

Abstract
Idiopathic pulmonary fibrosis (IPF) is an aging-associated interstitial lung disease resulting from repeated epithelial injury and inadequate epithelial repair. Alveolar type II cells (AEC2s) are progenitor cells that maintain epithelial homeostasis and repair the lung after injury. In the current study, we assessed lipid metabolism in AEC2s from human lungs of patients with IPF and healthy donors, as well as AEC2s from bleomycin-injured young and old mice. Through single-cell RNA sequencing, we observed that lipid metabolism–related genes were downregulated in IPF AEC2s and bleomycin-injured mouse AEC2s. Aging aggravated this decrease and hindered recovery of lipid metabolism gene expression in AEC2s after bleomycin injury. Pathway analyses revealed downregulation of genes related to lipid biosynthesis and fatty acid β-oxidation in AEC2s from IPF lungs and bleomycin-injured, old mouse lungs compared with the respective controls. We confirmed decreased cellular lipid content in AEC2s from IPF lungs and bleomycin-injured, old mouse lungs using immunofluorescence staining and flow cytometry. Futhermore, we show that lipid metabolism was associated with AEC2 progenitor function. Lipid supplementation and PPARγ (peroxisome proliferator activated receptor γ) activation promoted progenitor renewal capacity of both human and mouse AEC2s in three-dimensional organoid cultures. Lipid supplementation also increased AEC2 proliferation and expression of SFTPC in AEC2s. In summary, we identified a lipid metabolism deficiency in AEC2s from lungs of patients with IPF and bleomycin-injured old mice. Restoration of lipid metabolism homeostasis in AEC2s might promote AEC2 progenitor function and offer new opportunities for therapeutic approaches to IPF.
Keywords: aging, alveolar progenitor cells, idiopathic pulmonary fibrosis, lipid metabolism, three-dimensional organoid culture
Idiopathic pulmonary fibrosis (IPF) is an aging-related, fatal form of interstitial lung disease. IPF results from sustained epithelial injury and inadequate alveolar epithelial repair that leads to excessive fibroblast activation and distortion of the normal lung architecture (1–5). Type 2 alveolar epithelial cells (AEC2s) function as progenitor cells that maintain epithelial homeostasis and repair damaged epithelium after lung injury (3, 6–9). AEC2 progenitor cell exhaustion and decreased renewal capacity of AEC2s in IPF lungs have been recognized as a causal event for the disease (3, 10).
Aging is a critical risk factor in IPF (11–13). The incidence, prevalence, and mortality of IPF increase with age (14, 15). We know that aging delays lung repair (16). Although phenotypes of cellular aging in AEC2s have been well described in IPF (17–21), the mechanisms of AEC2 aging are less clear. We recently reported that synergistic effects of aging and AEC2 injury promote lung fibrosis (22).
Lipid metabolism becomes dysfunctional with aging (23–25). Furthermore, dysregulated lung lipid metabolism is involved in multiple pulmonary diseases, including IPF (5, 26–29). PPARγ (peroxisome proliferator activated receptor γ) plays a role in mediating myofibroblast differentiation (30). A PPARγ ligand inhibited TGF-β–stimulated differentiation of human lung fibroblasts to myofibroblasts (31) and bleomycin-induced lung inflammation and fibrosis (32). However, the demonstration of the lipid metabolism in AEC2 progenitor function is scarce. AEC2s are the most active lipid metabolic cells in the lung, with tightly regulated surfactant biosynthesis and recycling (26, 33, 34). Altered lipid metabolism and disrupted surfactant homeostasis have been reported in lung fibrosis (24, 26, 27, 33, 35, 36). Single-cell RNA sequencing (scRNA-seq) studies showed downregulation of multiple lipid metabolic pathways in whole lung and AEC2s from patients with IPF (37). Inhibition of the lipid synthesis enzyme SCD1 (stearoyl-Coenzyme A desaturase 1) induces endoplasmic reticulum stress in AEC2s and promotes fibrotic responses to injury (38). Elovl6 (elongation of long-chain fatty acids family member 6) was found to be downregulated after bleomycin lung injury and in IPF lungs (39). Mice deficient in Elovl6 exhibited a severe fibroproliferative response and derangement of fatty acid profiles after bleomycin injury compared with wild-type mice (39). Targeted deletion of the lipid synthesis enzyme Fasn in mouse AEC2s exacerbated bleomycin-induced lung fibrosis (40). These studies highlighted the crucial role of lipid metabolism in lung fibrosis. Lipid metabolic dysregulation has been recognized as a new player contributing to lung fibrosis. Therefore, we undertook a comprehensive analysis of lipid metabolism in AEC2s in the context of both aging and fibrotic lung injury.
In the current study, we performed scRNA-seq of flow cytometry–enriched lung epithelial cells and systematically investigated lipid metabolism in AEC2s from IPF and healthy donor lungs, as well as AEC2s from young and old mouse lungs under homeostasis and after experimental fibrotic lung injury. We identified downregulated expression of lipid metabolism–related genes in AEC2s from lungs of patients with IPF and from bleomycin-injured old mouse lungs. Aging aggravated this decrease and hindered recovery of lipid metabolism–related gene expression after bleomycin treatment. We confirmed decreased cellular lipid in AEC2s from bleomycin-injured old mouse lungs and IPF lungs with immunofluorescence staining and flow cytometry. Most importantly, our data showed that impaired lipid metabolism was associated with AEC2 progenitor cell failure. Lipid supplementation and PPARγ activation promoted progenitor renewal capacity of both human and mouse AEC2s in three-dimensional (3D) organoid cultures, whereas inhibition of lipid metabolism by a PPARγ antagonist suppressed AEC2 renewal.
Methods
See the data supplement for detailed materials and methods.
Study Approvals
The use of human tissues for research was approved by the Institutional Review Board (IRB) of Cedars-Sinai Medical Center and was in accordance with the guidelines outlined by the IRB (IRB number: Pro00032727). Informed consent was obtained from each subject. Mouse experiments were conducted under the guidance of the Cedars-Sinai Medical Center Institutional Animal Care and Use Committee (IACUC008529) in accordance with institutional and regulatory guidelines.
Mouse Lung Dissociation and Flow Cytometry
Ten- to 12-week-old young mice and 20- to 24-month-old mice were used for experiments. Mouse lung tissue dissociation and single-cell isolation were described previously (3, 10). Mouse AEC2s were gated as EpCAM+CD31−CD34−CD45−CD24− Sca-1− population for flow sorting. Detailed methods can be found in the data supplement.
Human Lung Dissociation and Flow Cytometry
Human lung dissociation and single-cell isolation were performed as described previously (3, 41). Human AEC2s were gated as EpCAM+HTII-280+CD31−CD45− cells in flow cytometry analysis and AEC2 sorting. Staining of PLIN2 (perilipin 2) and lipids in gated AEC2s were analyzed with FlowJo software. Detailed methods can be found in the data supplement.
Immunofluorescence of Human and Mouse Lung Sections
Cryosections and immunostaining followed standard protocols. Human lung sections were stained with mouse anti–HTII-280 IgM monoclonal antibody, and mouse lung sections were stained with rabbit anti-proSPC (catalog no. AB3786, Sigma-Aldrich) for AEC2s followed by secondary antibody. Lipid droplets were stained with LipidSpot (Biotium catalog no. 70069).
scRNA-Seq Data Analysis
The datasets of scRNA-seq of mouse and human epithelial cells are deposited under GSE157995 and GSE157996, respectively, from our laboratory (22, 41). Data analysis was performed as previously described (10, 22, 41, 42). Other datasets were additionally used: GSE122960 (37), GSE132771 (43), GSE132915 (19), GSE135893 (44), and GSE128033 (45).
3D Matrigel Cultures of Human and Mouse AEC2s
Flow-sorted human or mouse AEC2s (3 × 103) were cultured in a Matrigel/medium (1:1) mixture in the presence of lung fibroblast MLg2908 cells (2 × 105, catalog CCL-206, ATCC) as we described previously (3, 6, 46). The cells were cultured with 2% or 4% (vol/vol) chemically defined lipid mixture 1 (catalog no. L0288) and medium control or cultured with 10 μM rosiglitazone (RGZ) and 10 μM GW9662. The same volume of DMSO was used as control for RGZ and GW9662 treatment.
Statistics
The statistical difference between groups in the bioinformatics analysis was calculated using the Wilcoxon signed-rank test. For the scRNA-seq data, the lowest P values calculated in Seurat were P < 2.2 × 10−16. For cell treatment data, the statistical difference between groups was calculated using Prism (version 9.5.0, GraphPad). Data are expressed as the mean ± SEM. Differences in measured variables between experimental and control groups were assessed by using unpaired two-sided Student’s t tests. One-way ANOVA followed by Bonferroni’s multiple comparison test was used for multiple comparisons. Results were considered statistically significant at P < 0.05.
Results
Downregulated Lipid Metabolism Gene Expression in IPF AEC2s
To gain insights into lipid metabolism in AEC2s from IPF lungs, we analyzed lipid metabolism–related gene expression in AEC2s in our scRNA-seq dataset (GSE157996) of freshly isolated flow cytometry–enriched epithelial cells (Lin−EPCAM+ cells) from six healthy donor and six IPF lungs (10, 41). IPF AEC2s showed dysregulated lipid biosynthesis and metabolism, as evidenced by lower activation scores of fatty acid biosynthesis, phosphatidylcholine biosynthesis, and surfactant homeostasis (Figure 1A). Multiple lipid biosynthesis and metabolism related genes, including CHKA, SCD, FASN, CAT, ACOXL, ELOVL6, LPCAT1, and LPCAT3, were downregulated in IPF AEC2s compared with healthy AEC2s (Figure 1B). At the same time, the expression of genes for fatty acid β-oxidation enzymes, including ACAT1 and multiple ACSL genes, was all downregulated in IPF AEC2s (Figure 1C). There were fewer CPT1B- and CPT1C-expressing cells in IPF AEC2s compared with healthy AEC2s (see Figure E1A in the data supplement). IPF AEC2s showed a lower fatty acid β-oxidation score relative to healthy AEC2s (Figure 1A). The expression levels of the genes for lipid droplet, including PLIN2 (also known as adipose differentiation-related protein, ADRP) (Figure 1C), MGLL, and HILPDA (Figure E1B), were lower in IPF AEC2s. The expression of lipid uptake–related genes, including CD36, SLC27A4, and SLC27A5, were also significantly decreased in IPF AEC2s compared with healthy AEC2s (Figure 1C).
Figure 1.

Dysregulated lipid metabolism of idiopathic pulmonary fibrosis (IPF) alveolar type II cells (AEC2s). (A) Activation scores of fatty acid biosynthesis (FAB), phosphatidylcholine biosynthesis (PCB), β-oxidation (BO), and surfactant homeostasis (SH) of AEC2s from healthy and IPF lungs (red, healthy; blue, IPF) analyzed from dataset GSE157996. (B and C) Violin plots of expression of lipid metabolism–related genes (B) and fatty acid β-oxidation, lipid droplet, and lipid uptake–related genes (C) in healthy and IPF AEC2s (red, healthy; blue, IPF). (D) Violin plots of expression of FASN and PLIN2 (perilipin 2) with published single-cell RNA sequencing (scRNA-seq) datasets. (E) IPA pathway analysis of human AEC2s from IPF versus healthy lungs analyzed with dataset GSE157996. PPARγ = peroxisome proliferator activated receptor γ. IPA = ingenuity pathway analysis.
To confirm that our scRNA-seq dataset aligned with the scRNA-seq datasets generated by other studies, we analyzed publicly available scRNA-seq datasets GSE122960 (37), GSE132771 (43), GSE132915 (19), GSE135893 (44), and GSE128033 (45). We found that the same group of lipid metabolism–related genes downregulated in IPF AEC2s in our dataset were also downregulated in IPF AEC2s compared with those of healthy AEC2s in all of the datasets analyzed. Here, we listed the expression of FASN and PLIN2 as examples that are significantly downregulated in IPF AEC2s compared with those in healthy AEC2s (Figure 1D).
Pathway analysis showed downregulation of lipid biosynthesis pathways including PPAR signaling, PPARα/RXRα activation, fatty acid β-oxidation I and fatty acid activation, cholesterol biosynthesis pathways, and superpathway of cholesterol biosynthesis in IPF AEC2s, whereas triacylglycerol biosynthesis was upregulated in IPF AEC2s (Figure 1E). We further verified that the cholesterol biosynthesis genes including SQLE, DHCH7, FDFT1, and DHCR24 were downregulated in IPF AEC2s, whereas some triacylglycerol biosynthesis-related genes, including MGAT2, MGAT5, TMX1, and TMEM68, were with higher expression levels in IPF AEC2s relative to healthy AEC2s (Figures E1C and E1D). These data suggest dysregulated lipid metabolism in IPF AEC2s.
Decreased Cellular Lipid Levels in AEC2s from IPF Lungs
Next, we performed immunofluorescence costaining of lipid droplets and the human AEC2 marker HTII-280 with lung sections from IPF explants and healthy donors. We showed that in healthy lungs, the majority of HTII-280+ AEC2s contain high levels of lipid, whereas the lipid levels in AEC2s of IPF lung sections were much lower (Figure 2A). Some bright lipid staining appeared in IPF lung sections, but they were not colocalized with the AEC2 marker HTII-280 (Figure 2A), suggesting it might be in other cell types.
Figure 2.
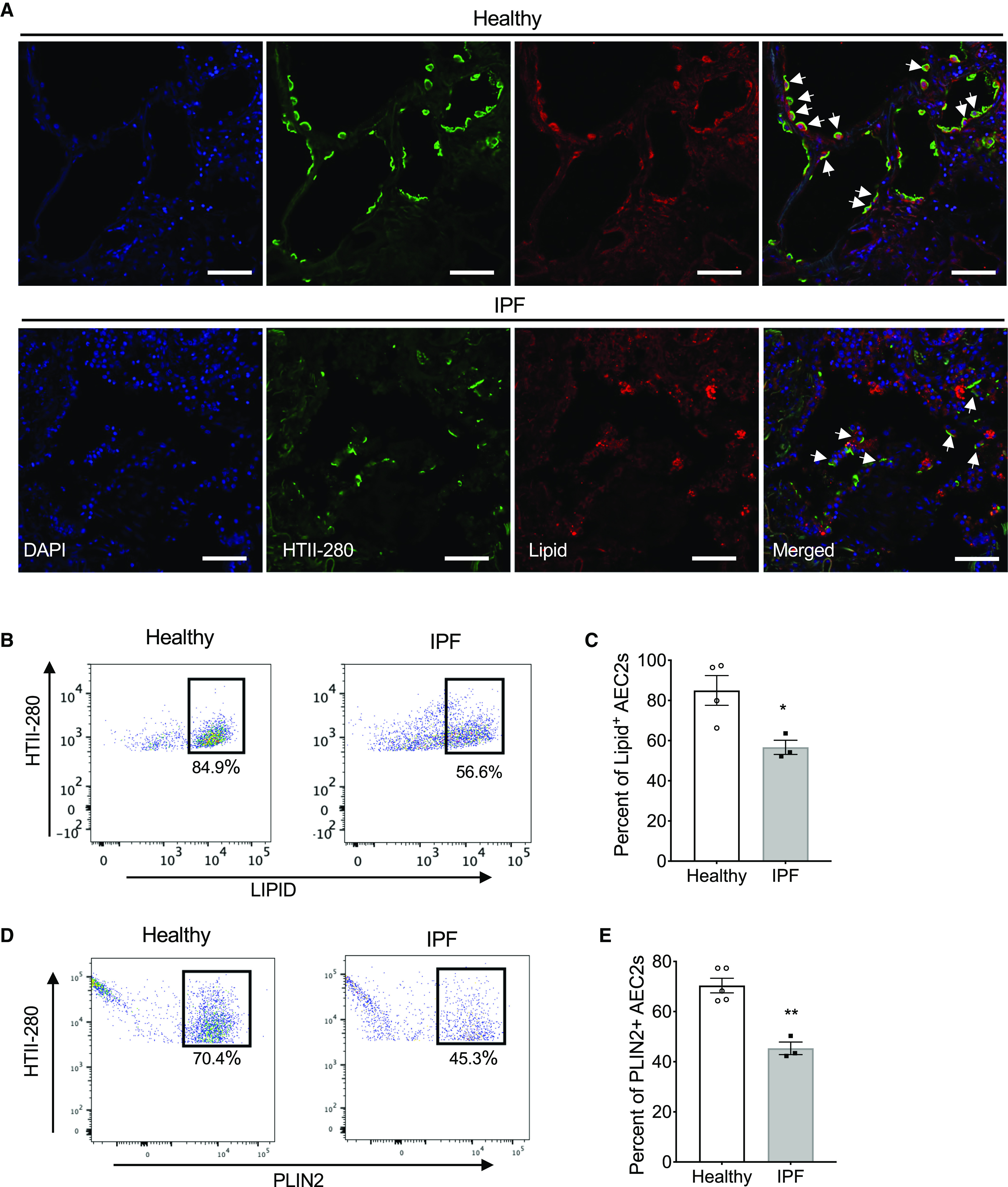
Decreased cellular lipid in AEC2s from lungs of patients with IPF. (A) Representative images of immunofluorescence costaining of HTII-280 and lipid, DAPI for nuclear staining, of lung sections from lung explants of patients with IPF and healthy donors (healthy, n = 3; IPF, n = 5). Arrows indicate AEC2s. Scale bars, 50 μm. (B–E) Flow cytometry analysis of AEC2s isolated from healthy and IPF lungs. Lipid staining (B) and the percentage of lipid+ cells in gated HTII-280+ AEC2s (C; n = 3–4; *P < 0.05 by unpaired Student’s t test). PLIN2 (perilipin 2) staining (D) and the percentage of PLIN2+ cells in gated HTII-280+ AEC2s (E; n = 3–5; **P < 0.01 by unpaired Student’s t test).
To further confirm the decreased lipid levels in IPF AEC2s, we performed flow cytometry analysis of AEC2s from patients with IPF and healthy donor lungs stained with AEC2 markers and for lipid droplets or an antibody against PLIN2. IPF AEC2s showed a reduced percentage of lipid-high cells (Figures 2B and 2C) and reduced PLIN2 expression (Figures 2D and 2E) compared with healthy AEC2s. Human AEC2s were gated as EPCAM+HTII280+CD31−CD45−, as described in our previous studies (3, 10) and as shown in Figure E2A. We also showed decreased lipid staining of SP-C+CD31−CD45− cells from IPF lungs relative to the cells from healthy lungs (Figures E2B and E2C).
Lipid Replenishment Promoted Human AEC2 Renewal
We next investigated if the lipid metabolic dysregulation could contribute to the impaired renewal capacities of IPF AEC2s. As proof-of-principle, we applied exogenous lipid treatments to 3D organoid cultures of AEC2s from both healthy and IPF lungs. Interestingly, 2% lipid treatment promoted renewal capacity of AEC2s from both healthy and IPF lungs (Figures 3A and 3B). However, the effect of lipid treatment on promoting AEC2 renewal was less significant with IPF AEC2s than with healthy AEC2s (Figures 3A and 3B). Lipid at 4% concentration showed a better effect on promoting colony-forming efficiency (CFE) of IPF AEC2s than with 2% lipid (Figure 3C). Lipid treatment increased 5-ethynyl-2′-deoxyuridine (EdU) incorporation in both healthy and IPF AEC2s derived from 3D cultured organoids, and the effect was less significant with IPF AEC2s (Figures 3D and 3E). The flow cytometry gating strategy for EdU-labeled cells is shown in Figure E3. AEC2s with lipid treatment also showed increased expression of SFTPC (Figure 3F) and type I alveolar epithelial cell marker genes, including PDPN, AGER, and AQP5, by quantitative PCR (Figure 3G). These data suggest lipid supplementation increased AEC2 renewal. To further demonstrate the role of lipid metabolism in regulating AEC2 progenitor function, we applied the PPARγ agonist RGZ and the PPARγ antagonist GW9662 to the 3D organoid cultures of healthy AEC2s. Our results showed that RGZ increased and GW9662 decreased colony formation of AEC2s from healthy lungs (Figure 3H). Importantly, RGZ treatment was able to increase colony formation of AEC2s from IPF lungs (Figure 3I).
Figure 3.

Lipid promoted human AEC2 renewal. (A and B) CFE of flow-sorted AEC2s (EpCAM+HTII-280+CD31−CD45−) from healthy (A) (n = 6; ***P < 0.001) and IPF lungs (B) (n = 3–4; **P < 0.01) in the absence or presence of 2% exogenous lipid. (C) CFE fold increase of 2% and 4% lipid treatment versus medium of IPF AEC2s (n = 3–4; **P < 0.01). (D and E) Percentage of 5-ethynyl-2’-deoxyuridine (EdU)+ AEC2s in in total AEC2s derived from three-dimensional (3D) cultured organoids of healthy (D; n = 3; *P < 0.05) and IPF (E; n = 3; P = 0.08; NS = not significant) AEC2s with and without 2% lipid. (F and G) Gene expression of SFTPC (F), PDPN, AGER, and AQP5 (G) in healthy AEC2s from 3D cultured organoids with and without 2% lipid and assessed with RT-PCR (n = 4–5; **P < 0.01). (H) CFE of healthy AEC2s treated with rosiglitazone (RGZ) and GW9662 (n = 4; ****P < 0.0001 by one-way ANOVA). (I) CFE of IPF AEC2s treated with RGZ (n = 5; ****P < 0.0001). P values were calculated by b unpaired Student’s t test (A–G and I) and by one-way ANOVA (H). CFE = colony-forming efficiency.
Bleomycin Injury Downregulated Lipid Metabolism Genes in Mouse AEC2s
Next, we investigated lipid metabolism in AEC2s in the bleomycin lung injury mouse model. We analyzed lipid metabolism gene expression in AEC2s with our mouse epithelial scRNA-seq dataset, which contains flow-sorted epithelial cells (EpCAM+CD31−CD34−CD45−) from uninjured and multiple time points of bleomycin-injured mice from dataset GSE157995 (10, 22, 41). Because Day 4 after bleomycin injury is a time point with maximum AEC2 injury (3, 22), we first analyzed the expression of lipid metabolism–related genes at Day 4 after bleomycin treatment and compared with their baseline expression in AEC2s from uninjured (Day 0) mice. Focusing on lipid metabolism, we found multiple lipid metabolism–related genes were downregulated in AEC2s from Day 4 bleomycin-injured mouse lungs (Figures 4A–4C). The downregulated genes include genes related to lipid biosynthesis and metabolism: Apoe, Apoec1, Fapb5, Soat1, Scd1, Scd2, Cat, Fasn, Acly, and Elovl1 (Figure 4A); genes related to phosphatidylcholine biosynthesis and transport: Chka, Lpcat1, and Abca3 (Figure 4B); and genes encoding enzymes related to fatty acid β-oxidation: Acsl4, Acsl5, and Echs1 (Figure 4C).
Figure 4.

Downregulated lipid metabolism gene expression in bleomycin-injured mouse AEC2s. (A–C) Violin plots of gene expression in AEC2s from uninjured (D0) and Day 4 (D4) bleomycin-injured mice from dataset GSE157995. (A) Lipid biosynthesis– and metabolism-related genes. (B) Genes related to phosphatidylcholine biosynthesis and transport. (C) Genes encoding enzymes related to fatty acid β-oxidation.
Aging Aggravated Decreases and Hindered Recovery of Lipid Metabolism–Related Gene Expression in AEC2s after Bleomycin Injury
To gain insight into how aging affects lipid metabolic changes in AEC2s after lung injury, we performed pathway analyses with gene expression in AEC2s from bleomycin-injured young and old mouse lungs harvested at Day 4 after bleomycin treatment from dataset GSE157995 (10, 22, 41). We observed that multiple lipid biosynthesis pathways, including the superpathway of cholesterol biosynthesis, oleate biosynthesis II, fatty acid β-oxidation I, LXR/RXR activation, and multiple cholesterol biosynthesis pathways, were all downregulated in AEC2s from old mice relative to AEC2s from young mice (Figure 5A). These data suggest that aging had an important impact on lipid metabolism of AEC2s after lung injury. We compared the expression levels of lipid metabolism–related genes in AEC2s between young and old mice at baseline (Day 0) and following a time course after bleomycin injury and found that the genes related to lipid metabolism were highly expressed in AEC2s from Day 0 intact lungs, and their expression was sharply decreased at Day 4 after bleomycin injury in AEC2s from both young and old mice (Figure 5B). AEC2s from Day 4 bleomycin-injured old mice suffered a more severe loss of expression of most of the genes (Figure 5B). Interestingly, at Day 14 after injury, the expression of lipid biosynthesis– and metabolism-related genes was largely restored in AEC2s from young mice. However, the expression of lipid metabolism–related genes continued decreasing in AEC2s from old mouse lungs compared with their baseline levels at Day 0. At Day 28 after bleomycin injury, the expression of these genes was not completely restored in AEC2s from old mice as what we observed in AEC2s from young mice (Figure 5B). Gene expression of phosphatidylcholine conversion enzymes Chka and Lpcat1 showed the same pattern. Their expression was significantly reduced in AEC2s from old mouse lungs at Day 14 and Day 28 after injury relative to that of AEC2 from young mice killed at the same time points (Figure 5C).
Figure 5.
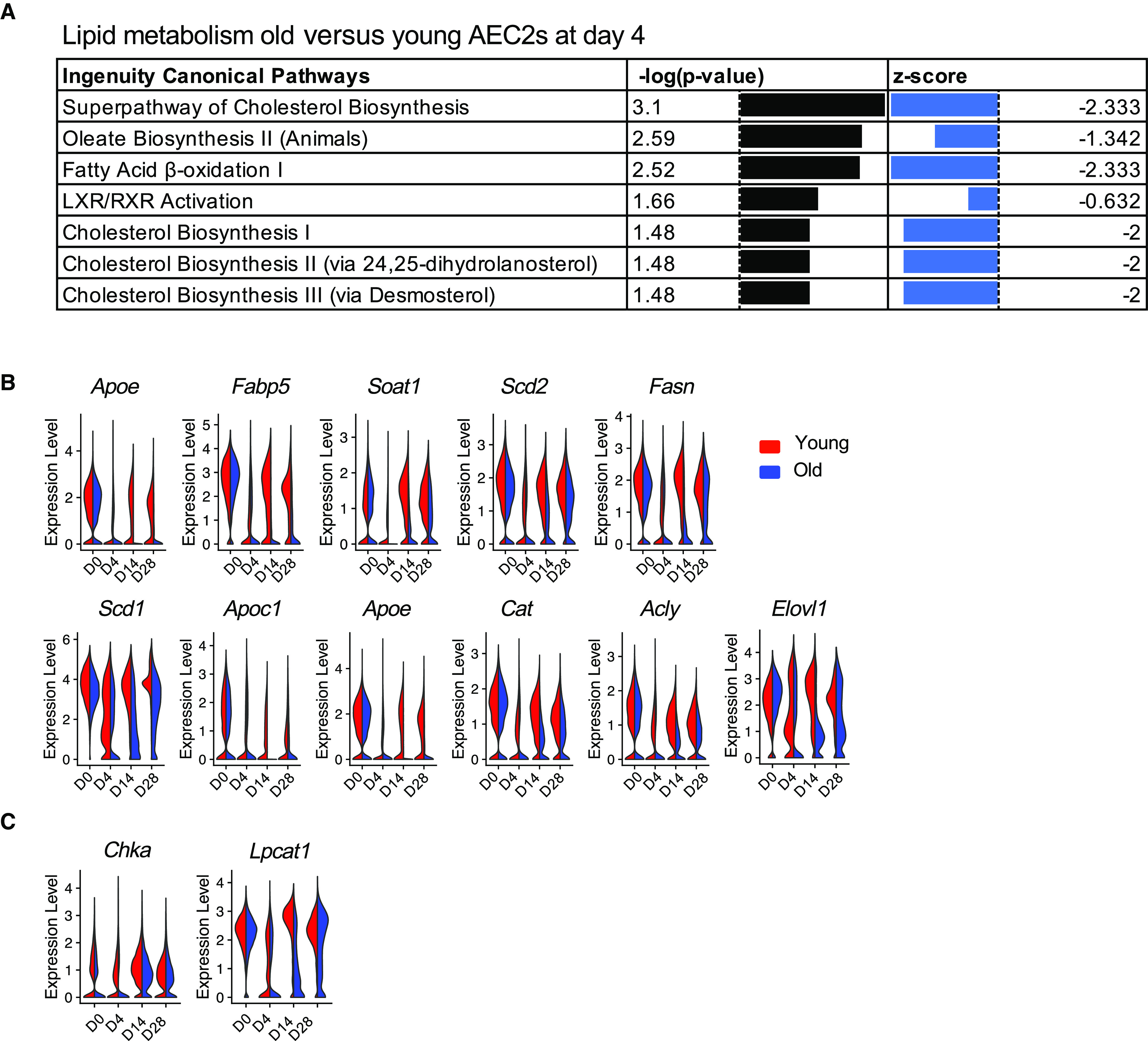
Aging aggravated decreases and hindered recovery of lipid metabolism gene expression in AEC2s after bleomycin injury. (A) IPA pathway analysis of mouse AEC2s from Day 4 bleomycin-injured lungs, old versus young from dataset GSE157995. (B) Violin plots of expression of fatty acid biosynthesis– and metabolism-related genes in AEC2s grouped by age and days after injury (red, young; blue, old). (C) Violin plots of expression of phosphatidylcholine biosynthesis–related genes of AEC2s grouped by age and days after injury (red, young; blue, old). IPA = ingenuity pathway analysis.
Decrease of Cellular Lipid in AEC2s from Bleomycin-injured Old Mice
To visualize the translational effects of downregulation of lipid metabolism in AEC2s from bleomycin-injured old mice, we performed immunofluorescence studies for cellular lipid in AEC2s. We challenged 10-week-old mice and 20-month-old mice with 2.5 U/kg bleomycin, harvested the lungs at Day 14 after bleomycin treatment, and performed immunofluorescence costaining of pro-SPC and lipid with the lung sections. We showed decreased lipid levels in AEC2s from bleomycin-injured old mouse lungs (Figures 6A and E4). The fluorescence intensities of lipid staining in AEC2s in old mouse lungs were much lower than those of AEC2s in young mouse lung sections (Figure 6B).
Figure 6.

Decreased cellular lipid in AEC2s from old and bleomycin-injured mouse lungs. (A) Representative images of immunofluorescence costaining of proSP-C and lipid, DAPI for nuclear staining, with lung sections from Day 14 bleomycin-treated young and old mice. Arrows indicate representative AEC2s. Scale bars: A, (left panels) 100 μm; (right panels) 20 μm. (B) Lipid staining intensity was quantified as ratio of red (lipid) intensity over blue (DAPI) intensity of each cell measured (n = young 100, old 64; *P < 0.05 by unpaired Student’s t test).
Lipid Replenishment Promoted Mouse AEC2 Renewal in 3D Organoid Cultures
We have reported that AEC2s from the lungs of old mice showed decreased renewal capacity compared with AEC2s from young mice (10). We reasoned that lipid deficiency might contribute to the decreased progenitor function of old AEC2s. To test this hypothesis, we applied exogenous lipid treatment to 3D organoid cultures of AEC2s isolated from both young and old mice. The lower concentration (2%) of exogenous lipid treatment was able to promote renewal of AEC2s from young mice (Figure 7A) but had no effect on AEC2s from old mice (Figure 7B). A higher concentration (4%) of lipid increased CFE of both young and old mouse AEC2s (Figure 7C). Lipid treatment increased the colony size (Figure 7D) and proliferation of AEC2s from old mice, as shown by increased EdU incorporation (Figure 7E), in 3D organoid cultures. Next, we isolated single cells from both young and old mice and cultured the cells in control medium or medium containing 4% lipid for 48 hours before flow cytometry analysis. Lipid treatment increased surfactant protein C expression of gated AEC2s compared with that of AEC2s cultured in control medium (Figure 7F). Lipid treatment increased expression of type I alveolar epithelial cell marker genes, including Ager (Figure 7G) and Aqp5 (Figure 7H), in 3D cultured mouse AEC2s. Immunofluorescence staining showed lipid treatment increased T1α expression in 3D cultured mouse AEC2 organoids (Figure 7I). These data indicated that lipid sufficiency is crucial for surfactant homeostasis and progenitor function of AEC2s.
Figure 7.
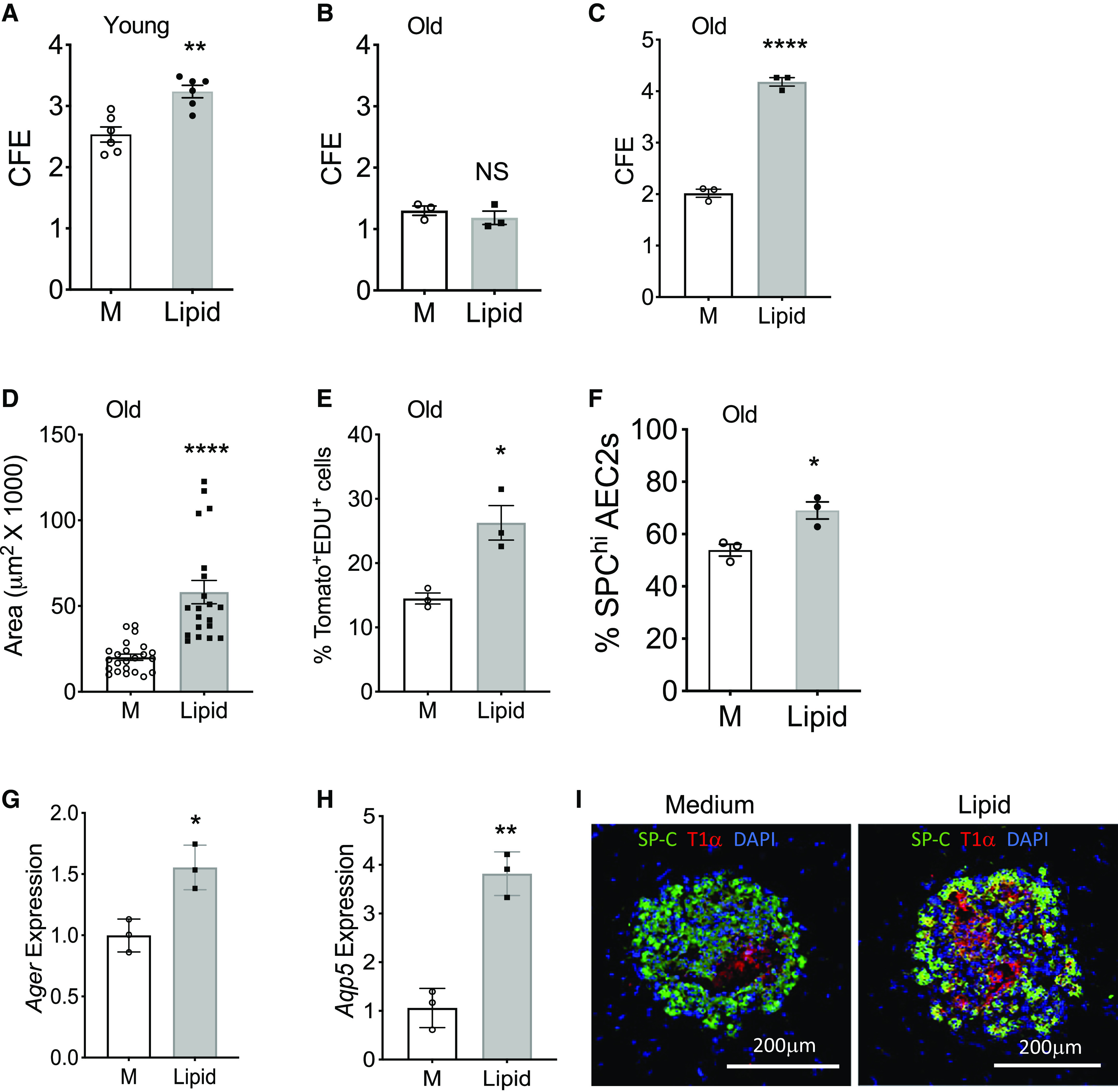
Lipid supplementation promoted renewal capacity of mouse AEC2s. (A and B) CFE of AEC2s from young (A; n = 6; **P < 0.01) and old mice (B; n = 3) with and without 2% lipid treatment. M = medium control. (C–E) 3D organoid cultures of AEC2s from 20-month-old tamoxifen-treated SFTPC-CreER+ Rosa-Tomatofl/fl mice with and without 4% lipid treatment. CFE (C; n = 3; ****P < 0.0001); Sizes of colonies (D; n = 20–23; ****P < 0.0001), and the percentage of EdU+ AEC2s in gated total Tomato+ AEC2s derived from 3D cultured organoids by flow cytometry (E; n = 3; *P < 0.05). (F) Freshly isolated lung single cells from 24-month-old mice were cultured with and without lipid supplementation for 48 hours. The percentage of SPChi cells in total gated AEC2s was determined by flow cytometry (n = 3; *P < 0.05). (G and H) Expression levels of Ager and Aqp5 in young mouse AEC2s after 3D culture with and without 2% lipid were assessed with RT-PCR (n = 3; **P < 0.01 and *P < 0.05). P values were calculated by unpaired two-tailed Student’s t test. (I) Representative images of 3D organoids derived from young mouse AEC2s and stained with SFTPC and T1α antibodies (n = 6). Scale bars, 200 μm.
Discussion
Lipid metabolism has been suggested to play a role in aging (23–25) and in multiple aging-associated lung diseases, including IPF (5, 26–28). AEC2 dysfunction has been recognized as a causal event for IPF (3, 10), and the maintenance of AEC2 functions relies on lipid metabolism homeostasis for surfactant biosynthesis and recycling. In this study, we assessed lipid metabolism changes in AEC2s with aging and lung fibrosis. We observed significant downregulation of lipid metabolism–related gene expression in IPF AEC2s compared with AEC2s from healthy donor lungs by scRNA-seq data analysis. We further confirmed lipid deficiency in IPF AEC2s using immunofluorescence histology and flow cytometry studies. With the lung fibrosis mouse model, we found that bleomycin injury decreased lipid metabolism gene expression in AEC2s, and this effect was aggravated with aging. Aging also hindered the recovery of lipid metabolism gene expression after bleomycin injury. Most importantly, we demonstrated that lipid deficiency impairs AEC2 progenitor renewal. Lipid supplementation and PPARγ activation promoted AEC2 colony formation in 3D organoid cultures.
Previous studies have suggested that lipid metabolism plays important roles in lung development and lung fibrosis. Mice with Elovl1 deficiency died shortly after birth because of epidermal barrier defects (47). Mice with Apoe deletion showed impaired alveologenesis, low lung function, and shorter lifespan compared with wild-type mice (48). Elovl6 was found downregulated after bleomycin lung injury and in IPF lungs, and mice with Elovl6 deficiency exhibited a severe fibroproliferative response to bleomycin injury compared with wild-type mice (39). Inhibition of lipid biosynthesis by targeted deletion of Fasn (fatty acid synthase) in AEC2s and inhibitor of the lipid synthesis enzyme SCD1 resulted in AEC2 mitochondrial dysfunction, epithelial cell endoplasmic reticulum stress, and worsened lung fibrosis after injury (38, 40). Enhancing lipid synthesis by overexpressing Fasn in mice or administration of an LXR agonist in vivo attenuated lung fibrosis in mouse lung fibrosis models (38, 40, 49). These studies demonstrated the importance of lipid metabolism in maintaining AEC2 function and limiting lung fibrosis. However, most of these studies focused on individual lipid metabolism enzymes and did not provide a complete picture of lipid metabolism dysregulation in lung injury and fibrosis. In this study, we comprehensively analyzed lipid metabolism of AEC2s in IPF and in a time course of the bleomycin injury mouse model. We demonstrated that multiple lipid metabolism genes and pathways were downregulated after bleomycin-induced lung injury and in IPF. Furthermore, we analyzed and compared lipid metabolism in AEC2s from young and old mice and demonstrated that aging aggravated the downregulation of lipid metabolism gene expression and enhanced intracellular lipid deficiency in AEC2s after bleomycin injury. To the best of our knowledge, this is the first study that comprehensively investigates the effect of aging on lipid metabolism dysregulation specifically in AEC2s with lung fibrosis.
Lipid metabolism is complicated, as different lipid species may have different roles in both physiological conditions and in diseases, whereas different cell types may also react differently to lipids in vivo. The detrimental effects of lipid metabolism in lung injury and fibrosis have also been reported. Accumulation of oxidized phospholipids within alveolar macrophages after bleomycin injury contributes to fibrogenesis (50). A high-fat diet increased bleomycin-induced lung fibrosis in mice by modulating endoplasmic reticulum stress (51). The plasma levels of lysophosphatidic acid species were found to be positively associated with IPF disease progress (52). Lipid metabolism might have diverse biological functions in lung fibrosis under different circumstances with different lipid species, lipid metabolites, or in different cell types (53). To make matters even more complicated, lipid metabolism is also associated with glucose metabolism and mitochondrial function. Therefore, further studies are needed to define the optimal balance of lipid profiles with different cell types in maintaining healthy lung function and the potential to limit fibrosis initiation and progression.
Our data indicate that lipid deficiency is one of the major defects of AEC2s in IPF and bleomycin-induced lung fibrosis in old mice. We further tested the role of lipid metabolism in AEC2 progenitor function with 3D organoid cultures. Our results showed that the lipid supplementation and PPARγ activation promoted renewal of both human and mouse AEC2s, and PPARγ antagonist inhibited AEC2 colony formation. Previous studies suggested that PPARγ in lung epithelial cells is essential for normal lung development (29), and PPARγ plays a role in myofibroblast differentiation (31, 32). In this study, we showed that PPARγ may play an important role in epithelial repair in fibrotic lungs by promoting AEC2 renewal. The molecular mechanisms that regulate lipid deficiency in AEC2s in fibrotic lungs are not clear and need further investigation. Our current findings provide important new data resources to the research community to further investigate lipid metabolism in lung fibrosis, lung progenitor cell renewal, and aging.
In summary, we have performed comprehensive scRNA-seq analysis of lung alveolar epithelial cells in mice and humans under homeostatic conditions, after lung injury in both old and young mice, and in human disease. We showed dysregulated lipid metabolism–related gene expression in injured mouse AEC2s and IPF AEC2s. The aberrant metabolism of injured AEC2s was manifested as decreased fatty acid and phospholipid biosynthesis–related gene expression in both injured mouse AEC2s and IPF AEC2s, resulting in lipid deficiency in AEC2s. Importantly, aging enhanced the lipid deficiency in injured AEC2s. We then linked lipid metabolism with AEC2 progenitor function and found that enhancing lipid metabolism with PPARγ activation and lipid replenishment promoted AEC2 renewal. These data suggest that efforts to restore the lipid metabolic balance in AEC2s with chemical or pharmaceutical reagents might promote AEC2 cell renewal and offer new opportunities for therapeutic approaches for diseases such as IPF.
Acknowledgments
Acknowledgment
The authors thank the members of the Noble and Jiang laboratories for support and helpful discussion during the course of the study. They also thank Genomics Core and Flow Cytometry Core at Cedars for their technical support.
Footnotes
Supported by National Heart, Lung, and Blood Institute grants P01-HL108793 and R35-HL150829, and National Institute on Aging grant R01-AG078655.
Author Contributions: J.L., P.W.N., and D.J. conceived the study. J.L. performed most of the experiments and analyzed the data. G.H. analyzed single-cell RNA transcriptome data and prepared figures. J.L., G.H., X.L., X.Z., and D.J. analyzed single-cell RNA transcriptome data. X.L., X.Z., A.R., N.L., K.F., F.T., K.D., and V.K. took part in mouse, cell culture, and biological experiments. J.L., P.W.N., and D.J. wrote the paper. All authors read and reviewed the manuscript.
This article has a data supplement, which is accessible at the Supplements tab.
Originally Published in Press as DOI: 10.1165/rcmb.2023-0290OC on April 24, 2024
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Noble PW, Barkauskas CE, Jiang D. Pulmonary fibrosis: patterns and perpetrators. J Clin Invest . 2012;122:2756–2762. doi: 10.1172/JCI60323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Borok Z, Horie M, Flodby P, Wang H, Liu Y, Ganesh S, et al. Grp78 loss in epithelial progenitors reveals an age-linked role for endoplasmic reticulum stress in pulmonary fibrosis. Am J Respir Crit Care Med . 2020;201:198–211. doi: 10.1164/rccm.201902-0451OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liang J, Zhang Y, Xie T, Liu N, Chen H, Geng Y, et al. Hyaluronan and TLR4 promote surfactant-protein-C-positive alveolar progenitor cell renewal and prevent severe pulmonary fibrosis in mice. Nat Med . 2016;22:1285–1293. doi: 10.1038/nm.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Xu Y, Mizuno T, Sridharan A, Du Y, Guo M, Tang J, et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight . 2016;1:e90558. doi: 10.1172/jci.insight.90558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Katzen J, Beers MF. Contributions of alveolar epithelial cell quality control to pulmonary fibrosis. J Clin Invest . 2020;130:5088–5099. doi: 10.1172/JCI139519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest . 2013;123:3025–3036. doi: 10.1172/JCI68782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Desai TJ, Brownfield DG, Krasnow MA. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature . 2014;507:190–194. doi: 10.1038/nature12930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hogan BL, Barkauskas CE, Chapman HA, Epstein JA, Jain R, Hsia CC, et al. Repair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell . 2014;15:123–138. doi: 10.1016/j.stem.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang D, Liang J, Noble PW. In: Encyclopedia of respiratory medicine. 2nd ed. Janes SM, editor. Cambridge, MA: Elsevier; 2020. Stem cells and progenitor cells in interstitial lung disease. [Google Scholar]
- 10. Liang J, Huang G, Liu X, Taghavifar F, Liu N, Wang Y, et al. The ZIP8/SIRT1 axis regulates alveolar progenitor cell renewal in aging and idiopathic pulmonary fibrosis. J Clin Invest . 2022;132:e157338. doi: 10.1172/JCI157338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thannickal VJ. Mechanistic links between aging and lung fibrosis. Biogerontology . 2013;14:609–615. doi: 10.1007/s10522-013-9451-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hecker L, Logsdon NJ, Kurundkar D, Kurundkar A, Bernard K, Hock T, et al. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci Transl Med . 2014;6:231ra47. doi: 10.1126/scitranslmed.3008182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hecker L. Mechanisms and consequences of oxidative stress in lung disease: therapeutic implications for an aging populace. Am J Physiol Lung Cell Mol Physiol . 2018;314:L642–L653. doi: 10.1152/ajplung.00275.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Raghu G, Chen SY, Hou Q, Yeh WS, Collard HR. Incidence and prevalence of idiopathic pulmonary fibrosis in US adults 18-64 years old. Eur Respir J . 2016;48:179–186. doi: 10.1183/13993003.01653-2015. [DOI] [PubMed] [Google Scholar]
- 15. Rojas M, Mora AL, Kapetanaki M, Weathington N, Gladwin M, Eickelberg O. Aging and lung disease: clinical impact and cellular and molecular pathways. Ann Am Thorac Soc . 2015;12:S222–S227. doi: 10.1513/AnnalsATS.201508-484PL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liang J, Ligresti G. Aging delays lung repair: insights from omics analysis in mice with pulmonary fibrosis. Am J Respir Cell Mol Biol . 2023;69:376–377. doi: 10.1165/rcmb.2023-0171ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bueno M, Brands J, Voltz L, Fiedler K, Mays B, St Croix C, et al. ATF3 represses PINK1 gene transcription in lung epithelial cells to control mitochondrial homeostasis. Aging Cell . 2018;17:e12720. doi: 10.1111/acel.12720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jiang C, Liu G, Luckhardt T, Antony V, Zhou Y, Carter AB, et al. Serpine 1 induces alveolar type II cell senescence through activating p53-p21-Rb pathway in fibrotic lung disease. Aging Cell . 2017;16:1114–1124. doi: 10.1111/acel.12643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yao C, Guan X, Carraro G, Parimon T, Liu X, Huang G, et al. Senescence of alveolar type 2 cells drives progressive pulmonary fibrosis. Am J Respir Crit Care Med . 2021;203:707–717. doi: 10.1164/rccm.202004-1274OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Burman A, Tanjore H, Blackwell TS. Endoplasmic reticulum stress in pulmonary fibrosis. Matrix Biol . 2018;68-69:355–365. doi: 10.1016/j.matbio.2018.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chilosi M, Carloni A, Rossi A, Poletti V. Premature lung aging and cellular senescence in the pathogenesis of idiopathic pulmonary fibrosis and COPD/emphysema. Transl Res . 2013;162:156–173. doi: 10.1016/j.trsl.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 22. Liang J, Huang G, Liu X, Liu N, Taghavifar F, Dai K, et al. Reciprocal interactions between alveolar progenitor dysfunction and aging promote lung fibrosis. eLife . 2023;12:e85415. doi: 10.7554/eLife.85415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Angelidis I, Simon LM, Fernandez IE, Strunz M, Mayr CH, Greiffo FR, et al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat Commun . 2019;10:963. doi: 10.1038/s41467-019-08831-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Anisimova AS, Meerson MB, Gerashchenko MV, Kulakovskiy IV, Dmitriev SE, Gladyshev VN. Multifaceted deregulation of gene expression and protein synthesis with age. Proc Natl Acad Sci USA . 2020;117:15581–15590. doi: 10.1073/pnas.2001788117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Johnson AA, Stolzing A. The role of lipid metabolism in aging, lifespan regulation, and age-related disease. Aging Cell . 2019;18:e13048. doi: 10.1111/acel.13048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Agudelo CW, Samaha G, Garcia-Arcos I. Alveolar lipids in pulmonary disease: a review. Lipids Health Dis . 2020;19:122. doi: 10.1186/s12944-020-01278-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mulugeta S, Nureki S, Beers MF. Lost after translation: insights from pulmonary surfactant for understanding the role of alveolar epithelial dysfunction and cellular quality control in fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol . 2015;309:L507–L525. doi: 10.1152/ajplung.00139.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fitzgerald ML, Xavier R, Haley KJ, Welti R, Goss JL, Brown CE, et al. ABCA3 inactivation in mice causes respiratory failure, loss of pulmonary surfactant, and depletion of lung phosphatidylglycerol. J Lipid Res . 2007;48:621–632. doi: 10.1194/jlr.M600449-JLR200. [DOI] [PubMed] [Google Scholar]
- 29. Reddy RC, Rehan VK, Roman J, Sime PJ. PPARs: regulators and translational targets in the lung. PPAR Res . 2012;2012:342924. doi: 10.1155/2012/342924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Burgess HA, Daugherty LE, Thatcher TH, Lakatos HF, Ray DM, Redonnet M, et al. PPARgamma agonists inhibit TGF-beta induced pulmonary myofibroblast differentiation and collagen production: implications for therapy of lung fibrosis. Am J Physiol Lung Cell Mol Physiol . 2005;288:L1146–L1153. doi: 10.1152/ajplung.00383.2004. [DOI] [PubMed] [Google Scholar]
- 31. Ferguson HE, Kulkarni A, Lehmann GM, Garcia-Bates TM, Thatcher TH, Huxlin KR, et al. Electrophilic peroxisome proliferator-activated receptor-gamma ligands have potent antifibrotic effects in human lung fibroblasts. Am J Respir Cell Mol Biol . 2009;41:722–730. doi: 10.1165/rcmb.2009-0006OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kulkarni AA, Thatcher TH, Hsiao HM, Olsen KC, Kottmann RM, Morrissette J, et al. The triterpenoid CDDO-Me inhibits bleomycin-induced lung inflammation and fibrosis. PLoS One . 2013;8:e63798. doi: 10.1371/journal.pone.0063798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wong BH, Mei D, Chua GL, Galam DL, Wenk MR, Torta F, et al. The lipid transporter Mfsd2a maintains pulmonary surfactant homeostasis. J Biol Chem . 2022;298:101709. doi: 10.1016/j.jbc.2022.101709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Andreeva AV, Kutuzov MA, Voyno-Yasenetskaya TA. Regulation of surfactant secretion in alveolar type II cells. Am J Physiol Lung Cell Mol Physiol . 2007;293:L259–L271. doi: 10.1152/ajplung.00112.2007. [DOI] [PubMed] [Google Scholar]
- 35. Kim K, Shin D, Lee G, Bae H. Loss of SP-A in the lung exacerbates pulmonary fibrosis. Int J Mol Sci . 2022;23:5292. doi: 10.3390/ijms23105292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Whitsett JA, Wert SE, Weaver TE. Diseases of pulmonary surfactant homeostasis. Annu Rev Pathol . 2015;10:371–393. doi: 10.1146/annurev-pathol-012513-104644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med . 2019;199:1517–1536. doi: 10.1164/rccm.201712-2410OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Romero F, Hong X, Shah D, Kallen CB, Rosas I, Guo Z, et al. Lipid synthesis is required to resolve endoplasmic reticulum stress and limit fibrotic responses in the lung. Am J Respir Cell Mol Biol . 2018;59:225–236. doi: 10.1165/rcmb.2017-0340OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sunaga H, Matsui H, Ueno M, Maeno T, Iso T, Syamsunarno MR, et al. Deranged fatty acid composition causes pulmonary fibrosis in Elovl6-deficient mice. Nat Commun . 2013;4:2563. doi: 10.1038/ncomms3563. [DOI] [PubMed] [Google Scholar]
- 40. Chung KP, Hsu CL, Fan LC, Huang Z, Bhatia D, Chen YJ, et al. Mitofusins regulate lipid metabolism to mediate the development of lung fibrosis. Nat Commun . 2019;10:3390. doi: 10.1038/s41467-019-11327-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huang G, Liang J, Huang K, Liu X, Taghavifar F, Yao C, et al. Basal cell-derived WNT7A promotes fibrogenesis at the fibrotic niche in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol . 2023;68:302–313. doi: 10.1165/rcmb.2022-0074OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu X, Rowan SC, Liang J, Yao C, Huang G, Deng N, et al. Categorization of lung mesenchymal cells in development and fibrosis. iScience . 2021;24:102551. doi: 10.1016/j.isci.2021.102551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tsukui T, Sun KH, Wetter JB, Wilson-Kanamori JR, Hazelwood LA, Henderson NC, et al. Collagen-producing lung cell atlas identifies multiple subsets with distinct localization and relevance to fibrosis. Nat Commun . 2020;11:1920. doi: 10.1038/s41467-020-15647-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Habermann AC, Gutierrez AJ, Bui LT, Yahn SL, Winters NI, Calvi CL, et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci Adv . 2020;6:eaba1972. doi: 10.1126/sciadv.aba1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Morse C, Tabib T, Sembrat J, Buschur KL, Bittar HT, Valenzi E, et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur Respir J . 2019;54 doi: 10.1183/13993003.02441-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen H, Matsumoto K, Brockway BL, Rackley CR, Liang J, Lee JH, et al. Airway epithelial progenitors are region specific and show differential responses to bleomycin-induced lung injury. Stem Cells . 2012;30:1948–1960. doi: 10.1002/stem.1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sassa T, Ohno Y, Suzuki S, Nomura T, Nishioka C, Kashiwagi T, et al. Impaired epidermal permeability barrier in mice lacking elovl1, the gene responsible for very-long-chain fatty acid production. Mol Cell Biol . 2013;33:2787–2796. doi: 10.1128/MCB.00192-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Massaro D, Massaro GD. Apoetm1Unc mice have impaired alveologenesis, low lung function, and rapid loss of lung function. Am J Physiol Lung Cell Mol Physiol . 2008;294:L991–L997. doi: 10.1152/ajplung.00013.2008. [DOI] [PubMed] [Google Scholar]
- 49. Shin H, Park S, Hong J, Baek AR, Lee J, Kim DJ, et al. Overexpression of fatty acid synthase attenuates bleomycin induced lung fibrosis by restoring mitochondrial dysfunction in mice. Sci Rep . 2023;13:9044. doi: 10.1038/s41598-023-36009-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Romero F, Shah D, Duong M, Penn RB, Fessler MB, Madenspacher J, et al. A pneumocyte-macrophage paracrine lipid axis drives the lung toward fibrosis. Am J Respir Cell Mol Biol . 2015;53:74–86. doi: 10.1165/rcmb.2014-0343OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chu SG, Villalba JA, Liang X, Xiong K, Tsoyi K, Ith B, et al. Palmitic acid-rich high-fat diet exacerbates experimental pulmonary fibrosis by modulating endoplasmic reticulum stress. Am J Respir Cell Mol Biol . 2019;61:737–746. doi: 10.1165/rcmb.2018-0324OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Neighbors M, Li Q, Zhu SJ, Liu J, Wong WR, Jia G, et al. Bioactive lipid lysophosphatidic acid species are associated with disease progression in idiopathic pulmonary fibrosis. J Lipid Res . 2023;64:100375. doi: 10.1016/j.jlr.2023.100375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mamazhakypov A, Schermuly RT, Schaefer L, Wygrecka M. Lipids: two sides of the same coin in lung fibrosis. Cell Signal . 2019;60:65–80. doi: 10.1016/j.cellsig.2019.04.007. [DOI] [PubMed] [Google Scholar]