

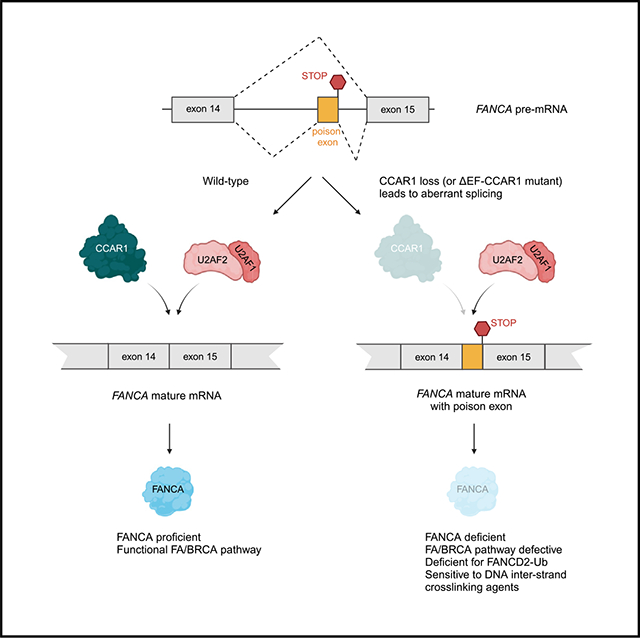
SUMMARY
The twenty-three Fanconi anemia (FA) proteins cooperate in the FA/BRCA pathway to repair DNA interstrand cross-links (ICLs). The cell division cycle and apoptosis regulator 1 (CCAR1) protein is also a regulator of ICL repair, though its possible function in the FA/BRCA pathway remains unknown. Here, we demonstrate that CCAR1 plays a unique upstream role in the FA/BRCA pathway and is required for FANCA protein expression in human cells. Interestingly, CCAR1 co-immunoprecipitates with FANCA pre-mRNA and is required for FANCA mRNA processing. Loss of CCAR1 results in retention of a poison exon in the FANCA transcript, thereby leading to reduced FANCA protein expression. A unique domain of CCAR1, the EF hand domain, is required for interaction with the U2AF heterodimer of the spliceosome and for excision of the poison exon. Taken together, CCAR1 is a splicing modulator required for normal splicing of the FANCA mRNA and other mRNAs involved in various cellular pathways.
Graphical Abstract
In brief
The Fanconi anemia (FA)/BRCA pathway plays a critical role in genome maintenance, and its loss leads to FA and cancer predisposition. Here, Harada et al. show that CCAR1 regulates FANCA expression by acting as a splicing modulator, ensuring accurate splicing of the FANCA transcript.
INTRODUCTION
The DNA repair system is a critical tumor suppressor mechanism required for maintaining the integrity of the human genome, and genome instability is a hallmark of cancer cells.1 Although defects in DNA repair confer survival advantages to cancer cells by increasing their adaptability, these defects are also a vulnerability that can be therapeutically exploited.2 Due to their reduced DNA repair, cancer cells are hypersensitive to conventional chemotherapy, which causes DNA damage. Some tumors depend on compensatory DNA repair pathways, resulting in their sensitivity to inhibitors of these alternative pathways.3–8
Fanconi anemia (FA) is a complex genetic disorder caused by inherited pathogenic variants in any of twenty-three genes (designated as complementation groups FANCA-FANCW).9,10 FA is characterized by bone marrow failure, congenital defects, and cancer predisposition. The inability to repair DNA interstrand cross-links (ICLs) is a key cellular feature of FA, and cells deficient in the FA/BRCA pathway are hypersensitive to ICL-inducing chemotherapeutic agents such as platinum compounds, nitrogen compounds, and mitomycin C (MMC).11 The FA pathway plays a central role in ICL repair, involving the detection of ICLs, the monoubiquitination of the FANCI-FANCD2 complex (ID2 complex) by the FA core complex, and the incision of ICLs by using the ubiquitinated ID2 complex and its binding partners. Somatic mutations in FA genes occur in malignancies, and loss of FA pathway activity renders cancer cells sensitive to inhibitors of DNA repair pathways, such as poly(ADP-ribose) polymerase (PARP) inhibitors.12 Therefore, understanding the FA pathway status in tumors is important for the rational development of precision medicine cancer therapies.
Recently, “knockout” of the cell division cycle and apoptosis regulator 1 (CCAR1) gene was shown to cause cellular hypersensitivity to ICL damage,13 suggesting that CCAR1 may be directly involved in the FA/BRCA pathway. CCAR1 was first discovered as a regulator of apoptosis signaling in breast cancer cells and hence named cell cycle and apoptosis regulator protein-1 (CARP-1).14 In breast cancer cells, increased expression of CCAR1 caused elevated levels of cyclin-dependent kinase inhibitor p21WAF1/CIP1 and reduced the levels of proliferative genes such as c-Myc and Cyclin B1.14 The reduced expression of c-Myc was shown to sensitize breast cancer cell lines to cisplatin.15,16 Paradoxically, loss of CCAR1 also enhanced sensitivity to ICL-inducing agents, and thus the role of CCAR1 in ICL repair is still controversial. Moreover, CCAR1 is thought to have multiple physiologic functions that extend beyond ICL repair, including roles in Wnt signaling, nuclear receptor function, adipogenesis, and apoptosis.17–21 Despite the importance of CCAR1 expression in these various cellular pathways, a precise cellular function of CCAR1 protein has remained elusive.
In this study, we explored the role of CCAR1 in the context of ICL repair based on the observation that CCAR1 appears in the same cluster as FA genes in DNA damage CRISPR knockout screens.13 We demonstrate that CCAR1 is a direct regulator of splicing of the FANCA mRNA. CCAR1 knockout results in FA phenotype characterized by deficiency in FANCD2 monoubiquitination, hypersensitivity to MMC, and increased radial chromosomes upon MMC exposure. Loss of FANCA protein expression is the key driver of the FA phenotype in CCAR1 knockout cells. Interestingly, CCAR1 co-immunoprecipitates with the FANCA pre-mRNA and regulates mRNA splicing through the removal of a poison exon. Moreover, CCAR1 interacts functionally with the U2AF1/2 subunits of the spliceosome to aid in exclusion of the poison exon from the FANCA transcript. Loss of CCAR1 results in inclusion of the poison exon in the FANCA transcript and reduced FANCA protein expression, leading to a defect in the FA/BRCA pathway.
RESULTS
CCAR1 knockout results in an FA phenotype
In the Genotoxic Screens app developed by the Durocher lab,13 CCAR1 was identified in the same cluster as FA genes (Figure 1A), indicating its possible direct function in the FA/BRCA pathway. To explore the biological function of CCAR1, CCAR1 knockout clones were generated in retinal pigment epithelial (RPE) p53−/−, K562, and HEK293T cell lines (Figures 1B, S1A, and S1B). All the CCAR1 knockout clones used in this study showed at least 85% knockout in the CCAR1 gene and hence have been referred to as knockout throughout the study (see STAR Methods and Table S2 for clone details). Since FANCA is one of the FA genes, and FANCA mutations are frequently found in FA patients, FANCA knockout clones were generated as positive control FA cells (Figures S1D and S1E). CCAR1 knockout cells showed a deficiency in FANCD2 monoubiquitination after MMC treatment, consistent with the FA phenotype (Figure 1C). Moreover, FANCD2 foci were decreased in CCAR1 knockout cells compared with parental RPE p53−/− cells, and levels of FANCD2 foci were restored by transduction of wildtype (WT) CCAR1 cDNA (Figures S1F and S1G). The sensitivity of CCAR1 knockout clones to MMC, olaparib, topotecan, and X-ray irradiation was evaluated in colony formation assays. Interestingly, similar to a FANCA knockout clone, CCAR1 knockout clones were sensitive to MMC and showed slightly increased sensitivity to olaparib, topotecan, and X-ray irradiation (Figures 1D, S1C, and S1H). Re-expression of WT CCAR1 in CCAR1 knockout clones restored MMC resistance (Figures 1E, S1I, and S1J). A chromosomal breakage assay in the presence of MMC further confirmed that CCAR1 knockout cells have a specific FA phenotype, similar to FANCA knockout cells (Figure 1F). CRISPR knockout of the related gene, CCAR2, did not result in increased MMC sensitivity (Figures S1K and S1L). Taken together, we found that CCAR1 knockout cells had a bona fide FA phenotype characterized by deficiency in FANCD2 monoubiquitination, high sensitivity to interstrand cross-linking agents, and chromosome instability.
Figure 1. CCAR1 knockout cells display FA-like phenotypes.
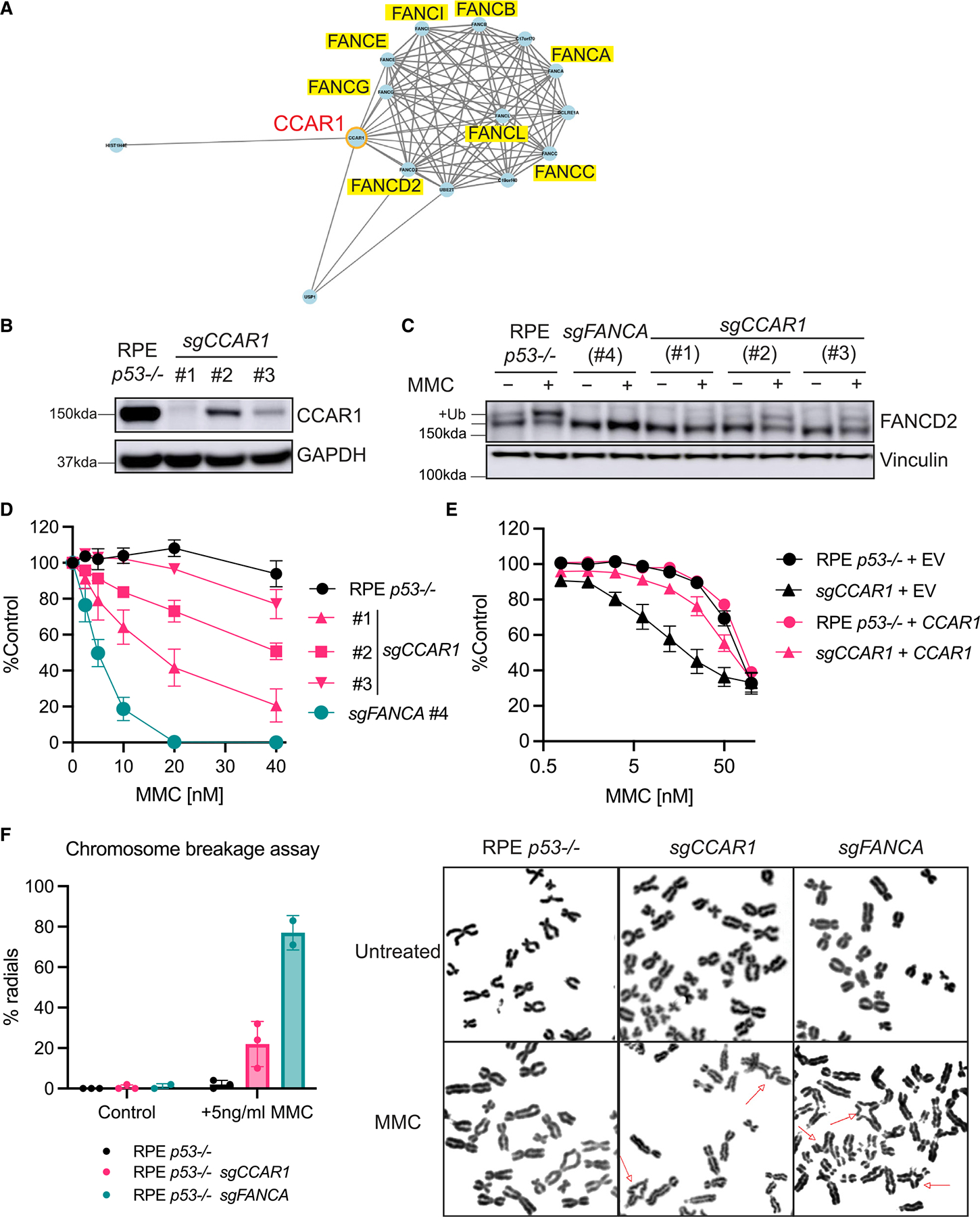
(A) CCAR1 was found in the same cluster as FA genes in CRISPR screens for DNA damage response. This figure was created using Genotoxic Screens app developed by the Durocher lab. Network correlation cutoff was set at 0.7.
(B) Western blot (WB) of CCAR1 in RPE p53−/− CCAR1 knockout clones used in this study.
(C) WB of FANCD2 in CCAR1 knockout clones (n = 2). The cells were treated with 100 ng/mL MMC for 24 h.
(D) Colony formation assay data of RPE p53−/− CCAR1 knockout clones treated with MMC. Mean and SEM from n = 3 independent experiments are plotted.
(E) CCAR1 transduced RPE p53−/− CCAR1 knockout clone #1 is resistant to MMC treatment. The cells were treated with MMC for 6 days, and then cell viability was measured by CellTiter-Glo assay. Mean and SEM from n = 3 independent experiments are plotted.
(F) Chromosome breakage analysis in RPE p53−/− CCAR1 knockout clone #1. The cells were treated with 5 ng/mL MMC for 48 h. Mean and SD from n = 3 independent experiments (left) and representative images of radial chromosomes (right) are shown. The chromosome spreads were imaged using 100× objective. Red arrows indicate radial chromosome.
See also Figure S1.
FANCA loss is a driver for the FA phenotype of CCAR1 knockout cells
We hypothesized that CCAR1 loss may result in dysfunction of the FA core complex, a multi-subunit complex required for monoubiquitination of FANCD2. Initially, the mRNA expression levels of FA core-complex genes were evaluated by reverse transcription quantitative polymerase chain reaction (RT-qPCR). CCAR1 knockout cells exhibited a decrease in the FANCA mRNA expression (Figures 2A and S2A). The decrease in FANCA mRNA level was also observed by RNA sequencing (RNA-seq) analysis (Figure 2B). FANCA protein expression was also decreased in RPE p53−/− CCAR1 knockout clones (Figure 2C). Since FANCG is a known direct interactor of FANCA,22 FANCG protein expression was evaluated in the CCAR1 knockout clones, and decreased expression of FANCG was also observed (Figure 2C). Although FANCE and FANCL transcripts were slightly decreased in CCAR1 knockout cells (Figures 2A and S2A), the protein expression level of FANCE and FANCL was not changed (Figure S2B).
Figure 2. FANCA loss is a key mechanism for the FA phenotypes of CCAR1 knockout cells.
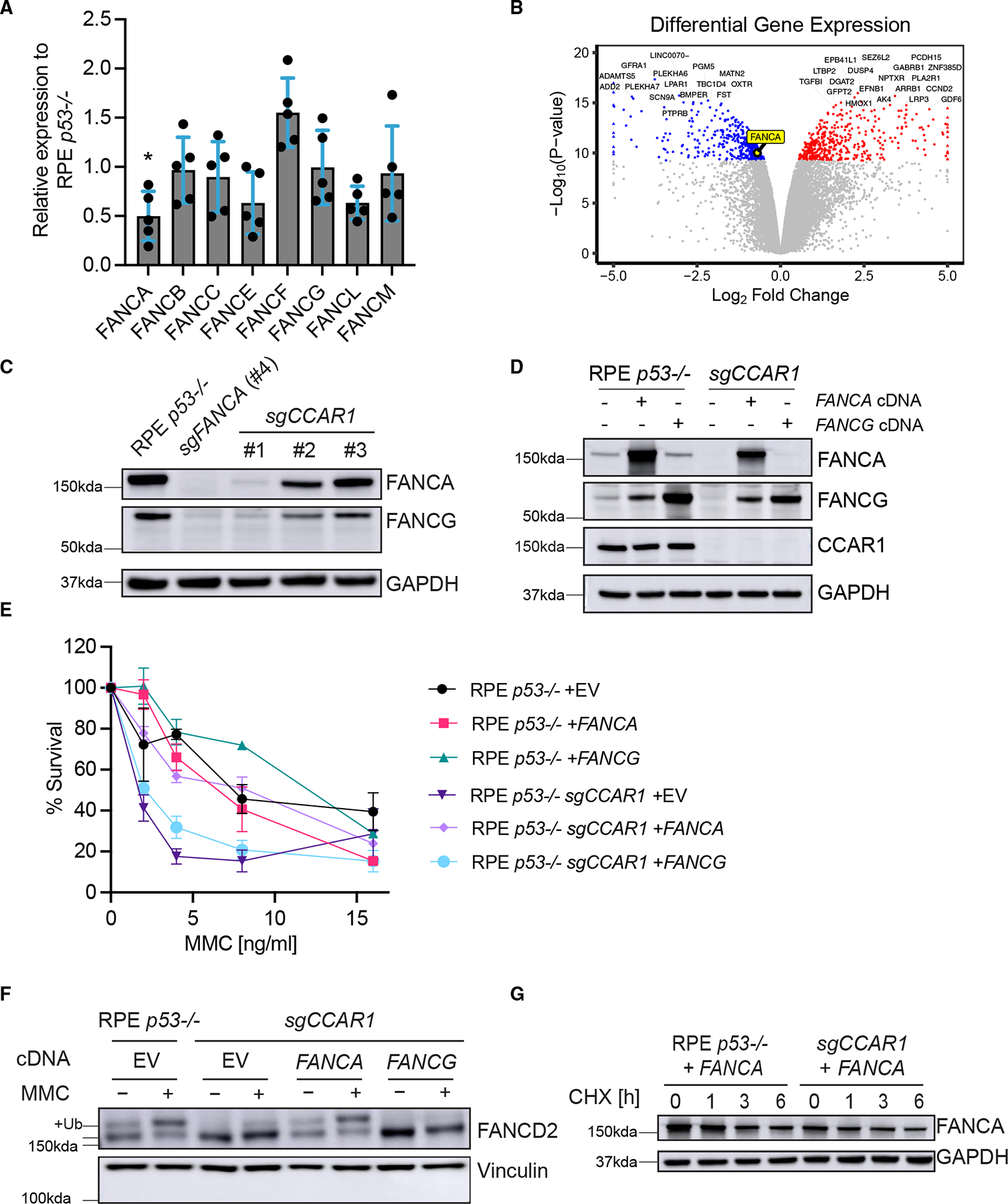
(A) mRNA expression levels of FA core-complex genes in RPE p53−/−CCAR1 knockout clone #1. Mean and SEM from n = 5 independent experiments are plotted. *: p < 0.05 (two-way ANOVA, Šídák’s multiple comparisons test).
(B) Volcano plots showing differential gene expression between control RPE p53−/− and CCAR1 knockout cells.
(C) WB of FANCA and FANCG in RPE p53−/− CCAR1 knockout clones (n = 2).
(D) WB showing complementation of RPE p53−/− and p53−/−CCAR1 knockout cells with FANCA or FANCG cDNA (n = 3).
(E) Colony formation assay data for FANCA or FANCG cDNA transduced RPE p53−/− or p53−/−CCAR1 knockout cells treated with MMC. The cells were treated with MMC for 7 days. Mean and SD from n = 3 independent experiments are plotted.
(F) WB of FANCD2 in RPE p53−/− CCAR1 knockout cells transduced with control (EV), FANCA, or FANCG cDNA. The cells were treated with 100 ng/mL MMC for 24 h (n = 2).
(G) WB of FANCA in RPE p53−/− cells and p53−/−CCAR1 knockout cells transduced with FANCA cDNA. Cells were treated with cycloheximide (CHX) for 1–6 h (n = 3).
See also Figure S2.
To correct the lack of FANCA and FANCG in CCAR1 knockout cells, we transduced them with the FANCA or FANCG cDNA. Interestingly, transduction of FANCA restored FANCG protein expression, while FANCG transduction did not change FANCA protein expression (Figures 2D and S2C). MMC sensitivity and FANCD2 monoubiquitination (Figures 2E and 2F) in these cells were evaluated to determine whether the transduction with FANCA or FANCG cDNA could rescue the FA/BRCA pathway defect in CCAR1 knockout cells. FANCA cDNA transduction, but not FANCG cDNA transduction, resulted in resistance to MMC (Figures 2E and S2D) and proficient FANCD2 monoubiquitination (Figure 2F). Loss of CCAR1 expression in K562 and HEK293T cells also resulted in reduced FANCA protein expression and MMC sensitivity (Figures S1A–S1C). Taken together, loss of FANCA expression is the cause of the FA phenotype in CCAR1 knockout cells, and loss of FANCG protein expression is secondary to the loss of FANCA.
We reasoned that CCAR1 loss might promote the degradation of the FANCA protein since CCAR1 interacts with the anaphase-promoting complex 2 (APC2), a component of the multi-subunit E3 ubiquitin ligase.23 However, no significant degradation of FANCA protein was observed in CCAR1 knockout cells transduced with the FANCA cDNA and exposed to protein synthesis inhibitor cycloheximide (Figures 2G and S2E). Moreover, recovery of FANCA protein was not observed in CCAR1 knockout cells exposed to the proteasome inhibitor MG-132 (Figure S2F). Furthermore, a protein-protein interaction between CCAR1 and FANCA was not observed upon co-immunoprecipitation (coIP) (Figure S2G). Therefore, while FANCA protein loss is the key phenotype of CCAR1 knockout cells, CCAR1 does not appear to directly or indirectly regulate FANCA protein stability.
CCAR1 binds to the FANCA pre-mRNA and promotes the excision of a poison exon from the FANCA transcript
We next hypothesized that CCAR1 loss may cause a defect in the processing of the FANCA transcript based on previous reports in C. elegans and mice showing a role for CCAR1 in alternative splicing24,25 (Figure 3). Initially, we performed RT-qPCR on RPE p53−/− CCAR1 knockout cells, which had been transduced with either empty vector (EV) or the WT CCAR1 cDNA, using primer sets corresponding to various FANCA transcript regions. Interestingly, longer PCR products were observed for the exon10–15 and exon14–22 PCR reactions in CCAR1 knockout cells (Figures 3A and 3B). The longer PCR products were decreased in CCAR1 knockout cells transduced with the WT CCAR1 cDNA. Sanger sequencing of the longer PCR products from CCAR1 knockout clones revealed a 36-bp retained cassette exon sequence between exon14 and exon15, which contained an in-frame STOP codon at the 3′ end (Figures 3B, 3C, and S3A). Knockout of CCAR1 in either K562 or HEK293T cells also resulted in retention of the poison exon in FANCA mRNA transcript (Figure S3B). Moreover, we found that the 36-bp insertion was observed in the full-length FANCA mRNA transcript derived from K562 CCAR1 knockout clones (data not shown).
Figure 3. CCAR1 regulates the splicing of the FANCA transcript.
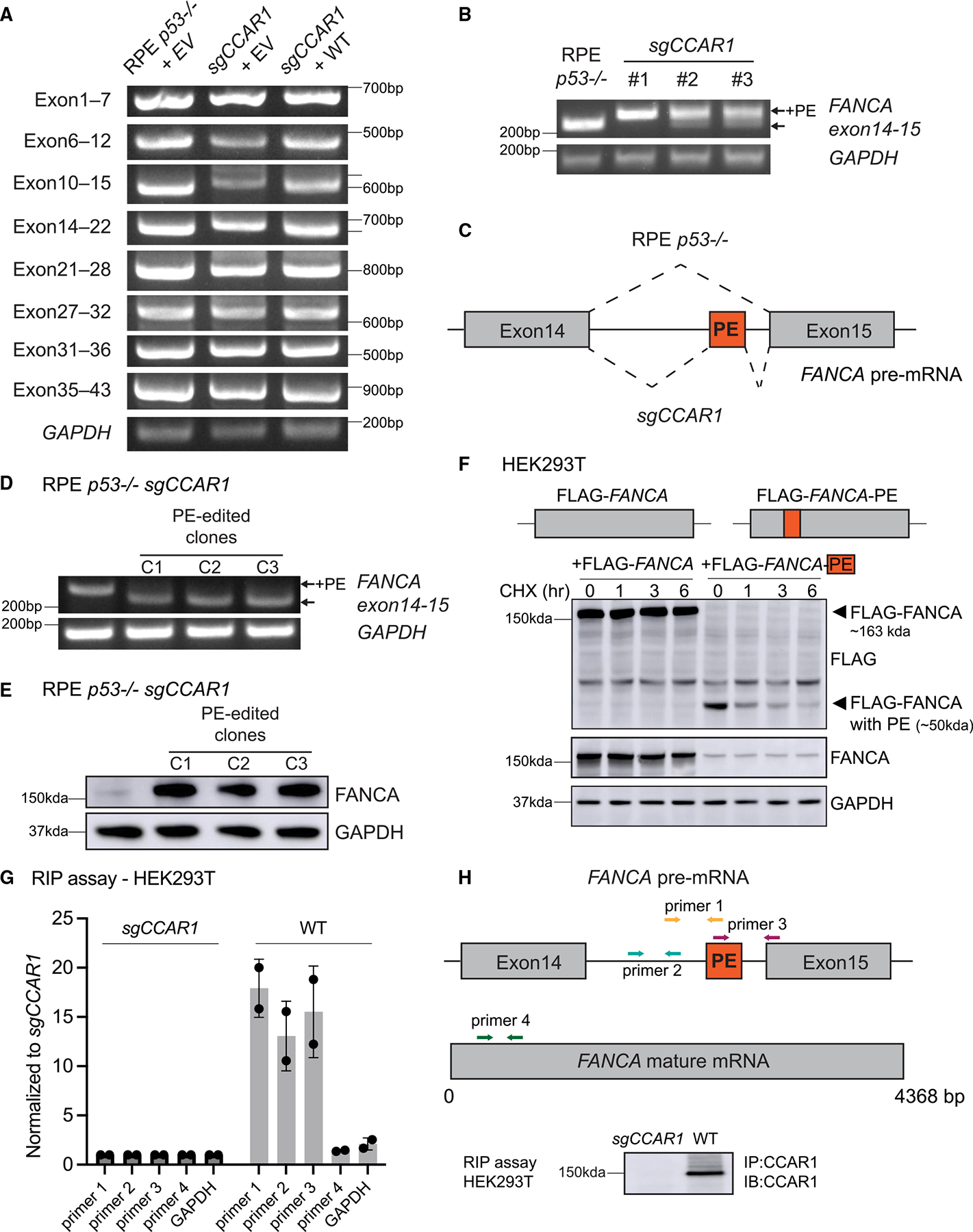
(A) RT-qPCR evaluation of alternative splicing of the FANCA transcript in RPE p53−/− cells transduced with empty vector (EV), p53−/−CCAR1 knockout cells transduced with EV or WT CCAR1. Each fragment was amplified using a primer set indicated in the figure (n = 2).
(B) RT-qPCR showing the poison exon (PE) in the FANCA transcript in CCAR1 knockout clones (n = 2).
(C) Schematic of the alternative splicing of the FANCA transcript in CCAR1 knockout cells. The PE was retained between exon14 and 15.
(D) RT-qPCR showing elimination of the FANCA PE in clones derived from RPE p53−/−CCAR1 knockout cells edited with poison exon targeting sgRNA (n = 2).
(E) WB of FANCA in PE-edited CCAR1 knockout clones (n = 2).
(F) FANCA mRNA containing the poison exon produces truncated FANCA, which is unstable. Blot showing HEK293T cells transfected with FLAG-FANCA cDNA containing WT FANCA CDS or FLAG-FANCA-PE cDNA containing FANCA CDS with the poison exon inserted between exon14 and 15.
(G) Binding of CCAR1 to FANCA pre-mRNA evaluated using the RNA immunoprecipitation (RIP) assay, performed using anti-CCAR1 Ab. Mean and SD from n = 2 are plotted. The assay was performed in HEK293T WT, and CCAR1 knockout was used as a negative control. qPCR was performed with primers targeting FANCA pre-mRNA, FANCA mature mRNA, and GAPDH.
(H) Schematic showing primers used for the RIP assay and blot showing immunoprecipitation of CCAR1 in the RIP assay samples.
See also Figure S3.
We next determined whether this retained poison exon provides a mechanism for the loss of the FANCA protein in CCAR1 knockout cells. We edited the poison exon by using the CRISPR-Cas9 method by designing two sgRNAs targeting the poison exon region (Figure S3A). One of these sgRNAs (sgPoison2) efficiently eliminated the poison exon from FANCA transcripts (Figure S3C). Next, we generated poison exon edited clones from CCAR1 knockout cells and confirmed elimination of the poison exon from FANCA transcript (Figure 3D). Sanger sequencing of these clones revealed an insertion of a thymidine after the GT donor sequence neighboring the poison exon. The poison exon edited clones exhibited normal FANCA protein expression, MMC resistance, and FANCA transcript levels comparable to the parental RPE p53−/− cells (Figures 3E, S3D, and S3E). Therefore, inclusion of the poison exon in the FANCA transcript is the key driver for the FANCA protein loss in CCAR1 knockout cells.
Next, we designed an antisense oligonucleotide (ASO) targeting the FANCA poison exon (Figure S3A). The ASO has complementary sequence to the first 10 bp of the poison exon and 10 bp of the neighboring intron. ASO transfection in RPE p53−/− CCAR1 knockout cells corrected the splicing defect in FANCA transcript and restored FANCA protein levels comparable to parental RPE p53−/− cells (Figures S3F and S3G).
Although we cannot detect full-length FANCA protein in CCAR1 knockout cells, we tested whether the FANCA transcript containing the poison exon is capable of producing a truncated FANCA protein. To check for truncated FANCA, we transfected HEK 293T cells with FLAG-FANCA cDNA containing WT FANCA coding sequence (CDS) or FLAG-FANCA-PE cDNA, which contains the poison exon inserted between exon14 and 15 of the FANCA CDS and performed a cycloheximide chase assay (Figure 3F). As expected, the FLAG-FANCA-PE construct produces a 50 kDa product due to the presence of the in-frame STOP codon in the poison exon. This truncated 50 kDa FANCA product is extremely unstable compared with the full-length FANCA (~163 kDa). Taken together, inclusion of the poison exon in FANCA mRNA leads to generation of truncated FANCA protein that is extremely unstable.
Our data demonstrate that CCAR1 plays a role in regulating splicing of FANCA mRNA. Using a previously published RNA immunoprecipitation (RIP) assay protocol,26 we determined whether CCAR1 interacts with the FANCA pre-mRNA. Briefly, we cross-linked samples with paraformaldehyde and performed immunoprecipitation (IP) with anti-CCAR1 Ab in HEK-WT and CCAR1 knockout cells. We reversed the cross-links from IP samples and purified the RNA and evaluated it by RT-qPCR, using the indicated PCR primers corresponding to sequences in the FANCA pre-mRNA, FANCA mature mRNA, and GAPDH (Figure 3H). Indeed, CCAR1 pull-down specifically co-immunoprecipitated the FANCA pre-mRNA as compared with GAPDH (Figure 3G). We also verified that the RT-qPCR signal is from RNA converted to cDNA and not from genomic DNA contamination by performing the RT-qPCR without the reverse transcriptase enzyme (Figure S3H). Interestingly, we observed specific enrichment for the FANCA pre-mRNA and not the mature mRNA in the CCAR1-IP complexes, suggesting a key role for CCAR1 binding and processing of the FANCA pre-mRNA (Figure 3G).
The EF hand domain of CCAR1 is required for removal of the poison exon from the FANCA transcript
To identify the functional domain of CCAR1 protein required for suppressing the FA phenotype, we generated CCAR1 mutants lacking either the S1-like domain (dS1) or the SAP motif (dSAP) (Figure 4A). For the dS1, an interaction with RNA polymerase II has been reported for CCAR2, a known paralog of CCAR1.27,28 The dSAP is a DNA-binding motif found in proteins, such as PARP and Ku70, involved in chromosomal organization and DNA damage repair pathways.29–31 Surprisingly, both dS1 and dSAP mutants restored FANCA splicing partially (Figure 4B); however, FANCA protein expression was restored fully, suggesting limited functionality in these domains (Figure S4A). Thus, neither the dS1 nor the dSAP is essential for regulating FANCA expression.
Figure 4. The EF hand domain of CCAR1 is essential for FANCA expression.
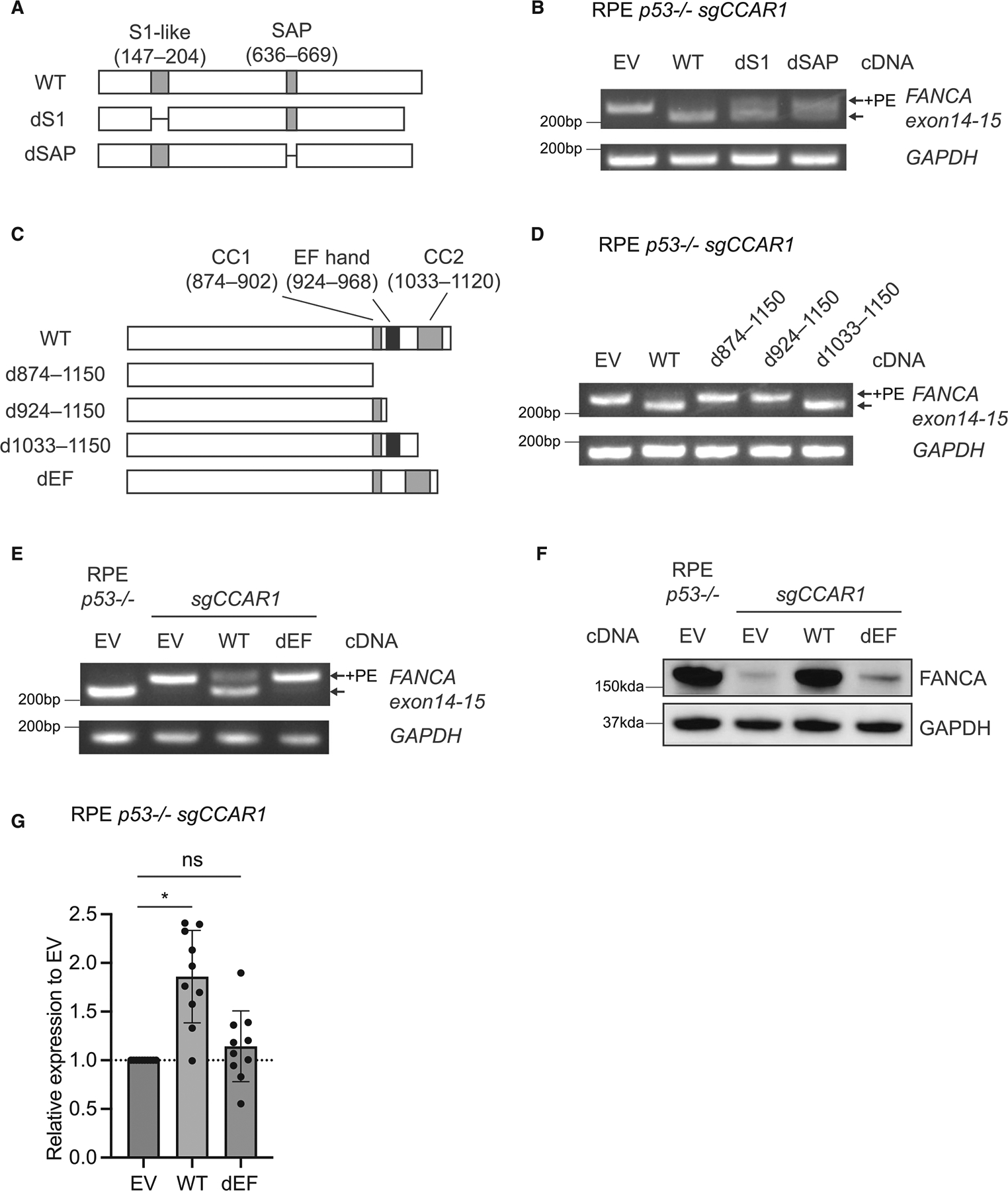
(A) Schematics of dS1 and dSAP mutants used in this study.
(B) RT-qPCR evaluating inclusion of the FANCA PE in RPE p53−/−CCAR1 knockout cells transduced with empty vector (EV), WT CCAR1, dS1, or dSAP mutants (n = 2).
(C) Schematics of d874–1,150, d924–1,150, d1033–1,150, and dEF mutants.
(D) RT-qPCR evaluating inclusion of the FANCA PE in RPE p53−/−CCAR1 knockout cells transduced with d874–1,150, d924–1,150, or d1033–1,150 mutants (n = 2).
(E) RT-qPCR evaluating inclusion of the FANCA PE in RPE p53−/−CCAR1 knockout cells transduced with the dEF mutant (n = 2).
(F) WB of FANCA in RPE p53−/−CCAR1 knockout cells transduced with the dEF mutant (n = 2).
(G) mRNA expression levels of FANCA in RPE p53−/−CCAR1 knockout cells transduced with the dEF mutant. Mean and SEM from n = 10 independent experiments are plotted. *p < 0.0001 (one-way ANOVA, Dunnett’s multiple comparisons test). Statistical analysis was performed by comparing to EV-transduced CCAR1 knockout cells.
See also Figure S4.
We next generated three N-terminal truncated mutants (Figures S4B and S4C) and three C-terminal truncated CCAR1 mutants (Figure 4C) to identify the functional domain required for normal FANCA splicing. Among the N-terminal truncations, all three truncated mutants were deficient in correcting the aberrant FANCA splicing (Figure S4D) and FANCA protein expression (Figure S4E). Since d2–380 and d2–477 lack the nuclear localizing signal (NLS) domain (322–357), a region presumably needed for nuclear localization of CCAR1, we considered that deletion of the NLS domain of CCAR1 might affect its localization. Indeed, deletion of this domain in CCAR2, a paralog of CCAR1, exclusively changes the localization of CCAR2 protein from the nucleus to the cytoplasm.32 For d2–321, we tested whether amino acids 2–146 or amino acids 205–321 were essential for CCAR1 function; however, both d2–146 and d205–321 mutants (Figure S4B) restored FANCA splicing and FANCA protein expression in CCAR1 knockout cells (Figures S4F and S4G). Thus, individual deletion of amino acids d2–146, d147–204 (S1 domain), or d205–321 yields minimal loss in FANCA protein expression (Figures S4A, S4F, and S4G). However, combined large deletion of d2–321 leads to a functional defect in FANCA expression (Figure S4E). Taken together, although the N-terminal region is required for CCAR1 function, a minimal functional domain in this region, except for the NLS, was not identified.
For the C-terminal truncated CCAR1 mutants, d1033–1,150 mutants restored FANCA splicing and FANCA protein expression, but the d874–1,150 and d924–1,150 deletion mutants did not (Figures 4D and S4H). These results suggested that the critical region could reside between Glu924 to Val1032 and that the EF hand domain could be the functional domain of CCAR1. To determine whether the EF hand domain is essential for FANCA protein expression, a dEF mutant lacking this domain was generated (Figure 4C). WT CCAR1 restored splicing of the FANCA transcript, FANCA protein expression, and FANCA transcript levels, but the dEF mutant did not (Figures 4E–4G). Therefore, the EF hand domain of CCAR1 is essential for proper splicing of the FANCA mRNA.
CCAR1 binds to subunits of the spliceosome and functions as a splicing factor
To identify interactors of CCAR1 and potential pathways in which CCAR1 functions, we performed mass spectrometry and pathway analysis of CCAR1-3xFlag IP complexes from chromatin fractions of HEK293T WT cells transfected with CCAR1-3xFlag (Figure S5A). Interestingly, we observed an enrichment for spliceosome pathway in CCAR1-IP complexes (Figure S5C). Next, to identify factors specifically enriched in IP complexes of WT-CCAR1 but not in the functionally defective mutant dEF-CCAR1, we performed mass spectrometry analysis of CCAR1-IP complexes from HEK CCAR1 knockout cells transfected with EV, WT-CCAR1-3xFlag, or dEF-CCAR1-3xFlag (Figure S5A). Interestingly, the WT-CCAR1, but not the dEF-CCAR1 mutant, co-immunoprecipitates with the U2AF1/U2AF2 heterodimer (Figures 5A, S5A; Table S1). We next validated the binding between CCAR1 and the U2AF1/U2AF2 heterodimer in the chromatin fraction by FLAG-IP in CCAR1 knockout cells transfected with WT CCAR1 or the dEF-CCAR1 mutant. WT CCAR1 co-immunoprecipitated with U2AF2 and U2AF1, while the dEF-CCAR1 mutant showed little interaction with this heterodimer (Figures 5B and S5B).
Figure 5. CCAR1 is a component of the spliceosome and regulates splicing events.
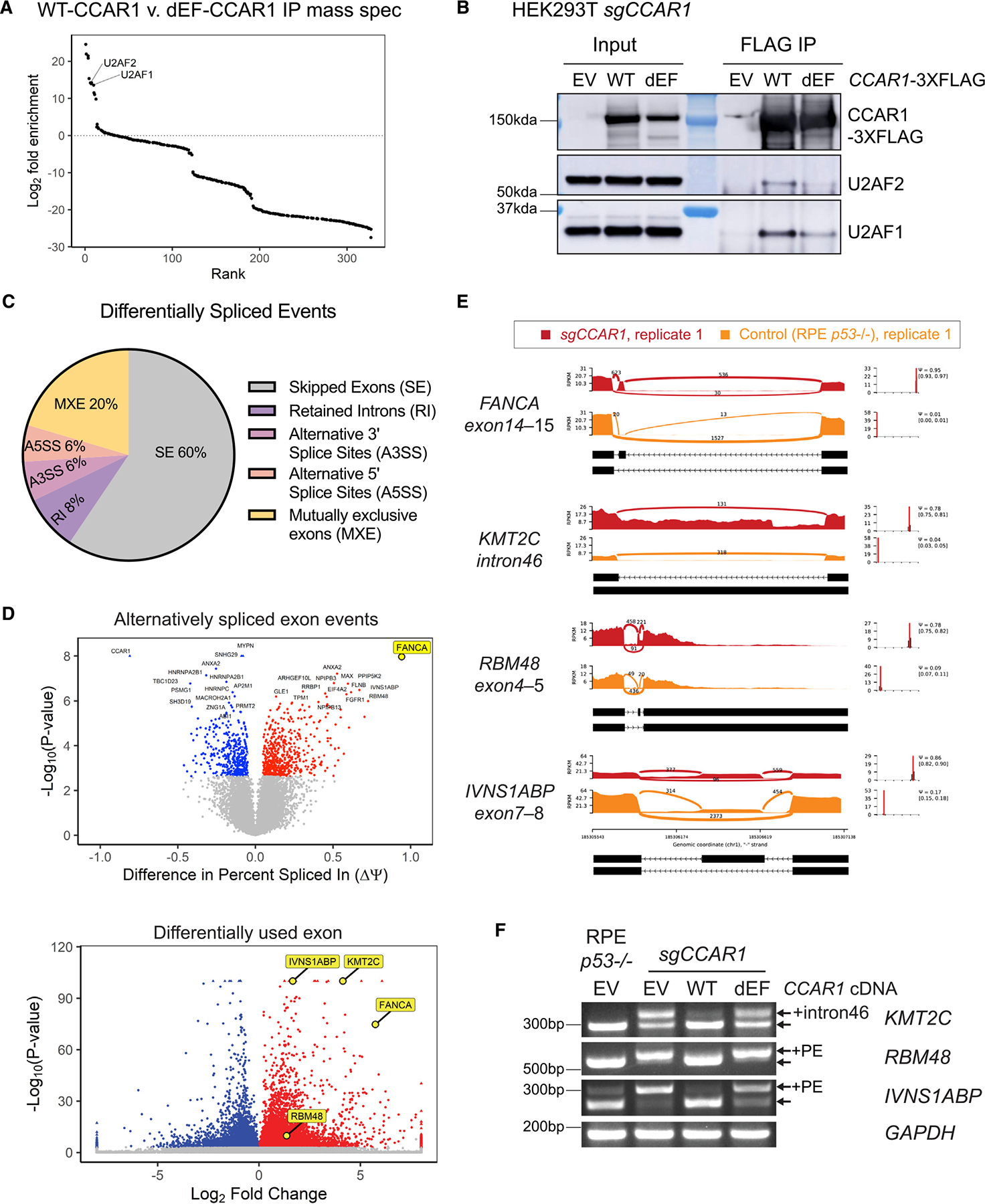
(A) FLAG IP-mass spectrometry analysis from HEK CCAR1 knockout transfected with EV, WT-CCAR1-3xFlag, or dEF-CCAR1-3xFlag. Rank plot shows log2 fold enrichment of factors in WT-CCAR1 as compared with dEF-CCAR1 pull-down (n = 2). Proteins with positive log2 fold enrichment have increased abundance in WT-CCAR1-3xFlag compared with dEF-CCAR1-3xFlag, and vice versa.
(B) WT-CCAR1 interacts with U2AF1/2 complex strongly as compared with dEF-CCAR1.
(C) Pie chart summarizing alternative splicing event types in RPE p53−/− CCAR1 knockout cells obtained by MISO analysis.
(D) Volcano plots showing alternatively spliced skipped exon (SE) events (upper) and differentially used exons (lower) in RPE p53−/− CCAR1 knockout cells compared with RPE p53−/− cells.
(E) Sashimi plots of the alternative splicing events in control and CCAR1 knockout cells: the region between exon14 and exon15 of FANCA transcript, retained intron46 of KMT2C transcript, the region between exon4 and exon5 of RBM48 transcript, and the region between exon7 and exon8 of IVNS1ABP transcript.
(F) RT-qPCR showing the intron retention of KMT2C and the poison exons of RBM48 and IVNS1ABP in CCAR1 knockout cells transduced with empty vector (EV), WT CCAR1, or dEF mutant (n = 2).
See also Figure S5.
We next evaluated the possible genome-wide effect of CCAR1 knockout using deep RNA-seq (Figures 5C and 5D). Alternative splicing events were plotted and analyzed using the mixture of isoforms (MISO) framework33 as well as the replicate multivariate analysis of transcript splicing (rMATs) framework34 (Figure S5D). Based on this analysis, in CCAR1 knockout cells, skipped exon was the most common alternative splicing event type (Figure 5C). Please note that in MISO and rMATS framework of analysis, skipped exon includes both type of events—inclusion or exclusion of exons in the transcripts of CCAR1 knockout relative to that in the parental RPE p53−/−cells. The poison exon of FANCA was identified as the top hit of the skipped exon events in CCAR1 knockout cells (Figures 5D and S5D). Differential expression of exons in CCAR1 knockout cells was also analyzed using DEXSeq analysis.35 Interestingly, the poison exon of FANCA was significantly increased in CCAR1 knockout compared with the parental RPE p53−/−cells (Figure 5D, lower). Thus, analysis of alternative splicing in CCAR1 knockout cells using three different algorithms all yield FANCA as the top target. Also, there were a variety of alternative splicing events other than FANCA in CCAR1 knockout cells (Figures 5E and S5E). For example, a retention of intron46, a poison exon between exon4 and exon5, a poison exon between exon7 and exon8 were increased in the KMT2C, RBM48, and IVNS1ABP transcripts, respectively. A CCAR1-specific consensus sequence motif was not evident in the transcripts regulated by CCAR1 based on the MISO and rMATs framework of analysis.
Next, we validated these alternative splicing events by RT-qPCR using p53−/−CCAR1 knockout cells transduced with WT CCAR1 or the dEF-CCAR1 mutant (Figure 5F). The retained intron46 was observed in the KMT2C transcript in p53−/−CCAR1 knockout cells but not in parental RPE p53−/−cells. For RBM48 transcripts, alternatively spliced transcripts with the poison exon were predominantly expressed in p53−/−CCAR1 knockout cells. For IVNS1ABP, though the poison exon was observed in both parental RPE p53−/−cells and p53−/−CCAR1 knockout cells, the poison exon was increased in p53−/−CCAR1 knockout cells. Moreover, these alternative splicing events were corrected by transduction with WT CCAR1 but not with the dEF-CCAR1 mutant. Taken together, CCAR1 interacts with multiple members of the spliceosome complex, including U2AF1 and U2AF2. These interactions regulate splicing events, and the EF hand domain is essential for the function of CCAR1 as a regulator of pre-mRNA splicing.
We next focused on skipped exon events since they are the most abundant type of alternative splice event in the dataset. Throughcomprehensive analysis using the rMATS algorithm, we identified both annotated and unannotated skipped exon events (Figure S5F). We observed a significant number of unannotated events in the dataset. Using stringent cut-offs for calling alternative splice events, we observed a skew toward inclusion of exons rather than exclusion in CCAR1 knockout, suggesting that CCAR1 functions as a repressor of splicing for these exons (Figure S5F). However, when we consider the entire dataset, we observed different types of alternative splicing events in CCAR1 knockout cells (Figure 5C). Thus, CCAR1 may function as an enhancer or a repressor of splicing, depending on the transcript.
The U2AF1/2 heterodimer is essential for exclusion of the FANCA poison exon by CCAR1
Since our mass spectrometry revealed that WT CCAR1, but not the dEF-CCAR1 mutant, binds to the U2AF1/2 heterodimer, we hypothesized that the inclusion of the FANCA poison exon upon CCAR1 depletion may result from the loss of the interaction between CCAR1 and the U2AF1/2 heterodimer. Because U2AF1/2 loss would affect a broad range of splicing events beyond the FANCA poison exon, we generated a mNeongreen-based reporter in which the mNeongreen is split into two exons with a synthetic intron derived from the intron 14 of FANCA inserted in between (Figures 6A and S6A). The mNeongreen cannot be expressed unless the intronic sequence is spliced out due to presence of the in-frame STOP codon in the poison exon. We normalized the mNeongreen signal to that of mScarlet, which is translated together with mNeongreen from the same mRNA. To determine whether this mini-gene reporter recapitulates the splicing of the endogenous FANCA poison exon, we transduced the reporter into K562 WT and CCAR1 knockout clones. As expected, the poison exon is retained in the mNeongreen mRNA in CCAR1 knockout clones but not in WT cells (Figure S6B). Hence, the ratio of mNeongreen to mScarlet is high in WT cells and low in CCAR1 knockout clones (Figure 6B). Utilizing this reporter, we confirmed that transduction of the WT CCAR1, but not the dEF-CCAR1 mutant, rescued the splicing defect in the CCAR1 knockout cells (Figures 6C and S6C–S6E). Interestingly, the dS1-CCAR1 and d205-321-CCAR1 mutant also showed a moderate reduction in the reporter signal compared with WT CCAR1, suggesting that these CCAR1 domains also play a role in the exclusion of the FANCA poison exon (Figure 6C), consistent with our results from Figure S4.
Figure 6. The CCAR1-U2AF1/2 axis is critical for exclusion of the FANCA poison exon.
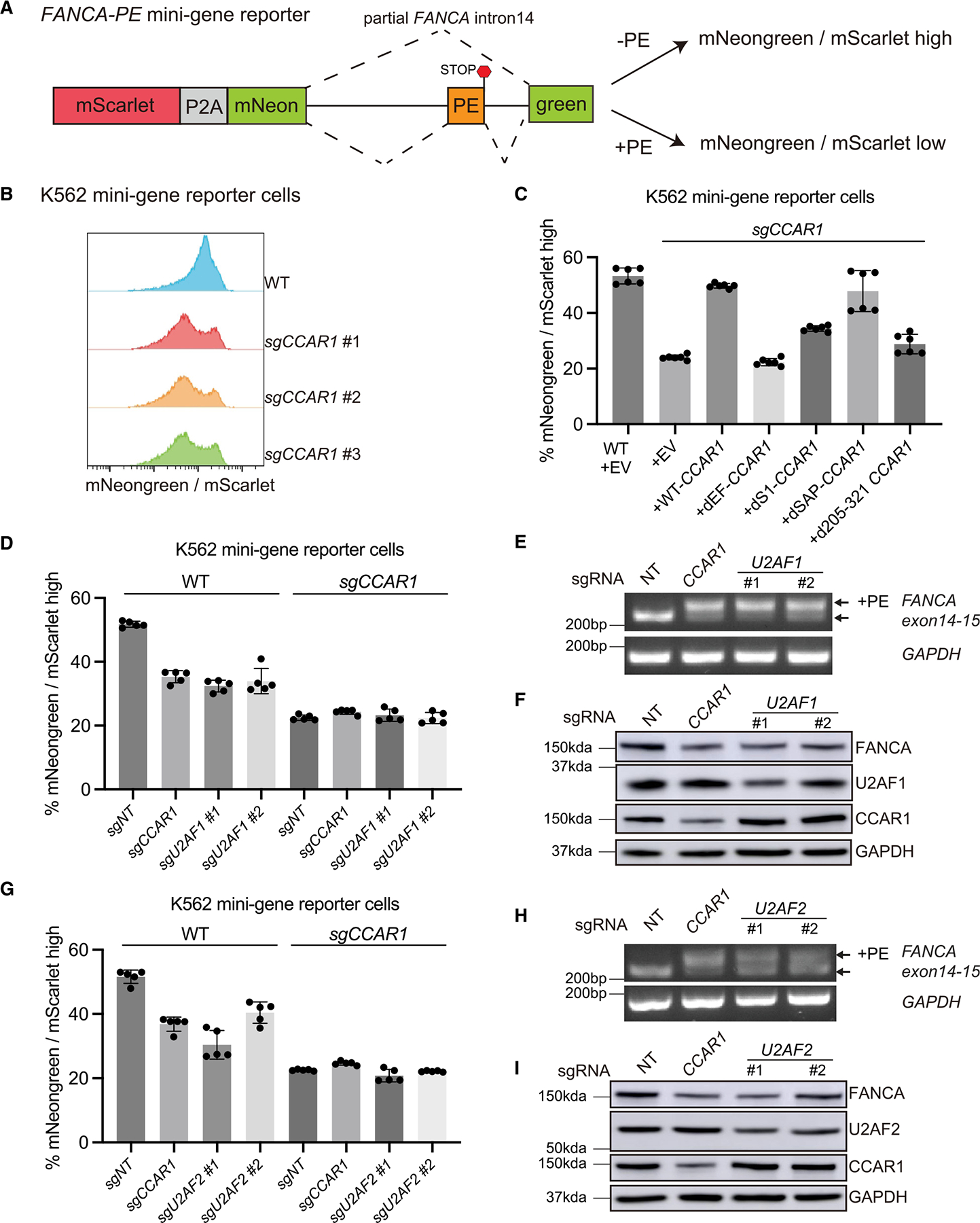
(A) Schematic representation of the fluorescence-based FANCA PE mini-gene reporter. See also Figure S6A.
(B) Histograms showing the ratio of mNeongreen to mScarlet (mNG/mSC) signal from K562 WT and CCAR1 knockout clones carrying the mini-gene reporter.
(C) K562 WT and CCAR1 knockout mini-gene reporter cells were transduced with the indicated lentiviral constructs. Mean and SD of mNG/mSC high cells are plotted (n = 6).
(D and G) Loss of CCAR1, U2AF1, or U2AF2 leads to reduced mNG/mSC indicating splicing defect. K562 WT or CCAR1 knockout mini-gene reporter cells were lentivirally transduced with Cas9 and either non-targeting control sgRNA (sgNT), sgCCAR1, or sgU2AF1 (D), or sgU2AF2 (G). 96 h after puromycin selection, the pool cells were analyzed by flow cytometry, and the mean and SD of mNG/mSC ratio was plotted (n = 5).
(E and H) Loss of U2AF1 or U2AF2 leads to inclusion of FANCA poison exon. RT-qPCR showing evaluation of FANCA poison exon (PE) inclusion in the K562 minigene reporter cells transduced with sgNT, sgCCAR1, sgU2AF1(E), or sgU2AF2(H).
(F and I) Loss of CCAR1, U2AF1, or U2AF2 leads to reduction in FANCA protein levels. Evaluation of FANCA protein levels in K562 mini-gene reporter cells transduced with sgNT, sgCCAR1, sgU2AF1(F), or sgU2AF2(I).
See also Figure S6.
Next, we performed individual depletion of U2AF1 (Figure 6D) or U2AF2 (Figure 6G), via CRISPR-Cas9, in K562 cells carrying the mini-gene reporter. Similar to CCAR1 depletion, loss of U2AF1 or U2AF2 reduced the reporter signal in WT cells but not in CCAR1 knockout cells (Figures 6D and 6G). We confirmed that loss of U2AF1 or U2AF2 causes an inclusion of the poison exon in FANCA mRNA (Figures 6E and 6H). Finally, we observed that the FANCA protein level was decreased after loss of U2AF1 or U2AF2 (Figures 6F and 6I). Importantly, CCAR1 loss did not affect the protein level of U2AF1 or U2AF2, and vice versa. Thus, the mechanism by which CCAR1 loss causes inclusion of the poison exon is not due to reduction in the protein level of spliceosome components such as the U2AF1/2 heterodimer. Collectively, the CCAR1-U2AF1/2 axis plays a pivotal role in the exclusion of the poison exon from the FANCA transcript.
DISCUSSION
CCAR1 showed similar characteristics to FA genes in DNA damage CRISPR knockout screens, suggesting that it may also impact ICL repair via the FA/BRCA pathway.13 FA is generally characterized by deficiency of FANCD2 monoubiquitination, high sensitivity to DNA interstrand cross-linking agents, and radial chromosomes. As predicted by the CRISPR screens, we observed FA phenotypes in CCAR1 knockout cells. Interestingly, the expression of FANCA and FANCG protein was decreased in CCAR1 knockout cells. As FANCA and FANCG proteins interact and stabilize each other,22,36 loss of at least one of these proteins may cause destabilization of the other protein. Transduction of FANCA cDNA in CCAR1 knockout cells restored FANCD2 monoubiquitination and MMC resistance, while transduction of FANCG cDNA did not. Therefore, FANCA loss is the primary outcome induced by CCAR1 loss, and the loss of FANCA protein subsequently triggers the destabilization of the FANCG protein.
Previous studies have reported interactions of CCAR1 with the spliceosome A complex37,38 and other splicing factors.24 Based on these findings, we hypothesized that CCAR1 loss caused a splicing error in FANCA transcript, and indeed, we found a poison exon with a stop codon between exon14 and exon15. We reasoned that inclusion of this poison exon could decrease FANCA protein levels in CCAR1 knockout cells, and we tested this hypothesis using two approaches, splice-switching ASO and CRISPR-mediated editing of the poison exon. ASO is a class of US Food and Drug Administration (FDA) approved RNA-based therapeutics that binds to reverse complementary regions of target pre-mRNA and alters splicing or induces degradation of pre-mRNA.39 The ASO was designed to bind the region around the 5′ edge of the FANCA poison exon to alter splicing of FANCA transcript in CCAR1 knockout cells. Targeting FANCA poison exon via ASO or CRISPR gRNA in CCAR1 knockout cells resulted in exclusion of the poison exon from FANCA transcript and restoration of FANCA protein expression. Hence, the inclusion of the FANCA poison exon is the critical mechanism for the loss of FANCA protein expression and FA phenotype in CCAR1 knockout cells.
We next identified a previously uncharacterized domain, the EF hand domain, as a functional splicing modulator. Complementation of CCAR1 knockout cells with WT CCAR1 restored FANCA protein expression; however, CCAR1 mutants lacking the EF hand domain were deficient in restoring FANCA protein expression. EF hand is a short motif of 45 amino acids and is involved in binding intracellular calcium.40 In CCAR1, the EF hand domain is predicted to be inactive and unlikely to bind calcium ions.32,41,42 Since the EF hand domain is conserved in CCAR1 orthologs from C. elegans (LST-3) to humans, it has been suggested that the EF hand domain might hetero-dimerize with other proteins.41 However, no functional interactions via the EF hand domain of CCAR1 have been identified so far, and we now report a significant function for the EF hand domain of CCAR1.
By mass spectrometric analysis of FLAG-CCAR1 IP complexes, we found an enrichment of factors involved in RNA splicing. Among the splicing factors, the U2AF1/2 heterodimer showed specific interaction with the WT CCAR1 but not the dEF-CCAR1 mutant. Interestingly, a recent C. elegans article also reported that CCAR1 affects alternative splicing via its interaction with UAF1/2 heterodimer, the worm homolog of human U2AF1/2.43 Importantly, a mini-gene reporter assay showed that depletion of U2AF1/2 yields a similar phenotype as loss of CCAR1, resulting in FANCA poison exon inclusion and loss of FANCA expression.
CCAR1 binds efficiently to the FANCA pre-mRNA, and loss of CCAR1 is sufficient for inclusion of the FANCA poison exon. Hence, we reasoned that CCAR1 directly cooperates with U2AF1/2 in splicing out the FANCA poison exon. U2AF1/2 has a defined consensus sequence for modulating splicing44,45; however, we did not find a novel CCAR1-specific consensus motif in the transcripts spliced in a CCAR1-dependent manner. Overall, our findings suggest that CCAR1 plays an important role in regulating FANCA poison exon splicing in concert with the U2AF1/2 heterodimer.
Comprehensive analysis based on RNA-seq revealed that CCAR1 plays a multi-faceted role in maintaining faithful splicing. Apart from skipped exon events, CCAR1 loss also triggers other types of alternative splicing events such as intronic retention, alternative 5′ splice site, alternative 3′ splice site, and mutually exclusive exons. Indeed, we also identified alternative splicing events in KMT2C, RBM48, and IVNS1ABP transcripts upon CCAR1 loss. KMT2C has been reported as a mediator of the estrogen dependence of breast cancer.46 RBM48 was recently purified biochemically as part of the minor spliceosome and reported as a regulator of U12-type intron splicing, whose defect in human hematopoietic stem cells is associated with myelodys-plastic syndrome, predisposing individuals to acute myeloid leukemia.47 It was recently reported that IVNS1ABP protein levels are impaired by inactivation of NSUN7 followed by CCDC9B loss, resulting in sensitivity to bromodomain inhibitors in liver cancer cells.48 Therefore, CCAR1 functions as a modulator of splicing and regulates splicing events not only of the FANCA transcript but also of other transcripts, indicating the potential of biological phenotypes other than FA in CCAR1-deficient cells. Future studies are needed to clarify how CCAR1 specifically regulates the various alternative splicing events.
In conclusion, CCAR1 regulates the FA pathway by modulating splicing via its EF hand domain. The loss of CCAR1 results in the inclusion of a poison exon in the FANCA transcript and thus results in loss of FANCA protein expression and high sensitivity to DNA interstrand cross-linking drugs. Our findings provide valuable information for further investigation of CCAR1 as a regulator of splicing events and a candidate target for cancer therapy.
Limitations of the study
In this study, our primary focus was the role of CCAR1 in FANCA mRNA splicing and how its loss could lead to FANCA loss. However, we observed that CCAR1 loss also results in mis-splicing of several other transcripts, the biological impact of which is yet to be determined. Further, the biological cues that trigger CCAR1-mediated splicing remain unclear at this point. Under what context does a cell choose to turn ON or OFF CCAR1-mediated splicing? Since the FANCA poison exon sequence is only found in primates, CCAR1-mediated fine-tuning may regulate specific developmental and differentiation processes. Also, how loss of CCAR1 can lead to diverse alternative splicing patterns, such as intronic retention, alternative 5′ splice sites, and alternative 3′ splice sites, remains unknown. Indeed, CCAR1 may interact with multiple spliceosomal components, and distinct CCAR1-containing complexes could modulate different aspects of splicing. Future studies are needed to identify the various spliceosomal components that work in concert with CCAR1 to modulate splicing.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Alan D. D’Andrea (alan_dandrea@dfci.harvard.edu).
Materials availability
The knock-out cell lines generated in this study are available from the lead contact without restrictions.
Data and code availability
The RNA-seq datasets discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GEO: GSE242781. All raw data files and images have been deposited in Mendeley Data and are accessible using Mendeley Data: https://doi.org/10.17632/drsrg8xtdm.2
The paper does not report any original code.
Any additional information required to reanalyze the data reported in this study is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell cultures and generation of CRISPR engineered cell lines
RPE p53−/− cells generated previously49 were grown in DMEM/F12 containing GlutaMAX (Gibco) and supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich). HEK293T (ATCC, CRL-3216) cells were grown in DMEM (Gibco) supplemented with 10%FBS. K562 (ATCC, CCL-243) cells were grown in RPMI 1640 supplemented with 10%FBS, 1% P/S. All cells were cultured in an incubator maintained at 37°C, 5% CO2 and a relative humidity of 95%.
All sgRNAs used in this study were manufactured by Synthego or Integrated DNA Technologies. TrueGuide sgRNA Negative Control, non-targeting 1 (Thermo Fisher Scientific) or Negative Control sgRNA (mod) #1 (Synthego) were used as a negative control sgRNA. Each sgRNA along with TrueCut Cas9 Protein v2 (Thermo Fisher Scientific) was transfected into RPE p53−/− cells using Lipofectamine CRISPRMAX (Thermo Fisher Scientific) in accordance with manufacturer’s instructions. Media was changed the following day, and then 2 days after the transfection the knockout pool cells were seeded into a 15 cm cell culture dish. Single colonies were picked up for genomic editing analysis via immuno-blotting.
For knockdown experiments, cells were cultured for at least 2 days after the transfection of each sgRNA, and then pool cells were used for further evaluations.
For generating CCAR1 knockout clones in HEK293T and K562 cells, a ribonucleoprotein (RNP) complex was formed by Alt-R™ S.p. HiFi Cas9 and CRISPR-Cas9 sgRNA targeting CCAR1 (Integrated DNA Technologies). Cells were resuspended in SF Nucleofector Solution with supplement (Lonza) and then mixed with the Cas9/sgRNA RNP complex and Alt-R™ Cas9 Electroporation Enhancer (Integrated DNA Technologies). The Cas9/sgRNA RNP complex was delivered to HEK293T and K562 cells by 4D-Nucleofector™ (Lonza). Media was changed the following day, and then 2 days after cells were seeded in a 96-well plate to isolate single clones.
Individual clones were tested for genomic editing analyses using immunoblotting and genomic PCR with subsequent Sanger sequencing.
All the CCAR1 “knockout” clones used in this study showed at least 85% knockout in the CCAR1 gene and hence they are referred to as “knockout” throughout the study (Table S2). We note that some of our knockout clones are polyclonal and also in some rare instances the CRISPR-mediated editing could lead to an in-frame disruption in the CCAR1 gene, thereby leading to residual expression of the CCAR1 protein (Table S2). We do see residual CCAR1 expression in some of our RPE p53−/− CCAR1 knockout clones. To mitigate the impact of residual CCAR1 expression on the phenotypic readouts, we have used at least three knockout clones from three different cell lines (RPE1, HEK293T and K562). The biological phenotypes are consistent across all the CCAR1 knockout clones used in this study suggesting that all our clones are likely functionally deficient for CCAR1.
METHOD DETAILS
Colony formation assay
Cells were seeded in 6-well plates at 1000–2000 cells/well. The day after seeding, cells were exposed to each drug or x-ray. X-ray irradiation was performed using RS-2000 Irradiator (Rad Source Technologies). For MMC treatment, the media containing MMC was removed after 24 h exposure, and fresh media without MMC was added, and the plates were incubated for 6–8 days. After incubation, plates were washed with PBS and fixed in a mixture of 50% methanol, 10% acetic acid, and 40% water for 20 minutes. The fixed cells were stained with 0.5% crystal violet (Sigma-Aldrich) in 20% methanol for 2–6 h and then washed with water twice. The plates were imaged on GE Amersham Imager 600 using colorimetric transillumination setting. The area fraction of the colonies in each well was estimated using ImageJ. Colony formation assays were performed in technical duplicate.
Plasmids
psPAX2 and pMD2.G (Addgene #12260 and #12259) were gifts from Dr. Didier Trono. pLV-EF1a-IRES-Blast (pLV-Blast) (Addgene #85133) was a gift from Dr. Tobias Meyer.51 pCMV6-CCAR1-Myc-DDK was purchased from Origene (RC224293). CCAR1 was cloned into pLV-Blast construct using BamHI/MluI cloning sites or pOZ construct using XhoI/NotI cloning sites, respectively. Deletion CCAR1 mutants and pMMP-FLAG-FANCA-PE were generated by site-directed mutagenesis. sgRNAs were cloned into lentiCRISPR-v2 construct as previously described.52 pMMP-puro-FANCA, pMMP-puro-FANCG and pMMP-FLAG-FANCA were generated previously in our laboratories.50
Plasmid transfection into HEK293T cells was performed using Lipofectamine LTX with Plus reagent (Thermo Fisher Scientific). For RPE p53−/− and K562 cells, plasmids were transduced using lentiviral or retroviral particles.
Virus generation
For all virus generations and transductions media supplemented with heat inactivated FBS was used. Retrovirus was generated by co-transfecting retroviral constructs, pMMP-puro, with packaging vectors, pCG-Gag-pol and pCG-VsVg, into HEK293T cells using CalPhos Mammalian Transfection Kit (Takara Bio). Two days after transfection, supernatant containing virus was harvested and filtered through 0.45 μm filter. Viral transductions were performed in the presence of 8 μg/mL polybrene (Sigma-Aldrich).
Lentivirus was generated by co-transfecting lentiviral constructs (pLV-Blast or lentiv2-CRISPR) with packaging vectors, psPAX2 and pMD2.G, into HEK293T cells using CalPhos Mammalian Transfection Kit (Takara Bio). Two days after transfection, supernatant containing virus was harvested and filtered through 0.45 μm filter. Viral transductions were performed in the presence of 8 μg/ml (for RPE p53−/− cells) or 4 μg/ml (for K562 cells) polybrene (Sigma-Aldrich).
Puromycin dihydrochloride (Sigma-Aldrich) or Blasticidin S HCl (Thermo Fisher Scientific) was used after infection of retrovirus with pMMP-puro vectors or lentivirus with pLV-Blast vectors, respectively. For antibiotic selection, cells were cultured with 12 μg/mL Puromycin dihydrochloride for 3 days or 10 μg/mL Blasticidin S HCl for 6–7 days.
CellTiter-Glo assay
To test MMC sensitivity, cells were seeded in 96-well plates at 500–1000 cells/well. The day after seeding, MMC was added to the wells, and then the plates were incubated for 6 days. After incubation, ATP in each well was quantified using CellTiter-Glo Luminescent Cell Viability Assay (Promega). CellTiter-Glo assays were performed in technical duplicate if not otherwise specified in a figure legend.
Immunofluorescence
Cells were seeded on glass coverslips (Fisher Scientific) in 6 well plates. Cells were treated with MMC the next day after seeding. After the MMC treatment, cells were washed with PBS three times. Next, cells were pre-extracted on ice for 5 minutes with the pre-extraction buffer (20 mM HEPES pH7.4, 3 mM MgCl2, 50 mM NaCl, 300 mM sucrose, 0.5% triton) supplemented with protease and phosphatase inhibitors (Cell Signaling Technology). Cells were then fixed in 3.7% paraformaldehyde for 15 minutes at room temperature, and then washed with PBS three times for 5 minutes each. Cells were blocked and permeabilized in blocking buffer (PBS containing 10% goat serum, 1% BSA and 0.5% Triton). Coverslips were incubated at 4°C overnight with primary antibody diluted in blocking buffer. Next day, the coverslips were washed three times in PBS for 5 minutes each. The coverslips were then incubated with secondary antibody diluted in blocking buffer for 45 minutes, washed with PBS three times for 5 minutes each, rinsed with milliQ water once, and mounted with ProLong Gold or ProLong Diamond containing DAPI (Thermo Fisher Scientific). The slides were allowed to cure in the dark for 24 h and then sealed with nail paint. Slides were imaged on Zeiss Axio Observer under 63X objective. For each field of view, images were captured with 5–7 Z-stacks and 5 phases of Apotome sectioning. Orthogonal projection was used to get the maximum intensity projections and the number of foci per cell was counted using Cell Profiler (Broad Institute). More than 80 cells were counted for each condition.
Immunoblotting
Whole cell lysate was prepared by lysing cells in ice-cold RIPA buffer (Cell Signaling Technology) supplemented with Protease/Phosphatase inhibitor Cocktail (Cell Signaling Technology). Cell lysate was cleared by centrifuging the samples at 12000 rpm, 4°C for 20 minutes. Supernatant was recovered and protein concentration was estimated using BCA assay kit (Thermo Fisher Scientific). Protein samples were boiled in 2X Laemmli sample buffer (Bio-rad) supplemented with 5% 2-mercaptoethanol or NuPAGE LDS sample buffer (Thermo Fisher Scientific) supplemented with NuPAGE sample reducing agent (Thermo Fisher Scientific). Samples were run in NuPAGE 4–12% Bis-Tris gels (Thermo Fisher Scientific) using NuPAGE MES SDS Running Buffer (Thermo Fisher Scientific) or NuPAGE 3–8% Tris-Acetate Gel (Thermo Fisher Scientific) using NuPAGE Tris-Acetate SDS Running Buffer (Thermo Fisher Scientific). After electrophoresis, samples were transferred onto a PVDF membrane (Amersham). Membranes were blocked for 1 h with Blocking Buffer, which was prepared by diluting Fish Serum Blocking Buffer (Thermo Fisher Scientific) with Tris-buffered saline containing 0.1% Tween20 (TBST). Membranes were incubated with primary antibodies diluted in Blocking Buffer overnight at 4°C. Membranes were washed with TBST three times, 5 minutes each, incubated with secondary antibodies conjugated to HRP diluted in Blocking Buffer for 1 h. After secondary antibody incubation, membranes were washed with TBST and imaged using chemiluminescence setting on GE Amersham Imager 600.
For ubiquitinated FANCD2 blotting, cells were treated with 100 ng/mL MMC for 24 h prior to harvesting cells. For testing FANCA stability, cells were treated with 100 μg/mL cycloheximide (CHX) (Sigma-Aldrich) for 1–6 h or 10 μM MG-132 for 6 h, respectively, prior to harvesting cells.
Chromosomal breakage analysis
Cells were exposed to 5 ng/mL MMC for 48 h. Cells were treated with 100 ng/mL of colcemid for 2 h, followed by a hypotonic solution (0.075 M KCl) for 20 minutes and fixed with 3:1 methanol/acetic acid. Slides were stained with Wright’s stain and 50 metaphase spreads were scored for aberrations.
Reverse transcription qPCR
Total RNA was extracted using RNeasy Mini Kit (QIAGEN). Complementary DNA synthesis was performed using SuperScript IV First-Strand cDNA Synthesis Reaction (Thermo Fisher Scientific). For analyzing FANCA transcript, a target region was amplified using each primer set and Q5 High-Fidelity 2X Master Mix (New England Biolabs). PCR products were separated in 2% agarose gel stained with SYBR Safe (Thermo Fisher Scientific) and imaged using ChemiDoc MP Imaging System (Bio-rad). Quantitative PCR (qPCR) was performed using Power SYBR Green PCR Master Mix (Thermo Fisher Scientific) and Quant Studio 7 flex Real-Time PCR System (Thermo Fisher Scientific). ΔCt was calculated using GAPDH as a control and normalized to RPE p53−/− cell line if not otherwise specified in a figure legend. RT-qPCR assays were performed in technical triplicate.
ASO transfection
ASO was designed to target near the 5’edge of the FANCA poison exon. The ASO was synthesized using 2’-O-methoxyethyl-phosphorothioated bases by Integrated DNA Technologies (IDT). Prior to ASO transfection, cells were seeded in a 6-well plate and cultured overnight. ASO was transfected at a final concentration of 0.1 μM using Lipofectamine LTX with Plus reagent (Thermo Fisher Scientific). After 48 h incubation, cells were harvested and analyzed by RT-PCR and western blotting.
Mass spectrometry analysis
HEK293T WT or CCAR1 knockout cells were seeded into 15cm dishes and cultured overnight. HEK293T WT cells were transfected with pOZ empty vector (EV) or pOZ-Flag-HA-CCAR1 plasmid. HEK293T CCAR1 knockout cells were transfected with pLV-EV, pLV-CCAR1-3xFLAG, or pLV-dEF-CCAR1-3xFLAG. Cells were transfected with 30 μg of vector /dish using Lipofectamine LTX with Plus reagent and harvested 48 h after transfection. Cells were lysed with 40 mM Tris-HCl pH 8 buffer supplemented with 0.5% NP-40, 200 mM NaCl, 2 mM EDTA, Protease/Phosphatase inhibitor Cocktail (Cell Signaling Technology), and MG-132. The lysate was centrifuged, and the pellet was resuspended with 20 mM Tris-HCl pH 7.5 supplemented with 100 mM KCl, 2 mM MgCl2, 1 mM CaCl2, and Protease/Phosphatase inhibitor Cocktail and treated with Micrococcal Nuclease (Thermo Fisher Scientific) for approximately 30 minutes, followed by addition of 5 mM EGTA (final concentration) to stop digestion. After three pulses of sonication (30 s each), samples were centrifuged to collect the supernatants as chromatin fractions. Immunoprecipitation was performed using Anti-FLAG M2 antibody (Sigma-Aldrich) conjugated with Dynabeads Protein G for Immunoprecipitation (Thermo Fisher Scientific). Beads were washed with TGN-150 wash buffer containing 20 mM Tris-HCl pH 7.6, 150 mM NaCl, 3 mM MgCl2, 10% glycerol, and 0.01% NP-40. Beads were then boiled at 70°C for 20 minutes in 1:1 diluted NuPAGE LDS Sample buffer (4X) (Thermo Fisher Scientific) to elute the proteins. The eluted proteins were applied to SDS-PAGE, and Coomassie stained gel band samples were used for the analyses. Two independent biological replicate samples were analyzed by mass spectrometry.
The gel bands were cut into approximately 1 mm3 pieces. Gel pieces were then subjected to a modified in-gel trypsin digestion procedure.62 Gel pieces were washed and dehydrated with acetonitrile for 10 minutes and completely dried in a SpeedVac (Thermo Fisher Scientific). The gel pieces were rehydrated with 50 mM ammonium bicarbonate containing 12.5 μg/mL modified sequencing-grade trypsin (Promega) at 4°C. After rehydration, the excess trypsin solution was removed and replaced with 50 mM ammonium bicarbonate solution. Samples were then incubated at 37°C overnight. Peptides were later extracted with a solution containing 50% acetonitrile and 1% formic acid and dried in a SpeedVac. The samples were reconstituted in 5–10 μL of HPLC solvent A (2.5% acetonitrile, 0.1% formic acid). A nano-scale reverse-phase HPLC capillary column was created by packing 2.6 μm C18 spherical silica beads into a fused silica capillary (100 μm inner diameter x ~30 cm length) with a flame-drawn tip.63 After equilibrating the column each sample was loaded via an EASY-nLC (Thermo Fisher Scientific). A gradient was formed, and peptides were eluted with increasing concentrations of solvent B (90% acetonitrile, 0.1% formic acid). As peptides eluted, they were subjected to electrospray ionization and then entered into a Orbitrap Exploris480 mass spectrometer (Thermo Fisher Scientific). Peptides were detected, isolated, and fragmented to produce a tandem mass spectrum of specific fragment ions for each peptide. Peptide sequences were determined by matching protein databases with the acquired fragmentation pattern by the software program, Sequest (Thermo Fisher Scientific).53 All databases include a reversed version of all the sequences and the data was filtered to between a one and two percent peptide false discovery rate.
For the IP-mass spectrometry samples from HEK293T WT cells transfected with empty vector or WT-CCAR1, the enriched proteins were determined for network and pathway analyses as follows. First, proteins that did not have at least 3 total counts in at least 2 samples were filtered out. Second, the log2 fold enrichment or change (LFC) values were calculated by 1) replacing empty intensities with ones, 2) log2 transforming the intensities, 3) subtracting the mean log2 values of EV-transfected cells from the mean log2 values of CCAR1-transfected cells to obtain the LFCs. Finally, proteins with positive LFCs were selected as enriched proteins. Intensity fold enrichment was plotted by calculating 2 to the power of LFC. Network and pathway analyses were performed using the Reactome Functional Interaction Plugin (version 8.0.6) in Cytoscape (version 3.10).54,55
For IP-mass spectrometry samples from HEK293T CCAR1 knockout cells transfected with empty vector, WT-CCAR1-3XFLAG, or dEF-CCAR1-3XFLAG, data analysis was performed similarly with the following exceptions. Proteins that did not have at least 2 total counts in at least 2 samples were filtered out. Also, proteins with negative LFC between WT-CCAR1-3xFlag and EV, or between dEF-CCAR1-3xFlag and EV, were filtered out. The rank plot of the LFC between WT-CCAR1-3xFlag and dEF-CCAR1-3XFLAG was generated using ggplot2.
RNA immunoprecipitation assay
Protocol for RIP assay was adapted from Nicholson-Shaw et al.26 HEK293T WT and CCAR1 knockout cells were cultured in 15 cm dishes and 2–3 plates were used per cell line for each assay. Cells were harvested, washed with ice-cold PBS 2X times, cross-linked with 0.2% PFA for 15 min. and then quenched by adding glycine to a final concentration of 125mM. Cells were lysed in iCLIP buffer (50mM Tris-HCl pH 7.4, 100mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate) supplemented with protease, phosphatase and RNase inhibitor cocktail. The samples were rotated at 4°C for 10 min. and then sonicated on ‘light’ setting twice. The lysate was incubated with Protein A beads on a rotor for 10 min. at 4°C to pre-clear the extract. The lysate was centrifuged at max speed for 10 min. and the supernatant was recovered and incubated for 2 hrs on a rotor at 4°C with Protein A beads coupled to CCAR1 Ab (Bethyl). A part of the lysate (0.4%–0.6%) was kept aside as input. After 2 hrs the beads were washed twice with iCLIP buffer. The input and the IP samples were all subjected to TURBO DNase treatment (Thermo Fisher Scientific) for 30 min. at 37°C to get rid of the genomic DNA and the cross-links were reversed by digesting with Proteinase K (NEB) for 30 min. at 37°C and further digestion and denaturation of proteins was achieved by adding urea to a final concentration of 2.5M and incubating the samples at 37°C for additional 20 min. After digestion of the DNA and protein from the samples, the RNA was purified by adding Trizol and processing the samples through Directzol RNA mini-prep kit (Zymo Research). The eluted RNA from the input and IP samples were converted to cDNA using the High-capacity RNA-to-cDNA kit (Thermo Fisher Scientific). The reverse transcription was performed with and without adding the reverse transcriptase enzyme. qPCR was set up with primers targeting FANCA pre-mRNA, FANCA mature mRNA, and GAPDH in technical replicates for each assay. Ct values from HEK293T WT samples were normalized to Ct values from HEK293T CCAR1 knockout cells.
Mini-gene reporter assay
A mini-gene reporter carrying mNeongreen split by synthetic intronic sequence derived from FANCA intron 14 was generated as previously described.64 The synthetic intron was made up of 200 bp of the start of the FANCA intron 14, 250 bp upstream of the poison exon, the 36bp poison exon and 159 bp downstream of the poison exon. The synthetic intron so generated was placed between the sequences encoding the N terminus and C terminus of split mNeongreen. The mScarlet-P2A-splitmNeongreen (N terminus)-synthetic intron-mNeongreen (C terminus) construct was integrated into a pLV-EF1a-IRES-Neo vector (Addgene #85139) to generate the mini-gene reporter. K562 cells were lentivirally transduced with the mini-gene reporter. 48hr after lentiviral transduction, the cells were enriched by using G418 (0.8mg/ul) for seven days. For the CCAR1 complementation experiments, the K562 CCAR1 knockout cells carrying the mini-gene reporter were lentivirally transduced with empty vector, CCAR1-wildtype cDNA, or cDNA encoding various mutant forms of CCAR1 expressed by a pLV-EF1a-IRES-Blast vector (addgene #85133). 48hr after transduction, cells were cultured with 10ug/ul blasticidin for 120hr. For the U2AF1 or U2AF2 depletion experiments, a sgRNA targeting either control, CCAR1, U2AF1, or U2AF2, together with Cas9 encoded by a lentiCRISPR-v2 vector (addgene #52961), were lentivirally transduced into the K562 minigene reporter cells. 48hr after transduction, cells were cultured with 1ug/ul puromycin for 96hr. After antibiotic selection, mScarlet and mNeongreen signals in the reporter cells were analyzed by CytoFLEX (Beckman). For the flow cytometry analyses, mScarlet positive fractions within living cells were gated. Thereafter, the ratio of mNeongreen to mScarlet was plotted by using Flowjo ver.10.9.0.
Next-generation sequencing
Library preparation and sequencing reactions were conducted at GENEWIZ, Lin/Azenta US, Inc. Standard RNA-seq (60 million reads/sample, N = 3 for each cell line) and Deep RNA-seq (300 million reads/sample, N = 2 for each cell line) were performed for RPE p53−/− (parental control) and p53−/−CCAR1 knockout cells. RNA samples were prepared using RNeasy Mini Kit and RNase-Free DNase Set (QIAGEN). RNA samples were quantified using Qubit 2.0 Fluorometer, and the RNA integrity was checked with 4200 TapeStation. NEBNext Ultra II RNA Library Prep Kit for Illumina was used following the manufacturer’s recommendations (New England BioLabs). Briefly, mRNAs were initially enriched with Oligo d(T) beads. Enriched mRNAs were fragmented for 15 minutes at 94°C. First strand and second strand cDNA were subsequently synthesized. cDNA fragments were end repaired and adenylated at 3’ends, and universal adapters were ligated to cDNA fragments, followed by index addition and library enrichment by PCR with limited cycles. The sequencing libraries were validated using Agilent TapeStation and quantified using Qubit 2.0 Fluorometer as well as by quantitative PCR (KAPA Biosystems). The sequencing libraries were clustered on flowcell lanes. After clustering, the flowcell was loaded on the Illumina NovaSeq instrument according to manufacturer’s instructions. The samples were sequenced using a 2×150 Paired End configuration. Image analysis and base calling were conducted by the Control Software (NCS). Raw sequence data (.bcl files) generated from the Illumina instrument was converted into fastq files and de-multiplexed using Illumina’s bcl2fastq (version 2.17) software. One mis-match was allowed for index sequence identification.
Several bioinformatics analyses were performed on the RNA-seq data to identify differentially expressed genes, differentially used exons, and differentially spliced events between RPE p53−/− control cells and p53−/−CCAR1 knockout cells.
For differential gene expression analysis, Kallisto (version 0.48.0)57 was used to map the raw RNA-seq reads against the GENCODE human reference gene annotation (Release 44)65 and generate raw transcript abundance estimates, and the tximport Bioconductor R package58 was used to summarize the transcript abundance estimates to produce gene-level estimated counts. Differentially expressed genes between control and CCAR1 knockout cell samples (N = 5) were determined using both edgeR (version 3.40.2)59 and DESeq2 (version 1.38.3)60 Bioconductor packages. The high concordance between the estimated log2 foldchange of the two algorithms were verified.
For both differential exon usage and differential splicing analyses, the two replicate sample pairs with deep RNA-seq, as well as the third replicate sample pair derived from combining the replicate samples with standard RNA-seq, were used. Before combining the samples with standard RNA-seq, Principal Component Analysis was performed to confirm that they clustered well. STAR (version 2.7.10b)56 was used to map RNA-seq reads and generate BAM files for these two analyses. Specifically, the 2-pass STAR was used to map the raw RNA-seq reads against the GRCh38/hg38 human reference genome, with the GENCODE human reference gene annotation (Release 44).65 The first round of the 2-pass STAR was run on only the deep RNA-seq reads to identify splice junctions. More confidence junctions were filtered by requiring them to have at least 5 total mapped reads. The second round of the 2-pass STAR was run using the filtered junctions to generate BAM files.
For differential exon usage analysis, DEXSeq (version 1.46.0)35 was used. In brief, a list of unique exonic regions was initially generated using the Python script dexseq_prepare_annotation.py. This script “collapsed” exonic regions from different transcripts to be used as exon counting bins. Next, the dexseq_count.py Python script was used to count reads for each sample. Finally, differentially used exons between control and CCAR1 knockout cells (n = 3) were determined using DEXSeq.
For the differential splicing analysis, MISO (version 0.5.4)33and other tools were used as discussed below. The exon-centric splicing event annotations were generated using the rnaseqlib Python package (http://github.com/yarden/rnaseqlib). These annotations were derived considering all transcripts from Ensembl, UCSC RefSeq and NCBI RefSeq gene annotations, which were obtained from https://hgdownload.soe.ucsc.edu/goldenPath/hg38/database, in files knownGene.txt, refGene.txt, and ncbiRef-Seq.txt. Each annotation event belongs to one of these 5 types: skipped exons (SE), alternative 3’ splice sites (A3SS), alternative 5’ splice sites (A5SS), mutually exclusive exons (MXE), and retained introns (RI). These annotated events were linked to representative overlapping exons or introns of the GENCODE human reference gene annotation (Release 44)65 using the intersect function of the bedtools toolset (version 2.31.0). The representative exons and introns were prioritized with high overlapping percentage (95%), followed by good transcript quality information including whether they were annotated as canonical and basic, and with better transcript support level. MISO was performed to quantify the isoforms and compute percent-spliced-in values (PSIs) of each alternative splicing event from the sorted BAM of each sample. Lower quality events were filtered out, for each replicate sample pair, first comparing the results of the control and the CCAR1 knockout samples using the compare_miso program, and then filtering the comparison results, using the filter_events program, by requiring each sample to have at least 35 (20 for the combined standard RNA-seq samples) total isoform-identifying reads. Finally, limma analysis (version 3.54.2)61 was performed on the PSI values of the filtered events to determine differential splicing events between control and CCAR1 knockout cells (N = 3). Significant differential alternative splicing events were called if they had FDR smaller than 0.05 and absolute delta PSI of at least 0.05. Events that had significant FDR (<0.05) but small absolute delta PSI (<0.05) were likely to be artifacts and thus were excluded from limma results. Volcano plots were generated using ggplot2. Sashimi plots were generated using the sashimi_plot program. The sashimi_plot program was modified to allow reads with multiple junctions, since these reads were excluded from events whose target regions were smaller than read length.
To have more confidence in the MISO results, we also independently analyzed differential splicing using rMATS (version v4.2.0).34 Since the FANCE PE skipped exon (SE) event was the most important and by far strongest among all different types of events, and SE events were the most frequent event type, we focused on analyzing the concordance of SE events between MISO and rMATS. First, rMATS was run using all sorted BAMs as inputs and the GENCODE human reference gene annotation (Release 44).65 Lower quality events were filtered out by requiring each sample to have at least 35 (20 for the combined standard RNA-seq samples) total isoform-identifying reads. Then, the shared events that were highly similar between MISO and rMATS were identified if their target regions overlapped by at least 95%. Since one MISO event could be linked to multiple slightly different rMATS events, for each MISO event, a representative rMATS event was chosen that had the largest absolute difference in inclusion level. Finally, we assessed the concordance between MISO and rMATS by comparing their inclusion differences (delta PSI values and differences in inclusion levels, respectively) from these shared events. We observed that a great majority (88.1%) of significant MISO events had a shared rMATS event. Furthermore, we observed that the inclusion differences of MISO and rMATS were highly positively correlated with R2 of 0.79. The scatterplot comparing the inclusion differences of the two algorithms was generated using ggplot2.
For Figure S5F to identify unannotated skipped exon events, the deep RNASeq data were additionally analyzed using rMATS with the “novelSS” option. Low quality events were filtered out as described above. Unannotated events were identified using those listed in the “fromGTF.novelSpliceSite.SE.txt” output file. Annotated skipped exon events were identified from the remainder events after excluding those listed in “fromGTF.novelJunction.SE.txt”. The annotated and unannotated events were filtered to include, and to exclude, the shared MISO (annotated) SE events, respectively. Significant skipped exon events were counted for different thresholds of FDR and difference in inclusion level (or delta PSI).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are represented as mean and SD or SEM for n = 2 or more independent experiments if not otherwise specified in a figure legend. For RT-qPCR, significance was calculated using one-way ANOVA or two-way ANOVA tests. All statistical analysis was performed using GraphPad Prism 10 software (GraphPad Software).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Vinculin (H-10) | Santa Cruz Biotechnology | Cat# sc-25336; RRID:AB_628438 |
| Rabbit monoclonal anti-Actinin (D6F6) | Cell Signaling Technology | Cat# 6487; RRID: AB_11179206 |
| Mouse monoclonal anti-GAPDH (D16H11) | Cell Signaling Technology | Cat# 5174; RRID: AB_10622025 |
| Rabbit monoclonal anti-Ku70 (D10A7) | Cell Signaling Technology | Cat# 4588; RRID: AB_11179211 |
| Rabbit polyclonal anti-CCAR1 | Bethyl | Cat# A300-270A; RRID:AB_155903 |
| Rabbit monoclonal anti-FANCA (D1L2Z) | Cell Signaling Technology | Cat# 14657; RRID:AB_2798558 |
| Rabbit polyclonal anti-FANCD2 | Novus | Cat# NB100-182; RRID AB_10002867 |
| Mouse monoclonal anti-FANCD2 (FI17) | Santa Cruz Biotechnology | Cat# sc-20022; RRID:AB_2278211 |
| Rabbit monoclonal anti-Histone H2A.X, phospho (Ser139) (20E3) | Cell Signaling Technology | Cat# 9718; RRID: AB_2118009 |
| Rabbit polyclonal anti-FANCE | Bethyl | Cat# 302-125A; RRID:AB_1720357 |
| Sheep polyclonal anti-FANCG | This study | N/A |
| Goat polyclonal anti-FANCL | GeneTex | Cat# GTX88033; RRID:AB_10720542 |
| Mouse monoclonal anti-FLAG (M2) | Sigma-Aldrich | Cat# F3165; RRID: AB_259529 |
| Rabbit polyclonal anti-U2AF35/U2AF1 | Abcam | Cat# ab86305; RRID:AB_1925546 |
| Rabbit polyclonal anti-U2AF2 | Cell Signaling Technology | Cat# 70471; RRID: N/A |
| Goat anti-rabbit IgG, HRP-linked | Cell Signaling Technology | Cat# 7074; RRID: AB_2099233 |
| Horse anti-mouse IgG, HRP-linked | Cell Signaling Technology | Cat# 7076; RRID: AB_330924 |
| Donkey anti-goat IgG (H+L), Secondary antibody, HRP | Thermo Fisher Scientific | Cat# A15999; RRID:AB_2534673 |
| Rabbit anti-sheep IgG-H&L HRP Conjugated | Abcam | Cat# ab6747; RRID: AB_955453 |
| Goat anti-mouse IgG (H+L) Highly adsorbed, Secondary antibody, Alexa Flour 488 | Thermo Fisher Scientific | Cat# A11029; RRID:AB_2534088 |
| Goat anti-rabbit IgG (H+L) Highly adsorbed, Secondary antibody, Alexa Flour 568 | Thermo Fisher Scientific | Cat# A11036; RRID:AB_10563566 |
| Chemicals, peptides, and recombinant proteins | ||
| Olaparib | Selleckchem | Cat# S1060 |
| Mitomycin C | Sigma-Aldrich | Cat# M0503 |
| Topotecan HCl | Selleckchem | Cat# S1231 |
| Cycloheximide | Sigma-Aldrich | Cat# 01810 |
| MG-132 | Selleckchem | Cat# S2619 |
| Lipofectamine CRISPRMAX Cas9 Transfection Reagent | Thermo Fisher Scientific | Cat# CMAX00003 |
| Lipofectamine LTX Reagent with PLUS Reagent | Thermo Fisher Scientific | Cat# 15338030 |
| TrueCut Cas9 Protein v2 | Thermo Fisher Scientific | Cat# A36499 |
| Alt-R™ S.p. HiFi Cas9 Nuclease V3 | Integrated DNA Technologies | Cat# 1081060 |
| Alt-R™ Cas9 Electroporation Enhancer | Integrated DNA Technologies | Cat# 1075915 |
| SF Cell Line 4D-Nucleofector™ X Kit | Lonza | Cat# V4XC-2012 |
| CalPhos Mammalian Transfection Kit | Takara Bio | Cat# 631312 |
| Polybrene | Sigma-Aldrich | Cat# TR-1003-G |
| Puromycin dihydrochloride from Streptomyces alboniger | Sigma-Aldrich | Cat# P7255 |
| Blasticidin S HCl | Thermo Fisher Scientific | Cat# A1113903 |
| GENETICIN (G418 Sulfate) | Thermo Fisher Scientific | Cat# 10131027 |
| Glycine | Sigma-Aldrich | Cat# G7126 |
| Urea | Sigma-Aldrich | Cat# U5378 |
| Dithiothreitol | Cell Signaling Technology | Cat# 7016L |
| Ultrapure 1M Tris-HCl pH 7.5 | Thermo Fisher Scientific | Cat# 15567027 |
| 5M NaCl | Thermo Fisher Scientific | Cat# AM9760G |
| IGEPAL CA-630 | Sigma-Aldrich | Cat# I8896 |
| Sodium deoxycholate | Sigma-Aldrich | Cat# D6750 |
| UltraPure SDS Solution, 10% | Thermo Fisher Scientific | Cat# 15553027 |
| 1M MgCl2 | Thermo Fisher Scientific | Cat# AM9530G |
| Crystal Violet certified by the Biological Stain Commission | Sigma-Aldrich | Cat# C0775 |
| ProLong Diamond Antifade Mountant with DAPI | Thermo Fisher Scientific | Cat# P36966 |
| ProLong Gold Antifade Mountant with DNA Stain DAPI | Thermo Fisher Scientific | Cat# P36941 |
| Triton X-100 | Sigma-Aldrich | Cat# T8787 |
| TWEEN-20 | Sigma-Aldrich | Cat# P9416 |
| RIPA Buffer (10X) | Cell Signaling Technology | Cat# 9806S |
| Protease/Phosphatase Inhibitor Cocktail (100X) | Cell Signaling Technology | Cat# 5872S |
| 2x Laemmli Sample Buffer | Bio-Rad Laboratories | Cat# 1610737 |
| 2-Mercaptoethanol | Sigma-Aldrich | Cat# M3148 |
| NuPAGE 4X LDS Sample Buffer | Thermo Fisher Scientific | Cat# NP0008 |
| NuPAGE Sample Reducing Agent (10X) | Thermo Fisher Scientific | Cat# NP0009 |
| Fish Serum Blocking Buffer | Thermo Fisher Scientific | Cat# 37527 |
| RNeasy Mini Kit | QIAGEN | Cat# 74104 |
| Direct-zol RNA MiniPrep | Zymo Research | Cat# R2050 |
| SuperScript IV First-Strand cDNA Synthesis Reaction | Thermo Fisher Scientific | Cat# 18091050 |
| High-Capacity RNA-to-cDNA kit | Thermo Fisher Scientific | Cat# 4387406 |
| RNase Inhibitor | Thermo Fisher Scientific | Cat# N8080119 |
| Q5 High-Fidelity 2X Master Mix | New England Biolabs | Cat# M0494S |
| SYBR Safe DNA Gel Stain | Thermo Fisher Scientific | Cat# S33102 |
| Cell Lysis Buffer (10X) | Cell Signaling | Cat# 9803S |
| cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | Cat# 11836170001 |
| Dynabeads Protein G for Immunoprecipitation | Thermo Fisher Scientific | Cat# 10004D |
| Dynabeads Protein A for Immunoprecipitation | Thermo Fisher Scientific | Cat# 10002D |
| Micrococcal Nuclease Solution | Thermo Fisher Scientific | Cat# 88216 |
| Sequencing Grade Modified Trypsin | Promega | Cat# V5113 |
| 16% Formaldehyde, Methanol-Free | Cell Signaling Technology | Cat# 12606S |
| Paraformaldehyde 16% solution EM Grade | Electron Microscopy Sciences | Cat#15710-S |
| Proteinase K | New England Biolabs | Cat# P8107S |
| TURBO DNase | Thermo Fisher Scientific | Cat# AM2238 |
| TRIzol LS reagent | Thermo Fisher Scientific | Cat# 10296028 |
| SUPERaseIn RNase Inhibitor (20U/ul) | Thermo Fisher Scientific | Cat# AM2694 |
| RNaseZap | Thermo Fisher Scientific | Cat# AM9780 |
| RNase-Free DNase Set | QIAGEN | Cat# 79254 |
| Critical commercial assays | ||
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat# G7573 |
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23227 |
| Power SYBR Green PCR Master Mix | Thermo Fisher Scientific | Cat# 4367659 |
| NEBNext Ultra II RNA Library Prep Kit for Illumina | New England Biolabs | Cat# E7770 |
| Deposited data | ||
| Human Reference Genome GRCh38/hg38 | The Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/grc/human |
| GENCODE Human Reference Gene Annotation Release 44 | GENCODE | https://www.gencodegenes.org/human/release_44.html |
| Ensembl GENCODE V43 Gene Annotation | UCSC Genome Browser | https://hgdownload.soe.ucsc.edu/goldenPath/hg38/database/knownGene.txt.gz |
| NCBI RefSeq Gene Annotation | UCSC Genome Browser | https://hgdownload.soe.ucsc.edu/goldenPath/hg38/database/ncbiRefSeq.txt.gz |
| UCSC RefSeq Gene Annotation | UCSC Genome Browser | https://hgdownload.soe.ucsc.edu/goldenPath/hg38/database/refGene.txt.gz |
| RNA-seq data | This study | GEO: GSE242781 |
| Raw data files and images | This study | Mendeley Data: https://doi.org/10.17632/drsrg8xtdm.2 |
| Experimental models: Cell lines | ||
| Human: RPE p53−/− | Lim et al.49 | N/A |
| Human: RPE p53−/−, CCAR1 knockout | This study | Table S2 |
| Human: RPE p53−/−, FANCA knockout | This study | N/A |
| Human: RPE p53−/−, CCAR1 knockout, FANCA poison exon-eliminated | This study | N/A |
| Human: RPE CCAR2 knockout | Iyer et al.28 | N/A |
| Human: HEK293T | ATCC | CRL-3216 |
| Human: HEK293T, CCAR1 knockout | This study | Table S2 |
| Human: K562 | ATCC | CCL-243 |
| Human: K562, CCAR1 knockout | This study | Table S2 |
| Human: K562, mini-gene reporter | This study | N/A |
| Human: K562, CCAR1 knockout, mini-gene reporter | This study | N/A |
| Oligonucleotides | ||
| Oligonucleotides | This study | Table S3 |
| Recombinant DNA | ||
| Plasmid: pMMP-puro (empty) | Kupfer et al.50 | N/A |
| Plasmid: pMMP-puro-FANCA | Kupfer et al.50 | N/A |
| Plasmid: pMMP-puro-FANCG | Garcia-Higuera et al.36 | N/A |
| Plasmid: pMMP-FLAG-FANCA | Kupfer et al.50 | N/A |
| Plasmid: pMMP-FLAG-FANCA-PE | This study | N/A |
| Plasmid: pLV-EF1a-IRES-Blast (pLV-Blast, EV) | Hayer et al.51 | Addgene# 85133 |
| Plasmid: pLV-EF1a-IRES-Neo | Hayer et al.51 | Addgene# 85139 |
| Plasmid: pLV-EF1a-IRES-Neo FANCA-PE mini-gene reporter | This study | N/A |
| Plasmid: CCAR1 (Myc-DDK-tagged) | Origene | Cat# RC224293 |
| Plasmid: pOZ-Flag-HA-CCAR1 | This study | N/A |
| Plasmid: pLV-Blast-CCAR1 | This study | N/A |
| Plasmid: pLV-Blast-CCAR1-3xFlag | This study | N/A |
| Plasmid: pLV-Blast-CCAR1 (ΔS1-like) | This study | N/A |
| Plasmid: pLV-Blast-CCAR1 (ΔSAP) | This study | N/A |
| Plasmid: pLV-Blast-CCAR1-3xFlag (Δ2–321) | This study | N/A |
| Plasmid: pLV-Blast-CCAR1-3xFlag (Δ2–380) | This study | N/A |
| Plasmid: pLV-Blast-CCAR1-3xFlag (Δ2–477) | This study | N/A |
| Plasmid: pLV-Blast-CCAR1 (Δ874–1150) | This study | N/A |
| Plasmid: pLV-Blast-CCAR1 (Δ924–1150) | This study | N/A |
| Plasmid: pLV-Blast-CCAR1 (Δ1033–1150) | This study | N/A |
| Plasmid: pLV-Blast-CCAR1-3xFlag (Δ2–146) | This study | N/A |
| Plasmid: pLV-Blast-CCAR1-3xFlag (Δ205–321) | This study | N/A |
| Plasmid: pLV-Blast-CCAR1-3xFlag (ΔEF) | This study | N/A |
| Plasmid: lentiCRISPR-v2 | Sanjana et al.52 | Addgene# 52961 |
| Plasmid: VsVg | Iyer et al.28 | N/A |
| Plasmid: Gag-pol | Iyer et al.28 | N/A |
| Plasmid: psPAX2 | Trono Lab | Addgene# 12260 |
| Plasmid: pMD2.G | Trono Lab | Addgene# 12259 |
| Software and algorithms | ||
| Sequest software | Eng et al.53 | https://www.thermofisher.com/order/catalog/product/OPTON-31014?SID=srch-srp-OPTON-31014 |
| Reactome Functional Interaction Plugin version 8.0.6 | Wu et al.54 | https://reactome.org/userguide/reactome-fiviz |
| Cytoscape version 3.10 | Shannon et al.55 | https://cytoscape.org/ |
| bcl2fastq version 2.17 | Illumina | https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html |
| DEXSeq version 1.46.0 | Anders et al.35 | https://bioconductor.org/packages/release/bioc/html/DEXSeq.html |
| dexseq_prepare_annotation.py program | Anders et al.35 | https://bioconductor.org/packages/release/bioc/html/DEXSeq.html |
| dexseq_count.py program | Anders et al.35 | https://bioconductor.org/packages/release/bioc/html/DEXSeq.html |
| STAR version 2.7.10b | Dobin et al56 | https://github.com/alexdobin/STAR |
| MISO version 0.5.4 | Katz et al.33 | https://miso.readthedocs.io/en/fastmiso/ |
| rMATS version v4.2.0 | Shen et al.34 | https://rnaseq-mats.sourceforge.io/ |
| rnaseqlib Python package | Katz et al.33 | https://miso.readthedocs.io/en/fastmiso/ |
| compare_miso program | Katz et al.33 | https://miso.readthedocs.io/en/fastmiso/ |
| filter_events program | Katz et al.33 | https://miso.readthedocs.io/en/fastmiso/ |
| sashimi_plot program | Katz et al.33 | https://miso.readthedocs.io/en/fastmiso/ |
| Kallisto version 0.48.0 | Bray et al.57 | https://pachterlab.github.io/kallisto/ |
| tximport Bioconductor R package | Soneson et al.58 | https://bioconductor.org/packages/release/bioc/html/tximport.html |
| edgeR version 3.40.2 | Robinson et al.59 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| DESeq2 version 1.38.3 | Love et al.60 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| FlowJo™ Software version 10.9.0 | Becton, Dickinson and Company | https://www.flowjo.com/ |
| bedtools toolset version 2.31.0 | N/A | https://bedtools.readthedocs.io/ |
| limma version 3.54.2 | Ritchie et al.61 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| Other | ||
| RS-2000 Irradiator | Rad Source Technologies | N/A |
| GE Amersham Imager 600 | GE HealthCare Technologies | N/A |
| CytoFLEX S | Beckman Coulter Life Sciences | Cat# B75442 |
| 4D-Nucleofector™ Core Unit | Lonza | Cat# AAF-1002B |
| 4D-Nucleofector™ X Unit | Lonza | Cat# AAF-1002X |
| ImageJ | U. S. National Institutes of Health | https://imagej.nih.gov/ij/ |
| CLARIOstar Plus Plate Reader | BMG Labtech | N/A |
| Zeiss Axio Observer | Zeiss | N/A |
| Cell Profiler | Broad Institute | https://cellprofiler.org/ |
| Auant Studio 7 Flex Real-Time PCR System | Thermo Fisher Scientific | N/A |
| Bioruptor UCD-200 | Diagenode | N/A |
| SpeedVac Vacuum Concentrators | Thermo Fisher Scientific | N/A |
| EASY n-LC | Thermo Fisher Scientific | N/A |
| Ortitrap Explors480 Mass Spectrometer | Thermo Fisher Scientific | N/A |
| Qubit 2.0 Fluorometer | Thermo Fisher Scientific | N/A |
| Illumina NovaSeq | Illumina | N/A |
| GraphPad Prism 10 | Graphpad | https://www.graphpad.com/ |
Highlights.
Loss of CCAR1 leads to Fanconi anemia (FA) phenotype
CCAR1 knockout cells are deficient in FANCA protein expression
CCAR1 modulates the splicing of FANCA pre-mRNA via the EF hand domain
CCAR1 acts in concert with the U2AF1/2 spliceosome complex to fine-tune splicing
ACKNOWLEDGMENTS
We thank members of the D’Andrea laboratory for their helpful suggestions and comments. This research was supported by R01 HL052725, the Dana-Farber/Harvard Cancer Center (DF/HCC) Specialized Program of Research Excellence (SPORE) in gastrointestinal (GI) cancer, a Lustgarten Foundation/Stand Up To Cancer Pancreatic Cancer Challenge grant, the Breast Cancer Research Foundation, the Ludwig Center at Harvard, and the Smith Family Foundation. DF/HCC is supported in part by NCI Cancer Center Support grant NIH P30 CA 006516. The work was also supported by Daiichi Sankyo Co, Ltd (to N.H.), the SENSHIN Medical Research Foundation and the Japan Society for the Promotion of Science (JSPS) Overseas Research Fellowships (to S.A.), and the TESARO-funded Foundation for Women’s Cancer post-doctoral fellowship award (to D.R.I.). Research in the Adelman lab is supported by the Ludwig Center at Harvard. The graphical abstract was created using BioRender.com.
DECLARATION OF INTERESTS
A.D.D. reports consulting for AbbVie, Deerfield Management Company, Impact Therapeutics, Moderna Therapeutics, PrimeFour Therapeutics, Schrödinger Inc., Servier BioInnovation LLC, and Tango Therapeutics; is a Scientific Advisory Board Member and Stockholder for Impact Therapeutics and Covant Therapeutics. K.A. is a member of the Advisory Board of Molecular Cell, the SAB of CAMP4 Therapeutics, consults for Syros Pharmaceuticals and Odyssey Therapeutics, and received research funding from Novartis not related to this work.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.molcel.2024.06.031.
REFERENCES
- 1.Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Yap TA, Plummer R, Azad NS, and Helleday T (2019). The DNA Damaging Revolution: PARP Inhibitors and Beyond. Am. Soc. Clin. Oncol. Educ. Book 39, 185–195. 10.1200/EDBK_238473. [DOI] [PubMed] [Google Scholar]
- 3.Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MIR, O’Connor KW, Konstantinopoulos PA, Elledge SJ, Boulton SJ, et al. (2015). Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 518, 258–262. 10.1038/nature14184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ceccaldi R, Rondinelli B, and D’Andrea AD (2016). Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 26, 52–64. 10.1016/j.tcb.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jo U, and Kim H (2015). Exploiting the Fanconi Anemia Pathway for Targeted Anti-Cancer Therapy. Mol. Cells 38, 669–676. 10.14348/molcells.2015.0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patterson-Fortin J, Bose A, Tsai WC, Grochala C, Nguyen H, Zhou J, Parmar K, Lazaro JB, Liu J, McQueen K, et al. (2022). Targeting DNA Repair with Combined Inhibition of NHEJ and MMEJ Induces Synthetic Lethality in TP53-Mutant Cancers. Cancer Res. 82, 3815–3829. 10.1158/0008-5472.CAN-22-1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quinet A, Tirman S, Jackson J, Šviković S, Lemaçon D, Carvajal-Maldonado D, González-Acosta D, Vessoni AT, Cybulla E, Wood M, et al. (2020). PRIMPOL-Mediated Adaptive Response Suppresses Replication Fork Reversal in BRCA-Deficient Cells. Mol. Cell 77, 461–474.e9. 10.1016/j.molcel.2019.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stok C, Kok YP, van den Tempel N, and van Vugt MATM (2021). Shaping the BRCAness mutational landscape by alternative double-strand break repair, replication stress and mitotic aberrancies. Nucleic Acids Res. 49, 4239–4257. 10.1093/nar/gkab151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ceccaldi R, Sarangi P, and D’Andrea AD (2016). The Fanconi anaemia pathway: new players and new functions. Nat. Rev. Mol. Cell Biol 17, 337–349. 10.1038/nrm.2016.48. [DOI] [PubMed] [Google Scholar]
- 10.Niraj J, Färkkilä A, and D’Andrea AD (2019). The Fanconi Anemia Pathway in Cancer. Annu. Rev. Cancer Biol 3, 457–478. 10.1146/annurev-cancerbio-030617-050422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kottemann MC, and Smogorzewska A (2013). Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 493, 356–363. 10.1038/nature11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nalepa G, and Clapp DW (2018). Fanconi anaemia and cancer: an intricate relationship. Nat. Rev. Cancer 18, 168–185. 10.1038/nrc.2017.116. [DOI] [PubMed] [Google Scholar]
- 13.Olivieri M, Cho T, Álvarez-Quilón A, Li K, Schellenberg MJ, Zimmermann M, Hustedt N, Rossi SE, Adam S, Melo H, et al. (2020). A Genetic Map of the Response to DNA Damage in Human Cells. Cell 182, 481–496.e21. 10.1016/j.cell.2020.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rishi AK, Zhang L, Boyanapalli M, Wali A, Mohammad RM, Yu Y, Fontana JA, Hatfield JS, Dawson MI, Majumdar APN, and Reichert U (2003). Identification and characterization of a cell cycle and apoptosis regulatory protein-1 as a novel mediator of apoptosis signaling by retinoid CD437. J. Biol. Chem 278, 33422–33435. 10.1074/jbc.M303173200. [DOI] [PubMed] [Google Scholar]
- 15.Muthu M, Cheriyan VT, and Rishi AK (2015). CARP-1/CCAR1: a biphasic regulator of cancer cell growth and apoptosis. Oncotarget 6, 6499–6510. 10.18632/oncotarget.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Venkatesh J, Rishi AK, and Reddy KB (2020). Novel strategies to target chemoresistant triple-negative breast cancer. Genes Cancer 11, 95–105. 10.18632/genesandcancer.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson GS, Rajendran P, and Dashwood RH (2020). CCAR1 and CCAR2 as gene chameleons with antagonistic duality: Preclinical, human translational, and mechanistic basis. Cancer Sci. 111, 3416–3425. 10.1111/cas.14579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim JH, Yang CK, Heo K, Roeder RG, An W, and Stallcup MR (2008). CCAR1, a key regulator of mediator complex recruitment to nuclear receptor transcription complexes. Mol. Cell 31, 510–519. 10.1016/j.molcel.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ou CY, Chen TC, Lee JV, Wang JC, and Stallcup MR (2014). Coregulator cell cycle and apoptosis regulator 1 (CCAR1) positively regulates adipocyte differentiation through the glucocorticoid signaling pathway. J. Biol. Chem 289, 17078–17086. 10.1074/jbc.M114.548081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ou CY, Kim JH, Yang CK, and Stallcup MR (2009). Requirement of cell cycle and apoptosis regulator 1 for target gene activation by Wnt and beta-catenin and for anchorage-independent growth of human colon carcinoma cells. J. Biol. Chem 284, 20629–20637. 10.1074/jbc.M109.014332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sekhar SC, Venkatesh J, Cheriyan VT, Muthu M, Levi E, Assad H, Meister P, Undyala VV, Gauld JW, and Rishi AK (2019). A H2AX(−) CARP-1 Interaction Regulates Apoptosis Signaling Following DNA Damage. Cancers (Basel) 11, 221. 10.3390/cancers11020221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garcia-Higuera I, Kuang Y, Denham J, and D’Andrea AD (2000). The fanconi anemia proteins FANCA and FANCG stabilize each other and promote the nuclear accumulation of the Fanconi anemia complex. Blood 96, 3224–3230. [PubMed] [Google Scholar]
- 23.Puliyappadamba VT, Wu W, Bevis D, Zhang L, Polin L, Kilkuskie R, Finley RL Jr., Larsen SD, Levi E, Miller FR, et al. (2011). Antagonists of anaphase-promoting complex (APC)-2-cell cycle and apoptosis regulatory protein (CARP)-1 interaction are novel regulators of cell growth and apoptosis. J. Biol. Chem 286, 38000–38017. 10.1074/jbc.M111.222398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raj B, Irimia M, Braunschweig U, Sterne-Weiler T, O’Hanlon D, Lin ZY, Chen GI, Easton LE, Ule J, Gingras AC, et al. (2014). A global regulatory mechanism for activating an exon network required for neurogenesis. Mol. Cell 56, 90–103. 10.1016/j.molcel.2014.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu R, Zhu Y, Jiang X, Li Y, Zhu M, Dong M, Huang Z, Wang C, Labouesse M, and Zhang H (2018). CCAR-1 affects hemidesmosome biogenesis by regulating unc-52/perlecan alternative splicing in the C. elegans epidermis. J. Cell Sci 131, jcs214379. 10.1242/jcs.214379. [DOI] [PubMed] [Google Scholar]
- 26.Nicholson-Shaw AL, Kofman ER, Yeo GW, and Pasquinelli AE (2022). Nuclear and cytoplasmic poly(A) binding proteins (PABPs) favor distinct transcripts and isoforms. Nucleic Acids Res. 50, 4685–4702. 10.1093/nar/gkac263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Close P, East P, Dirac-Svejstrup AB, Hartmann H, Heron M, Maslen S, Chariot A, Söding J, Skehel M, and Svejstrup JQ (2012). DBIRD complex integrates alternative mRNA splicing with RNA polymerase II transcript elongation. Nature 484, 386–389. 10.1038/nature10925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iyer DR, Harada N, Clairmont C, Jiang L, Martignetti D, Nguyen H, He YJ, Chowdhury D, and D’Andrea AD (2022). CCAR2 functions downstream of the Shieldin complex to promote double-strand break end-joining. Proc. Natl. Acad. Sci. USA 119, e2214935119. 10.1073/pnas.2214935119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aravind L, and Koonin EV (2000). SAP - a putative DNA-binding motif involved in chromosomal organization. Trends Biochem. Sci 25, 112–114. 10.1016/s0968-0004(99)01537-6. [DOI] [PubMed] [Google Scholar]
- 30.Hnízda A, Tesina P, Nguyen TB, Kukačka Z, Kater L, Chaplin AK, Beckmann R, Ascher DB, Novák P, and Blundell TL (2021). SAP domain forms a flexible part of DNA aperture in Ku70/80. FEBS Journal 288, 4382–4393. 10.1111/febs.15732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weinheimer AS, Paung Y, Rageul J, Khan A, Lo N, Ho B, Tong M, Alphonse S, Seeliger MA, and Kim H (2022). Extended DNA-binding interfaces beyond the canonical SAP domain contribute to the function of replication stress regulator SDE2 at DNA replication forks. J. Biol. Chem 298, 102268. 10.1016/j.jbc.2022.102268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sundararajan R, Chen G, Mukherjee C, and White E (2005). Caspase-dependent processing activates the proapoptotic activity of deleted in breast cancer-1 during tumor necrosis factor-alpha-mediated death signaling. Oncogene 24, 4908–4920. 10.1038/sj.onc.1208681. [DOI] [PubMed] [Google Scholar]
- 33.Katz Y, Wang ET, Airoldi EM, and Burge CB (2010). Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods 7, 1009–1015. 10.1038/nmeth.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, Zhou Q, and Xing Y (2014). rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 111, E5593–E5601. 10.1073/pnas.1419161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anders S, Reyes A, and Huber W (2012). Detecting differential usage of exons from RNA-seq data. Genome Res. 22, 2008–2017. 10.1101/gr.133744.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garcia-Higuera I, Kuang Y, Naf D, Wasik J, and D’Andrea AD (1999). Fanconi anemia proteins FANCA, FANCC, and FANCG/XRCC9 interact in a functional nuclear complex. Mol. Cell. Biol 19, 4866–4873. 10.1128/MCB.19.7.4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Agafonov DE, Deckert J, Wolf E, Odenwälder P, Bessonov S, Will CL, Urlaub H, and Lührmann R (2011). Semiquantitative proteomic analysis of the human spliceosome via a novel two-dimensional gel electrophoresis method. Mol. Cell. Biol 31, 2667–2682. 10.1128/MCB.05266-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hegele A, Kamburov A, Grossmann A, Sourlis C, Wowro S, Weimann M, Will CL, Pena V, Lührmann R, and Stelzl U (2012). Dynamic protein-protein interaction wiring of the human spliceosome. Mol. Cell 45, 567–580. 10.1016/j.molcel.2011.12.034. [DOI] [PubMed] [Google Scholar]
- 39.Lieberman J (2018). Tapping the RNA world for therapeutics. Nat. Struct. Mol. Biol 25, 357–364. 10.1038/s41594-018-0054-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lewit-Bentley A, and Réty S (2000). EF-hand calcium-binding proteins. Curr. Opin. Struct. Biol 10, 637–643. 10.1016/s0959-440x(00)00142-1. [DOI] [PubMed] [Google Scholar]
- 41.Anantharaman V, and Aravind L (2008). Analysis of DBC1 and its homologs suggests a potential mechanism for regulation of sirtuin domain deacetylases by NAD metabolites. Cell Cycle 7, 1467–1472. 10.4161/cc.7.10.5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brunquell J, Yuan J, Erwin A, Westerheide SD, and Xue B (2014). DBC1/CCAR2 and CCAR1 Are Largely Disordered Proteins that Have Evolved from One Common Ancestor. BioMed Res. Int 2014, 418458. 10.1155/2014/418458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lugano DI, Barrett LN, Chaput D, Park MA, and Westerheide SD (2024). CCAR-1 works together with the U2AF large subunit UAF-1 to regulate alternative splicing. RNA Biol. 21, 1–11. 10.1080/15476286.2023.2289707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ilagan JO, Ramakrishnan A, Hayes B, Murphy ME, Zebari AS, Bradley P, and Bradley RK (2015). U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 25, 14–26. 10.1101/gr.181016.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maji D, Glasser E, Henderson S, Galardi J, Pulvino MJ, Jenkins JL, and Kielkopf CL (2020). Representative cancer-associated U2AF2 mutations alter RNA interactions and splicing. J. Biol. Chem 295, 17148–17157. 10.1074/jbc.RA120.015339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gala K, Li Q, Sinha A, Razavi P, Dorso M, Sanchez-Vega F, Chung YR, Hendrickson R, Hsieh JJ, Berger M, et al. (2018). KMT2C mediates the estrogen dependence of breast cancer through regulation of ERalpha enhancer function. Oncogene 37, 4692–4710. 10.1038/s41388-018-0273-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Siebert AE, Corll J, Paige Gronevelt J, Levine L, Hobbs LM, Kenney C, Powell CLE, Battistuzzi FU, Davenport R, Mark Settles A, et al. (2022). Genetic analysis of human RNA binding motif protein 48 (RBM48) reveals an essential role in U12-type intron splicing. Genetics 222, iyac129. 10.1093/genetics/iyac129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ortiz-Barahona V, Soler M, Davalos V, García-Prieto CA, Janin M, Setien F, Fernández-Rebollo I, Bech-Serra JJ, De La Torre C, Guil S, et al. (2023). Epigenetic inactivation of the 5-methylcytosine RNA methyltransferase NSUN7 is associated with clinical outcome and therapeutic vulnerability in liver cancer. Mol. Cancer 22, 83. 10.1186/s12943-023-01785-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lim KS, Li H, Roberts EA, Gaudiano EF, Clairmont C, Sambel LA, Ponnienselvan K, Liu JC, Yang C, Kozono D, et al. (2018). USP1 Is Required for Replication Fork Protection in BRCA1-Deficient Tumors. Mol. Cell 72, 925–941.e4. 10.1016/j.molcel.2018.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kupfer G, Naf D, Garcia-Higuera I, Wasik J, Cheng A, Yamashita T, Tipping A, Morgan N, Mathew CG, and D’Andrea AD (1999). A patient-derived mutant form of the Fanconi anemia protein, FANCA, is defective in nuclear accumulation. Exp Hematol 27, 587–593. 10.1016/s0301-472x(99)00022-3. [DOI] [PubMed] [Google Scholar]
- 51.Hayer A, Shao L, Chung M, Joubert LM, Yang HW, Tsai FC, Bisaria A, Betzig E, and Meyer T (2016). Engulfed cadherin fingers are polarized junctional structures between collectively migrating endothelial cells. Nat. Cell Biol 18, 1311–1323. 10.1038/ncb3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eng JK, McCormack AL, and Yates JR (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom 5, 976–989. 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 54.Wu G, Feng X, and Stein L (2010). A human functional protein interaction network and its application to cancer data analysis. Genome Biol. 11, R53. 10.1186/gb-2010-11-5-r53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol 34, 525–527. 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 58.Soneson C, Love MI, and Robinson MD (2015). Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 4, 1521. 10.12688/f1000research.7563.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. 10.1093/bio-informatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shevchenko A, Wilm M, Vorm O, and Mann M (1996). Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem 68, 850–858. 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 63.Peng J, and Gygi SP (2001). Proteomics: the move to mixtures. J. Mass Spectrom 36, 1083–1091. 10.1002/jms.229. [DOI] [PubMed] [Google Scholar]
- 64.North K, Benbarche S, Liu B, Pangallo J, Chen S, Stahl M, Bewersdorf JP, Stanley RF, Erickson C, Cho H, et al. (2022). Synthetic introns enable splicing factor mutation-dependent targeting of cancer cells. Nat. Biotechnol 40, 1103–1113. 10.1038/s41587-022-01224-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Frankish A, Diekhans M, Jungreis I, Lagarde J, Loveland JE, Mudge JM, Sisu C, Wright JC, Armstrong J, Barnes I, et al. (2021). Gencode 2021. Nucleic Acids Res. 49, D916–D923. 10.1093/nar/gkaa1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq datasets discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GEO: GSE242781. All raw data files and images have been deposited in Mendeley Data and are accessible using Mendeley Data: https://doi.org/10.17632/drsrg8xtdm.2
The paper does not report any original code.
Any additional information required to reanalyze the data reported in this study is available from the lead contact upon request.