

Abstract
Viral genomes are poorly annotated in metagenomic samples, representing an obstacle to understanding viral diversity and function. Current annotation approaches rely on alignment-based sequence homology methods, which are limited by the paucity of characterized viral proteins and divergence among viral sequences. Here, we show that protein language models can capture prokaryotic viral protein function, enabling new portions of viral sequence space to be assigned biologically meaningful labels. When applied to global ocean virome data, our classifier expanded the annotated fraction of viral protein families by 29%. Among previously unannotated sequences, we highlight the identification of an integrase defining a mobile element in marine picocyanobacteria, and a capsid protein that anchors globally widespread viral elements. Furthermore, improved high-level functional annotation provides a means to characterize similarities in genomic organization among diverse viral sequences. Protein language models thus enhance remote homology detection of viral proteins, serving as a useful complement to existing approaches.
Introduction
Viruses of microbes, hereafter, ‘viruses’, are abundant in the environment and have wide-ranging impacts on microbial communities. Much of what we know about viral diversity, ecology, and function comes from analysis of sequences obtained from environmental samples, yet viruses are difficult to identify, classify, and annotate. Thus, we make statements about viral biology and their impacts on microbial community structure and function based on a tiny fraction of viral sequences with sufficient similarity to existing references. In recent years, next-generation sequencing and increasing computational resources have been applied to catalogue the world’s virome1–7. While there has been substantial methodological progress in identifying viral DNA in whole community metagenomic sequence data8–16, sequence feature annotation and overall taxonomic assignment of identified uncultivated virus genomes (UViGs) has lagged considerably. Viruses have no universal conserved marker genes to enable broad, unified, taxonomic analysis and thus most of the hundreds of thousands of new viruses uncovered in viral catalogue studies remain unclassified1–7. Viral taxonomic classification is generally based on using predicted UViG proteins as features for clustering-based17–19 or machine learning-based20 taxonomic classification. Yet, as many as 86% of environmental viral protein clusters match uncharacterized protein families or have no hits at all6,7,16,21,22. Though detailed manual investigation of these sequence clusters may be able to yield hints of potential functions in some cases, such labor-intensive efforts do not readily scale to the amount of data being generated. Improved annotation of viral protein families (VPFs) is thus a necessary, unrealized step towards understanding the roles of viruses in microbial ecology.
Viral protein annotation currently relies on sequence homology using state-of-the-art profile Hidden Markov Model (pHMM)-based approaches. For viral metagenomics, sequence homology methods suffer from two fundamental limitations: (1) the limited library of annotated viral protein sequences from which to construct probabilistic sequence models and (2) the rate at which viral proteins change, quickly diverging beyond recognition by traditional sequence homology metrics. An alignment-free method that does not depend on constructing sequence profiles for statistical sequence homology and that can leverage functional homology between proteins could overcome both challenges.
Advances in the field of natural language processing have increasingly been utilized to identify viral sequences in whole community sequencing data, including k-mer frequency9,11 and learned vector representation10,16,23,24 methods. In natural language processing, current state-of-the-art large language models are trained in an unsupervised manner on gigantic corpora of text to predict sequences of words. Recently, this approach has been used to train protein language models (PLMs) on billions of protein sequences. PLMs capture physico-chemical properties of amino acids and can resolve protein structural and functional information from sequence input alone25–32. Unlike sequence, structure and function of viral proteins are better maintained over evolutionary time due to biochemical and fitness constraints33,34. We hypothesized that annotating VPFs based on functional homology captured in PLM-based protein representations, rather than strict protein sequence homology, would improve VPF annotation. Therefore, we developed a PLM-based viral protein function classifier and asked if it could improve the viral protein annotation problem.
Using curated VPF databases and recently published PLMs, we show that PLM-based representations of viral protein sequences can capture viral functional homology beyond remote sequence homology. Our analysis focuses on two aspects of viral sequence annotation: systematic labeling of protein families, and specific function identification for biologic discovery. First, we utilize our PLM-based classifier to expand the annotated fraction of VPFs collected from the ocean virome by 29%. To highlight the utility of this approach in biological discovery, we use the classifier to identify previously unannotated and globally widespread viral-like integrases and major capsid proteins (MCPs). Additionally, we demonstrate that the PLM-based representations capture function groupings specific to viral biology. Finally, we show that a high-level functional classification approach enables the discovery of shared organizations in diverse sequences from the global oceans, patterns that are obscured by detailed annotations and lost to sequence homology-based approaches due to sequence diversity of viral proteins. PLMs capture features of viral proteins that aid in detecting remote homology and are thus a powerful discovery tool and a complementary method to alignment-based approaches for understanding the functions of viral populations across the world.
Results
Protein language models capture viral protein function
We first asked whether PLMs can capture properties of viral protein function that are invisible to state-of-the-art approaches such as pHMMs. Given the extensive resources required to train PLMs, we based our work upon existing resources, including VPF databases and pre-trained PLMs (Figure 1). Our reference annotations were based on the Prokaryotic virus Remote Homologous Groups (PHROGs) database, a curated library of VPFs constructed to capture remote sequence homology and manually annotated to high-level functional categories21. PHROGs contains 868,340 protein sequences clustered into 38,880 families, of which 5,088 are annotated to 9 functional classes (Figure 2a). The database was constructed to maximize remote sequence homology captured by each family, though intra-category profile similarities differed between functions (Figure 2b). To evaluate the performance of PLM-based representations for function annotation, PHROGs sequences were embedded with a PLM and a multi-class function classifier was trained on VPFs to predict the functional category of sequences from held out VPFs. We then carried out five-fold cross validation over the entire annotated set with proteins embedded using four pre-trained PLMs28–30 (Supplemental Table 1). The PLM trained on the largest protein dataset (Transformer BFD28) performed the best of the PLMs evaluated (Extended Data Figure 1), with an average area under the receiver operating characteristic curve of 0.90 (Figure 2c) and average area under the precision-recall curve of 0.62 across all classes and folds (Figure 2d). The Transformer BFD model was used for all subsequent analyses.
Figure 1: Viral protein family (VPF) function prediction using protein language models (PLMs) uncovers novel biology.
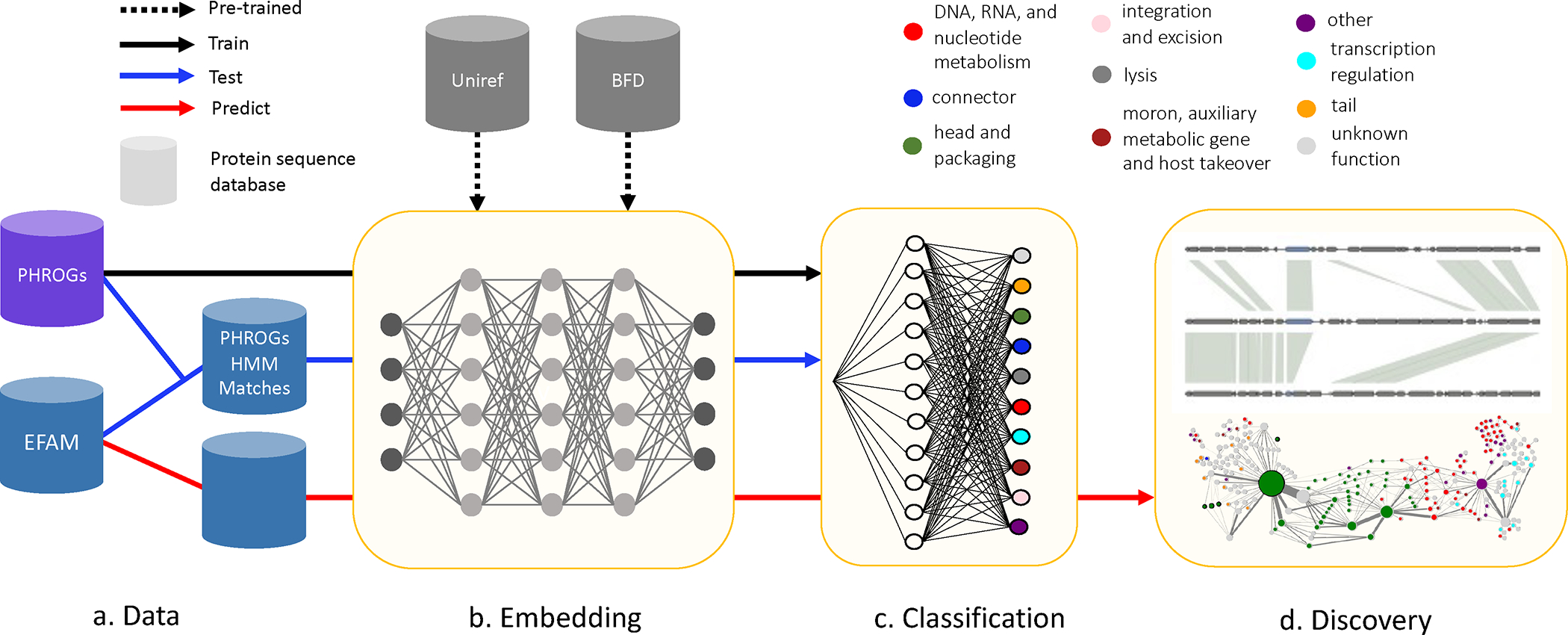
(a) VPFs were collected from the curated databases Prokaryotic Virus Remote Homologous Groups (PHROGs) and EFAM. (b) Protein sequences are embedded using pre-trained PLMs. (c) Embeddings are used as input to a multi-class classifier for high-level function prediction. (d) Classifier predictions of unannotated VPFs lead to biologic discovery. HMM-hidden Markov model; BFD-Big Fantastic Database.
Figure 2: Functional category classification of PHROG VPFs with PLM-based protein embeddings.
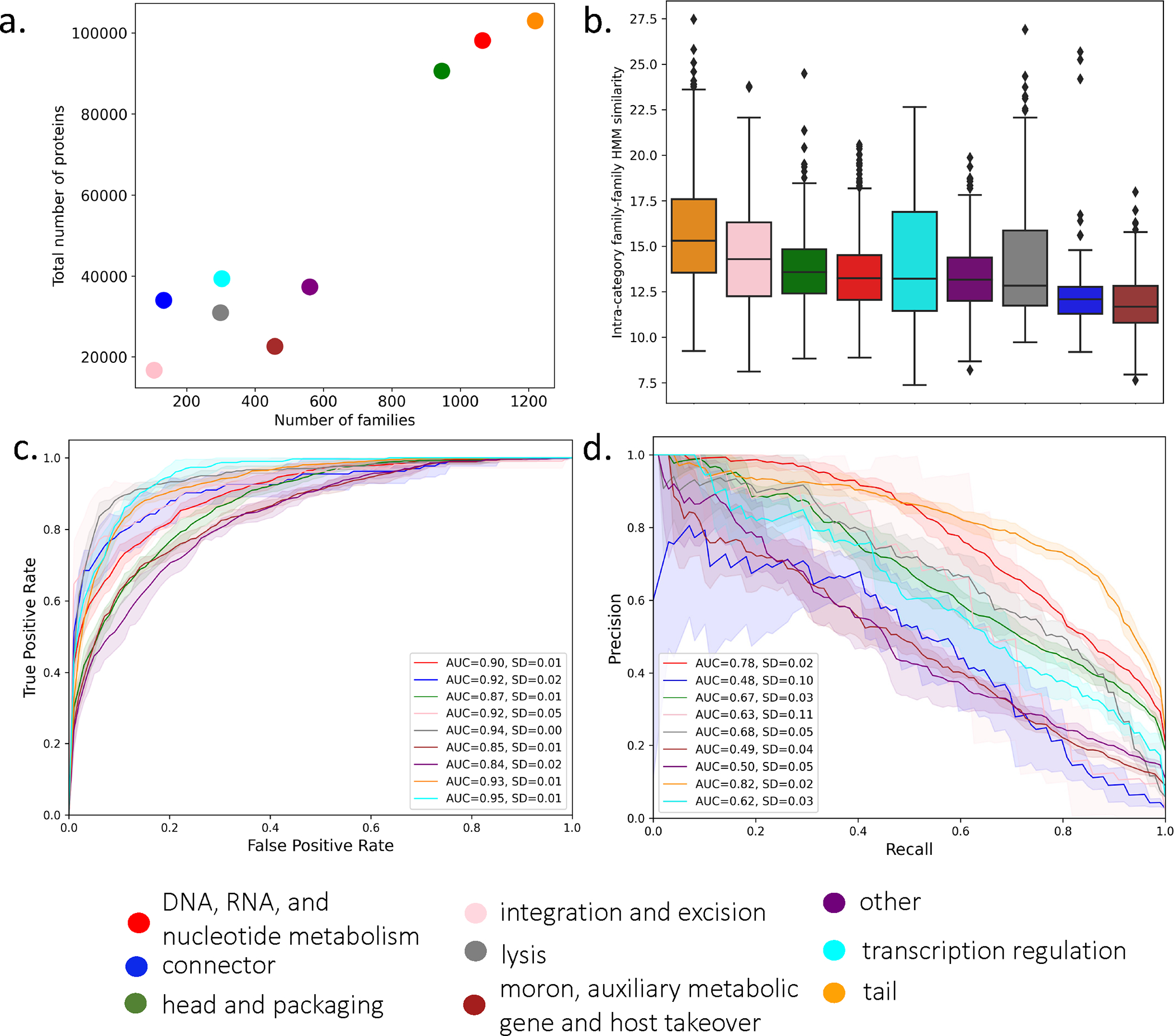
(a) PHROG category family and total protein numbers. (b) Distribution of pairwise profile similarity of families in a functional category (DNA, RNA, and nucleotide metabolism- n=1,065; connector- n=133; head and packaging=946; integration and excision- n=105; lysis- n=299; moron, auxiliary metabolic gene and host takeover- n=458; other- n=560; tail- n=1,219; transcription regulation- n=303). Boxes represent interquartile range; whiskers represent the entire distribution with the exception of outliers (diamonds); horizontal line indicates median. (c-d) Multi-class function classifier performance for five-fold stratified splits of annotated PHROGs families. (c) Receiver operating characteristic curve with average area under curve (AUC) and standard deviation (SD) over five folds. (d) Precision-recall curve with AUC and SD over five folds. Per fold, training is performed over all proteins in a family and testing is performed on a random single sequence from test families. Protein sequences were embedded using the Transformer BFD PLM and the classifier consists of a three hidden layer dense neural network and an output layer with softmax activation.
A second multi-class classification model, which we used for subsequent analyses, was then trained on all annotated families as well as families of the unknown function category in order to capture sequences that do not match the functional categories. After classifier training, a new version of the PHROGs database (v4) was released, in which 57 PHROG families were reclassified. The classifier correctly predicted the re-annotation of 38/57 families (66.6%) despite being trained on the previous incorrect annotation for those families (Supplemental Table 2). The performance on the re-annotated families serves as a validation of the classifier’s ability to capture function.
Language model protein embeddings capture viral biology
Having determined that PLM-based representations of viral proteins can predict function, we investigated the viral protein embeddings to understand what enables the PLM to detect differences between functions. Because a PLM can produce a dense vector representation for any protein sequence, VPFs were represented as the centroid of sequence embeddings for constituent proteins, and were visualized for the functionally annotated PHROGs subset (Figure 3a). We first interrogated the similarity of sequences in a family, and families in a functional category, using vector similarity. While the sequence-sequence vector similarity in families across all categories is high (Extended Data Figure 2a), the intra-category family-family similarity varied between functional categories (Extended Data Figure 2b) but higher similarity did not correspond to better classification performance. We then asked if there are groupings of categories in the embedding space. We measured the category-category similarity as the average of the family-family vector similarity for all pairs of families between two categories (Extended Data Figure 3). We spectrally clustered the category-category distance matrix (Figure 3b), revealing a biologically meaningful partition of functional categories into those relating to virion structure and infection (cluster1) and those relating to viral genome replication and other host derived genes (cluster2). The partition was apparent when the embedding space is relabeled with cluster assignment (Figure 3c). We grouped functional categories into the two clusters identified and trained a binary classifier using five-fold cross validation (Figure 3d) that resulted in better performance compared to random partitions of the categories into groups of two (Figure 3e).
Figure 3: Investigation of PLM-based embedding of PHROG VPFs.
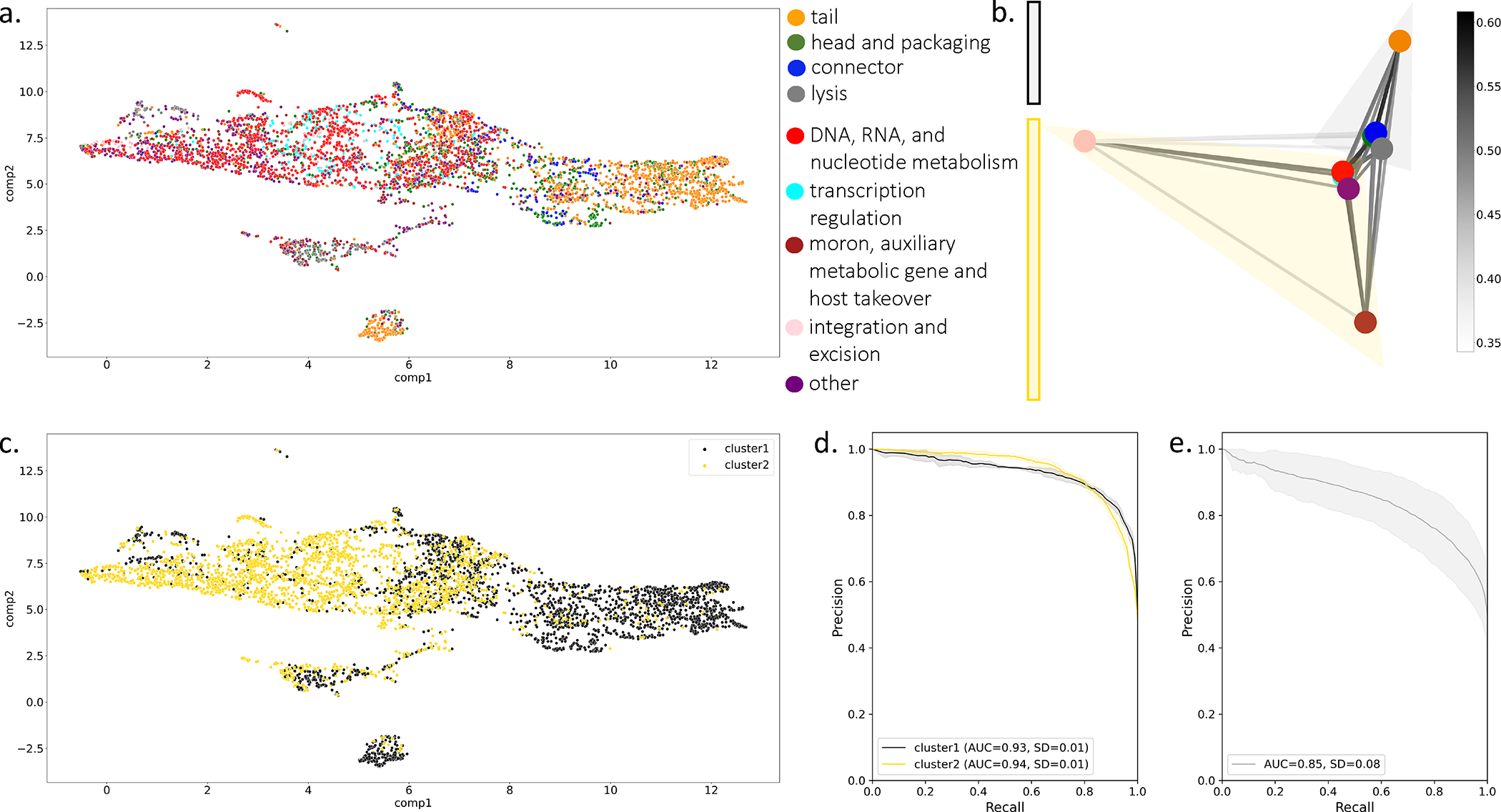
(a) umap projection of PHROG VPFs. VPFs were represented as the centroid of sequence vectors. (b) Spectral network visualization of the inter-category family-family similarity (edge weight), which is measured as the mean family-family centroid similarity across all family pairs between two categories. The category-category similarity matrix is clustered with n=2 into two groups (black and yellow). (c) Spectral clusters are used to color PHROGs VPF umap projection. (d) Clusters are used as binary classes for PHROGs VPF classifier as in 2B. (e) Classifier performance on 10 random two group splits with AUPRC averaged over groups and splits. (d-e) Performance is reported as average AUC over five folds and error represents one standard deviation.
The ability to classify structural proteins, termed phage virion proteins (PVPs) in bacteriophages, is important for identifying and grouping viral sequences, and several methods have recently been developed to tackle this problem35,36. We compared PLM-based classification with existing methods for PVP prediction. Using a PVP identification task designed previously36,37, our method achieved performance on par with state-of-the-art approaches (Supplemental Table 3). Thus, the clustering of functions in the embedding space, and partitioning of viral protein sequences among different functional groupings (whether due to primary sequence, structure, or other properties) may reflect some of the types of information captured in PLM pre-training which enables function prediction from PLM-based representations of viral proteins.
Improved classification of proteins from the ocean virome
To further test the capabilities of the trained function classifier, we evaluated its performance against pHMMbased annotation of the largest pan-ecosystem viral protein family database, EFAM, which was curated from uncultivated virus genomes identified in the global oceans22. Viral genomes in EFAM are not present in the PHROGs training sequences, making this dataset well-suited for an external validation of our classifier. To assign ‘true’ functional categories to the EFAM VPFs, we first used profile-profile HMM matching based on HMMs provided by the PHROGs database. 88,605/240,311 (36.9%) of EFAM VPFs matched PHROGs VPFs, of which 66,137 (74.7%) had functional annotation. These PHROGs-annotated EFAM VPFs were also predicted using our PLM-based functional classifier. We used the F1-score, a measure of classification performance that combines precision and recall, to evaluate our predictions. A F1-score of 1 indicates perfect recall and precision, and a score of 0 means either precision or recall is 0. All categories had strong performance (Figure 4a) and the weighted F1-score across all functional categories was 0.85. Using the validation set, we performed a per-class calibration analysis (Extended Data Figure 4) and determined a classification decision boundary for each class with a maximum false discovery rate (FDR) of 10% (Supplemental Table 4, Extended Data Figure 5). Next, we used the calibrated classifier to predict the functional category of EFAM VPFs not captured by the PHROGs HMMs (Figure 4b). In total we expanded the annotated fraction of EFAM by 26,770 families, a 29.4% increase over the number annotated within the EFAM database supplemented with annotation by PHROGs (91,156 families). The largest increases in annotated functions were for the ‘head and packaging’ and ‘tail’ categories, which contain VPFs that retain pairwise sequence embedding similarity for lower pairwise sequence identity in the PHROGs database (Extended Data Figure 6). This result indicates that PLM-based classification can supplement pHMM representations for remote homology detection.
Figure 4: Functional category classifier validation and discovery with the EFAM database of VPFs curated from the ocean virome.
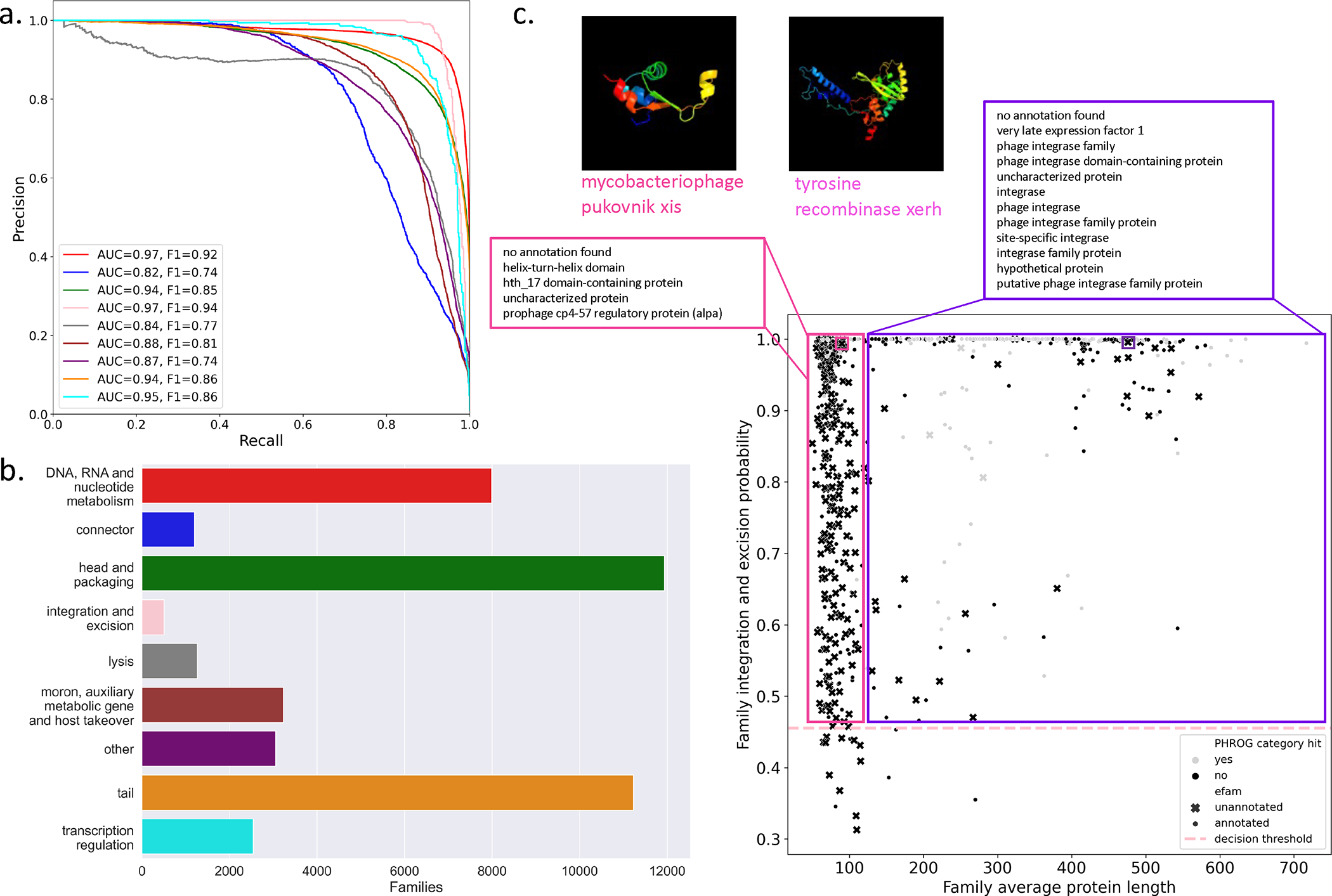
(a) Precision-recall curve for EFAM VPFs labeled with PHROGs HMMs and predicted with the PLM-based function classifier. Performance is measured with AUPRC and F1-score. (b) Number of VPFs in EFAM that are labeled to each functional category based on the category-specific optimal threshold and not captured by PHROGs HMMs. (c) EFAM VPFs predicted ”integration and excision” class probability by average protein length in the VPF. Annotation of excisionase (pink) and integrase/recombinase (purple) terms are for VPFs annotated in EFAM (°§). Structural prediction for two EFAM VPFs that do not match PHROGs HMMs and are unannotated in EFAM (x) are shown with predicted structure, one excisionase (cluster122519) and one integrase (cluster86903). Decision probability is the FDR-based threshold for ”integration and excision”.
PLMs enable identification of a tyrosine integrase family
To determine whether PLM-based functional classifications can accurately identify genes of biological interest from large datasets, we first examined predictions from the ‘integration and excision’ category. This group was chosen for having the best prediction performance, and detection of viral integrases within host genomes is of biological interest for identifying temperate bacteriophage. EFAM VPFs predicted in this category can be stratified based on their annotation in the EFAM database itself, with VPFs having average protein lengths >120 matching annotation to known integrase/recombinase proteins and VPFs with average protein lengths <120 matching known excisionases (Figure 4c). We validated our integration and excision prediction for EFAM VPFs that were not annotated in EFAM or by PHROGs HMM matching using both structure and domain predictions (Supplemental Table 5). Further investigation of predicted EFAM integrase families led to the annotation of an integrase (EFAM cluster86903) on a previously reported putative prophage in uncultured Alphaproteobacteria38, supporting the utility of this approach.
Our method was also able to annotate related genes in non-viral contexts. The PLM model predicted a previously unannotated VPF, EFAM cluster158946, as a putative integrase. This cluster caught our attention as the sequences were located not within viral sequences but rather marine picocyanobacterial genomes, including members of the globally abundant cyanobacteria Prochlorococcus and Synechococcus. Phylogenetic analysis revealed these enzymes as an unidentified subgroup within the tyrosine integrase/recombinase family of sitespecific integrases. Cyanobacterial integrases in this sequence cluster are distinct from others commonly seen in bacteriophages and bacterial mobile genetic elements, or those associated with Tycheposon mobile elements in Prochlorococcus39; their closest relatives were to a few members of the diverse group of tyrosine recombinases associated with VEIME bacteriophage satellites40 (Figure 5a). The predicted integrases have a different domain structure than is typical of many tyrosine integrases41, yet structural modeling confirmed that this enzyme retains the key catalytic residues required for activity42 (Extended Data Figure 7). These enzymes are only found within a subset of available Prochlorococcus and Synechococcus genomes, where they are typically located upstream of one of two specific tRNAs, either tRNA-Phe or tRNA-Cys. tRNAs are frequent integration sites for mobile genetic elements43 and phylogenetic groupings of these enzymes correlate with their respective tRNA (Figure 5b), suggesting that these may represent the integration site. The integrases are located within genomic islands of variable genetic content and are also frequently, though not exclusively, found near a small serine recombinase (Figure 5c–d). Together, these properties suggest that this enzyme defines a mobile genetic element within marine picocyanobacteria.
Figure 5: Identification of an integrase/recombinase within marine picocyanobacteria.
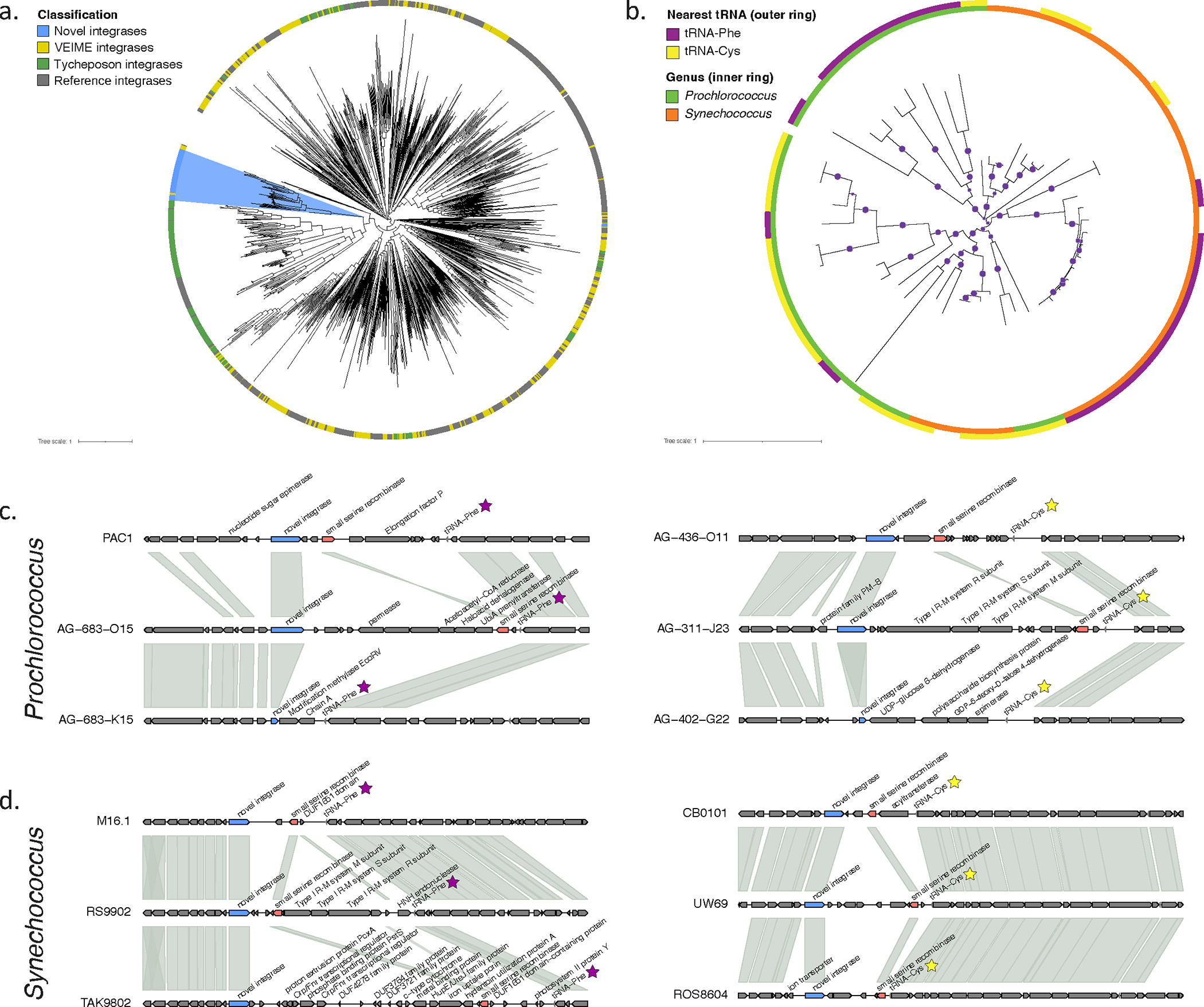
(a) Phylogenetic relationship of the identified integrases (blue) in comparison with tyrosine recombinases described in marine viral parasites (VEIMES; yellow), cyanobacterial mobile element integrases (green) and classes commonly found among well-described phage and mobile elements (e.g. IS, PICIs, ICEs). (b) Phylogenetic groupings of full length (>350aa) integrases in Prochlorococcus and Synechococcus, in relationship to the closest downstream tRNA (outer ring) and genome taxonomy (inner ring). Gaps reflect unknown tRNA associations from limitations of genome assemblies. Purple dots indicate branch supports of 0.5 or greater. (c and d) Genomic context of the integrase in selected marine Prochlorococcus and Synechococcus genomes, respectively. Colored genes indicate the integrase (blue), a small serine recombinase frequently found near the integrase (red) and the downstream tRNA (purple or yellow). Shaded regions connect orthologous genes.
PLM-based annotation uncovers dispersed major capsid protein
As a further demonstration of using the PLM-based classifier to uncover biologically informative annotations, we next turned to viral MCPs. MCPs serve as the core element of the virion capsid and are frequently used to define viral lineages. To showcase the power of our approach to annotate unexplored regions of viral sequence space, we utilized the function classifier to identify unannotated MCPs. The classifier predicted 8,398 unannotated VPFs in the EFAM database as belonging to the ‘head and packaging’ category. We manually investigated one high confidence cluster (EFAM cluster41798) using structural homology and found evidence that it is a HK97-like MCP. The VPF has high sequence homology to sequences found throughout the oceans, and their global diversity is broadly divided into two clades (Figure 6a). This MCP is also found in other environments such as aquatic sediments and freshwater lakes, where it was also unannotated (Supplemental Table 6). The putative MCP is consistently found near other ‘head and packaging’ proteins (Figure 6b), a pattern that is widely observed among known MCPs in bacteriophage genomes44. Despite the sequence divergence among the MCP-containing genome scaffolds, the high-level functional annotations provided by the classifier revealed similarities in the genome organization of this viral element (Figure 6c). Together, these data are consistent with the identification of a previously unannotated MCP by the PLM classifier, and further highlight the potential for using patterns in high-level functional annotation as a tool for viral genome identification and/or characterization.
Figure 6: Discovery of a major capsid protein (MCP).
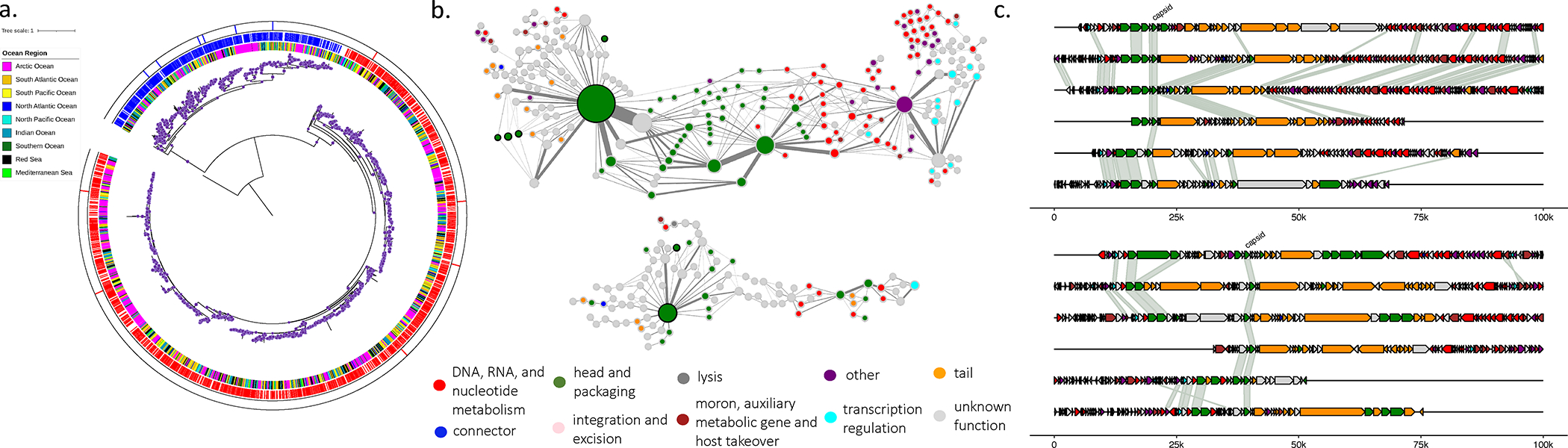
(a) Phylogenetic relationship of the MCP and distribution across the global oceans. The MCP has two major clades (red and blue). Purple dots indicate branch supports of 0.5 or greater. (b) Network depiction of MCP containing contigs where protein clusters (PCs, depicted as nodes) are constructed from all contigs that contain the MCP and the number of times two PCs are adjacent on a contig is counted (depicted as edges). For visualization, PCs are filtered for size >= 10 members. Node size reflects the number of proteins in the PC; edge width reflects the number of times PCs are adjacent; and black halo on node indicates the PC is a cluster of the MCP. (c) Genomic context of the MCP in selected contigs from the two MCP clades. Color of nodes (b) and genes (c) are predicted functional class by the PLM-based function classifier. Networks and genome maps represent the two clades from (a), top (red) and bottom (blue).
Discussion
While large-scale environmental metagenomic data have revealed an astounding amount of viral diversity, current approaches annotate on average less than 30% of viral protein families6,7,16,21,22. This limited understanding of global viral sequence space represents a clear barrier to our understanding of viral biology, restricting interpretation to those sequences with sufficient similarity to the small fraction of well-characterized viral genomes. Annotating viral proteins is also key to studies of viral evolution45, characterization of isolate genomes46, and to understand the role of viruses as disseminators of DNA in microbial populations47. Here, we demonstrate the utility of PLMs to improve classification of sequences within large-scale metagenomic datasets. Our work provides a proof of concept that high-level viral functions can be learned with PLM-based representations and extends existing capabilities for remote homology detection. These models thus represent a useful complement to widely used, state-of-the-art, alignment-based methods to provide novel insights into viral biology. The utility of incorporating PLM-based models into bioinformatic discovery workflows is highlighted by the above identifications of unannotated viral-like proteins from large-scale ocean datasets. The PLM classifier enabled the characterization of a previously unrecognized integrase that may define a mobile element in abundant marine picocyanobacteria, as well as that of an unannotated, HK97-like MCP found throughout the global oceans. These preliminary identifications, supported by contextual bioinformatic data, represent only two of thousands of annotations provided by this approach. Thus, high-level functional annotations can serve a useful role in biological discovery by helping to identify candidate proteins of interest for detailed study from vast sequence datasets.
As with all classifiers, the PLM model used here is highly dependent on the nature of the training data. PHROGs functional categories are aggregations that differ in their granularity and specificity, as well as in the number of VPFs and total sequences they contain. We have relied on the database category definitions and chosen to include all categories to maintain fidelity to their characterization of the functional space as a whole, as well as the relevance of all categories in our applied classifier. While the categories ‘other’ and ‘moron, auxiliary metabolic gene and host takeover’ are not functional descriptions, they contain groups of functions that make up substantial fractions of the categories that could be learned by the classifier, including transferases in the former and membrane proteins in the latter.
We show that across all nine categories in PHROGs, a single multi-class classifier was able to learn viral protein function across the annotated PHROG VPFs. ‘Tail’ and ‘DNA, RNA, and nucleotide metabolism’ had the highest predictive performance and the largest number of families. The heterogeneity of the ‘other’ and ‘moron, auxiliary metabolic gene and host takeover’ did result in worse performance for these classes, though both could be predicted. Even with total sequences and number of VPFs in the bottom third of categories, ‘lysis’ and ‘integration and excision’ both had high predictive capacity. ‘Head and packaging’ has similar counts to the highest performing classes but did not perform as well. Taken together, the number and diversity of sequences in a function are factors in the predictability of the function but do not fully explain the performance, highlighting an area for further investigation.
Of the PHROGs categories, the classifier was able to achieve the largest increase in annotations in the EFAM dataset for the ‘head and packaging’ and ‘tail’ categories. These groups had greater intra-family embedding similarity compared to other categories for families with low average sequence identity. These categories describe functions related to virion physical structure and represent one axis of the diversity of viruses. PLMs are hypothesized to perform best at capturing structural similarity in protein sequences27,28; that ‘head and packaging’ and ‘tail’ have VPFs that are relatively better captured by the PLM could indicate that strongly conserved structural features are a defining characteristic of these VPFs. The ability to better annotate proteins with these functions that are foundational for viral biology will contribute to cataloging viruses across environments.
PLM model training is computationally expensive, and one motivation of this work was to determine whether pre-trained PLMs can be effectively leveraged for challenges in metagenomics through transfer learning, or the application of knowledge learned in one task to another task. We evaluated four PLMs with different training corpora, architectures, and objectives. Utilization of a PLM trained on the largest existing protein sequence database48, including sequences from uncultivated genomes in metagenomic sequencing data, resulted in the best function classifier performance. Interestingly, supplemental supervision tasks in PLM training related to structure30 or function29 did not result in better classification performance. It is possible that this is due to the dearth of viral protein representation in protein structure and knowledge databases, and future work is necessary to determine if there are viral-specific supervised tasks that can enhance PLM training. However, our work demonstrates that transfer learning with pre-trained PLMs can be utilized for targeted biologic problems by researchers who cannot access the computational resources necessary to train large language models. We note that our classifier was trained on PHROGs and then calibrated on viral metagenomes from the EFAM global oceans database, thus making the final model particularly well-suited for discovery in marine metagenomes. For other ecosystems, such as soils or host-associated microbial communities, the initial PHROGs training could be augmented by calibrating on ground-truth datasets from the ecosystem under study.
As part of our efforts to explore and validate the classifier predictions, we bioinformatically identified a mobile genetic element defined by a previously unrecognized integrase related to the tyrosine integrase/recombinase family. The genomic context of these integrases indicates that their activity contributes to generating genomic diversity among globally abundant marine picocyanobacteria. We identified representative sequences of this integrase in cultured isolate and single-cell genomes of Prochlorococcus and Synechococcus and found that the region immediately surrounding the integrase represents a genomic island whose length, gene content, and gene orientation varies among individual genomes. Variable genes found near the integrase include putative restriction/modification systems, biosynthetic enzymes, and nutrient acquisition genes, indicating that the integraseassociated element can move genetic cargo of ecological relevance in the ocean. The consistent proximity of the integrase to two specific tRNAs suggests these as likely integration sites for the element. The integrases are also frequently, though not exclusively, found near a small serine recombinase which might contribute to resolving mobile element insertion into a target molecule49. However, the specific mechanism through which this element is mobilized or integrated is not yet known. Mobile genetic elements, frequently defined in part by their associated integrases, are widespread in environmental samples and are still being discovered and characterized39,40. As such, expanding the ability to rapidly identify such enzymes represents an important step in understanding the origins and dynamics of these elements. While not specifically investigated here, other proteins classified as being in the ‘integration and excision’ functional category may be of particular interest in viral profiling studies, where they are used to distinguish between lytic and temperate viral life-cycles13,16,50.
We next mined EFAM predictions in the ‘head and packaging’ category to seek unidentified capsid proteins, a key calling card of viral genomes. The high-throughput classifier annotations identified a strong hit with structural similarity to a HK97-like MCP and that appears to be widespread in the global oceans. With contigs containing the MCP in hand, we utilized our classifier to reveal conserved genomic organization across these sequence diverse contigs. A major benefit of the high-level functional annotations that is particularly important for viral sequences is their ability to highlight functional conservation that spans sequence-diverse proteins. We find that our newlyidentified MCP is in the neighborhood of other predicted ‘head and packaging’ proteins, as would be expected for capsid genes. In one of the two architectures we identified, this neighborhood is flanked on one side by ‘tail’ proteins and on the other side by ‘DNA, RNA, and nucleotide metabolism’ proteins, which are in turn flanked by ‘transcription regulation’ proteins and host-associated proteins. Together, this high-level functional annotation begins to paint a picture of what this viral element ‘looks’ like genomically in different ocean regions at a level of abstraction that may be appropriate for examining features of genome architecture that would be difficult to resolve from sequence similarity alone.
Our study must acknowledge several limitations. In attempting to systematically annotate VPF function and highlight the ability to label individual VPFs, we note that for experimentalists interested in annotating UViGs there are a plethora of methods, parameters, and thresholds to decide, and they may arrive at an annotation for a specific gene not annotated in large-scale approaches by thorough investigation. Annotation goals are projectspecific and may require different levels of annotation granularity; here we have focused on protein family level annotations. In selecting the PHROGs database for training the function classifier, we benefited from the highlevel functional category annotation which collapses a wide array of annotation terms into defined categories. However, the categories vary in their scope and while some are relatively narrow (e.g., ‘integration and excision’ and ‘lysis’) and their prediction can be relevant to experimentalists, the ones that are comparatively broad are limited in their ability to provide specific information when predicted.
In conclusion, our PLM-based classifier is trained on the same data that underlies the PHROG pHMMs yet can detect homology across a larger sequence space, identifying proteins that the original pHMMs and other annotation tools did not. This suggests that PLMs are accessing features of sequence space that alignment-based methods cannot and are thus a complementary approach to these existing, widely-used, methods. Using our approach, targeted hypotheses about protein function can be gleamed from PLM-based classification and then tested experimentally, providing a powerful method for directing study into currently hidden functions of interest.
Methods
Viral protein sequence data
The PHROGs VPF database v321 (https://phrogs.lmge.uca.fr/) was downloaded on 01/26/2022. Re-annotation data was downloaded after the v4 release. The EFAM VPF database was downloaded from its project repository on the CyVerse Data Commons on 09/07/202251. PHANNs protein sequences and annotations37 (https://phanns.com/downloads) was downloaded on 01/17/2023.
PHROGs intra-category family sequence similarity
PHROG VPF similarity was measured using hhsearch52 for each family against a HMM database of all families in a category and the average score was collected for each VPF. Category HMM databases were constructed by converting all category families multiple sequence files downloaded from PHROGs to a3m format and then constructing a hhm database using ffindex build, ffindex apply, and cstranslate as described in hhsuite v3.3.052.
Protein language models
Protein sequences were embedded to vectors using trained PLMs. The Transformer BFD PLM from the Prot-Trans28 project was used via the DeepChainBio/BioTransformers python package (https://github.com/DeepChainBio/biotransformers). Sequences were embedded with pool mode=‘mean’ and batch size=2. Sequences were cut off at 5,096 amino acids which is the limit of the Transformer BFD PLM. LSTM Uniref90 and LSTM Uniref90 MT from the ProSE30 project were download from the project GitHub repository and protein sequences were embedded with the embed sequences.py script with –pool avg. Transformer Uniref90 MT from the ProteinBERT29 project was downloaded from the project GitHub repository and protein sequences were embedded using the get model with hidden layers as outputs function in the proteinbert python package. All protein sequence embedding was performed on 2 NVIDIA TITAN V GPUs.
Classifier training and evaluation
To test the ability of a model to predict a functional category for a test sequence, all labeled PHROG families were split into five stratified sets for five-fold cross-validation. In each split, training was done on all sequences in the training families while testing was performed on a single randomly selected sequence from the testing families. Data preparation for model training was done using scikit-learn53 methods StratifiedKFold and LabelBinarizer. The same training-validation procedure was used for the five-fold cross-validation of virion structure and infection (cluster1) vs viral genome replication and other host derived genes proteins (cluster2).
The classifier architecture is a dense, feed-forward neural network, which has been shown to perform well with protein embeddings as input26, and was trained with tensorflow54. The network has three hidden layers of dimensions 512, 256, and 128 trained with 20% dropout and ReLU activation. The output layer is of dimension equal to the number of functional categories being predicted and has a softmax activation. Input dimension is equal to the embedding vector length output from the PLM. For PLMs with embedding dimension greater than 1,024, an additional hidden layer of dimension 1,024 was added as the first hidden layer. The model was fit with the following parameters: n _epoch=20, loss=categorical crossentropy, opt=Adam(0.0001), batch size=60. Class prediction is assigned based on the highest probability of the softmax layer. We did not perform hyper-parameter optimization, which could result in a higher performing model. For binary classifiers based on clusters of PHROGs functional categories and for the EFAM classifier, the same architecture and training parameters are used with the exception of n epochs=5.
For training the PHROGs function classifier used in the EFAM classification experiment, families from the ‘unknown function’ category were included as an additional functional category. However, because the unknown families may be missing annotation, any family that was predicted by the model trained without the unknown function category with a score >0.8 was removed from training (n=9,080), leaving 24,712 families for training.
Evaluation for the classifier was measured per-functional category using area under the receiver operating characteristic curve (AUROC), area under precision-recall curve (AUPRC), and the F1-score: , where TP, FP, and FN are the number of true positive, false positive, and false negatives predicted, respectively. ROC and PRC curves, AUC, and F1-score were all calculated using scikit-learn53 methods roc curve, precision recall curve, and auc. In the case of PHROGs five-fold cross-validation, true labels are known for holdout families. In the case of EFAM, true labels are assigned based on HMM matching of EFAM families to PHROG families. EFAM families were aligned using clustal omega v1.2.455 and searched against the PHROG HMM database using hhsearch52. PHROGs functional label assignment was made if an EFAM family matched a PHROGs HMM with e-value < 1E-10. The label of the PHROGs family with the lowest e-value is considered the true label unless that label is unknown function in which case the next lowest family label is assigned. For predicting EFAM category in the absence of PHROGs HMM hits, the decision threshold probability for category assignment in EFAM was identified by calculating the per-category maximum F1-score with FDR <= 0.1. Model calibration analysis was performed with scikit-learn53 calibration curve method and n bins=10. Our trained classifier is available for download (https://github.com/kellylab/viral-protein-function-plm). For EFAM VPFs with annotation in the EFAM database, annotation terms present > 10 times in families predicted by the classifier as ‘integration and excision’ are shown to highlight the split around proteins of length 120 in the category.
Viral protein family embedding space
PHROGs v4 annotation were used for interrogation of the embedding space. PHROGs families were collapsed to centroid vectors by taking the column average of the vector representation of all proteins in a family. Uniform Manifold Approximation and Projection for Dimension Reduction (UMAP) in python56 was used to visualize embedded VPFs. Cosine similarity is a measure of similarity between two vectors and is calculated:
where u and v are vectors of length n and xi is the i-th element of each vector. It is used to measure sequencesequence similarity and family-family similarity from protein vectors and family centroid vectors, respectively, and calculated using scikit-learn53. Families with vector similarities > 0.999 (n=312) were excluded from category median family mean sequence-sequence similarity calculation as some families have only duplicate sequences as PHROGs did not de-duplicate protein sequences. For intra-category similarity, pairwise similarity was calculated for all category families. For inter-category similarity, each family in one category was compared to each family in another category with the mean across all pairwise comparisons constituting the category-category similarity. Differences in the distribution of similarities between categories were evaluated with the independent student t-test with Bonferroni correction using statannotations57. The category-category similarity matrix was converted to a network using networkx58 and displayed with spectral layout. The distance matrix was clustered using scikitlearn53 SpectralClustering with n clusters=2.
PHROGs intra-family sequence similarity
Sequence identity was calculated using the Bio.Align.PairwiseAligner59 method to find the global alignment score with default scores: match score = 1, mismatch score = 0, and gap score = 0. Sequence identity was calculated for all pairwise sequence combinations in a family and averaged for a single family score. A linear regression for family sequence identity and embedding similarity was calculated using scipy60.
Phage virion protein classification
To compare the performance of PLM representations for PVP identification, we used the PHANNs37 database.
PHANNs protein sequences were embedded using the Transformer BFD PLM and a PVP vs ‘other’ classifier was trained with the same architecture and parameters as the cluster1 vs. cluster2 classifier. Training and testing sequence split is as described previously36. All sequences in the 10 PHANNs validation splits for all PVP classes are combined to a single PVP training set (n=154,183) and all 10 ‘other’ validation splits were combined to a single ‘other’ training set (n=336,151). Testing was done on the held PVP sequences for all classes (n=14,477) and the held out ‘other’ sequences (n=33,402).
Viral protein sequence annotation validation tools
Viral sequence predictions were manually validated using existing sequence and structural homology software. Individual sequence homology was performed with NCBI-hosted blastp61 using the nr database and default parameters. Domain prediction was performed using InterPro62. MPI bioinformatics suite63 was used for searching protein sequences against HMM databases using hhpred64 with default databases (PDB mmCIF30 10 Jan, UniProt-SwissProt-viral70 3 Nov 2021, COG KOG v1.0, PHROGs v4) and parameters and for searching sequence databases (nr30 17 jan) for HMM hits using HMMER v3.3.265 with default parameters. Phyre2 was used for protein structural fold prediction and 3D model prediction66.
Investigation of predicted integrase protein families
A putative integrase protein sequence (MAK08069.1) from cluster158946 was used to search MGniFY67 for similar sequences in metagenomic datasets. We took the first MGnify hit, MGYP000503484273 (e-value 3.3E-257), and used it as a seed to search for additional sequences using the IMG/VR68,69 Viral Protein Database using default cutoffs (1E-5). The search uncovered putative integrase homologs from Prochlorococcus and Synechococcus genomes, which were interrogated further.
Integrase family sequences originally identified in IMG/VR were used to query a custom database of Prochlorococcus genomes from cultured isolates and single cell genomes39 and additional sequences, such as those from Synechococcus, were retrieved through blastp searches of the NCBI nr database. The tyrosine integrase phylogeny was constructed from a set of tyrosine recombinases extracted from the UniRef50 database (http://www.uniprot.org/uniref) using HMM models from ref41; a set of integrases associated with Prochlorococcus Tycheposons and cryptic elements39; and representative sequences of VEIME-associated integrases40 (based on 40% identity clusters as generated by MMSeqs270). Sequences were aligned with Mafft v7.520 (options –maxiterate 1000 –genafpair)71, a maximum likelihood phylogeny was generated using FastTree v2.1.11 using default settings72, and the tree was plotted using iTOL73. Genome regions surrounding the integrases were plotted in R using gggenomes 0.9.7.9000 (https://github.com/thackl/gggenomes).
Major capsid protein analysis
EFAM VPFs classified as belonging to the ‘head and packaging’ category, and that were unannotated, were investigated for putative MCPs. Using structure homology searching with the aligned cluster proteins in hhpred64 as above and individual cluster members in foldseek74, we found that EFAM cluster41798, while most similar to unannotated proteins, also contained hits to HK97-like MCPs. We next looked for similar proteins encoded within the GOV2.0 dataset3, as predicted by prodigal75 v2.6.3 (options -p meta –c). The cluster41798 HMM was used with hmmsearch65 v3.3.2 to identify sequences at a 1E-100 cutoff, yielding a total of 2,203 candidate MCP sequences. Capsid sequence alignments and phylogeny were computed as above for the integrases. We use the best hhpred hit for an experimentally determined structure (PDB: 6WKK D, e-value=6E-7) as an out-group for the MCP sequence tree. To see if the MCP is found in other environments, cluster41798 was used to query the geNomad database (v1.3)76 of viral protein marker families using hmmscan65 v3.3.2 to identify families at a 1E-100 cutoff, yielding one family (GENOMAD.062939, e-value=9.7E-122). Genome and ecosystem annotation was pulled from IMG/VR68 where available.
All contigs containing the MCP were used to construct gene neighbor networks. Contigs were de-replicated at 95% identity and protein clusters (PCs) were constructed at 50% identity for all proteins on de-replicated contigs using MMSeqs2 v14.7e28470 (respective parameters: contig: -c 1.0 –cluster-mode 2 –cov-mode 1 –min-seq-id 0.95; protein: -s 6 -e 1e-5 -c 0.8 –cov-mode 0 –cluster-mode 2 –min-seq-id 0.5 –cluster-reassign 1). The number of times two PCs are immediately adjacent on contigs is used to construct a network where nodes represent PCs and edges represent instances of PCs being adjacent. Network visualization was done in cytoscape77. PC functional classification was assigned using the EFAM-calibrated function classifier. If two classes were predicted for a PC, the higher probability assignment was used for labeling. Genome regions surrounding the MCPs were plotted as above for integrase genomes.
Protein structure modeling of identified integrase sequence
Protein structure can be conserved among very distantly related sequences. We previously utilized homology modeling approaches to identify distantly related structural homologs to novel viral capsid protein sequences78. Here, we took a similar approach to identify structures related to sequences in our putative integrase family. We utilized the fully automated protein structure homology-modelling server SWISS-MODEL via the Expasy web server79 for template selection, target/template alignment, and model generation using default parameters for an integrase sequence from the Prochlorococcus PAC1 genome (WP 052038630). The top template, as identified by the Global Model Quality Estimate score, was PDB ID 1Z1B, the phage lambda integrase80. The target/template alignment has 13% sequence identity, consistent with our sequences not previously being identified as integrases. The MolProbity protein quality score, provided by SWISS-MODEL, which combines protein structure quality features that together reflect crystallographic resolution, was 2.281. The lambda integrase is a tyrosine recombinase with defined active site residues Arg 212, Lys 235, His 308, Arg 311, His 333, and Tyr 34280. In a study of catalysis requirements for tyrosine recombinases, the key residues strictly required for function were identified as the Tyr (Y) and Lys (K) residues42. The target/template alignment demonstrates that residues Arg 212, Lys 235, Arg 311, and Tyr 342 are conserved in our target sequence (Extended Data 4, panel A). The sequence is modeled as a homo-tetramer, consistent with the quaternary structure of the template (Extended Data Figure 4, panel B).
Study code, data, and visualizations
The trained classifier is available for download82 as well as PLM representations for PHROGs and EFAM protein sequences used in this study. Python packages numpy83 and pandas84 were used for analysis, matplotlib85 and seaborn86 were used for data visualization, and jupyter notebooks87 was used for analysis.
Extended Data
Extended Data Figure 1: Performance of four different PLM-based representations for viral VPF functional classification.
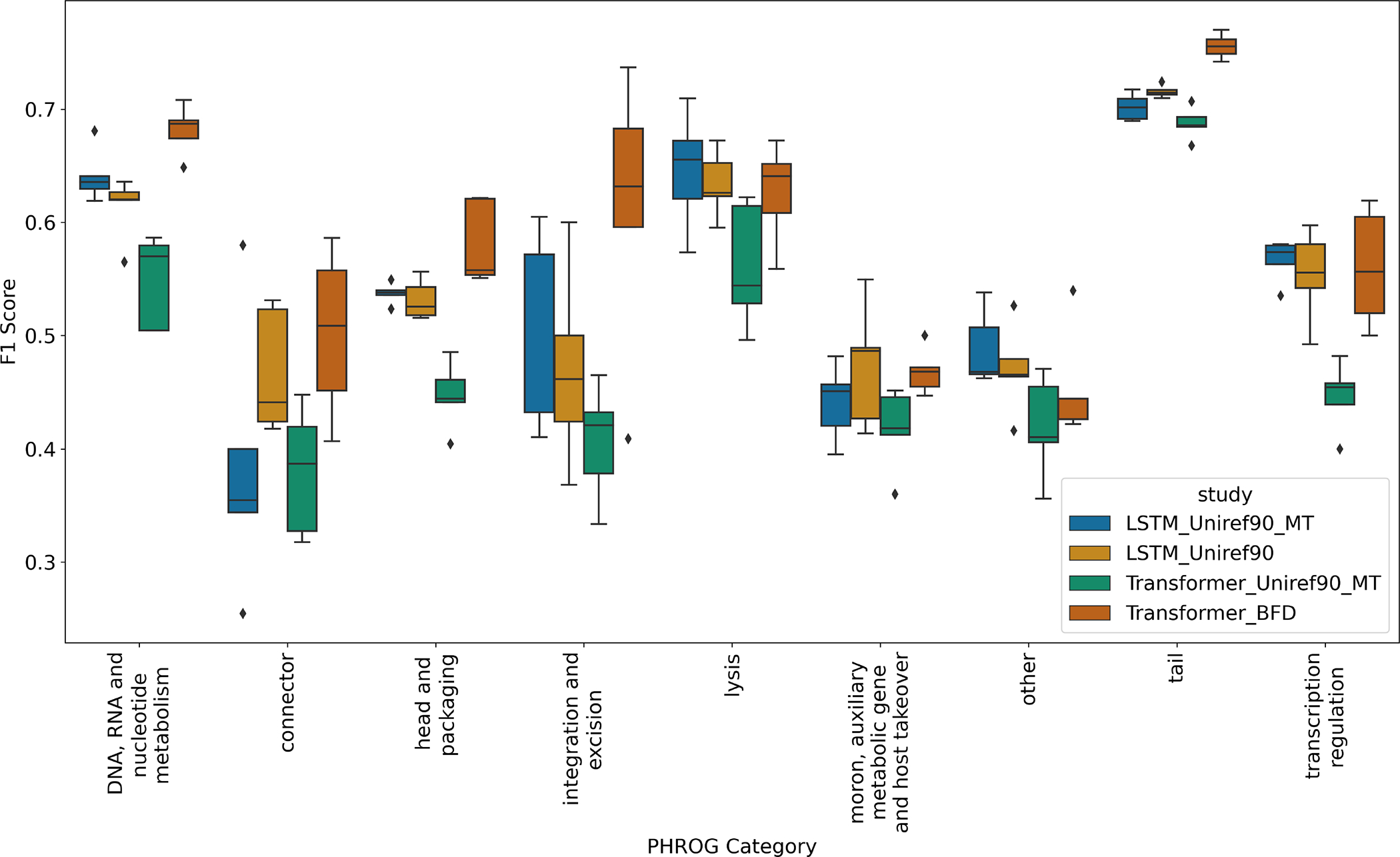
Embedded proteins were used to train and evaluate PHROGs functional annotation classification. Performance is measured as F1-score over five-fold training-testing splits of PHROGs VPFs (n=5). Study is described by the model architecture, protein source, and whether the PLM is trained with a multi-task training objective (MT). Boxes represent interquartile range; whiskers represent the entire distribution with the exception of outliers (diamonds); horizontal line indicates median. BFD-Big Fantastic Database; LSTM-long short-term memory.
Extended Data Figure 2: Evaluation of embedding similarities of constituent families between functional categories.
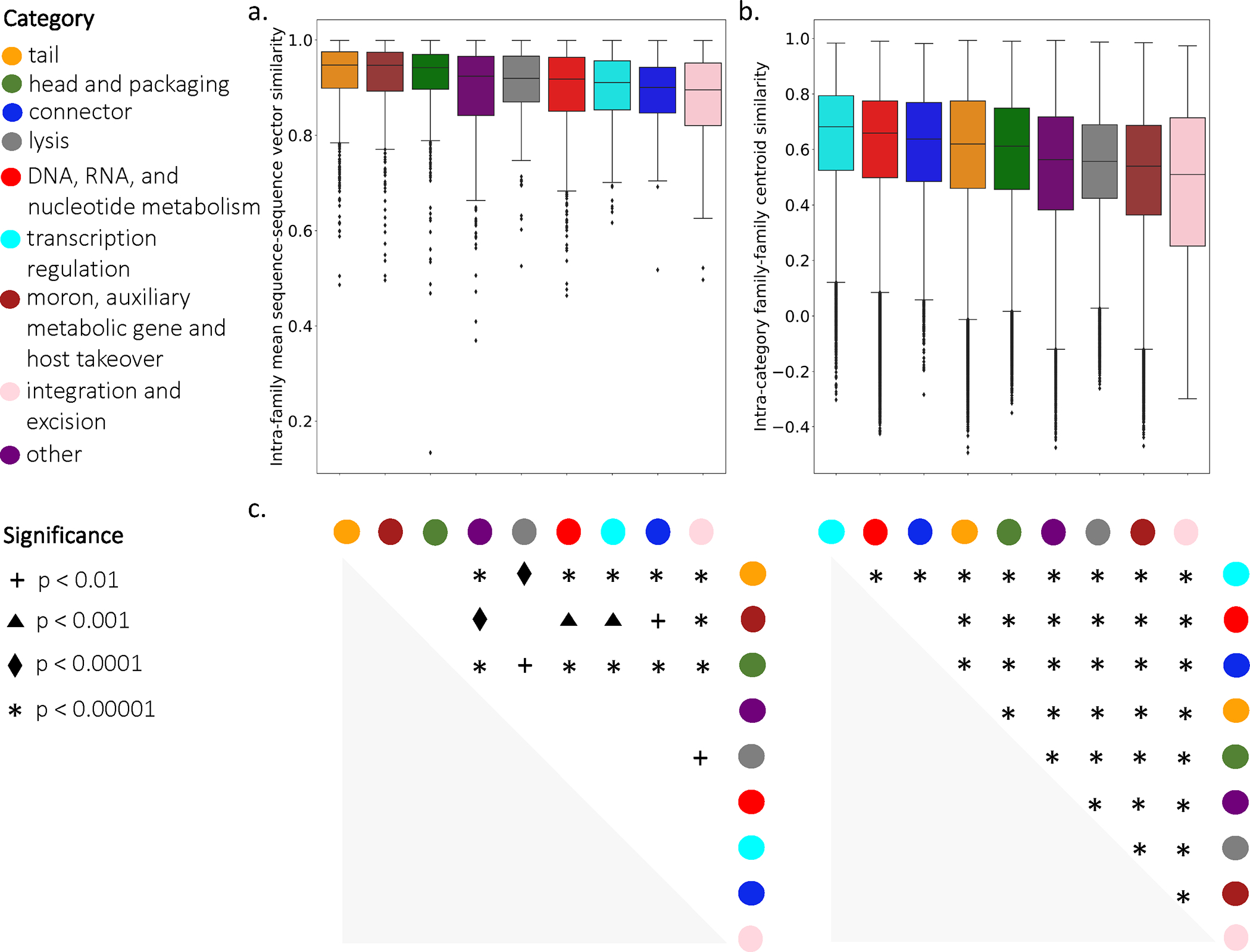
(a) Distribution of family average sequence-sequence similarity. (b) Distribution of family-family centroid similarity. (a-b: DNA- n=1,065; connector- n=133; head- n=946; integration- n=105; lysis- n=299; moronn=458; other- n=560; tail- n=1,219; transcription- n=303) (c) Significance of pairwise category distribution comparison using a two-sided independent t-test with Bonferroni correction (left-lysis vs. integration- p=1.493e-02; head vs. other- p=1.014e-11; head vs. lysis- p=1.096e-02; head vs. DNA- p=2.098e-10; head vs. transcriptionp=4.978e-07; head vs. connector- p=1.207e-05; head vs. integration- p=2.085e-09; moron vs. other- p=1.205e04; moron vs. DNA- p=2.084e-03; moron vs. transcription- p=9.269e-03; moron vs. connector- p=1.484e-02; moron vs. integration- p=5.970e-05; tail vs. other- p=4.449e-16; tail vs. lysis- p=4.479e-04; tail vs. DNAp=3.759e-15; tail vs. transcription- p=1.814e-09; tail vs. connector- p=3.121e-07; tail vs. integration- p=1.482e11, right-transcription vs. DNA- p=1.010e-111; transcription vs. connector- p=4.416e-22; transcription vs. tailp=7.299e-260; transcription vs. head- p=0.000e+00; transcription vs. other- p=0.000e+00; transcription vs. lysis- p=0.000e+00; transcription vs. moron- p=0.000e+00; transcription vs. integration- p=0.000e+00; DNA vs. tail- p=1.943e-208; DNA vs. head- p=0.000e+00; DNA vs. other- p=0.000e+00; DNA vs. lysis- p=0.000e+00; DNA vs. moron- p=0.000e+00; DNA vs. integration- p=0.000e+00; connector vs. tail- p=4.234e-06; connector vs. head- p=7.253e-30; connector vs. other- p=2.294e-228; connector vs. lysis- p=1.662e-196; connector vs. moron- p=0.000e+00; connector vs. integration- p=2.827e-271; tail vs. head- p=1.145e-243; tail vs. otherp=0.000e+00; tail vs. lysis- p=0.000e+00; tail vs. moron- p=0.000e+00; tail vs. integration- p=0.000e+00; head vs. other- p=0.000e+00; head vs. lysis- p=0.000e+00; head vs. moron- p=0.000e+00; head vs. integrationp=0.000e+00; other vs. lysis- p=9.303e-21; other vs. moron- p=1.099e-188; other vs. integration- p=6.600e-87; lysis vs. moron- p=4.171e-204; lysis vs. integration- p=5.199e-138; moron vs. integration- p=1.478e-25). Boxes represent interquartile range; whiskers represent the entire distribution with the exception of outliers (diamonds); horizontal line indicates median.
Extended Data Figure 3: Inter-category similarity for PHROGs functional categories.
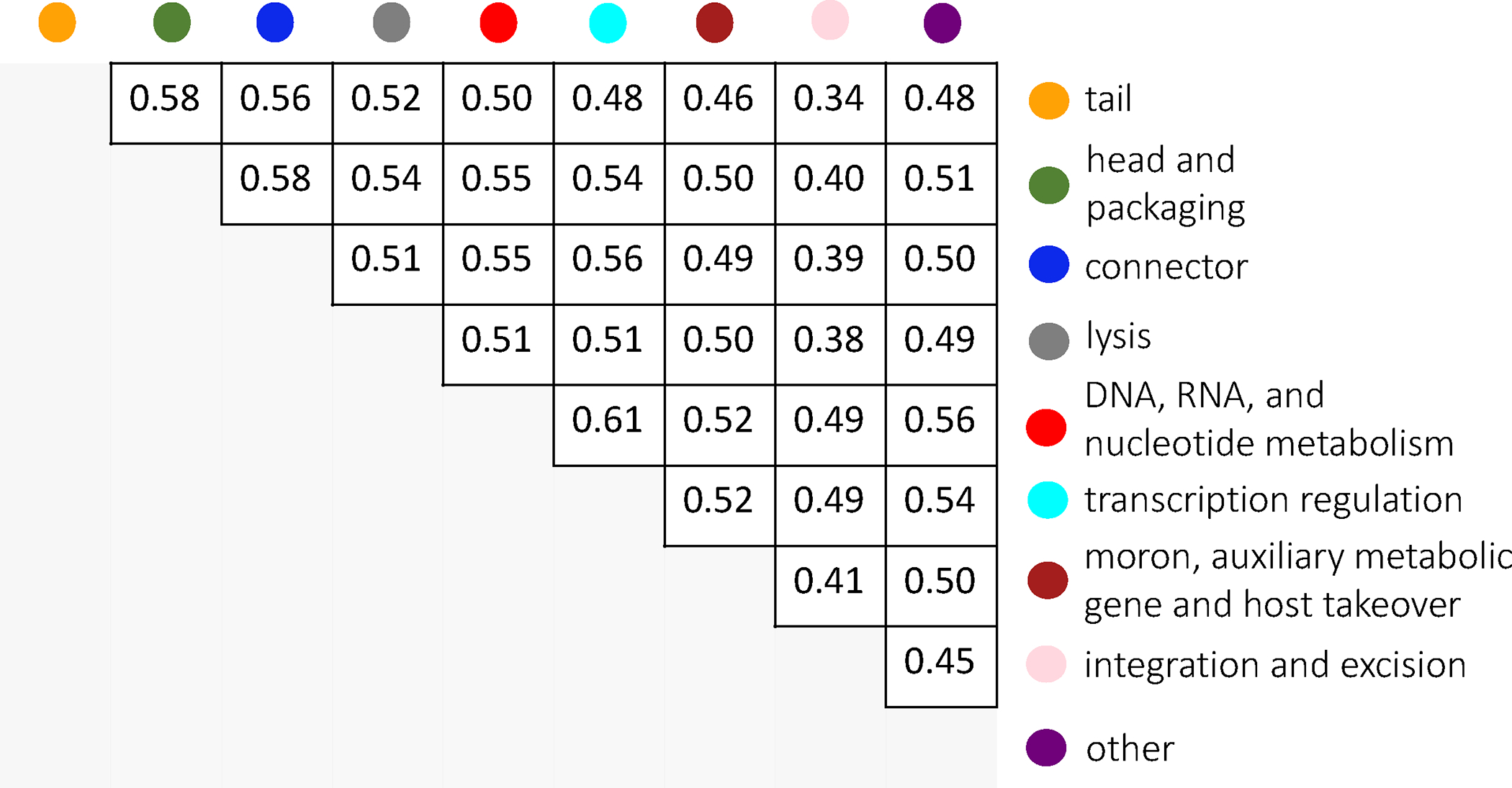
Pairwise family centroid similarities were calculated for every combination of families between the two categories. Score is the average over all comparisons.
Extended Data Figure 4: EFAM function classifier calibration analysis.
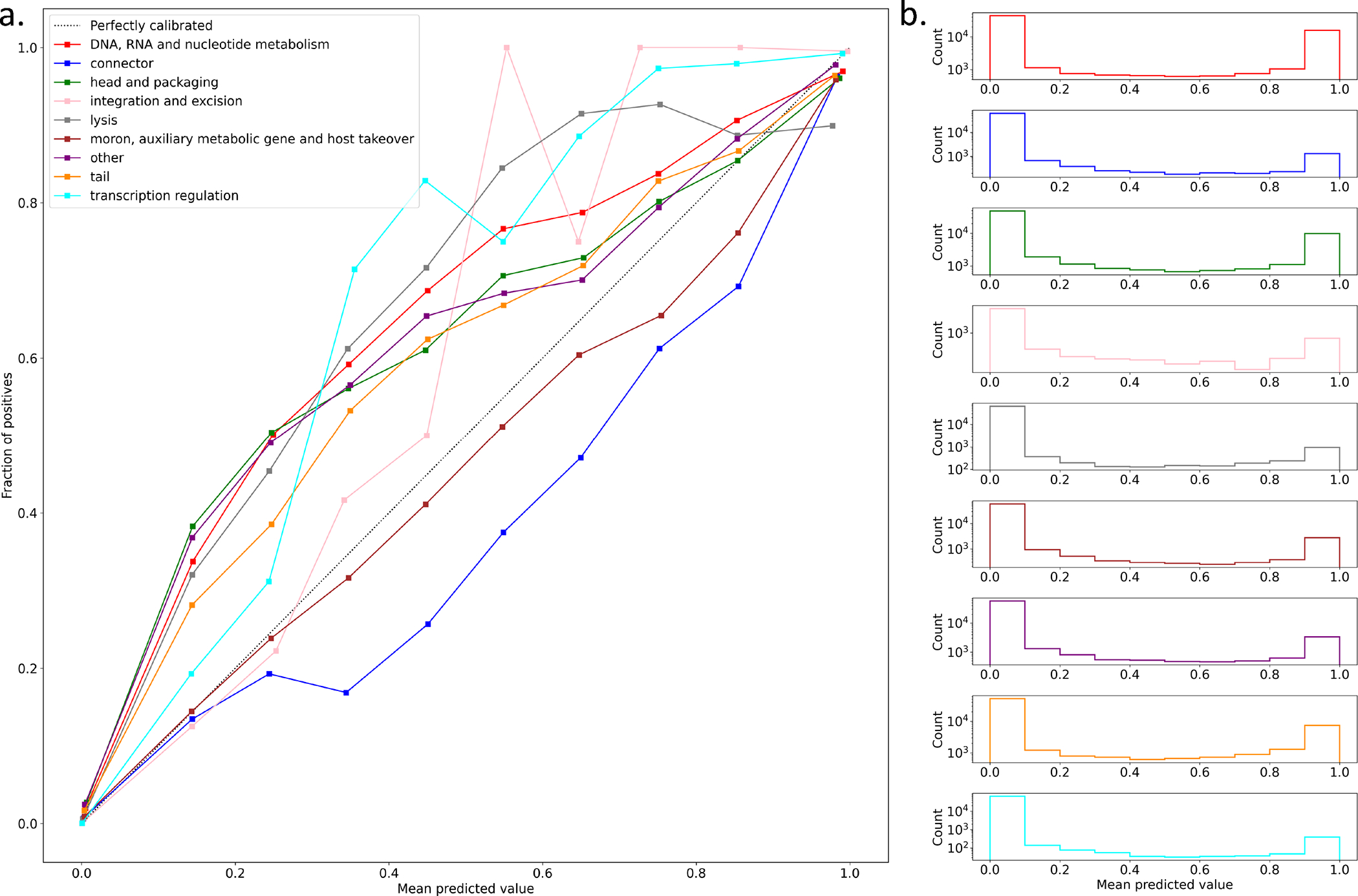
(a) EFAM VPFs that have hits to annotated PHROG HMMs (test set) are used to evaluate the model calibration for each category. For each class, probabilities across all VPFs in the test set are binned into 10 partitions and the fraction of true positives for each bin is calculated. A perfectly calibrated model (dotted line) has a true positive proportion equal to the mean predicted probability for each bin. Below the perfect model indicates overconfidence and under the perfect model indicates under confidence. (b) Histogram of the number of predictions across the test set for each probability bin.
Extended Data Figure 5: Decision threshold evaluation for function classifier predictions on EFAM VPFs.
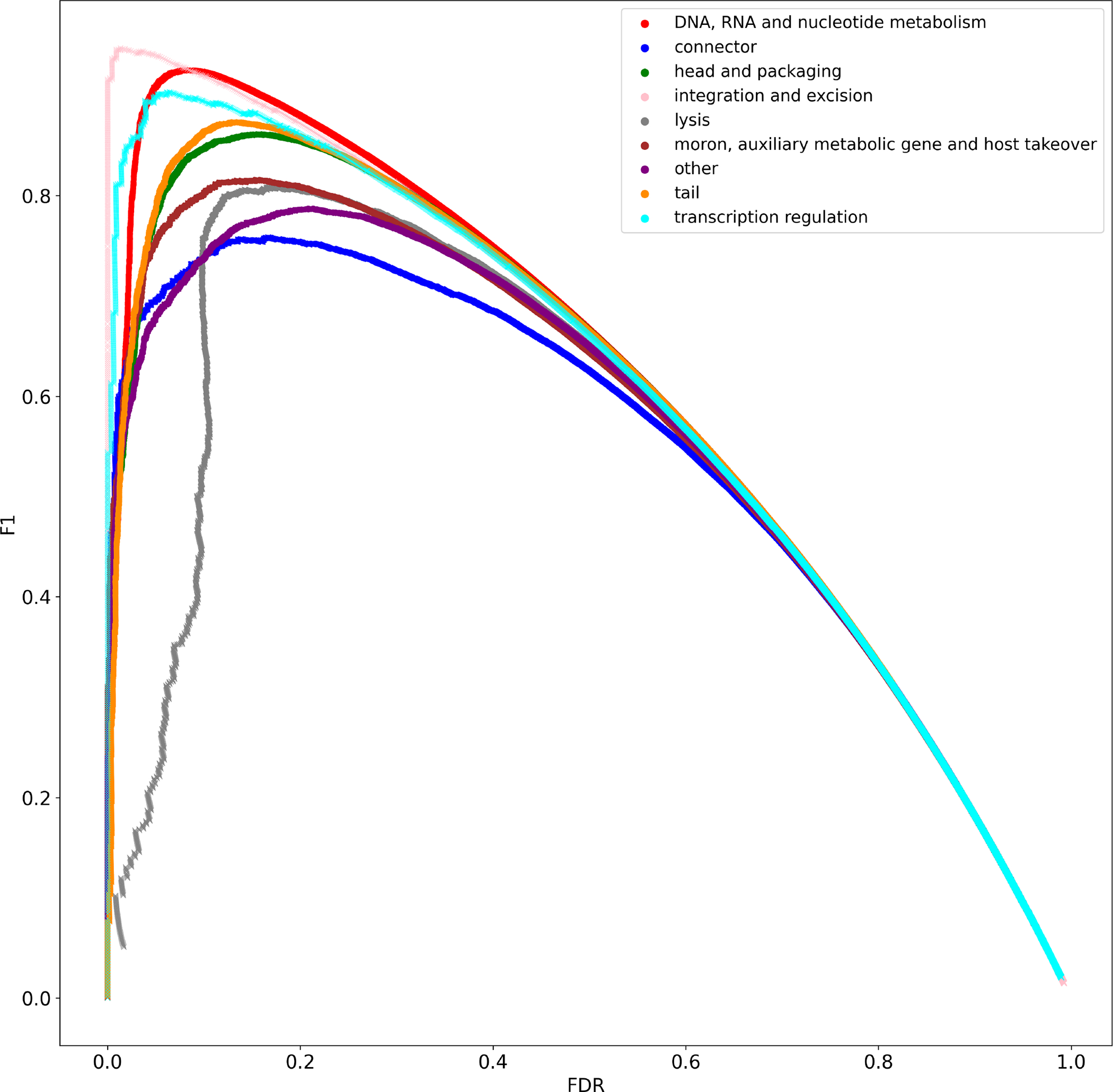
EFAM VPFs with PHROG hits were used as ground truth for prediction with the function classifier. Classifier thresholds are determined by considering false discovery rate (FDR) and F1-score (F1). The final decision threshold for each class is the decision boundary with maximal F1 with FDR <= 0.1.
Extended Data Figure 6: Comparison of PLM embedding similarity and sequence identity for PHROG VPFs.
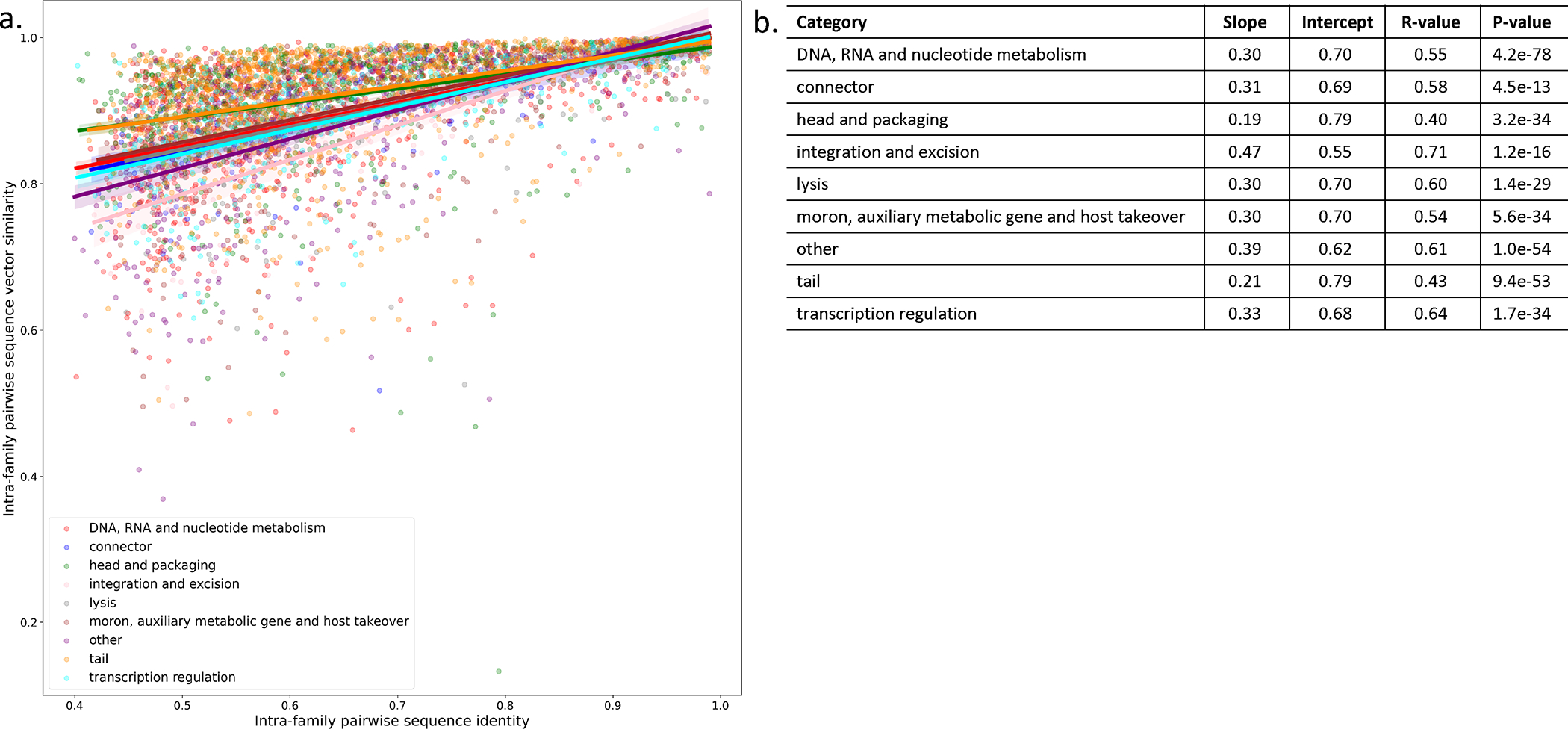
(a) The intra-family pairwise sequence embedding similarity, measured using cosine similarity, and sequence identity, measured using global alignment identity, were calculated for all annotated PHROG VPFs. Families are colored by functional category annotation. Solid line represents a linear regression for each function with shading representing a 95% bootstrapped confidence interval for the regression estimation. (b) Linear regression results for each category. R-value is measured using Pearson correlation coefficient. P-value is calculated using the Wald Test.
Extended Data Figure 7: Comparative protein structure modelling of an integrase family sequence supports annotation as a tyrosine recombinase.
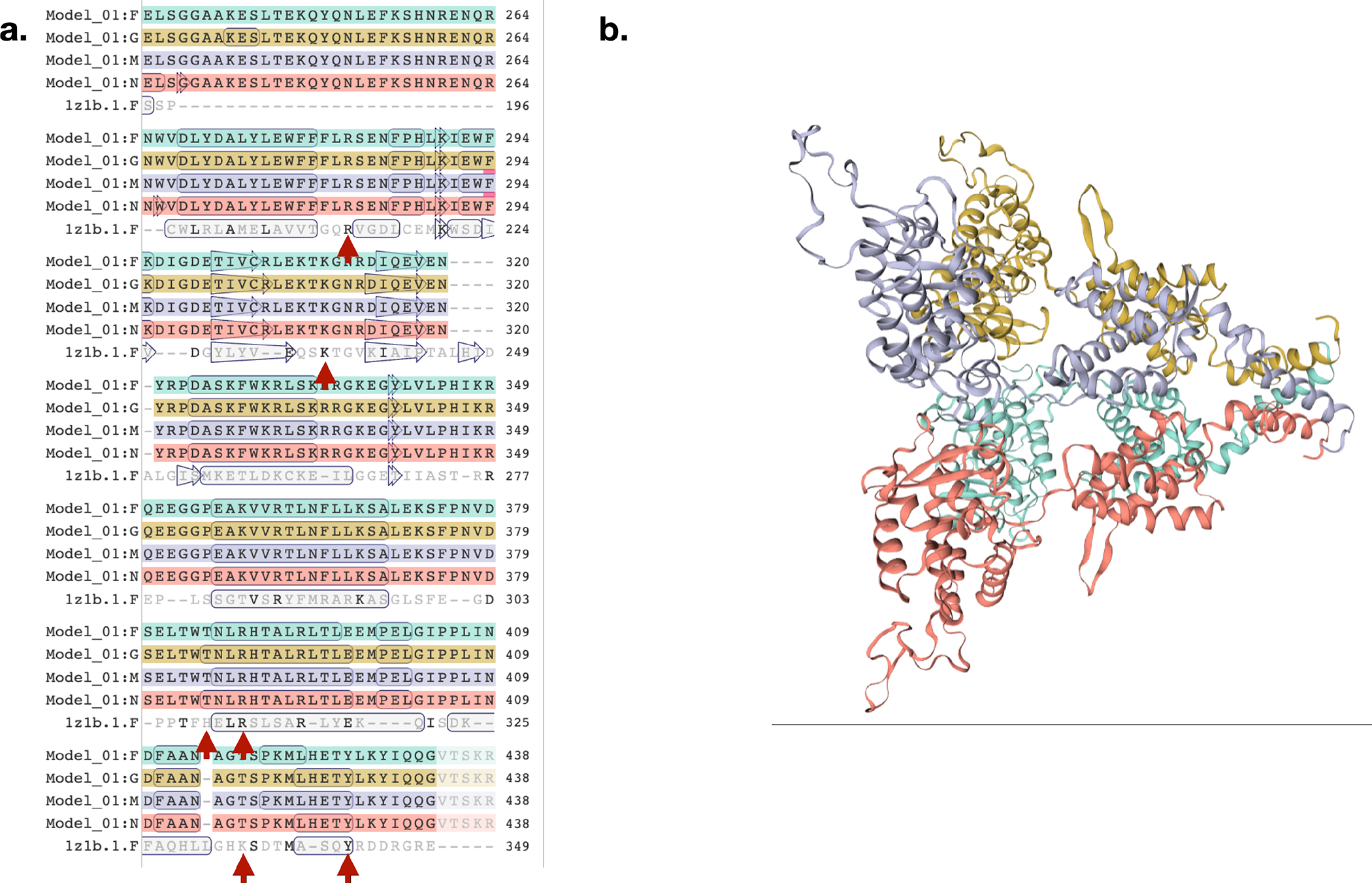
(a) Target/template alignment between the Prochlorococcus PAC1 sequence (indicated as Model _01), and the template sequence 1Z1B, the phage lambda integrase. Red arrows point to active site residues Arg 212, Lys 235, His 308, Arg 311, His 333, and Tyr 342. Boxed amino acid regions represent secondary structure. (b) Homology model of Prochlorococcus PAC1 sequence based on template 1Z1B. Colors indicate individual monomers of the homo-tetramer template protein structure in both a and b.
Supplementary Material
Acknowledgements
We thank Thomas Hackl, Kathryn Kauffman, and Cole Matrishin for helpful discussions. Z.N.F. was supported by the Einstein Medical Scientist Training Program (1T32GM149364). S.J.B. was supported by grants from the National Science Foundation (OCE-2049004 and OCE-2304066) and the Simons Foundation (Award ID 917971). L.K. is supported in part by NIH NHLBI grant R01HL069438. Computational resources were supported by an award from the Google Cloud Research Credits program (GCP19980904) to L.K. We thank the NVIDIA Academic Hardware Grant Program for GPUs used in this work.
Footnotes
Competing Interests
The authors declare no competing interests.
Code Availability
Code for generating embeddings and using the classifier is available on GitHub at https://github.com/kellylab/viral-protein-function-plm, DOI- 10.5281/zenodo.10182746. We made a no-code Google Colaboratory notebook available to utilize the classifier that is linked from the GitHub repository README. Code to produce figures and tables is available on GitHub at https://github.com/kellylab/viral-protein-function-annotation-with-protein-language-model, DOI- 10.5281/zenodo.10182750.
Data Availability
The PHROGs VPF database v3 (https://phrogs.lmge.uca.fr/) was downloaded on 01/26/2022. Reannotation data was downloaded after v4 release. EFAM VPF database was downloaded from project repository on CyVerse Data Commons on 09/07/2022. PHANNs protein sequences and annotations (https://phanns.com/downloads) were downloaded on 01/17/2023. geNomad HMM database and annotations (v1.3) were downloaded from zenodo (https://zenodo.org/record/7793532) on 08/10/2023. Protein sequences used for integrase and major capsid protein investigation were collected from the following databases: MGniFY, IMG-VR, NCBI nr, UniRef50. Protein sequence embeddings generated for PHROGs and EFAM sequences are available at https://doi.org/10.5281/zenodo.8339381. Additional data generated for this study is available in a public Google Cloud Platform bucket (http://storage.googleapis.com/viral protein family plm embeddings). See the README on the project repository https://github.com/kellylab/viral-protein-function-annotation-with-protein-language-model for details on downloading the data.
REFERENCES
- [1].Roux S et al. Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature, 537(7622):689–693, 2016. [DOI] [PubMed] [Google Scholar]
- [2].Paez-Espino D et al. Uncovering Earth’s virome. Nature, 536(7617):425–430, 2016. [DOI] [PubMed] [Google Scholar]
- [3].Gregory AC et al. Marine DNA Viral Macro- and Microdiversity from Pole to Pole. Cell, 177(5):1109–1123.e14, may 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].ter Horst AM et al. Minnesota peat viromes reveal terrestrial and aquatic niche partitioning for local and global viral populations. Microbiome, 9(1):233, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gregory AC et al. The gut virome database reveals age-dependent patterns of virome diversity in the human gut. Cell Host & Microbe, 28(5):724–740.e8, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Camarillo-Guerrero LF et al. Massive expansion of human gut bacteriophage diversity. Cell, 184(4):1098–1109.e9, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Nayfach S et al. Metagenomic compendium of 189,680 DNA viruses from the human gut microbiome. Nature Microbiology, 6(7):960–970, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Roux S et al. Virsorter: mining viral signal from microbial genomic data. PeerJ, 3:e985, May 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ren J et al. VirFinder: a novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome, 5(1):69, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ren J et al. Identifying viruses from metagenomic data using deep learning. Quantitative Biology, 8(1):64–77, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wood DE et al. Improved metagenomic analysis with Kraken 2. Genome Biology, 20(1):257, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Guo J et al. VirSorter2: a multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome, 9(1):37, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kieft K et al. VIBRANT: automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome, 8(1):90, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tisza MJ et al. Cenote-Taker 2 democratizes virus discovery and sequence annotation. Virus Evolution, 7(1):veaa100, jan 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Glickman C et al. Simulation study and comparative evaluation of viral contiguous sequence identification tools. BMC Bioinformatics, 22(1):329, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Camargo AP et al. Identification of mobile genetic elements with genomad. Nature Biotechnology, Sep 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Meier-Kolthoff JP and Goker M.¨ VICTOR: genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics, 33(21):3396–3404, 07 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bin Jang H et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by genesharing networks. Nature Biotechnology, 37(6):632–639, 2019. [DOI] [PubMed] [Google Scholar]
- [19].Moraru C. Virclustmdash;a tool for hierarchical clustering, core protein detection and annotation of (prokaryotic) viruses. Viruses, 15(4), 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pons JC et al. VPF-Class: taxonomic assignment and host prediction of uncultivated viruses based on viral protein families. Bioinformatics, 37(13):1805–1813, 01 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Terzian P et al. PHROG: families of prokaryotic virus proteins clustered using remote homology. NAR Genomics and Bioinformatics, 3(3), 08 2021. lqab067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zayed AA et al. efam: an expanded, metaproteome-supported HMM profile database of viral protein families. Bioinformatics, 37(22):4202–4208, 06 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Abdelkareem AO et al. Virnet: Deep attention model for viral reads identification. In 2018 13th International Conference on Computer Engineering and Systems (ICCES), pp. 623–626, 2018. [Google Scholar]
- [24].Tynecki P et al. Phageai - bacteriophage life cycle recognition with machine learning and natural language processing. bioRxiv, 2020. [Google Scholar]
- [25].Asgari E and Mofrad MRK. Continuous distributed representation of biological sequences for deep proteomics and genomics. PLOS ONE, 10(11):1–15, 11 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Heinzinger M et al. Modeling aspects of the language of life through transfer-learning protein sequences. BMC Bioinformatics, 20(1):723, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rives A et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proceedings of the National Academy of Sciences, 118(15):e2016239118, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Elnaggar A et al. Prottrans: Towards cracking the language of lifes code through self-supervised deep learning and high performance computing. IEEE Transactions on Pattern Analysis and Machine Intelligence, pp. 1–1, 2021.31331880 [Google Scholar]
- [29].Brandes N et al. ProteinBERT: a universal deep-learning model of protein sequence and function. Bioinformatics, 01 2022. btac020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bepler T and Berger B. Learning the protein language: Evolution, structure, and function. Cell Systems, 12(6):654–669.e3, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dohan D et al. Improving protein function annotation via unsupervised pre-training: Robustness, efficiency, and insights. In Proceedings of the 27th ACM SIGKDD Conference on Knowledge Discovery & Data Mining, KDD ‘21, pp. 2782–2791, New York, NY, USA, 2021. Association for Computing Machinery. [Google Scholar]
- [32].Gane A et al. Protnlm: Model-based natural language protein annotation. Preprint, 2022. [Google Scholar]
- [33].Nasir A and Caetano-Anolles G.´ A phylogenomic data-driven exploration of viral origins and evolution. Science Advances, 1(8):e1500527, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Balaji S and Srinivasan N. Comparison of sequence-based and structure-based phylogenetic trees of homologous proteins: Inferences on protein evolution. Journal of Biosciences, 32(1):83–96, 2007. [DOI] [PubMed] [Google Scholar]
- [35].Meng C et al. Review and comparative analysis of machine learning-based phage virion protein identification methods. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics, 1868(6):140406, 2020. [DOI] [PubMed] [Google Scholar]
- [36].Fang Z et al. DeePVP: Identification and classification of phage virion proteins using deep learning. GigaScience, 11, 08 2022. giac076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Cantu VA et al. Phanns, a fast and accurate tool and web server to classify phage structural proteins. PLOS Computational Biology, 16(11):1–18, 11 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Mizuno CM et al. Genomes of abundant and widespread viruses from the deep ocean. mBio, 7(4):e00805–16, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hackl T et al. Novel integrative elements and genomic plasticity in ocean ecosystems. Cell, 186(1):47–– 62.e16., 2023. [DOI] [PubMed] [Google Scholar]
- [40].Eppley JM et al. Marine viral particles reveal an expansive repertoire of phage-parasitizing mobile elements. Proceedings of the National Academy of Sciences, 119(43):e2212722119, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Smyshlyaev G et al. Sequence analysis of tyrosine recombinases allows annotation of mobile genetic elements in prokaryotic genomes. Molecular Systems Biology, 17(5):e9880, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Gibb B et al. Requirements for catalysis in the Cre recombinase active site. Nucleic Acids Research, 38(17):5817–5832, 05 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Williams KP. Integration sites for genetic elements in prokaryotic tRNA and tmRNA genes: sublocation preference of integrase subfamilies. Nucleic Acids Research, 30(4):866–875, 02 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hatfull GF and Hendrix RW. Bacteriophages and their genomes. Current Opinion in Virology, 1(4):298–303, 2011. Vaccines/Viral genomics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Koonin EV et al. The global virome: How much diversity and how many independent origins? Environmental Microbiology, 25(1):40–44, 2023. [DOI] [PubMed] [Google Scholar]
- [46].Shen A and Millard A. Phage genome annotation: Where to begin and end. PHAGE, 2(4):183–193, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Borodovich T et al. Phage-mediated horizontal gene transfer and its implications for the human gut microbiome. Gastroenterology Report, 10, 04 2022. goac012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Jumper J et al. Highly accurate protein structure prediction with alphafold. Nature, 596:583–589, 8 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Nicolas E et al. The tn3-family of replicative transposons. Microbiology Spectrum, 3(4):3.4.14, 2015. [DOI] [PubMed] [Google Scholar]
- [50].Mavrich TN and Hatfull GF. Bacteriophage evolution differs by host, lifestyle and genome. Nature Microbiology, 2, 7 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Mohamed Mohssen AZ, Dominik Lucking. efam, 2021. efam is an Expanded, metaproteome annotationssupported HMM profile database of viral protein families to aid the detection and characterization of viral sequences from any ecosystem. [Google Scholar]
- [52].Steinegger M et al. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinformatics, 20(1):473, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Pedregosa F et al. Scikit-learn: Machine learning in Python. Journal of Machine Learning Research, 12:2825–2830, 2011. [Google Scholar]
- [54].Abadi M et al. TensorFlow: Large-scale machine learning on heterogeneous systems, 2015. Software available from tensorflow.org.
- [55].Sievers F and Higgins DG. Clustal omega for making accurate alignments of many protein sequences. Protein Science, 27(1):135–145, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].McInnes L et al. Umap: Uniform manifold approximation and projection for dimension reduction, 2018. [Google Scholar]
- [57].Charlier F et al. Statannotations, October 2022. [Google Scholar]
- [58].Hagberg AA et al. Exploring network structure, dynamics, and function using networkx. In Varoquaux G et al. , editors, Proceedings of the 7th Python in Science Conference, pp. 11–15, Pasadena, CA; USA, 2008. [Google Scholar]
- [59].Cock PJA et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics, 25(11):1422–1423, 03 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Virtanen P et al. SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nature Methods, 17:261–272, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Altschul SF et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research, 25(17):3389–3402, 09 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Paysan-Lafosse T et al. InterPro in 2022. Nucleic Acids Research, 51(D1):D418–D427, 11 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Gabler F et al. Protein sequence analysis using the mpi bioinformatics toolkit. Current Protocols in Bioinformatics, 72(1):e108, 2020. [DOI] [PubMed] [Google Scholar]
- [64].Zimmermann L et al. A completely reimplemented mpi bioinformatics toolkit with a new hhpred server at its core. Journal of Molecular Biology, 430(15):2237–2243, 2018. Computation Resources for Molecular Biology. [DOI] [PubMed] [Google Scholar]
- [65].Potter SC et al. HMMER web server: 2018 update. Nucleic Acids Research, 46(W1):W200–W204, 06 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Kelley LA et al. The phyre2 web portal for protein modeling, prediction and analysis. Nature Protocols, 10:845–858, 6 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Mitchell AL et al. MGnify: the microbiome analysis resource in 2020. Nucleic Acids Research, 48(D1):D570–D578, 11 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Roux S et al. IMG/VR v3: an integrated ecological and evolutionary framework for interrogating genomes of uncultivated viruses. Nucleic Acids Research, 49(D1):D764–D775, 11 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Paez-Espino D et al. IMG/VR v.2.0: an integrated data management and analysis system for cultivated and environmental viral genomes. Nucleic Acids Research, 47(D1):D678–D686, 11 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Steinegger M and Soding J. Mmseqs2 enables sensitive protein sequence searching for the analysis of¨ massive data sets. Nature Biotechnology, 35:1026–1028, 11 2017. [DOI] [PubMed] [Google Scholar]
- [71].Katoh K and Standley DM. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Molecular Biology and Evolution, 30(4):772–780, 01 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Price MN et al. Fasttree 2 – approximately maximum-likelihood trees for large alignments. PLOS ONE, 5(3):1–10, 03 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Letunic I and Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Research, 49(W1):W293–W296, 04 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].van Kempen M et al. Fast and accurate protein structure search with foldseek. Nature Biotechnology, May 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Hyatt D et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics, 11(1):119, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Camargo A. genomad database. Zenodo. April 2023. [Google Scholar]
- [77].Shannon P et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research, 13(11):2498–2504, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Kauffman KM et al. Viruses of the Nahant Collection, characterization of 251 marine Vibrionaceae viruses. Scientific Data, 5(1):180114, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Waterhouse A et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Research, 46(W1):W296–W303, 05 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Biswas T et al. A structural basis for allosteric control of dna recombination by lambda integrase. Nature, 435:1059–1066, 6 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallographica Section D, 66(1):12–21, Jan 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Flamholz Z. kellylab/viral-protein-function-plm: v1.0, November 2023. [Google Scholar]
- [83].Harris CR et al. Array programming with NumPy. Nature, 585(7825):357–362, September 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].pandas development team T. pandas-dev/pandas: Pandas, February 2020. [Google Scholar]
- [85].Hunter JD. Matplotlib: A 2d graphics environment. Computing in Science & Engineering, 9(3):90–95, 2007. [Google Scholar]
- [86].Waskom ML. seaborn: statistical data visualization. Journal of Open Source Software, 6(60):3021, 2021. [Google Scholar]
- [87].Granger BE and Perez F. Jupyter: Thinking and storytelling with code and data. Computing in Science Engineering, 23(2):7–14, 2021.35939280 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The PHROGs VPF database v3 (https://phrogs.lmge.uca.fr/) was downloaded on 01/26/2022. Reannotation data was downloaded after v4 release. EFAM VPF database was downloaded from project repository on CyVerse Data Commons on 09/07/2022. PHANNs protein sequences and annotations (https://phanns.com/downloads) were downloaded on 01/17/2023. geNomad HMM database and annotations (v1.3) were downloaded from zenodo (https://zenodo.org/record/7793532) on 08/10/2023. Protein sequences used for integrase and major capsid protein investigation were collected from the following databases: MGniFY, IMG-VR, NCBI nr, UniRef50. Protein sequence embeddings generated for PHROGs and EFAM sequences are available at https://doi.org/10.5281/zenodo.8339381. Additional data generated for this study is available in a public Google Cloud Platform bucket (http://storage.googleapis.com/viral protein family plm embeddings). See the README on the project repository https://github.com/kellylab/viral-protein-function-annotation-with-protein-language-model for details on downloading the data.