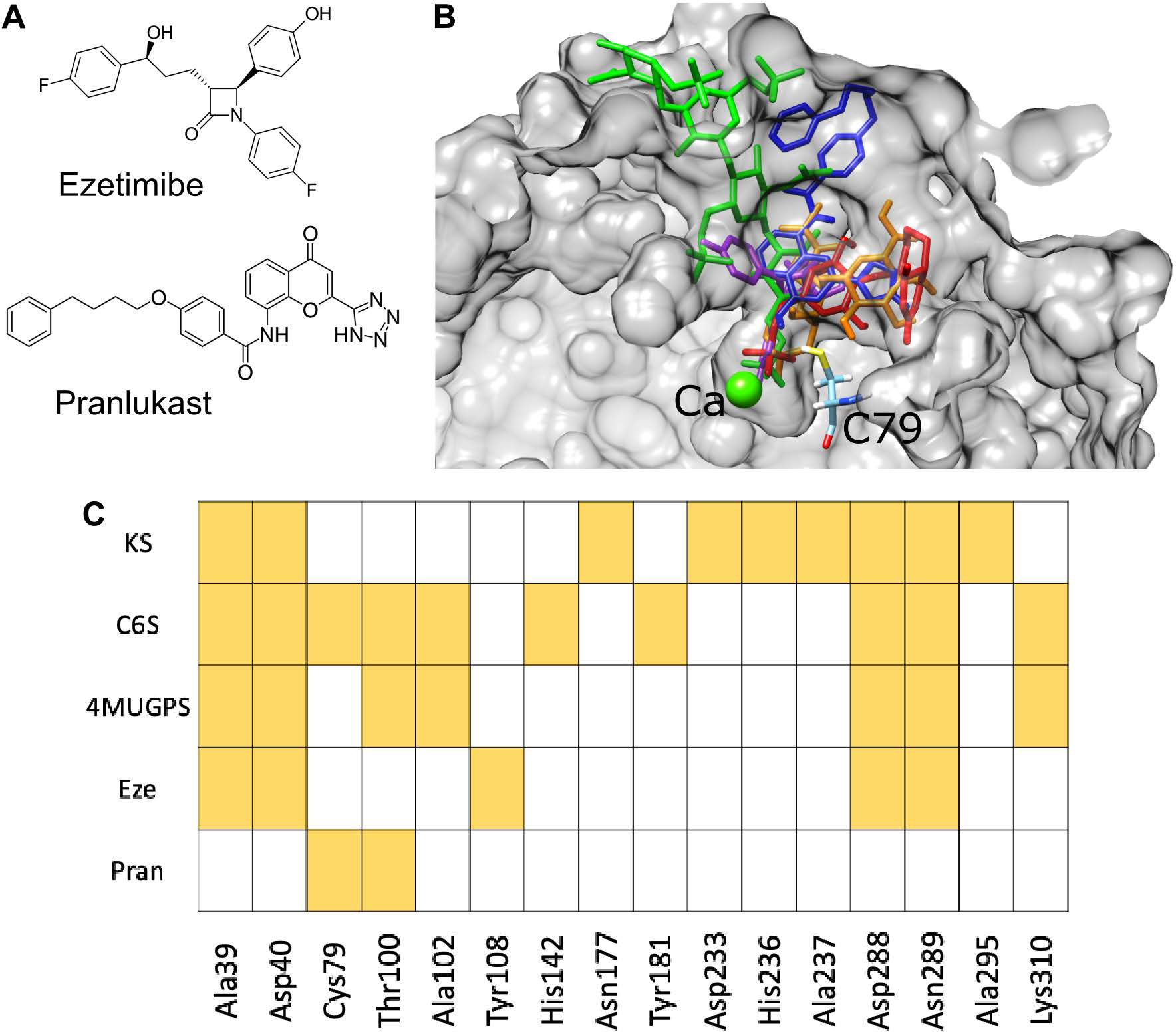
A) Structures of ezetimibe and pranlukast. B) In-silico molecular docking of chondroitin-6-sulfate (orange), keratan sulfate (green), 4-methylumbelliferyl-β-d-galactopyranoside-6-sulfate (red), ezetimibe (purple), and pranlukast (blue) within the active site cavity of human GALNS. Catalytic residue (C79) and calcium ion from GALNS are shown. GALNS tertiary structure, previously reported20, was modeled by using the human arylsulfatase A (PDB 1AUK) as a template for protein threading. C) Hydrogen bonds interactions between GALNS and ligands.
Docking results predicted that ezetimibe and pranlukast also interact with some of the amino acids predicted for the natural (KS and C6S) and the artificial (4MUGPS) substrates (Figure 1C and Supporting Information Figure S1). The Ca2+ ion present in the active cavity has been proposed as a cofactor for the GALNS enzyme and the interaction of a substrate with Ca2+ has been proposed to be required for enzyme activity14, 20. It is noteworthy that docking results also predicted an interaction of ezetimibe with Ca2+ present within the active cavity.
Molecular dynamics simulations showed that GALNS substrates (KS, C6S, and 4MUGPS) and the selected small molecules (ezetimibe and pranlukast) were stable within the active cavity during the simulation (Figure 2A). In addition, both ezetimibe (−41.19 ± 0.028 kJ/mol) and pranlukast (−58.15 ± 0.024 kJ/mol) showed higher Gibbs free energy of binding (meaning a lower binding affinity) than that observed for the enzyme substrates (KS: −112.75 ± 0.049 kJ/mol, CS: −84.70 ± 0.082 kJ/mol and 4MUGPS: −91.60 ± 0.073 kJ/mol) (Figure 2B). These results suggest that ezetimibe and pranlukast may be weak competitors of the enzyme substrates, KS and C6S, for GALNS in lysosomes. This data is consistent with the characteristics of a pharmacological chaperone, which often acts as a mild competitor with enzyme substrates.
Figure 2.
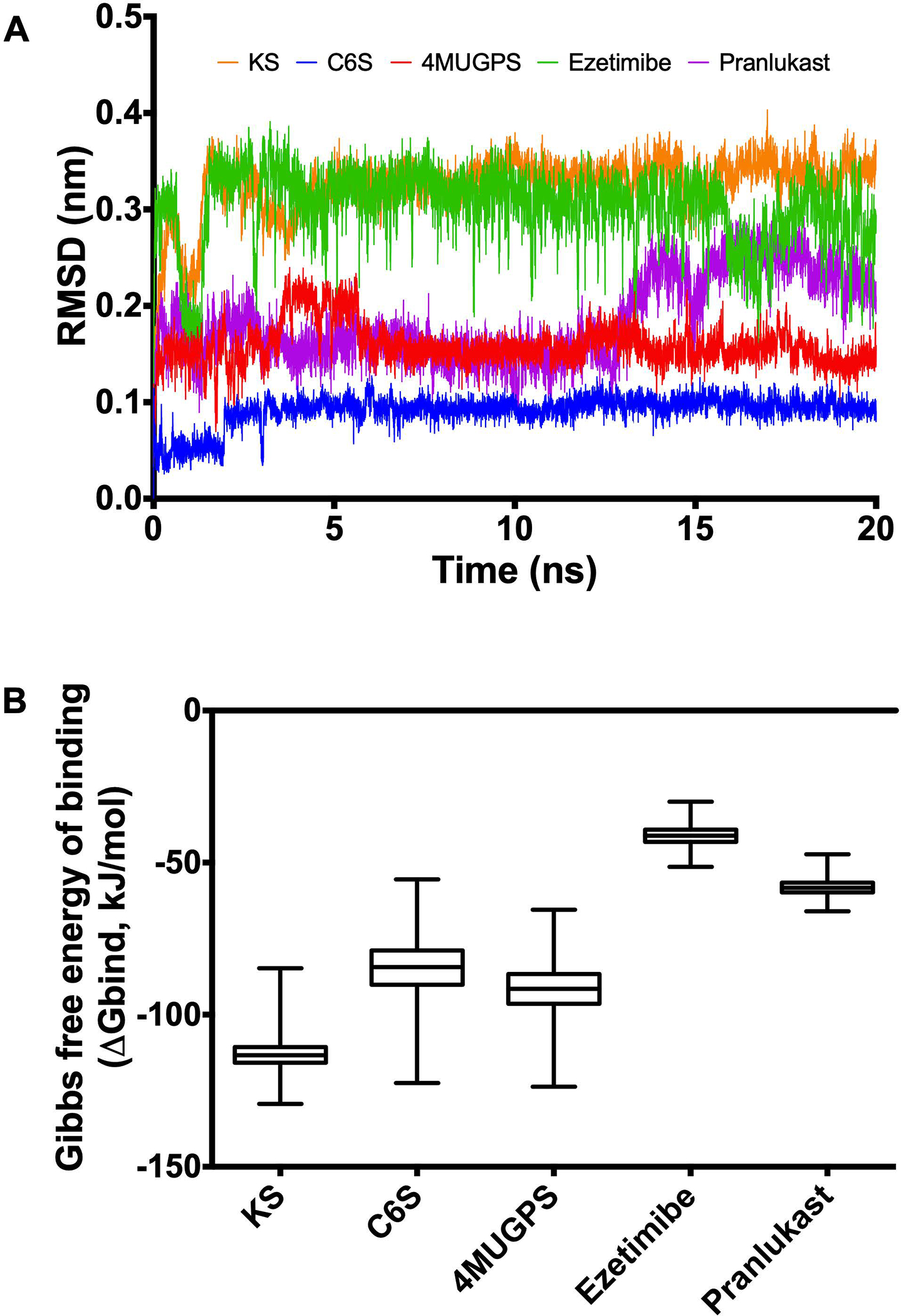
Molecular dynamics simulation for GALNS and ligands. Molecular dynamics simulations were carried out for 20 ns using GROMAC 4.5.5. The trajectories were analyzed by RMSD (A) and the affinity energy (B). KS: keratan sulfate, C6S: chondroitin-6-sulfate, 4MUGPS: 4-methylumbelliferyl-β-d-galactopyranoside-6-sulfate.
Ezetimibe and pranlukast weakly inhibited GALNS activity and increased its thermal stability
Previously, we reported the characterization of hrGALNS produced in the yeast Pichia pastoris32, 33. This enzyme showed similar post-translational processing as GALNS from human leucocytes with the maximum activity at pH 5.0 and a stability profile similar to that of hrGALNS produced in CHO cells32, 33. Both ezetimibe and pranlukast inhibited the enzyme activity of hrGALNS by 40% to 50% at a concentration of 10 μM (Figure 3A). In a thermal shift experiment, both ezetimibe and pranlukast increased the thermal stability of the hrGALNS by approximately 5 °C (Figure 3B). Together, the results revealed that ezetimibe and pranlukast are direct binders and weak inhibitors of GALNS, and thus may function as GALNS pharmacological chaperones.
Figure 3. Effects of ezetimibe and pranlukast on enzyme activity and thermal stability of hrGALNS.
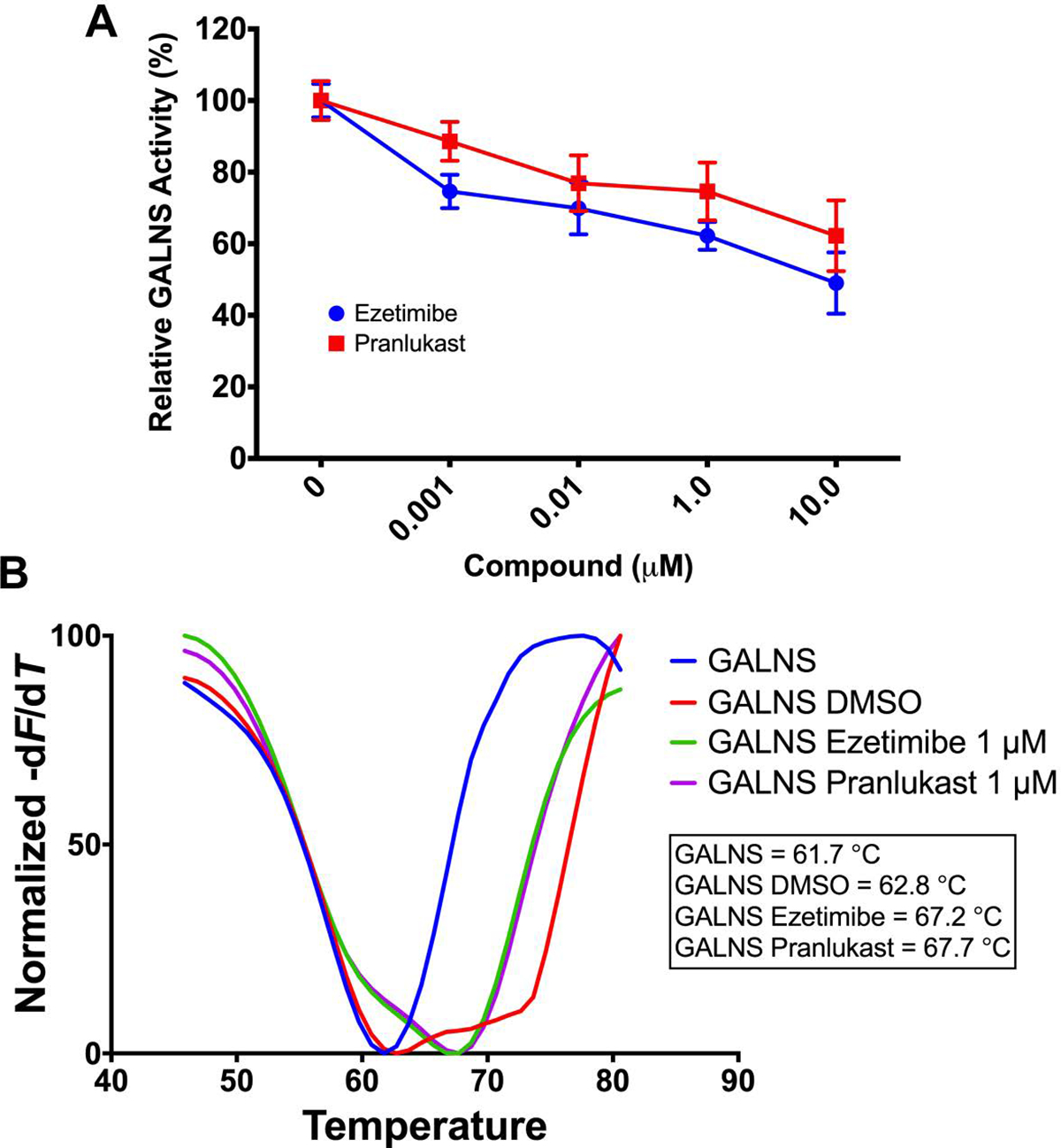
A) Recombinant GALNS was incubated with the fluorogenic substrate 4-methylumbelliferyl-β-D-galactopyranoside-6-sulfate with or without ezetimibe or pranlukast. GALNS activity was measured after an 18 h incubation as described in the Materials and Methods section. The results are reported as percentage of activity relative to the results observed without ezetimibe or pranlukast. B) Thermal stability was carried out by thermal shift assay. The hrGALNS was incubated with or without ezetimibe or pranlukast. The Tm was calculated by plotting the first derivative of the fluorescence emission as a function of temperature (−dF/dT). All assays were performed in triplicate.
Ezetimibe and pranlukast increased production of recombinant GALNS
Protein folding represents an important bottleneck in the production of recombinant proteins. Misfolded proteins not only reduce a protein’s biological activity, but also trigger cellular stress, which results in low production yields34, 35. We previously demonstrated that improvement of protein folding significantly increased GALNS activity36. In this sense, we hypothesized that ezetimibe and pranlukast may improve the proper folding of hrGALNS due to their pharmacological chaperone activity. Hence, we evaluated the effects of ezetimibe and pranlukast on production of hrGALNS in E. coli BL21(DE3), P. pastoris GS115, and HEK293 cells by a measurement of GALNS activity in cultures.
In the E. coli BL21(DE3) expression system, hrGALNS is detected both in the intra- and extracellular fractions36–38. After addition of ezetimibe or pranlukast to E. coli BL21(DE3) cultures, the GALNS activity in the intracellular fraction was not changed. In the extracellular fraction, the GALNS activity was increased up to 1.3- and 1.7-fold (p < 0.001), DMSO-treated cultures, in the presence of ezetimibe and pranlukast, respectively, as seen in Figure 4A.
Figure 4.
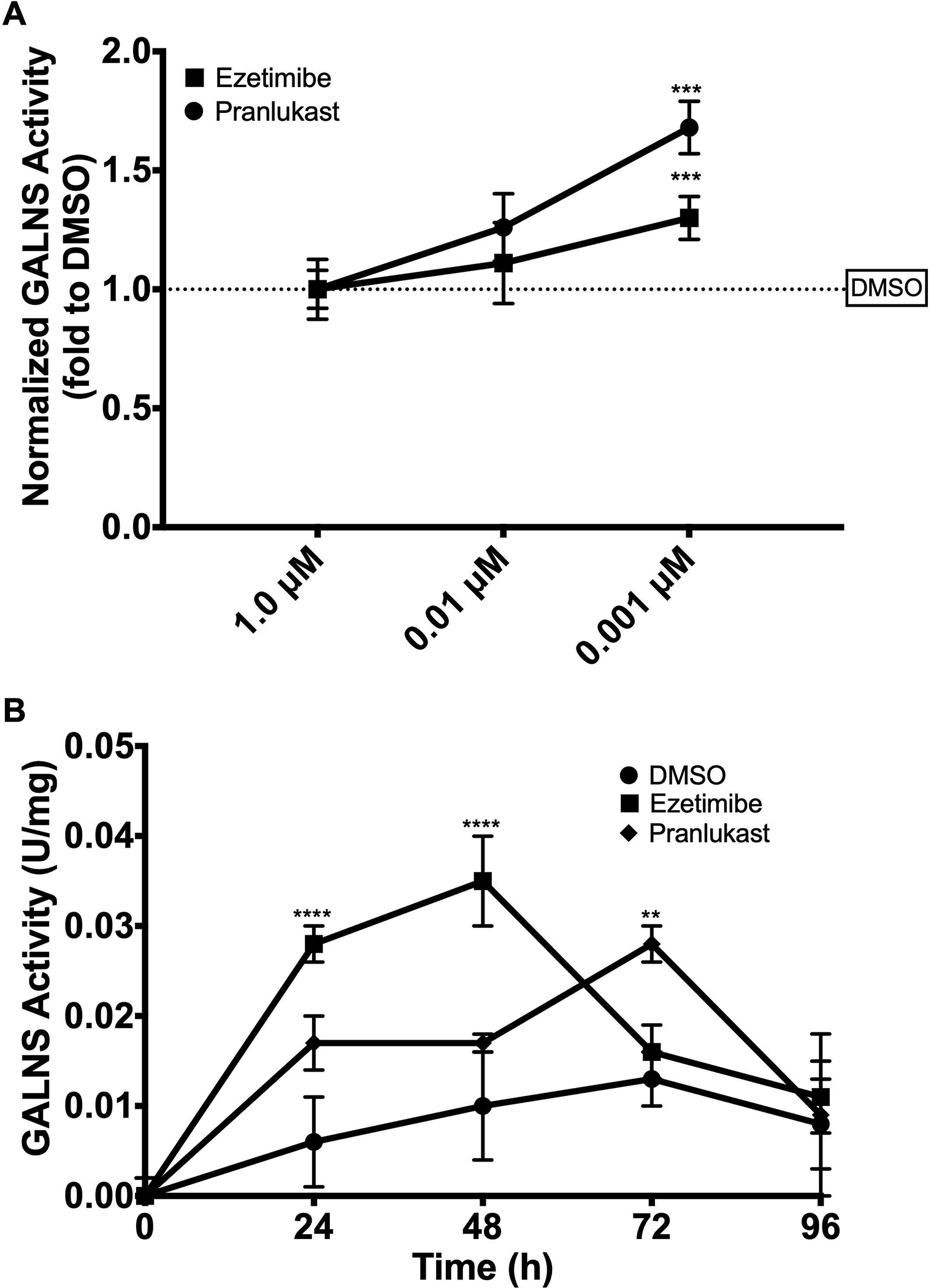
A) Effect of ezetimibe and pranlukast on the production of hrGALNS in E. coli. Enzyme activity was measured in the soluble fraction of the cell lysate after 24 a h induction. No effect of ezetimibe and pranlukast on GALNS activity was observed in the extracellular fraction. Results are reported as relative to those observed in the control (DMSO alone) treated cultures. B) Effect of ezetimibe and pranlukast (0.001 μM) on the production of hrGALNS in the yeast Pichia pastoris. GALNS activity was measured in the extracellular fractions at 24, 48, 72, and 96 h. No effect on GALNS activity was observed at higher concentrations of ezetimibe and pranlukast. The DMSO treated cultures were used as the control groups (n = 3, ANOVA Sidak t test ** p < 0.01, *** p < 0.001, **** p < 0.0001).
In the P. pastoris expression system, the hrGALNS was detected in the extracellular fraction due to the presence of a secretion signal peptide32. The addition of 0.001 μM ezetimibe or pranlukast every 24 h in the culture led to an increase of up to 5.0-fold of GALNS activity compared with DMSO-treated cultures (Figure 4B). The highest enzyme activity in the culture was observed after 48 h incubation with ezetimibe (p < 0.0001), compared to that enzyme activity in the control culture at 72 h. Similar results were observed during the production of hrGALNS in P. pastoris at bioreactor scale (Supporting Information Figure S2).
In the mammalian expression system with the plasmid pCXN-GALNS39, the transfected HEK293 cells showed a 9.3- and 10-fold increase in the intracellular and extracellular GALNS activity (Figure 5A). Treatment of 0.01 μM ezetimibe did not increase intracellular GALNS activity but induced a slight increase in the extracellular activity (1.16-fold); while 0.001 μM ezetimibe produced a marked increase in GALNS activity both intra- (1.5-fold) and extracellularly (1.6-fold). On the other hand, the treatment with 0.01 μM and 0.001 μM pranlukast produced a 1.4- and 1.3-fold increase in GALNS activity intracellularly, respectively, and 1.7- and 1.8-fold extracellularly, respectively (Figure 5A). Interestingly, co-treatment with ezetimibe and pranlukast did not induce any increase in GALNS activity at the extracellular fraction, and had a negative effect in the intracellular GALNS, inducing a 1.5-fold reduction in enzyme activity. These results might be due to the additive inhibitory activity of the two compounds. Immunostaining showed increases in GALNS protein levels in the transfected HEK293 cells treated with these two compounds (Figure 5B). Together, the results indicated that ezetimibe and pranlukast could increase GALNS production in all three-expression systems through the pharmacological chaperone activity by improving the proper folding and trafficking of newly synthesized GALNS proteins.
Figure 5.
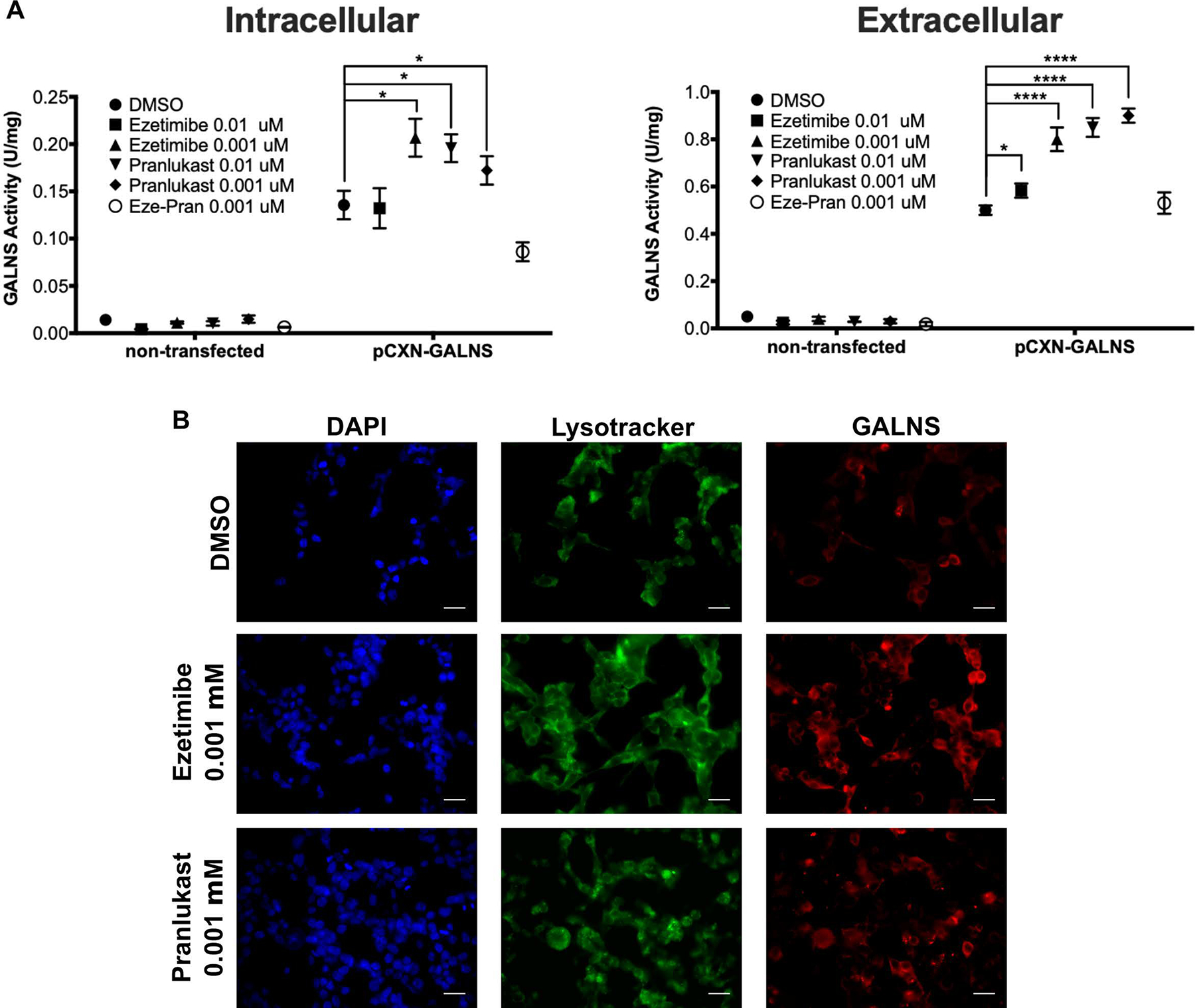
Effect of ezetimibe and pranlukast on the production of recombinant GALNS in HEK293 cells. The cells were transfected with plasmid pCXN-GALNS and treated with different concentrations of ezetimibe and pranlukast. A) Enzyme activities were measured in the cell lysates (intracellular) and culture media (extracellular) after compound treatments. DMSO treated cells were used as control (n=3, ANOVA Sidak t test * p < 0.05, **** p < 0.0001). B). HEK293 overexpressing human GALNS were immunostained to analyze the effect of pharmacological chaperones on the intracellular trafficking of hrGALNS. After 48 h treatment, cells were stained with Lysotracker® Green DND-26 and GALNS was detected with a rabbit polyclonal anti-GALNS antibody. Scale bar = 20 μm.
Ezetimibe and pranlukast acted as pharmacological chaperons in MPS IVA patient-derived fibroblasts
The mutations in the MPS IVA fibroblasts were identified and confirmed as p.R61W, p.R94C, p.F285del, p.A393S and p.W405_T406del (Table 1). All the fibroblasts, with the exception of GM00958 that is homozygous for the pA393S mutation, are heterozygous for missense mutations or amino acid deletions predicted as deleterious or damaging by at least one of the prediction tools. Bioinformatics analysis predicted that all mutations induced structural changes in the protein, as observed by changes in total energy and RMSD values that were more marked for the in-frame deletions p.F285del and p.W405_T406del (Table 2). Molecular docking against KS, C6S, and 4MUGPS predicted that mutations p.F285del and p.W405_T406del significantly changed the affinity energies for all of the evaluated substrates (Table 3), suggesting that the structural changes caused by these mutations may affect the protein-ligand interactions. For the remaining mutations, reductions in ligand affinity were observed for two of the three evaluated ligands, with C6S showing the most marked reductions. Overall, these results show the MPS IVA fibroblasts produce mutated GALNS susceptible to be treated by pharmacological chaperones.
Table 1.
Prediction analysis of MPS IVA fibroblasts used for evaluating ezetimibe and pranlukast. Fibroblasts were obtained from Coriell Institute. N.A., not applicable.
| Coriell ID | Mutation | Prediction Tool | ||
|---|---|---|---|---|
| PolyPhen-2 | PROVEAN | SIFT | ||
| GM00593 | p.R386C | Probably damaging | Deleterious | Damaging |
| p.F285del | N.A. | Deleterious | N.A. | |
| GM00958 | p.A393S | Benign | Neutral | Damaging |
| GM01259 | p.R94C | Probably damaging | Deleterious | Damaging |
| p.A393S | Benign | Neutral | Damaging | |
| GM01361 | p.R61W | Probably damaging | Deleterious | Damaging |
| p.W405_T406del | N.A. | Deleterious | N.A. | |
Table 2.
Bioinformatics analysis of wild type and mutated GALNS.
| Protein | Total Energy (kJ/mol) | Δ energy vs wild-type (kJ/mol) | RMSD (Å) | |
|---|---|---|---|---|
| Backbone | All atoms | |||
| Wild Type | −22,914.273 | --- | --- | --- |
| p.R61W | −22,408.814 | −505.5 | 0.043 | 0.051 |
| p.R94C | −22,492.967 | −421.3 | 0.043 | 0.051 |
| p.F285del | −20,120.439 | −2793.8 | 0.212 | 0.317 |
| p.R386C | −22,399.646 | −514.6 | 0.043 | 0.051 |
| p.A393S | −22,690.773 | −223.5 | 0.043 | 0.050 |
| p.W405_T406del | −20,905.438 | −2008.8 | 0.195 | 0.284 |
Table 3.
Molecular docking analysis of wild type and mutant GALNS against natural (KS and C6S) and artificial (4MUGPS) substrate.
| Protein | Affinity energy (kcal/mol) | ||
|---|---|---|---|
| KS | C6S | 4MUGPS | |
| Wild Type | 90.1 | 128.6 | 76.9 |
| p.R61W | 78.0 | 81.5 | 128.1 |
| p.R94C | 87.5 | 104.5 | 76.3 |
| p.F285del | 81.9 | 53.5 | 66.8 |
| p.R386C | 91.9 | 106.0 | 77.2 |
| p.A393S | 122.8 | 131.4 | 77.8 |
| p.W405_T406del | 81.7 | 64.5 | 45.6 |
As shown in Figure 6A, all evaluated patient fibroblasts had reduced GALNS enzyme activity, ranging from 20% to 64% of the wild type (WT) level. There was no clear genotype-phenotype correlation, as previously reported16, 20. In addition, western-blot analysis showed reduced GALNS levels in all the patient fibroblasts (Figure 6B). Ezetimibe treatment significantly increased GALNS activity by 1.6-, 1.4-, 2.5-, and 2.5-fold in the GM01361, GM01259, GM00958, and GM00593 fibroblasts compared to DMSO-treated cells, respectively (Figure 6A). With 0.001 μM ezetimibe treatment, the GALNS activity in GM01259 patient fibroblasts equaled that of WT fibroblasts. Western blot analysis showed that 0.01 or 0.001 μM ezetimibe treatment significantly increased the GALNS protein levels in all the MPS IVA fibroblasts (Figure 6B). Pranlukast treatment produced an increase in GALNS activity in GM01361 (p.R61W/p.W405_T406del) and GM00958 (p.A393S) patient fibroblasts, albeit less effectively than ezetimibe (Figure 6A). Western blot results showed that treatment with pranlukast significantly increased GALNS protein levels in all the evaluated patient fibroblasts (Figure 6B).
Figure 6. Effects of pharmacological chaperone compounds on GALNS enzyme activity.
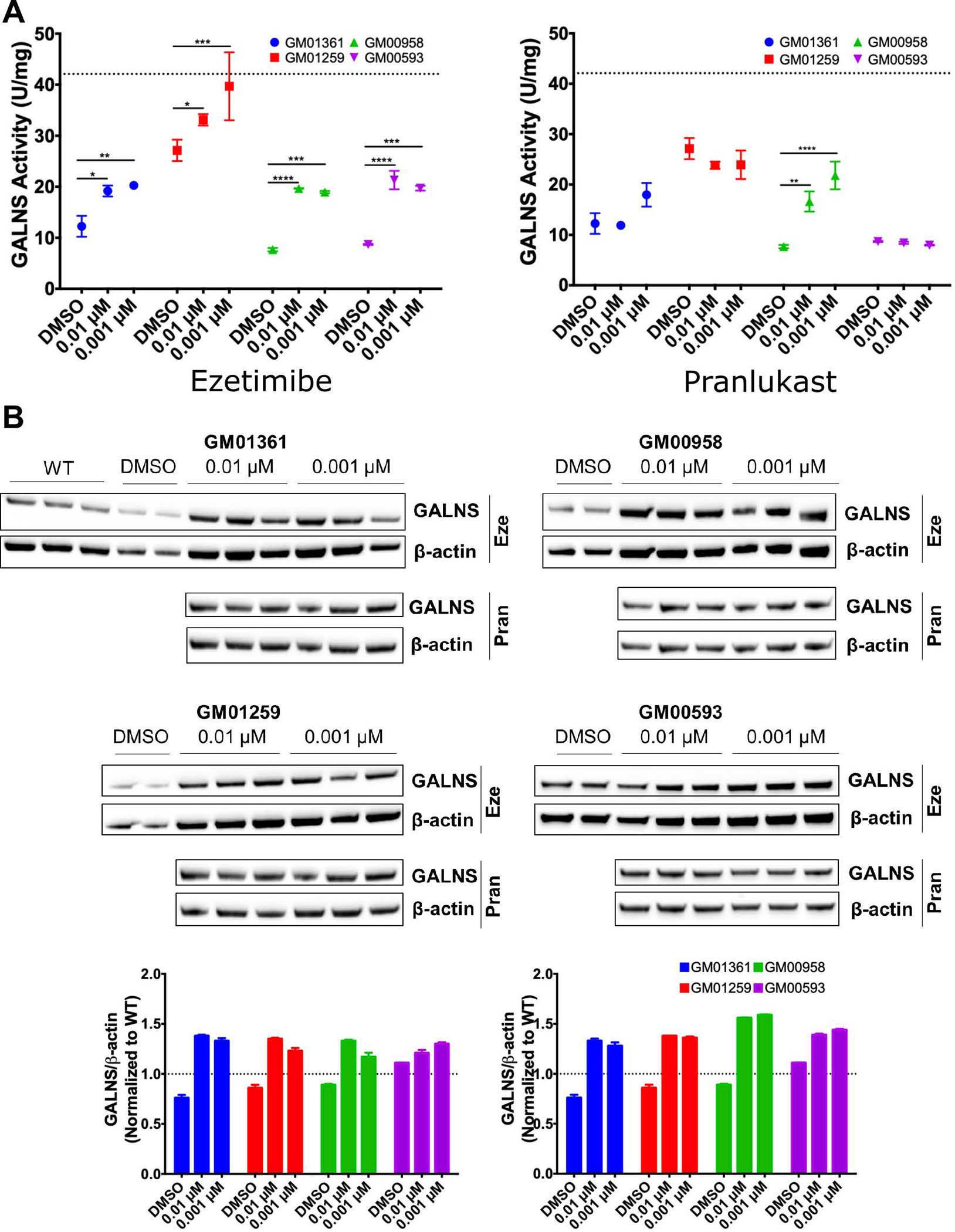
A) Effect of ezetimibe and pranlukast on GALNS activity in MPS IVA patient fibroblasts. The fibroblasts were treated with different concentrations of ezetimibe and pranlukast for 48 h after which the GALNS activity was measured in the cell lysate. Dotted line corresponds to WT activity levels. All assays were performed in triplicate (ANOVA Sidak t test * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001). B) Effect of ezetimibe (Eze) and pranlukast (Pran) on GALNS protein levels as determined by Western-blot analysis. The fibroblasts were treated with different concentrations of ezetimibe and pranlukast for 48 hrs after which the protein lysates from the fibroblasts were analyzed. The densities of specific bands were calculated by using NIH Image J 1.8.0. GALNS protein levels were normalized against β-actin density and reported as fold-change to WT levels (dotted line). All assays were performed in triplicate.
Lysosomal mass increased in patient-derived fibroblasts is ameliorated in ezetimbe- and pranlukast-treated cells.
LysoTracker staining, a phenotypic assay that detects enlarged lysosomes and acidic organelles40, 41, exhibited 1.2- to 2.4-fold increases in MPS IVA patient cells, indicating significant increases in lysosomal mass compared to WT fibroblasts (Figure 7). The LysoTracker staining was dose-dependently reduced in all the disease fibroblast lines after the ezetimibe treatment, demonstrating the reduction in enlarged lysosome mass (Figure 7A). In this sense, this study shows, for the first time, that this phenotypic assay can be used for drug screening in MPS IVA patient-derived cells. In addition, the data from Figures 6 and 7 suggest that the ezetimibe treatment significantly increased GALNS protein levels and activity in the MPS IVA patient cells, resulting in reduction of the enlarged lysosome mass. Results with pranlukast were slightly different wherein LysoTracker staining did not change in patient fibroblasts treated with pranlukast (Figure 7B). Together with information from Figure 6, these results suggest that pranlukast might increase the stability of the enzyme, but not its proper folding, thus limiting the recovery of the function of the mutated enzymes.
Figure 7.
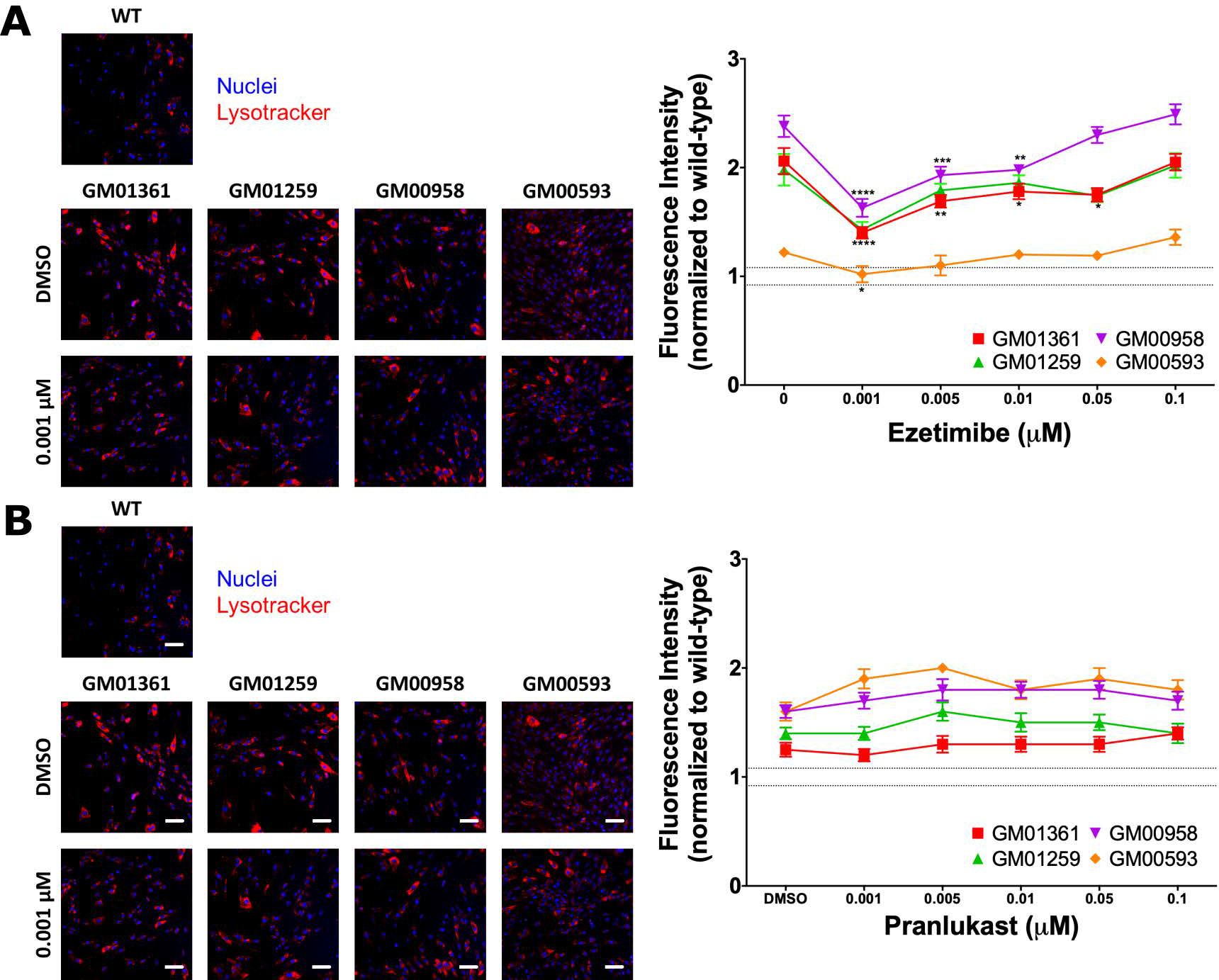
Effect of ezetimibe (A) or pranlukast (B) on reduction of lysosome mass in MPS IVA patient fibroblasts. Cells were treated with ezetimibe or pranlukast for 48 h, after which the cells were stained with LysoTracker Red DND-99 dye. The images (9 images/well) were acquired using the IN Cell Analyzer 2200 imaging system. Results are presented as cell intensity normalized to WT fluorescence level (dotted lines). All assays were performed in triplicate (ANOVA Sidak t test * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001). Scale bar: 100 μm.
Abnormal autophagy markers in the MPS IVA patient fibroblasts were ameliorated by ezetimibe and pranlukast
Prior to this study, the autophagy markers p62 and LC3B-II in MPS IVA cells have not been reported. We found a significant reduction of p62 levels in all MPS IVA cells and decrease in LC3B-II levels in the GM01259, GM00958, and GM00593 fibroblasts, as shown in Figure 8. The results suggested that autophagic processes might be disrupted in patient cells. To further evaluate the therapeutic potential of ezetimibe and pranlukast, we measured the changes in p62 and LC3B-II levels after treatment with these two pharmacological chaperones. The treatments of MPS IVA fibroblasts with ezetimibe and pranlukast at 0.01 μM and 0.001 μM concentrations, respectively, significantly increased the levels of p62 and LC3B-II as seen in Figure 8. Higher increases in p62 levels were observed in the ezetimibe treated cells than the increased levels in the pranlukast treated cells.
Figure 8.
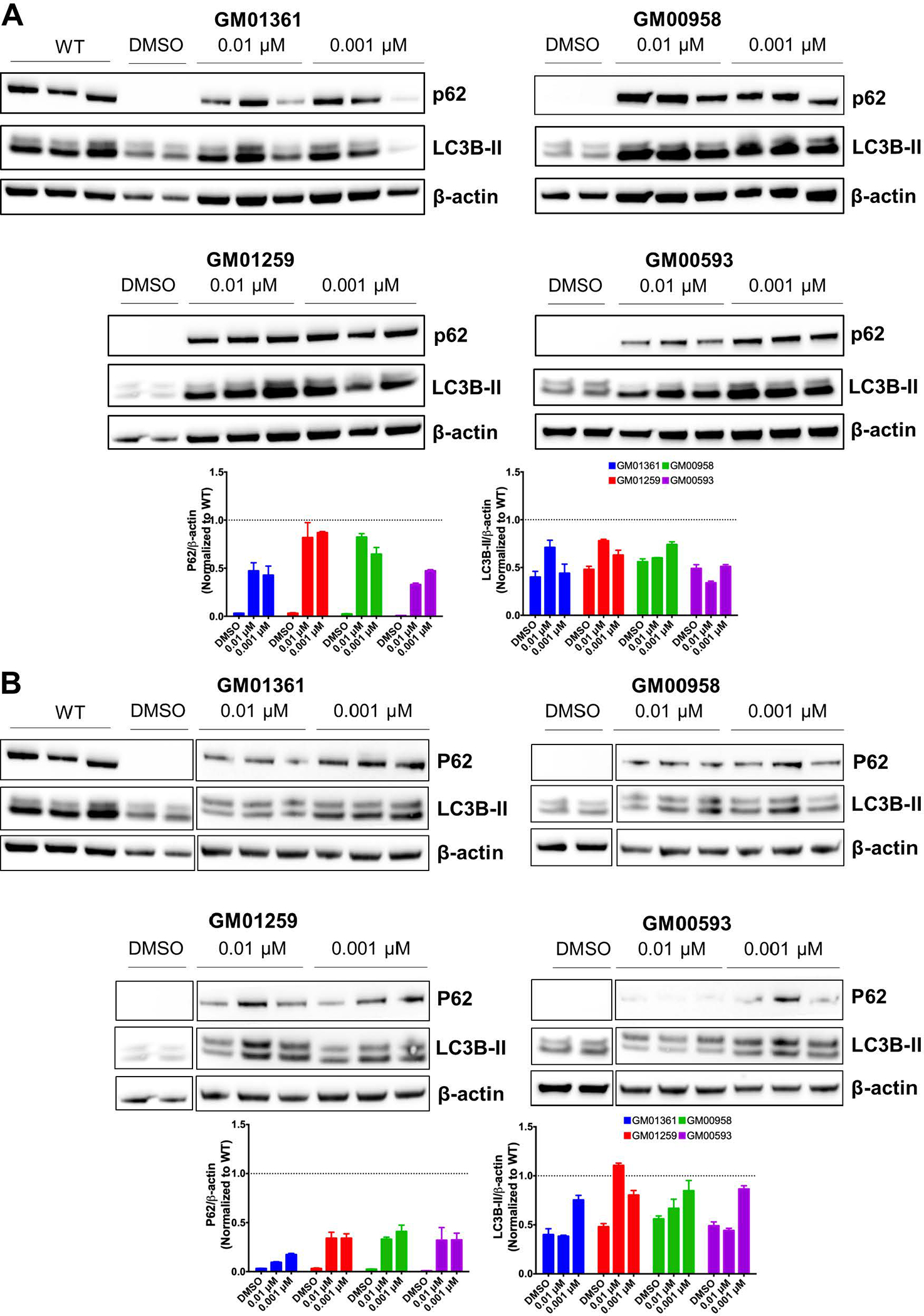
Evaluation of the autophagy markers p62 and LC3B-II in MPS IVA fibroblasts treated with ezetimibe (A) or pranlukast (B). The fibroblasts were treated with different concentrations of the pharmacological chaperones for 48 h after which the protein was analyzed in the cell lysates. The densities of specific bands were calculated by using NIH Image J 1.8.0. Results were normalized against β-actin density and reported as fold differences from the WT levels (dotted line).
Combination therapy of ERT with pharmacological chaperone
We previously found that the cellular uptake of hrGALNS produced in P. pastoris is mediated by both mannose and mannose-6-phosphate receptors and targeted to the lysosome32, 33. To evaluate the effect of the two pharmacological chaperones on ERT, we co-administrated hrGALNS enzyme with ezetimibe or pranlukast in the MPS IVA fibroblasts. As shown in Figure 9, the combination treatment of patient cells with hrGALNS and a pharmacological chaperone (ezetimibe or pranlukast) further decreased LysoTracker staining compared to those treated only with hrGALNS, indicating an additive effect with further reduction of lysosomal mass in the patient cells. The combination therapy with hrGALNS and pranlukast almost normalized the LysoTracker staining in all MPS IVA fibroblasts to the levels in WT control cells, although pranlukast did not reduce lysosomal mass when it was used by itself. Together, the results suggested that the combination therapy of ERT with ezetimibe or pranlukast might improve the clinical efficacy of ERT in MPS IVA patients.
Figure 9. Effects of combination treatment of hrGALNS with ezetimibe or pranlukast on the reduction of lysosome size in MPS IVA patient fibroblasts.
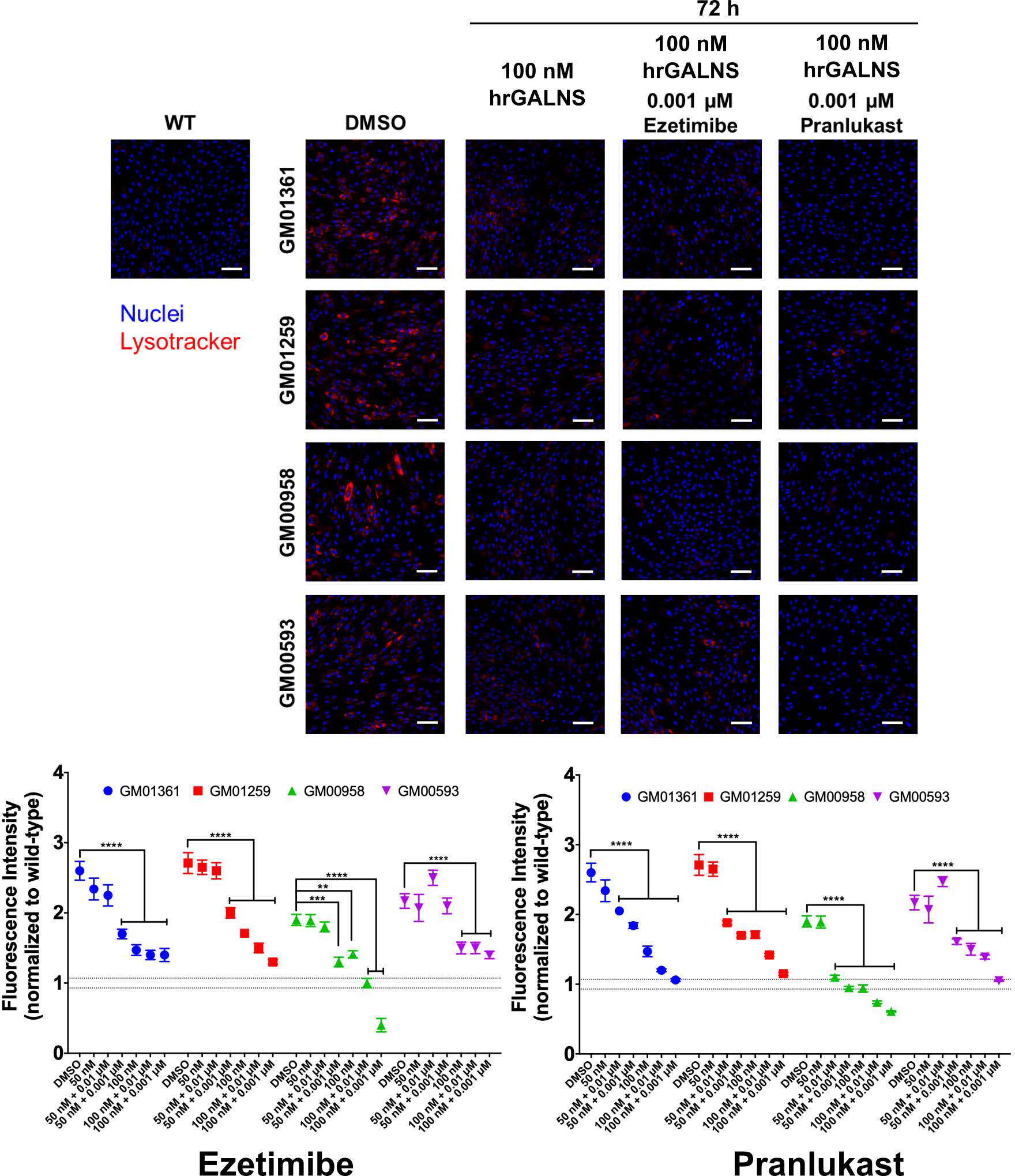
Co-administration of hrGALNS produced in the yeast P. pastoris and the pharmacological chaperones ezetimibe and pranlukast decreased the size of the lysosomes to WT levels. Cells were treated for 72 h with hrGALNS in the presence or absence of ezetimibe or pranlukast. The cells were stained with LysoTracker Red DND-99 dye and the images (9 images/well) were acquired using the IN Cell Analyzer 2200 imaging system. Results are presented as cell intensity normalized to the wild-type fluorescence level (dotted lines). All assays were performed in triplicate (ANOVA Sidak t test ** p < 0.01, *** p < 0.001, **** p < 0.0001). Scale bar: 100 μm.
3. DISCUSSION.
In this study, we describe, for the first time, two small molecules (ezetimibe and pranlukast) with pharmacological chaperone activity for GALNS. These two molecules bind to the active cavity of GALNS and act as chaperones by stabilizing the protein structure. They increased the productions of hrGALNS in E. coli, P. pastoris, and HEK293 cells. In addition, ezetimibe treatment increased the activity and protein level of GALNS in MPS IVA fibroblasts, leading to a reduction of lysosomal mass and normalization of autophagic function. Treatment of MPS IVA cells with a combination of hrGALNS and ezetimibe (or pranlukast) significantly reduced the lysosomal accumulation phenotype in MSP IVA cells, suggesting an additive effect of the combination treatment.
One type of pharmacological chaperone for LSDs emulates the structure of the enzyme’s substrate. For example, 1-deoxynojirimycin-based, isofagomine-based, and 1,5-dideoxy-1,5-imino-D-xylitol-based compounds act as pharmacological chaperones by binding to the active site of enzymes24, 25. In the case of MPS II, a sulfated disaccharide derived from heparin (Δ-unsaturated 2-sulfouronic acid-N-sulfoglucosamine) was reported as a pharmacological chaperone that mimics the structure of the natural substrate of iduronate-2-sulfatase (IDS)29. Non-substrate-like pharmacological chaperones have been described by Gaucher42, 43 and Krabbe44. Yilmazer et al.45 used a ligand-based and structure-based pharmacophore hypothesis for virtual screening of a ZINC database, resulting in the identification of chaperone compounds for β-glucocerebrosidase that have tricyclic pyridothieno-pyrimidine or dioxino quinolone scaffolds and include antiallergic, antibiotic, and antineoplastic drugs. In the present study, molecular docking and virtual screening identified ezetimibe and pranlukast as potential pharmacological chaperones for human GALNS. Ezetimibe is an approved drug that blocks intestinal cholesterol absorption by selectively inhibiting Niemann-Pick C1-like 1 protein and is indicated for treatment of disorders with elevated cholesterol levels46. Pranlukast is an orally administered, selective and competitive cysteinyl leukotriene type 1 receptor antagonist, indicated for the treatment of bronchial asthma in pediatric and adult patients47. In terms of safety, both ezetimibe and pranlukast are well tolerated and have adverse event profiles similar to placebo46, 47. Recently, it was reported an agonist activity of pranlukast over the farnesoid X receptor (FXR)48. This could represent a potential drawback of pranlukast use as a pharmacological chaperone, since activation of FXR may have an important adverse effect in the metabolic cholesterol elimination by repressing cholesterol 7α-hydroxylase (CYP7A1). Nevertheless, pranlukast was characterized as a moderately potent partial agonist of FXR with an EC50 of 15 μM48, which is 15,000-fold higher than the concentration that showed the best pharmacological chaperone effects in this study. In this sense, we do not expect any additional adverse effect for pranlukast to those previously reported. Medicinal chemistry optimization of pranlukast may provide better analogs with more less off-target liabilities.
Most pharmacological chaperones bind to the catalytic site of an enzyme and are reversible inhibitors that dissociate with the enzymes due to the high substrate concentrations in the lysosomal compartment28. Ezetimibe and pranlukast inhibited GALNS activity by 40% to 50% at 10 μM concentration, which confirmed the modeling prediction of partial and weak binding of these compounds to the active cavity of GALNS. Similarly, Δ-unsaturated 2-sulfouronic acid-N-sulfoglucosamine competed with the substrate of the human IDS leading to a reduction of approximately 40% in enzyme activity29. The pharmacological chaperones of the lysosomal enzyme aspartylglucosaminidase weakly inhibited the enzyme activity49.
Ezetimibe and pranlukast increased the GALNS thermal stability, indicating they bind directly to this enzyme. These results agreed with those reported for Δ-unsaturated 2-sulfouronic acid-N-sulfoglucosamine, which significantly increased the thermal stability of IDS29. Similarly, glycomimetic ligands for human glucocerebrosidase increased its thermal stability between 5.6 and 21.7 °C, depending on the ligands and the inhibition mechanism (competitive, semi-irreversible, or irreversible)50.
Several factors affect the production of a recombinant protein, including the host, vectors, culture conditions, and protein activation and folding51. Misfolded proteins can cause reduced activity and low production yields34, 35. Previously, we showed that activity of a hrGALNS expressed in E. coli was increased by improving protein folding through the control of gene expression, induction of osmoprotectants, and improvement in the formation of disulfide bonds36. In addition, it has been reported that protein translocation is a bottleneck in the production of properly folded proteins, and that several methods could be used to improve this process34. We observed that ezetimibe and pranlukast increased the activity of hrGALNS produced in bacteria, yeast, and mammalian cells. However, when a combination of ezetimibe and pranlukast was used during production of recombinant GALNS in HEK293 cells, it significantly decreased GALNS activity. This decrease might be a consequence of an additive effect of enzyme inhibition by the two molecules. Nevertheless, the results have shown potential applications for the small molecule chaperone compounds to improve production yield and activity of hrGALNS. The increase in activity of hrGALNS might help to reduce immunogenicity, as the amount of protein used in ERT can be reduced52.
Prior to the treatment with the pharmacological chaperones, we identified the mutations present in the MPS IVA fibroblasts. The MPS IVA patient fibroblasts used in this study have the mutations p.R61W, p.R94C, p.F285del, p.A393S, and p.W405_T406del in the GALNS protein. The mutations p.R386C and in-frame deletions p.F285del and p.W405_T406del are associated with rapid disease progression and a severe growth reduction phenotype53–55, while mutations p.R61W and p.R94C are associated with a mild/attenuated phenotype16, 55. On the other hand, mutation p.A393S was predicted to be benign or neutral by PolyPhen-2 and PROVEAN, respectively, and no reports for this mutation have been found in the literature. Another bioinformatics analysis showed that p.A393S induces structural changes in GALNS. All of these mutations can lead to a significant reduction in GALNS activity and protein level, resulting in autophagy disruption and an increase in lysosomal mass. In addition, since a mutated protein is synthetized, they could be susceptible to be treated by pharmacological chaperones.
Our results demonstrated that ezetimibe increased GALNS activity levels by 1.2- to 2.5-fold and reduced lysosomal mass in all of the MPS IVA patient cells we examined. Pranlukast only increased GALNS activity in patient cells with p.A393S or p.R61W/p.W405_T406del mutations without an obvious reduction in lysosomal mass. Recently, we showed that treatment of MPS IVA fibroblasts with a rhGALNS produced in P. pastoris reduced KS levels33, suggesting that this GAG is stored in MPS IVA fibroblasts and that the reduction in lysosomal mass observed after treatment with the pharmacological chaperones could be associated to a KS reduction. It has been reported that the effects of pharmacological chaperones may depend on the particular mutations of the enzyme and properties of the compound. For instance, treatment of MPS II fibroblasts with Δ-unsaturated 2-sulfouronic acid-N-sulfoglucosamine showed a 1.6- to 39.6-fold increase in IDS activity without a significant reduction of GAGs level29. Similarly, treatment of MPS IVB and GM1-gangliosidosis fibroblasts with the pharmacological chaperone (5aR)-5a-C-pentyl-4-epi-isofagomine increased β-galactosidase activity by 1.5- to 35-fold30. Treatment of Gaucher patient fibroblasts with different pharmacological chaperones resulted in up to a 2.5-fold increase in β-glucocerebrosidase activity, and a significant reduction in the stored substrate56, 57. Finally, pharmacological chaperones led to a 2- to 4-fold increase in aspartylglucosaminidase activity in fibroblasts derived from patients with aspartylglucosaminuria, as well as improvement of protein processing and reduction of lysosomal mass49. These previously reported findings suggest that a similar strategy could be employed to treat MPS IVA patients. However, it is important to evaluate the biodistribution of these drugs to the main affected tissues in MPS IVA patients, such as bone, heart, and cornea. Nevertheless, since circulation times increase the probability for a drug to reach the target disease sites58, it could be expected that ezetimibe and pranlukast reach those tissues due to: 1) longer half-life times (22h and 9 h, respectively)47, 59, than MPS IVA ERT (35 min)60, and 2) higher administration frequency (daily or every other day) than ERT (once a week). In case that these drugs do not shown an acceptable biodistribution profile; there are some targeting alternatives that could be implemented61, 62, in addition to the possibility of drug properties optimization through medicinal chemistry.
Autophagy malfunction has been widely reported in LSDs. For instance, inhibition or blockade of autophagic flux has been reported for neural ceroid lipofuscinoses, Gaucher disease, Niemann-Pick type C1 (NPC1), Pompe disease, and Danon disease, among others63. However, other studies showed activation of the autophagy pathway, as observed in NPC164 or mucolipidosis type IV65. In the case of mucopolysaccharidoses, MPS I and MPS VII mice showed an increase in protein degradation which was associated with an increase in autophagic flux66. On the other hand, LC3 levels were decreased in MPS IIIC mice with increased autophagy activity67. We found that both p62 and LC3B-II levels were reduced in the MSP IVA cells compared to the WT control. Treatment of MPS IVA fibroblasts with ezetimibe or pranlukast significantly increased the levels of p62 and LC3B-II, with higher p62 levels observed after the ezetimibe treatment. Recently, it was described that ezetimibe increases autophagy flux in mice through AMPK activation and subsequent increase of TFEB nuclear translocation68. In primary hepatocytes, ezetimibe increased LC3B-II levels; the number of both autolysosomes and autophagic vacuoles; and the expression of autophagy-related genes such as Tfeb, Atg7, Lc3b, Atg3, Atg5, Atg12, Lc3a, Ulk1, Benc1, Sqstm1 (p62), and Lamp168. Similarly, in-vivo administration of ezetimibe induced autophagy in the liver, as well as reducing the number of lipid droplets, hepatic triglycerides, and cholesterol levels in methionine- and choline-deficient diet-fed mice compared with controls68. Therefore, the effect of ezetimibe on MPS IVA patient fibroblasts could be the result of both pharmacological chaperone activity and normalization of defective autophagy.
Our results indicated that the efficacy of ERT for MPS IVA patients may be overcome by a combination therapy of hrGALNS with ezetimibe or pranlukast because the pharmacological chaperones can increase the activity of hrGALNS, reducing the dose needed for ERT. In addition, the effect of ezetimibe on autophagy can be functionally additive to ERT and may further improve the ERT’s therapeutic efficacy in MPS IVA patients. Our results are consistent with a previous report that showed a co-administration of recombinant human acid α-glucosidase (rhGAA) with the pharmacological chaperone duvoglustat increased the half-life and plasma levels of rhGAA resulting in a higher reduction of glycogen accumulation in heart and skeletal muscles compared to rhGAA used alone in the Pompe disease model69. Recently, a clinical evaluation of this therapeutic option revealed a significant increase in rhGAA activity and protein levels in plasma and muscle, demonstrating the benefit of combinational therapy of ERT with a pharmacological chaperone compound70. In this sense, Tomatsu et. al.71 showed that an increase in the ERT half-life, by using an engineered rhGALNS carrying a bone-targeting peptide, significantly improved the biodistribution to bone and heart, as well as the therapeutic efficacy. Taken together, we consider that the limited effects in correcting the skeletal and cardiac abnormalities of the current MPS IVA ERT could be improved by increasing the half-life and activity of the rhGALNS by combination therapy with a pharmacological chaperone.
4. CONCLUSIONS.
In this study, we have identified and characterized ezetimibe and pranlukast as the first pharmacological chaperones described for the lysosomal enzyme GALNS. Both compounds significantly increased the amounts and activities of hrGALNS produced in E. coli, P. pastoris, and HEK293 cells, indicating that both ezetimibe and pranlukast may be used to increase the production yields of hrGALNS. Ezetimibe significantly increased the protein level and activity of mutated GALNS in MPS IVA fibroblasts as well as significantly reducing lysosomal mass and ameliorating abnormal autophagy. The combination therapy of hrGALNS with ezetimibe or pranlukast additively reduced lysosomal mass in patient-derived fibroblasts. Therefore, ezetimibe has potential for use as a monotherapy for MPS IVA patients. The combination therapy of ezetimibe or pranlukast with the reduced dose of ERT may provide a new therapeutic strategy to improve the efficacy of ERT for MPS IVA patients.
5. EXPERIMENTAL SECTION.
Virtual docking.
Virtual docking was carried out using a previously reported 3D GALNS structure20 and AutoDock Vina72 against 11,421 compounds, called the ZINC In Man, a special subset of ZINC73. Docking for each ligand was run 20 times and constrained to the active cavity (specifically, the grid box was centered between formylglycine 79 and calcium ion). Results of the protein-ligand interactions are reported as the affinity energy (kcal mol−1)72. KS (1KES) and C6S (CID 24766) structures were retrieved from the RCSB-Protein Data Bank and PubChem, respectively. The structure of the artificial ligand 4MUGPS used in GALNS activity assay74 was built using MarvinSketch at Marvin Suite (ChemAxon Ltd., Budapest, Hungary). Molecular dynamics analysis was done using GROMAC 4.5.575. Topology of ligands was generated using the Automated Topology Builder (ATB) and Repository version 2.276. Simulations were carried out for 20 ns. The trajectories were analyzed by root mean squared distance (RMSD; g_rms_d) and the affinity energy (g_lie_d). All simulations were done at the High-Performance Computing Center (ZINE) of Pontificia Universidad Javeriana (Bogotá, Colombia).
Productions of human recombinant GALNS (hrGALNS).
Productions of hrGALNS in Escherichia coli BL21(DE3) and Pichia pastoris GS115 were carried out a shaken-flask scale (100 mL) as previously reported32, 36, 37, 77. The production of hrGALNS in mammalian cells was performed using the HEK293 cells (ATCC CRL1573) transfected with the pCXN-GALNS vector (kindly donated by Dr. Shunji Tomatsu)39. The HEK293 cells were cultured in Dulbecco’s modified medium (DMEM, Gibco, Thermo Fisher Scientific, Grand Island, NY, USA) supplemented with 15% fetal bovine serum, (Eurobio, Les Ulis, France), penicillin 100 U mL−1, and streptomycin 100 U mL−1 (Walkersville, MD, USA), at 37 °C in a 5% CO2 incubator. Lipofectamine 2000 was used in the transfection per the manufactureŕs instructions (Invitrogen, Thermo Fisher Scientific, San Jose, CA, USA). Ezetimibe and pranlukast (Sigma-Aldrich, St. Louis, MO, USA) were dissolved in DMSO and added at different concentrations during hrGALNS production. GALNS activity was measured in the cell lysate (HEK293 cells) or cell supernatant (E. coli and P. pastoris). Recombinant GALNS produced in P. pastoris was purified from culture medium following a previously reported protocol32. Briefly, culture medium (~1.7 L) was filtered sequentially through 0.45 and 0.22 μm using polyether sulphone membranes (Pall Corp, Port Washington, NY, USA). Permeate was ultra-filtered through a 30 kDa cut-off membrane (Millipore, Billerica, MA, USA). The retentate was dialyzed in acetate buffer (25 mM, pH 5.0). Finally, hrGALNS was purified by a two-step process using a cation exchange chromatography followed by size exclusion chromatography. Fractions with the highest GALNS activity were pooled, diafiltrated against 25 mM sodium acetate pH 5.0, and lyophilized.
Culture of fibroblasts.
WT and MPS IVA patient-derived skin fibroblasts were obtained from the Coriell Institute (Table 1). Cells were cultured in DMEM (Gibco, Thermo Fisher Scientific, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Eurobio, Les Ulis, France) at 37 °C in a CO2 incubator. Characterizations of mutations in the GALNS gene were performed by ACGT, Inc. (Wheeling, IL, USA) through exon sequencing. Gene variants were analyzed by using the Variant Annotation Integrator tool from the University of California Santa Cruz Genome Browser78, and further studied by PolyPhen279 and SIFT/PROVEAN80.
The point mutations (R386C, A393S, R61W and R94C) were modeled by site-directed mutagenesis using the reported crystal structure of human GALNS (PDB 4FDI)14 on PyMOL v2.0.7 (Molecular Graphics System, v2.0 Schrödinger LLC), followed by an energetic minimization using Chimera v1.1281. Deletions (F285del and W405_T406del) were modeled using the I-TASSER online server82. Formylglycine 79 was manually included within these models using PyMOL; a calcium ion was added between Asp39, Asp40, Asp288 and Asp28914, 20 using YASARA View v18.4.2483, followed by an energetic minimization using Chimera v1.1281. The comparison of the models against the WT protein crystal was done using PyMOL, which compared the RMSD of the protein backbone to all the other atoms in the protein. Molecular docking of the modeled GALNS structures and the natural and artificial ligands was done using AutoDock Vina72. The search space was centered between FGly79 and the calcium ion, as previously described20. Docking for each ligand was run 20 times and constrained to the active cavity. Protein-peptide interactions were reported as affinity energy (kcal mol-1)72 and were evaluated by using YASARA View v18.4.2483, and UCSF Chimera v1.12 81.
LysoTracker staining and immunofluorescence staining.
HEK293 cells overexpressing GALNS were treated with the chaperone compounds for 48 h followed by a cell wash and incubation with 50 nM Lysotracker® Green DND-26 (Molecular Probes, Thermo Fisher Scientific, San Jose, CA, USA) in complete DMEM media at 37 °C for 1 hour. For the immunofluorescence staining, cells (after the compound treatment) were washed twice with 1X PBS, fixed with 4% paraformaldehyde, and permeabilized with 0.1% Triton X-100 followed by blocking with 2% bovine serum albumin in 1X TBS. After blocking, the cells were incubated overnight at 4 °C with a rabbit polyclonal anti-GALNS antibody (ab187516, Abcam) in the blocking buffer. Goat anti-rabbit IgG (H+L) cross-adsorbed, Alexa Fluor® 633 antibody (A-21070, Thermo Fisher Scientific, San Jose, CA, USA) was added at a 1:1,000 dilution in the blocking buffer. The cellular nuclei were stained with 4´,6-diamidino-2-phenylindole dihydrochloride (DAPI, Thermo Fisher Scientific, San Jose, CA, USA). The cells were visualized using an Axio Observer Z1 microscope (ZEISS, Birkerød, Denmark) with a Filter set 109 HE LED (E), containing beam splitter TBS 405 + 493 + 575 and emission filter TBP 425/29 + 514/31 + 632/100; this configuration collected DAPI, Lysotracker® Green and anti-GANLS signals, respectively. Images were processed by using NIH Image J software84.
To evaluate the effects of ezetimibe and pranlukast on the reduction of lysosomal mass in 96-well plates, cells were seeded in black clear bottomed plates (Greiner Bio-One, #655090, Monroe, NC, USA) at 4000 cells/well and cultured overnight. On the day of the experiment, MPS IVA and WT fibroblasts were treated with different concentrations of ezetimibe or pranlukast in DMEM medium for 24 h. The cells were then stained with 50 nM LysoTracker Red DND-99 dye in the medium at 37 °C for 1 hour, followed by two plate washes with DPBS. The plates were fixed and stained simultaneously in 3.2% paraformaldehyde solution with Hoechst dye at a 1:5,000 dilution for 30 minutes at room temperature. The plates were washed twice using DPBS and stored with 100 μl/well DPBS at 4 °C until imaging. The images (9 images/well) were acquired using the INCell Analyzer 2200 imaging system (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA). INCell image analysis software (GE Healthcare Bio-Sciences) was used to quantify the fluorescence intensity (LysoTracker staining). The fluorescence intensity corresponds to the average of all the pixels in a cell and averaged across all the cells within an image. The DAPI (excitation = 350 ± 50 nm, emission = 455 ± 50 nm) and Texas Red (excitation = 545 ± 20 nm, emission = 593 ± 20 nm) filter sets were used to visualize Hoechst nuclear staining and LysoTracker Red DND-99 staining, respectively.
Thermal shift assay.
Thermal stability was carried out by thermal shift assay85. For this purpose, 5 μM hrGALNS, in 25 mM sodium acetate pH 5.0, was mixed with 1 μM of ezetimibe or pranlukast and 20X SYPRO orange (Molecular Probes, Thermo Fisher Scientific, Eugene, OR, USA). Thermal denaturation was carried out in an Applied Biosystems ViiA7 with ViiA7 RUO software (Applied Biosystems, Thermo Fisher Scientific, Carlsbad, CA, USA). Melting curve was set as follows: 1) 2:00 hold at 25°C, 2) ramp up in 1 °C/30 sec increments to a final temperature of 95 °C, and 3) 2:00 hold at 95°C. The Tm was calculated by plotting the first derivative of the fluorescence emission as a function of temperature (−dF/dT). All assays were performed in triplicate.
GALNS activity.
GALNS activity was assayed by using 4-methylumbelliferyl-β-d-galactopyranoside-6-sulfate (Toronto Chemicals Research, North York, ON, Canada) as a substrate74, 86. One unit (U) was defined as the amount of enzyme catalyzing 1 nmol substrate per hour. Specific GALNS activity was expressed as U/mg of protein as determined by Lowry assay. To evaluate the inhibitory effect of ezetimibe or pranlukast, hrGALNS was co-incubated with the substrate and various concentrations of compounds followed by detection of fluorescent product generation. To evaluate the effect of ezetimibe or pranlukast on GALNS activity in cells, 1×105 cells/well were seeded in a 6-well plate and cultured for one day. The compound dilutions were added to the cells and incubated for 48 h followed by cell lysis using 1% sodium deoxycholate (Sigma-Aldrich). The enzyme activities of cell lysates were determined as described above. MPS IVA and WT fibroblasts treated with DMSO were used as controls. All experiments were performed in triplicate.
Western-blot.
Western-blot against GALNS, p62, and LC3B were performed on ezetimibe or pranlukast treated MPS IVA and WT fibroblasts using DMSO as a control. Cells were seeded in 6-well plates as described above. Equivalent amounts of cell lysates (20 μg) were loaded and run on NuPAGE™ 10% Bis-Tris Protein Gels (Invitrogen, Thermo Fisher Scientific) and transferred to PVDF membranes using iBlot™ Transfer Stack and iBlot 2 Dry Blotting System (Invitrogen, Thermo Fisher Scientific). Membranes were blocked with StartingBlock (Invitrogen, Thermo Fisher Scientific) at room temperature for 1 h. After blocking, the membranes were incubated overnight at 4 °C with the primary antibodies including the mouse monoclonal anti-P62 (SQSTM1(D-3) antibody in the blocking buffer (SC-28359, Santa Cruz Biotechnology), rabbit monoclonal anti-LC3B antibody (#3668S, Cell Signaling Technology), rabbit polyclonal anti-GALNS antibody (ab187516, Abcam), or mouse monoclonal anti-β-actin antibody (#3700S, Cell Signaling Technology). The membranes were then washed and incubated with a secondary antibody of goat anti-mouse IgG (#7076S) or goat anti-rabbit IgG (#7074P2, Cell Signaling Technology, Danvers, MA, USA) conjugated with horseradish peroxidase (HRP, 1:3000 dilution in blocking buffer). The specific protein bands were visualized using a chemiluminescence reagent (Luminata™ Western HRP Chemiluminescence Substrate, EMD Millipore, Burlington, MA, USA) and were subsequently analyzed by using NIH Image J software84.
Statistical analysis.
The results are shown as the mean ± the standard deviation (S.D.) and were analyzed by an ANOVA, followed by the Sidak t test when appropriate. Differences between groups were considered significant when p < 0.05 on GraphPad PRISM 7.0.
Supplementary Material
Acknowledgments.
CJAD was supported by Pontificia Universidad Javeriana [Grant ID 7204 and 7520] and COLCIENCIAS [Grant ID 5174 - contract No. 120356933205, and Grant ID 5170 - contract No. 120356933427]. ARL received a doctoral scholarship from Pontificia Universidad Javeriana. CJAD received a scholarship from Fulbright Colombia. KG, RL and WZ were supported by the Intramural Research Program of the National Center for Advancing Translational Sciences, National Institutes of Health, USA. Icons on the Table of Contents Graphic were obtained from Smartline and Pixel Perfect from www.flaticon.com. The authors declare that they have no competing interests. The authors thank Dr. DeeAnn Visk, a medical writer and editor, for editing the manuscript.
Abbreviations.
- MPS IVA
Mucopolysaccharidosis type IV A
- ERT
enzyme replacement therapy
- GALNS
N-acetylgalactosamine-6-sulfate sulfatase
- hrGALNS
human recombinant GALNS
- LSD
lysosomal storage disease
- GAGs
glycosaminoglycans
- KS
keratan sulfate
- C6S
chondroitin-6-sulfate
- HSCT
hematopoietic stem cell transplantation
- 4MUGPS
4-Methylumbelliferyl-β-D-galactopyranoside-6-sulfate
- hrGALNS
human recombinant GALNS
- WT
wild-type
- IDS
iduronate-2-sulfatase
- NPC1
Niemann-Pick type C1
- rhGAA
recombinant human acid α-glucosidase
Footnotes
Ancillary Information
- Top 20 hits of the compounds interacting with hrGALNS after virtual screening against the ZINC In Man subset from ZINC; Protein-ligand interactions between GALNS, KS, C6S, 4MUGPS, ezetimibe, and pranlukast; and Production of hrGALNS in P. pastoris at bioreactor scale (1.7 L).
- PDB-formatted coordinates of GALNS docked with KS, C6S, 4MUGPS, ezetimibe, and pranlukast. Authors will release the atomic coordinates and experimental data upon article publication.
- Molecular Formula Strings.
7. References
- [1].Sawamoto K; Alméciga-Díaz CJ; Mason RW; Orii T; Tomatsu S, Mucopolysaccharidosis Type IVA: Clinical Features, Biochemistry, Diagnosis, Genetics, and Treatment. In Mucopolysaccharidoses Update (2 Volume Set), Tomatsu S; Lavery C; Giugliani R; Harmatz P; Scarpa M; Węgrzyn G; Orii T, Eds. Nova Science Publishers, Inc.: Hauppauge, NY, 2018; Vol. I, pp 235–272. [Google Scholar]
- [2].Leadley RM; Lang S; Misso K; Bekkering T; Ross J; Akiyama T; Fietz M; Giugliani R; Hendriksz CJ; Hock NL; McGill J; Olaye A; Jain M; Kleijnen J, A systematic review of the prevalence of Morquio A syndrome: challenges for study reporting in rare diseases. Orphanet J Rare Dis 2014, 9, 173–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Khan S; Almeciga-Diaz CJ; Sawamoto K; Mackenzie WG; Theroux MC; Pizarro C; Mason RW; Orii T; Tomatsu S, Mucopolysaccharidosis IVA and glycosaminoglycans. Mol. Genet. Metab. 2017, 120 (1–2), 78–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Taylor M; Khan S; Stapleton M; Wang J; Chen J; Wynn R; Yabe H; Chinen Y; Boelens JJ; Mason RW; Kubaski F; Horovitz DDG; Barth AL; Serafini M; Bernardo ME; Kobayashi H; Orii KE; Suzuki Y; Orii T; Tomatsu S, Hematopoietic stem cell transplantation for mucopolysaccharidoses: Past, present, and future. Biol. Blood Marrow Transplant. 2019, DOI: 10.1016/j.bbmt.2019.1002.1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tomatsu S; Sawamoto K; Shimada T; Bober MB; Kubaski F; Yasuda E; Mason RW; Khan S; Alméciga-Díaz CJ; Barrera LA; Mackenzie WG; Orii T, Enzyme replacement therapy for treating mucopolysaccharidosis type IVA (Morquio A syndrome): effect and limitations. Expert Opin Orphan Drugs 2015, 3 (11), 1279–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hendriksz CJ; Burton B; Fleming TR; Harmatz P; Hughes D; Jones SA; Lin SP; Mengel E; Scarpa M; Valayannopoulos V; Giugliani R; Slasor P; Lounsbury D; Dummer W; Investigators S, Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): a phase 3 randomised placebo-controlled study. J Inherit Metab Dis 2014, 37 (6), 979–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hendriksz CJ; Giugliani R; Harmatz P; Mengel E; Guffon N; Valayannopoulos V; Parini R; Hughes D; Pastores GM; Lau HA; Al-Sayed MD; Raiman J; Yang K; Mealiffe M; Haller C, Multi-domain impact of elosufase alfa in Morquio A syndrome in the pivotal phase III trial. Mol Genet Metab 2015, 114 (2), 178–185. [DOI] [PubMed] [Google Scholar]
- [8].Doherty C; Stapleton M; Piechnik M; Mason RW; Mackenzie WG; Yamaguchi S; Kobayashi H; Suzuki Y; Tomatsu S, Effect of enzyme replacement therapy on the growth of patients with Morquio A. J. Hum. Genet. 2019, 64, 625–635. [DOI] [PubMed] [Google Scholar]
- [9].Schweighardt B; Tompkins T; Lau K; Jesaitis L; Qi Y; Musson DG; Farmer P; Haller C; Shaywitz AJ; Yang K; O'Neill CA, Immunogenicity of elosulfase alfa, an enzyme replacement therapy in patients with Morquio A syndrome: results from MOR-004, a phase III trial. Clin. Ther. 2015, 37 (5), 1012–1021. [DOI] [PubMed] [Google Scholar]
- [10].Chinen Y; Higa T; Tomatsu S; Suzuki Y; Orii T; Hyakuna N, Long-term therapeutic efficacy of allogenic bone marrow transplantation in a patient with mucopolysaccharidosis IVA. Mol Genet Metab Rep 2014, 1, 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yabe H; Tanaka A; Chinen Y; Kato S; Sawamoto K; Yasuda E; Shintaku H; Suzuki Y; Orii T; Tomatsu S, Hematopoietic stem cell transplantation for Morquio A syndrome. Mol. Genet. Metab. 2016, 117 (2), 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wang J; Luan Z; Jiang H; Fang J; Qin M; Lee V; Chen J, Allogeneic hematopoietic stem cell transplantation in thirty-four pediatric cases of mucopolysaccharidosis-A ten-year report from the China children transplant group. Biol. Blood Marrow Transplant. 2016, 22 (11), 2104–2108. [DOI] [PubMed] [Google Scholar]
- [13].Masue M; Sukegawa K; Orii T; Hashimoto T, N-acetylgalactosamine-6-sulfate sulfatase in human placenta: purification and characteristics. J. Biochem. (Tokyo). 1991, 110 (6), 965–970. [DOI] [PubMed] [Google Scholar]
- [14].Rivera-Colon Y; Schutsky EK; Kita AZ; Garman SC, The structure of human GALNS reveals the molecular basis for mucopolysaccharidosis IV A. J. Mol. Biol. 2012, 423 (5), 736–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Stenson PD; Mort M; Ball EV; Evans K; Hayden M; Heywood S; Hussain M; Phillips AD; Cooper DN, The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136 (6), 665–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tomatsu S; Montaño A; Nishioka T; Gutierrez M; Peña O; Trandafirescu G; Lopez P; Yamaguchi S; Noguchi A; Orri T, Mutation and polymorphism spectrum of the GALNS gene in mucopolysaccharidosis IVA (Morquio A). Hum Mutat 2005, 26, 500–512. [DOI] [PubMed] [Google Scholar]
- [17].Sukegawa K; Nakamura H; Kato Z; Tomatsu S; Montaño AM; Fukao T; Toietta G; Tortora P; Orii T; Kondo N, Biochemical and structural analysis of missense mutations in N-acetylgalactosamine-6-sulfate sulfatase causing mucopolysaccharidosis IVA phenotypes. Hum Mol Genet 2000, 9 (9), 1283–1290. [DOI] [PubMed] [Google Scholar]
- [18].Montaño AM; Sukegawa K; Kato Z; Carrozzo R; Di Natale P; Christensen E; Orii KO; Orii T; Kondo N; Tomatsu S, Effect of 'attenuated' mutations in mucopolysaccharidosis IVA on molecular phenotypes of N-acetylgalactosamine-6-sulfate sulfatase. J Inherit Metab Dis 2007, 30 (5), 758–767. [DOI] [PubMed] [Google Scholar]
- [19].Sudhakar SC; Mahalingam K, Structural and functional analysis of N-acetylgalactosamine-6-sulfate sulfatase using bioinformatics tools: Insight into mucopolysaccharidosis IVA. J Pharm Res 2011, 4 (11), 3958–3962. [Google Scholar]
- [20].Olarte-Avellaneda S; Rodríguez-López A; Alméciga-Díaz CJ; Barrera LA, Computational analysis of human N-acetylgalactosamine-6-sulfate sulfatase enzyme: an update in genotype-phenotype correlation for Morquio A. Mol Biol Rep 2014, 41 (11), 7073–7088. [DOI] [PubMed] [Google Scholar]
- [21].Leidenheimer NJ, Pharmacological chaperones: Beyond conformational disorders. Handb Exp Pharmacol 2018, 245, 135–153. [DOI] [PubMed] [Google Scholar]
- [22].Tao YX; Conn PM, Pharmacoperones as novel therapeutics for diverse protein conformational diseases. Physiol. Rev. 2018, 98 (2), 697–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Parenti G; Moracci M; Fecarotta S; Andria G, Pharmacological chaperone therapy for lysosomal storage diseases. Future Med Chem 2014, 6 (9), 1031–1045. [DOI] [PubMed] [Google Scholar]
- [24].Valenzano KJ; Khanna R; Powe AC; Boyd R; Lee G; Flanagan JJ; Benjamin ER, Identification and characterization of pharmacological chaperones to correct enzyme deficiencies in lysosomal storage disorders. Assay Drug Dev. Technol. 2011, 9 (3), 213–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Pereira DM; Valentao P; Andrade PB, Tuning protein folding in lysosomal storage diseases: the chemistry behind pharmacological chaperones. Chem Sci 2018, 9 (7), 1740–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hughes DA; Nicholls K; Shankar SP; Sunder-Plassmann G; Koeller D; Nedd K; Vockley G; Hamazaki T; Lachmann R; Ohashi T; Olivotto I; Sakai N; Deegan P; Dimmock D; Eyskens F; Germain DP; Goker-Alpan O; Hachulla E; Jovanovic A; Lourenco CM; Narita I; Thomas M; Wilcox WR; Bichet DG; Schiffmann R; Ludington E; Viereck C; Kirk J; Yu J; Johnson F; Boudes P; Benjamin ER; Lockhart DJ; Barlow C; Skuban N; Castelli JP; Barth J; Feldt-Rasmussen U, Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J. Med. Genet. 2017, 54 (4), 288–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Parenti G; Andria G; Valenzano KJ, Pharmacological chaperone therapy: preclinical development, clinical translation, and prospects for the treatment of lysosomal storage disorders. Mol. Ther. 2015, 23 (7), 1138–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Boyd R; Lee G; Rybczynski P; Benjamin ER; Khanna R; Wustman B; Valenzano K, Pharmacological chaperones as therapeutics for lysosomal storage diseases. J. Med. Chem. 2013, 56 (7), 2705–2725. [DOI] [PubMed] [Google Scholar]
- [29].Hoshina H; Shimada Y; Higuchi T; Kobayashi H; Ida H; Ohashi T, Chaperone effect of sulfated disaccharide from heparin on mutant iduronate-2-sulfatase in mucopolysaccharidosis type II. Mol. Genet. Metab. 2017, 123 (2), 118–122. [DOI] [PubMed] [Google Scholar]
- [30].Front S; Biela-Banas A; Burda P; Ballhausen D; Higaki K; Caciotti A; Morrone A; Charollais-Thoenig J; Gallienne E; Demotz S; Martin OR, (5aR)-5a-C-Pentyl-4-epi-isofagomine: A powerful inhibitor of lysosomal beta-galactosidase and a remarkable chaperone for mutations associated with GM1-gangliosidosis and Morquio disease type B. Eur J Med Chem 2017, 126, 160–170. [DOI] [PubMed] [Google Scholar]
- [31].Olarte-Avellaneda S; Rodríguez-López A; Alméciga-Díaz C, In-silico Analysis of the Active Cavity of N-acetylgalactosamine-6-sulfate Sulfatase in Eight Species. In Advances in Computational Biology, Castillo LF; Cristancho M; Isaza G; Pinzón A; Corchado JM, Eds. Springer International Publishing: 2014; pp 141–146. [Google Scholar]
- [32].Rodríguez-López A; Alméciga-Díaz CJ; Sánchez J; Moreno J; Beltran L; Díaz D; Pardo A; Ramírez AM; Espejo-Mojica AJ; Pimentel L; Barrera LA, Recombinant human N-acetylgalactosamine-6-sulfate sulfatase (GALNS) produced in the methylotrophic yeast Pichia pastoris. Sci Rep 2016, 6, 29329–29342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Rodriguez-Lopez A; Pimentel-Vera LN; Espejo-Mojica AJ; Van HA; Tiels P; Tomatsu S; Callewaert N; Almeciga-Diaz CJ, Characterization of human recombinant N-acetylgalactosamine-6-sulfate sulfatase produced in Pichia pastoris as potential enzyme for mucopolysaccharidosis IVA treatment. J. Pharm. Sci. 2019, DOI: 10.1016/j.xphs.2019.1003.1034. [DOI] [PubMed] [Google Scholar]
- [34].Delic M; Gongrich R; Mattanovich D; Gasser B, Engineering of protein folding and secretion-strategies to overcome bottlenecks for efficient production of recombinant proteins. Antioxid Redox Signal 2014, 21 (3), 414–437. [DOI] [PubMed] [Google Scholar]
- [35].Gasser B; Prielhofer R; Marx H; Maurer M; Nocon J; Steiger M; Puxbaum V; Sauer M; Mattanovich D, Pichia pastoris: protein production host and model organism for biomedical research. Future Microbiol. 2013, 8 (2), 191–208. [DOI] [PubMed] [Google Scholar]
- [36].Reyes LH; Cardona C; Pimentel L; Rodriguez-Lopez A; Almeciga-Diaz CJ, Improvement in the production of the human recombinant enzyme N-acetylgalactosamine-6-sulfatase (rhGALNS) in Escherichia coli using synthetic biology approaches. Sci Rep 2017, 7 (1), 5844–5858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mosquera A; Rodríguez A; Soto C; Leonardi F; Espejo A; Sánchez OF; Alméciga-Díaz CJ; Barrera LA, Characterization of a recombinant N-acetylgalactosamine-6-sulfate sulfatase produced in E. coli for enzyme replacement therapy of Morquio A disease. Process Biochem. 2012, 47, 2097–2102. [Google Scholar]
- [38].Hernandez A; Velasquez O; Leonardi F; Soto C; Rodriguez A; Lizaraso L; Mosquera A; Bohorquez J; Coronado A; Espejo A; Sierra R; Sanchez OF; Almeciga-Diaz CJ; Barrera LA, Effect of culture conditions and signal peptide on production of human recombinant N-acetylgalactosamine-6-sulfate sulfatase in Escherichia coli BL21. J. Microbiol. Biotechnol. 2013, 23 (5), 689–698. [DOI] [PubMed] [Google Scholar]
- [39].Tomatsu S; Montaño A; Gutiérrez M; Grubb J; Oikawa H; Dung V; Ohashi A; Nishioka T; Yamada N; Tosaka Y; Trandafirescu G; Orii T, Characterization and pharmacokinetic study of recombinant human N-acetylgalactosamine-6-sulfate sulfatase. Mol Genet Metab 2007, 91 (1), 69–78. [DOI] [PubMed] [Google Scholar]
- [40].Xu M; Liu K; Swaroop M; Sun W; Dehdashti SJ; McKew JC; Zheng W, A phenotypic compound screening assay for lysosomal storage diseases. J. Biomol. Screen. 2014, 19 (1), 168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lloyd-Evans E; Morgan AJ; He X; Smith DA; Elliot-Smith E; Sillence DJ; Churchill GC; Schuchman EH; Galione A; Platt FM, Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 2008, 14 (11), 1247–1255. [DOI] [PubMed] [Google Scholar]
- [42].Marugan J; Huang W; Motabar O; Zheng W; Xiao J; Patnaik S; Southall N; Westbroek W; Lea WA; Simeonov A; Goldin E; Debernardi M; Sidransky E, Non-iminosugar glucocerebrosidase small molecule chaperones. Med Chem Comm 2012, 3 (1), 56–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zheng W; Padia J; Urban DJ; Jadhav A; Goker-Alpan O; Simeonov A; Goldin E; Auld D; LaMarca ME; Inglese J; Austin CP; Sidransky E, Three classes of glucocerebrosidase inhibitors identified by quantitative high-throughput screening are chaperone leads for Gaucher disease. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (32), 13192–13197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Berardi A; Pannuzzo G; Graziano A; Costantino-Ceccarini E; Piomboni P; Luddi A, Pharmacological chaperones increase residual beta-galactocerebrosidase activity in fibroblasts from Krabbe patients. Mol. Genet. Metab. 2014, 112 (4), 294–301. [DOI] [PubMed] [Google Scholar]
- [45].Yilmazer B; Yagci ZB; Bakar E; Ozden B; Ulgen K; Ozkirimli E, Investigation of novel pharmacological chaperones for Gaucher disease. J. Mol. Graph. Model. 2017, 76, 364–378. [DOI] [PubMed] [Google Scholar]
- [46].Phan BA; Dayspring TD; Toth PP, Ezetimibe therapy: mechanism of action and clinical update. Vasc Health Risk Manag 2012, 8, 415–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Keam SJ; Lyseng-Williamson KA; Goa KL, Pranlukast: a review of its use in the management of asthma. Drugs 2003, 63 (10), 991–1019. [DOI] [PubMed] [Google Scholar]
- [48].Schierle S; Schmidt J; Kaiser A; Merk D, Selective optimization of pranlukast to farnesoid X receptor modulators. ChemMedChem 2018, 13 (23), 2530–2545. [DOI] [PubMed] [Google Scholar]
- [49].Banning A; Gulec C; Rouvinen J; Gray SJ; Tikkanen R, Identification of small molecule compounds for pharmacological chaperone therapy of aspartylglucosaminuria. Sci Rep 2016, 6, 37583–37595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ben B; Kallemeijn W; Oussoren S; Scheij S; Bleijlevens B; Florea B; van Roomen C; Ottenhoff R; van Kooten M; Walvoort M; Witte M; Boot R; Ubbink M; Overkleeft H; Aerts J, Stabilization of glucocerebrosidase by active site occupancy. ACS Chem Biol. 2017, 12 (7), 1830–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Demain AL; Vaishnav P, Production of recombinant proteins by microbes and higher organisms. Biotechnol Adv 2009, 27 (3), 297–306. [DOI] [PubMed] [Google Scholar]
- [52].Maas C; Hermeling S; Bouma B; Jiskoot W; Gebbink MF, A role for protein misfolding in immunogenicity of biopharmaceuticals. J. Biol. Chem. 2007, 282 (4), 2229–2236. [DOI] [PubMed] [Google Scholar]
- [53].Tomatsu S; Dieter T; Schwartz IV; Sarmient P; Giugliani R; Barrera LA; Guelbert N; Kremer R; Repetto GM; Gutierrez MA; Nishioka T; Serrato OP; Montaño AM; Yamaguchi S; Noguchi A, Identification of a common mutation in mucopolysaccharidosis IVA: correlation among genotype, phenotype, and keratan sulfate. J. Hum. Genet. 2004, 49 (9), 490–494. [DOI] [PubMed] [Google Scholar]
- [54].Morrone A; Tylee KL; Al-Sayed M; Brusius-Facchin AC; Caciotti A; Church HJ; Coll MJ; Davidson K; Fietz MJ; Gort L; Hegde M; Kubaski F; Lacerda L; Laranjeira F; Leistner-Segal S; Mooney S; Pajares S; Pollard L; Ribeiro I; Wang RY; Miller N, Molecular testing of 163 patients with Morquio A (Mucopolysaccharidosis IVA) identifies 39 novel GALNS mutations. Mol. Genet. Metab. 2014, 112 (2), 160–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Tomatsu S; Fukuda S; Cooper A; Wraith JE; Ferreira P; Di NP; Tortora P; Fujimoto A; Kato Z; Yamada N; Isogai K; Yamagishi A; Sukegawa K; Suzuki Y; Shimozawa N; Kondo N; Sly WS; Orii T, Fourteen novel mucopolysaccharidosis IVA producing mutations in GALNS gene. Hum. Mutat. 1997, 10 (5), 368–375. [DOI] [PubMed] [Google Scholar]
- [56].Castilla J; Risquez R; Higaki K; Nanba E; Ohno K; Suzuki Y; Diaz Y; Ortiz MC; Garcia FJM; Castillon S, Conformationally-locked N-glycosides: exploiting long-range non-glycone interactions in the design of pharmacological chaperones for Gaucher disease. Eur J Med Chem 2015, 90, 258–266. [DOI] [PubMed] [Google Scholar]
- [57].Laigre E; Hazelard D; Casas J; Serra-Vinardell J; Michelakakis H; Mavridou I; Aerts JM; Delgado A; Compain P, Investigation of original multivalent iminosugars as pharmacological chaperones for the treatment of Gaucher disease. Carbohydr. Res. 2016, 429, 98–104. [DOI] [PubMed] [Google Scholar]
- [58].Gouveia VM; Lima SA; Nunes C; Reis S, Non-Biologic nanodelivery therapies for rheumatoid arthritis. J Biomed Nanotechnol 2015, 11 (10), 1701–1721. [DOI] [PubMed] [Google Scholar]
- [59].Cicero AFG; Bove M; Borghi C, Pharmacokinetics, pharmacodynamics and clinical efficacy of non-statin treatments for hypercholesterolemia. Expert Opin Drug Metab Toxicol 2018, 14 (1), 9–15. [DOI] [PubMed] [Google Scholar]
- [60].Qi Y; Musson DG; Schweighardt B; Tompkins T; Jesaitis L; Shaywitz AJ; Yang K; O'Neill CA, Pharmacokinetic and pharmacodynamic evaluation of elosulfase alfa, an enzyme replacement therapy in patients with Morquio A syndrome. Clin. Pharmacokinet. 2014, 53 (12), 1137–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Stapleton M; Sawamoto K; Alméciga-Díaz CJ; Mackenzie WG; Mason RW; Orii T; Tomatsu S, Development of bone targeting drugs. Int J Mol Sci 2017, 18 (7), 1345–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Madni A; Rahem MA; Tahir N; Sarfraz M; Jabar A; Rehman M; Kashif PM; Badshah SF; Khan KU; Santos HA, Non-invasive strategies for targeting the posterior segment of eye. Int. J. Pharm. 2017, 530 (1–2), 326–345. [DOI] [PubMed] [Google Scholar]
- [63].Seranova E; Connolly KJ; Zatyka M; Rosenstock TR; Barrett T; Tuxworth RI; Sarkar S, Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem. 2017, 61 (6), 733–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Pacheco CD; Elrick MJ; Lieberman AP, Tau deletion exacerbates the phenotype of Niemann-Pick type C mice and implicates autophagy in pathogenesis. Hum. Mol. Genet. 2009, 18 (5), 956–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Vergarajauregui S; Connelly PS; Daniels MP; Puertollano R, Autophagic dysfunction in mucolipidosis type IV patients. Hum. Mol. Genet. 2008, 17 (17), 2723–2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Woloszynek JC; Kovacs A; Ohlemiller KK; Roberts M; Sands MS, Metabolic adaptations to interrupted glycosaminoglycan recycling. J Biol Chem 2009, 284 (43), 29684–29691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Pshezhetsky AV, Lysosomal storage of heparan sulfate causes mitochondrial defects, altered autophagy, and neuronal death in the mouse model of mucopolysaccharidosis III type C. Autophagy 2016, 12 (6), 1059–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kim SH; Kim G; Han DH; Lee M; Kim I; Kim B; Kim KH; Song YM; Yoo JE; Wang HJ; Bae SH; Lee YH; Lee BW; Kang ES; Cha BS; Lee MS, Ezetimibe ameliorates steatohepatitis via AMP activated protein kinase-TFEB-mediated activation of autophagy and NLRP3 inflammasome inhibition. Autophagy 2017, 13 (10), 1767–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Khanna R; Flanagan JJ; Feng J; Soska R; Frascella M; Pellegrino LJ; Lun Y; Guillen D; Lockhart DJ; Valenzano KJ, The pharmacological chaperone AT2220 increases recombinant human acid alpha-glucosidase uptake and glycogen reduction in a mouse model of Pompe disease. PloS one 2012, 7 (7), e40776–40718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kishnani P; Tarnopolsky M; Roberts M; Sivakumar K; Dasouki M; Dimachkie MM; Finanger E; Goker-Alpan O; Guter KA; Mozaffar T; Pervaiz MA; Laforet P; Levine T; Adera M; Lazauskas R; Sitaraman S; Khanna R; Benjamin E; Feng J; Flanagan J; Barth J; Barlow C; Lockhart D; Valenzano K; Boudes P; Johnson F; Byrne B, Duvoglustat HCl increases systemic and tissue exposure of active acid alpha-glucosidase in Pompe patients co-administered with alglucosidase alpha. Mol. Ther. 2017, 25 (5), 1199–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Tomatsu S; Montaño AM; Dung V; Ohashi A; Oikawa H; Oguma T; Orii T; Barrera L; Sly W, Enhacement of drug delivery: Enzyme-replacement therapy for murine Morquio A syndrome. Mol. Ther. 2010, 18 (6), 1094–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Trott O; Olson AJ, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31 (2), 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Irwin J; Sterling T; Mysinger M; Bolstad E; Coleman R, ZINC: a free tool to discover chemistry for biology. J Chem Inf Model 2012, 52 (7), 1757–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].van Diggelen OP; Zhao H; Kleijer WJ; Janse HC; Poorthuis BJ; van Pelt J; Kamerling JP; Galjaard H, A fluorimetric enzyme assay for the diagnosis of Morquio disease type A (MPS IV A). Clin Chim Acta 1990, 187 (2), 131–139. [DOI] [PubMed] [Google Scholar]
- [75].Pronk S; Pall S; Schulz R; Larsson P; Bjelkmar P; Apostolov R; Shirts MR; Smith JC; Kasson PM; van der Spoel D; Hess B; Lindahl E, GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29 (7), 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Koziara KB; Stroet M; Malde AK; Mark AE, Testing and validation of the Automated Topology Builder (ATB) version 2.0: prediction of hydration free enthalpies. J. Comput. Aided Mol. Des. 2014, 28 (3), 221–233. [DOI] [PubMed] [Google Scholar]
- [77].Rodriguez A; Espejo AJ; Hernandez A; Velasquez OL; Lizaraso LM; Cordoba HA; Sanchez OF; Almeciga-Diaz CJ; Barrera LA, Enzyme replacement therapy for Morquio A: an active recombinant N-acetylgalactosamine-6-sulfate sulfatase produced in Escherichia coli BL21. J. Ind. Microbiol. Biotechnol. 2010, 37 (11), 1193–1201. [DOI] [PubMed] [Google Scholar]
- [78].Casper J; Zweig A; Villarreal C; Tyner C; Speir ML; Rosenbloom K; Raney B; Lee C; Lee B; Karolchik D; Hinrichs A; Haeussler M; Guruvadoo L; Navarro GJ; Gibson D; Fiddes I; Eisenhart C; Diekhans M; Clawson H; Barber G; Armstrong J; Haussler D; Kuhn R; Kent W, The UCSC Genome Browser database: 2018 update. Nucleic Acids Res. 2018, 46 (D1), D762–D769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Adzhubei IA; Schmidt S; Peshkin L; Ramensky VE; Gerasimova A; Bork P; Kondrashov AS; Sunyaev SR, A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7 (4), 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Choi Y; Sims GE; Murphy S; Miller JR; Chan AP, Predicting the functional effect of amino acid substitutions and indels. PloS one 2012, 7 (10), e46688–46621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE, UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25 (13), 1605–1612. [DOI] [PubMed] [Google Scholar]
- [82].Yang J; Yan R; Roy A; Xu D; Poisson J; Zhang Y, The I-TASSER Suite: protein structure and function prediction. Nat. Methods 2015, 12 (1), 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Krieger E; Vriend G, YASARA View - molecular graphics for all devices - from smartphones to workstations. Bioinformatics 2014, 30 (20), 2981–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Schneider CA; Rasband WS; Eliceiri KW, NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9 (7), 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Huynh K; Partch CL, Analysis of protein stability and ligand interactions by thermal shift assay. Curr Protoc Protein Sci 2015, 79, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Ullal AJ; Millington DS; Bali DS, Development of a fluorometric microtiter plate based enzyme assay for MPS IVA (Morquio type A) using dried blood spots. Mol Genet Metab Rep 2014, 1, 461–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.