

SUMMARY
Immunotherapy is a promising treatment for triple-negative breast cancer (TNBC), but patients relapse, highlighting the need to understand the mechanisms of resistance. We discovered that in primary breast cancer, tumor cells that resist T cell attack are quiescent. Quiescent cancer cells (QCCs) form clusters with reduced immune infiltration. They also display superior tumorigenic capacity and higher expression of chemotherapy resistance and stemness genes. We adapted single-cell RNA-sequencing with precise spatial resolution to profile infiltrating cells inside and outside the QCC niche. This transcriptomic analysis revealed hypoxia-induced programs and identified more exhausted T cells, tumor-protective fibroblasts, and dysfunctional dendritic cells inside clusters of QCCs. This uncovered differential phenotypes in infiltrating cells based on their intra-tumor location. Thus, QCCs constitute immunotherapy-resistant reservoirs by orchestrating a local hypoxic immune-suppressive milieu that blocks T cell function. Eliminating QCCs holds the promise to counteract immunotherapy resistance and prevent disease recurrence in TNBC.
In brief
Spatially resolved transcriptome analysis of tumors identifies quiescent cell niches that instruct T cells to be hyporesponsive, thus suggesting a route for therapeutic targeting in triple-negative breast cancer.
Graphical Abstract
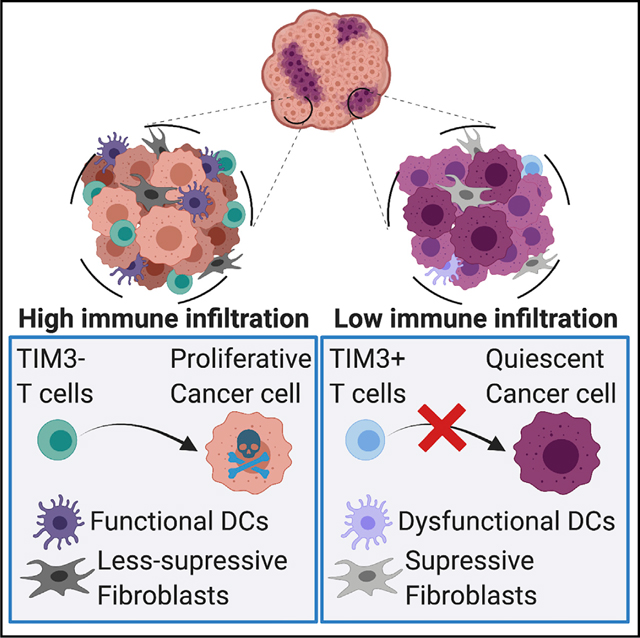
INTRODUCTION
Triple-negative breast cancer (TNBC) is the most aggressive form of breast cancer. It lacks expression of hormone receptors and HER2, preventing the use of targeted therapies. Thus, chemotherapy has long been the only systemic therapy for these patients, limiting their options. Recently, two phase III clinical trials found that immune checkpoint blockade (ICB) in combination with chemotherapy improved progression-free survival in metastatic PD-L1-positive TNBC, leading to their approval as a first line treatment (Cortes et al., 2020; Schmid et al., 2018). However, only a fraction of patients benefit from immunotherapy, raising questions about resistance mechanisms.
There is a major ongoing effort to identify the drivers of tumor microenvironment (TME) diversity, which correlates with immunotherapy resistance (Binnewies et al., 2018). Cancer cells are known to modulate the TME (Wellenstein and Visser, 2018), although the mechanisms are poorly understood. CRISPR screens have uncovered cancer-cell intrinsic resistance mechanisms (Pan et al., 2018; Patel et al., 2017) but cannot describe how cancer cells orchestrate a multicellular immunosuppressive TME. Alternatively, bulk tumor sequencing can identify signatures of ICB resistance but must contend with different populations of cancer cells within the tumor (Wolf et al., 2019). Different cancer-cell clones can evolve distinct strategies for immune evasion. Thus, tumor cells that lose the targeted antigen(s) do not need any other means to escape from T cell attack. These intermingled populations can enhance the difficulty of identifying other resistance mechanisms, mostly those involving modulation of the TME.
Here, we investigate resistance to T cell killing in cancer cells that unequivocally express the targeted antigen by using green fluorescent protein (GFP) as a visible tumor neo-antigen and exploiting our GFP-specific CD8+ Jedi T cells (Agudo et al., 2015). We found that antigen-expressing (GFP+) but T cell-resistant cancer cells were principally quiescent cancer cells (QCCs). These cells formed clusters with reduced immune infiltration and demonstrated a higher tumorigenic potential. We adapted a technique involving photo-labeling, FACS-sorting, and scRNA-seq (Medaglia et al., 2017) to a tumor setting, which we named photo-conversion of areas to dissect micro-environments (PADME-seq). PADME-seq was used to profile infiltrates inside these QCC clusters and, in parallel, other regions from the same tumor mass. We uncovered that QCCs form a niche containing immune-suppressive fibroblasts, dysfunctional dendritic cells, and highly exhausted T cells. Such ecosystems were orchestrated by QCCs through activation of a hypoxia-induced program.
RESULTS
Cancer cells that survive upon adoptive T cell therapy do not proliferate
To investigate how tumor cells that retain expression of targeted antigen(s) can escape T cell killing, we used anti-GFP-specific Jedi T cells (Agudo et al., 2015) in combination with GFP-expressing TNBC 4T07 cells. GFP as a visible tumor antigen allows one to distinguish cells that survive T cell attack and retain antigen expression. We orthotopically injected a mix of GFP+ and mCherry+ cells in mice. Since Jedi T cells do not recognize mCherry, mCherry+ cells constituted labeled controls that were embedded within the same milieu (Figure 1A). We generated PD1-deficient Jedi T cells (PD1−/− Jedi) by crossing with PD1−/− mice (Keir et al., 2007); so, PD1 inactivation was directed only against GFP+ cells. PD1−/− Jedi T cells were transferred at either day 7 or day 15, to capture different stages of tumor growth (Figures 1A and S1A). PD1−/− Jedi T cells effectively killed most but not all GFP+ cancer cells (Figures 1B, 1C, and S1B). Surviving GFP+ cancer cells formed clusters (Figure 1D) with two-fold reduced T cell infiltration compared with mCherry+ regions, indicating partial T cell exclusion (Figures 1E, 1F, and S1C–S1E). This result demonstrates that mammary carcinomas contain regions with restricted T cell infiltration likely contributing to their resistance to immunotherapy.
Figure 1. Breast tumor cells that escape from PD1−/− T cell killing are quiescent.

(A) Schematic of the experiment: mammary carcinoma was grown with mixed GFP+ and mCherry+ 4T07 cells (1:1). Mice were treated with 7M PD1−/− Jedi T cells at either day 7 or day 15 after tumor inoculation. All mice were analyzed 7 days post-T cell injection (n = 3–4 mice).
(B) Flow cytometry plots from tumors in (A), gated on DAPI— CD45.
(C) Mean ± SEM of data in (B).
(D) Tumor images from (A) at day 14.
(E and F) Number of T cells per tumor area from mice in (A). Bar marks the median (Multiple areas from 3 tumors).
(G) Principal component analysis (PCA) from RNA-seq data from mCherry+ and GFP+ tumor cells from (A) at day 14.
(H) H2-Kd (MHCI) in cancer cells from mice in (A) (n = 4 mice).
(I) Pathway enrichment analysis from (G) at day 14 ranked by lowest adjusted p value.
(J) EdU labeling in tumors from (A). EdU was injected 30 h before adoptive transfer of Jedi T cells and continued daily (n = 5 mice).
(K) EdU intensity from pictures in (J). Line marks the median (n = 15 images from 5 tumors).
(L) EdU in mVenus-p27K-expressing 4T07 tumors (n = 3 mice).
(M and N) GFP and tdTomato-p27K 4T07 cells mixed with H2B-miRFP670 and tdTomato-p27k 4T07 tumor-bearing mice were treated with the specified number of PD1−/− Jedi T cells (n = 5 mice). (M) Flow cytometry plots gating on GFP+ and miRFP670+ tumor cells. Percentages calculated over color coded populations.
(N) Mean ± SEM of percentage of tdTomato-p27KHigh cells.
(O and P) Genetic circuit to assess quiescence in cancer cells prior to injection of Jedi T cells. GFP+ 4T07 cells with the circuit were injected in BALB/c or NSG mice. All mice received doxycycline on day 6 post-tumor inoculation. 30 h later, 5M Jedi T cells were injected into the BALB/c group. (O) Schematic. (P) Mean ± SEM of the percentage of H2B-tdTomato+ cells (n = 4–5 mice).
(Q) Quantification of p27KHigh cells in Jedi T cell-resistant cancer cells upon loss of IFNAR1 signaling. Either Ifnar1−/− or WT GFP 4T07 cells were mixed with WT H2B-miRFP670 4T07 cells (1:1) and were injected into BALB/c mice. WT GFP+ and H2B-miRFP670+ 4T07 cells were injected into NSG mice as no T cell controls. All cancer cells carried tdTomato-p27K reporter. 5M Jedi T cells were injected into all BALB/c mice. Mean ± SEM of percentage of tdTomato-p27KHigh cells in each group is shown. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
We FACS-sorted GFP+ surviving antigen-expressing cancer cells along with control mCherry+ cells to perform RNAsequencing. Principal component analysis (PCA) revealed that GFP+ and mCherry+ cells from the same tumors did not cluster together. Instead GFP+ samples from the various tumors were closer to each other than to mCherry+ cells from the corresponding tumor (Figures 1G, S1F, and S1G). This provided a good framework to search for differentially expressed genes between GFP+ and mCherry+ cells to uncover mechanisms of resistance to T cell killing. Major Histocompatibility Complex I (MHCI) has been reported to decrease in resistant pancreatic tumor cells (Pommier et al., 2018), but we found no reduction in antigen presentation pathway genes (Figure S1H) or MHCI protein expression (Figures 1H and S1I). Instead, we found that genes related to resistance to chemotherapy, tumor initiation, and hypoxia were upregulated, such as Ngfr, Aldh1a1, and Car9 (Lee et al., 2017; Semenza, 2017; Wu et al., 2021b) (Figures S1J and S1K).
The most significant gene ontology pathways that differed between GFP+ and mCherry+ cells were all related to cell cycle (Figures 1I and S1L). Key genes involved in proliferation and DNA replication, such as Mki67 and Pcna, were all downregulated (Figures S1M and S1N). To confirm whether these resistant cells were in cell-cycle arrest, we treated tumor-bearing mice with the thymidine analog 5-ethynyl-2′-deoxyuridine (EdU) daily, starting before adoptive transfer of PD1−/− Jedi T cells. Six days post-Jedi treatment, most mCherry+ cancer cells were positive for EdU. Conversely, the EdU signal was low in T cell-resistant GFP+ tumor cells. This demonstrated that most T cell-resistant GFP+ cancer cells were not cycling prior to adoptive T cell transfer and did not cycle during T cell killing (Figures 1J and 1K). Therefore, cancer cells that escaped attack by CD8+ T cells formed distinct clusters of QCCs.
QCCs are more resistant to direct attack from cytotoxic T cells
To understand QCC resistance to T cell killing, we set up an approach to isolate and profile live non-cycling cancer cells. We used a published quiescence reporter (Oki et al., 2014), consisting of mVenus fused to an inactive p27-CDK binding domain (mVenus-p27K). We validated this mVenus-p27K reporter in 4T07 tumors and observed that all mVenus-p27KHigh cells were negative for Ki67 (Figures S1O and S1P). mVenus-p27KHigh cells also remained EdU-negative after 5 days of EdU treatment in vivo (Figures 1L and S1Q). These findings are consistent with previous studies reporting the existence of p27+ quiescent tumor cells in primary breast cancer (Fluegen et al., 2017). mVenus-p27KHigh cells were also observed in 4T07 tumors in immune-deficient NOS scid IL2Rgamma (NSG) mice (Figure S1R), demonstrating that these QCCs arise independently of T cells.
Next, we replaced mVenus with the red fluorescent protein tdTomato to prevent spectral overlap with GFP and then engineered 4T07 cells to express either GFP as an antigen or miRFP670 (far red) as a non-immunogenic control label. Mice with tumors that were mix of GFP:miRFP670 cells and carrying the tdTomato-p27K reporter received adoptive transfer of PD1−/− Jedi T cells. Surviving GFP+ cells had a 2-fold increase in tdTomato-p27KHigh cells compared with miRFP670+ cells (Figures 1M, 1N, and S1S). We repeated this experiment using a higher number of PD1−/− Jedi T cells to enable more stringent killing (Figure S1S). In this setting, the few resistant GFP+ cells were almost all quiescent (Figures 1M and 1N). Taken together, this suggests that intra-tumor QCCs are more resistant to T cell killing than actively cycling cancer cells.
Next, we assessed whether p27KHigh cells were enriched upon T cell killing or, conversely, surviving GFP+ cells became quiescent during T cell attack. We built a genetic circuit where doxycycline induces H2B-tdTomato only in p27K+ cells (Figure 1O). p27K+ tumor cells were labeled by one doxycycline injection. The next day, after drug clearance, Jedi T cells were adoptively transferred. Around 65% of the surviving GFP+ cells were tdTomato+, while only ~20% of the cells were tdTomato+ in tumors from NSG mice (with no T cell pressure) (Figure 1P). This indicates that most surviving GFP+ tumor cells after Jedi treatment were already p27KHigh prior to adoptive transfer of Jedi cells.
Since interferons can induce quiescence in disseminated tumor cells (Correia et al., 2021) and the top upregulated pathway in surviving GFP+ cells after T cell attack was “IFN-beta signaling” (Figure 1I), we generated knockout cells for the IFNB receptor Ifnar1 (Figure S1T). GFP+ Ifnar1−/− cancer cells were killed by Jedi T cells in a similar rate to their WT counterparts (Figures 1Q and S1U). Among the surviving cells, there was also a similar ratio of p27KHigh cells compared with WT GFP+ cells (Figure 1Q), suggesting that IFNB was not critical for the quiescent phenotype observed in surviving cells.
QCCs possess greater cancer initiation potential
Since QCCs did not proliferate for multiple days, we questioned whether these resistant cells could re-start a growth program after T cell therapy. We first tested the ability of surviving GFP+ cells to regrow tumors compared with that of bulk GFP cells. Jedi T cell-resistant GFP+ cells were able to regrow tumors more efficiently than non-selected GFP+ tumor cells (Figures S2A and S2B). Next, we investigated the ability of p27KHigh cells from untreated tumors to re-enter into cell cycle. First, we investigated the existence of p27KHigh QCCs in other models of TNBC: EMT6 and D2A1. In these models, mVenus-p27KHigh cells also formed clusters and did not proliferate for several days (Figures S2C and S2D). Once the presence of QCCs in multiple TNBC models was confirmed, we addressed whether their quiescent state was terminal. We FACS-sorted live mCherry+ mVenus-p27KHigh (QCCs) and mCherry+ mVenus-p27KNeg proliferative counterparts from these models and injected them in new mice (Figure 2A). In all cases, mVenus-p27KHigh cells regrew tumors, proving that they were not in a terminal state of quiescence. Remarkably, QCCs displayed a higher tumor initiation potential at these limiting numbers than mVenus-p27KNeg cells (Figures 2B and 2C). Thus, QCCs were not only able to re-enter the cell cycle and initiate tumor growth but did so more effectively than their proliferating counterparts.
Figure 2. Tumor cell quiescence leads to exclusion of T cells.

(A–C) Assessment of tumor initiation potential in QCCs. mCherry+ mVenus-p27KHigh and mCherry+ mVenus-p27KNeg cancer cells were isolated from 4T07, EMT6, and D2A1 tumors. Live sorted cells (500 for EMT6 and 1,000 for 4T07 and D2A1) were injected in new mice. (A) Schematic. (B) Representative picture 4T07 cells. (C) Percentage of tumor growth (4T07, n = 6; EMT6, n = 3; D2A1, n = 3).
(D) Immunofluorescence analysis of CD3+ cells in mCherry and mVenus-p27K-expressing 4T07, EMT6, and D2A1 mammary carcinomas (n = 4 mice).
(E) Quantification of T cell density in mVenus-p27KHigh versus mVenus-p27KNeg areas in 4T07 tumors. Bar marks the median (n = 28 images from 3 mice).
(F) CD4 and CD8 immunofluorescence in mVenus-p27K-expressing 4T07 tumors (n = 3 mice).
(G) CD45 immunofluorescence in mVenus-p27K-expressing 4T07 tumors (n = 5 mice). Yellow dashed line marks mVenus-p27KHigh area.
(H) Immunofluorescence analysis of CD3, p27, Ki67, and E-cadherin (E-cad) in tissue sections from TNBC de-identified patients (n = 10 patients).
(I) Density of p27+ and Ki67+ tumor cells surrounding T cells from patients in (H). Each dot represents the averaged density of the corresponding marker (p27 or Ki67) in all analyzed areas from each patient. Bar marks the median.
(J) Patient cohort treated with ICB at DFCI (n = 29 patients).
(K) Most significantly upregulated transcriptional program in durable responders compared with non-responders in (J). ****p < 0.0001.
Clusters of QCCs form a niche with limited immune infiltration
We performed immuno-staining of CD3 in untreated mVenus-p27K-expressing 4T07, EMT6, and D2A1 tumors (Figure 2D). Although T cells were found inside the clusters of QCCs, their numbers were always lower in these regions (Figures 2E and S2E–S2G). Staining for CD4 and CD8 confirmed that both types of T cells were reduced inside the clusters of QCCs (Figures 2F, S2H, and S2I). Further analysis with anti-CD45 staining revealed that regions with QCCs had poor immune infiltration compared with cycling areas (Figure 2G). These results demonstrated that QCCs reside in a niche with reduced numbers of infiltrating immune cells.
Proliferation correlates with T cell infiltration in patients
To dissect the relationship between QCCs and T cells in patients, we analyzed p27, Ki67, CD3, and E-cadherin on tumor tissue (Figure 2H). We measured the number of Ki67+ (proliferative) or p27+ (quiescent) tumor cells in contact with T cells in cancer biopsies from 10 de-identified treatment naive patients. This uncovered that Ki67+ cells were more frequently found in the vicinity of T cells than p27+ cancer cells (Figures 2I and S2J). Next, we investigated whether the abundance of quiescent versus proliferative cells in human TNBC could influence the response to ICB. We utilized bulk RNA-seq datasets from pre-treatment biopsies in a cohort of 29 patients with metastatic TNBC that were treated with PD-1/PD-L1 inhibitors (B.-S.R., J.F., K. Collie, et al., unpublished data). In this cohort, 5 patients presented with primary resistance to ICB; we defined them as non-responders. Among the 24 patients with benefit, 5 patients experienced a durable response (durable responders; progression-free survival ranging from 26 to 60 months) (Figure 2J). Gene-set enrichment analysis (GSEA) of these data comparing durable responders and non-responders showed “reactome E2F regulation of DNA replication” as the number one most enriched pathway (Figure 2K), suggesting that tumor proliferation may correlate with response to ICB. This result agreed with recent reports comparing chemotherapy+ICB to chemotherapy alone. RNA-seq analysis of >800 patients in a multi-center clinical trial revealed “cell proliferation” as a top pathway enriched in responders in the ICB arm (Emens et al., 2021). Although it is only an association, these data suggest that TNBC with abundant proliferative tumor cells could have a better response to ICB, while QCCs could lead to resistance.
QCCs express genes related to chemoresistance, hypoxia, and glycolysis
To investigate mechanisms of resistance in QCCs, we FACS-sorted live QCCs (mVenus-p27KHigh) and non-quiescent (mVenus-p27KNeg) tumor cells from 4T07, EMT6, and D2A1 tumors for RNA-seq. Unbiased gene ontology (GO) analysis from differentially expressed genes (Figure S3A) showed that the most downregulated genes in QCCs were involved in cell cycle, confirming their quiescent nature (Figure S3B). Among the most upregulated genes in QCCs, we identified genes linked to chemoresistance: Car9 (Lu et al., 2017), Kdm5a, and Kdm5b (Hinohara et al., 2018); and related to stemness: Runx1, Klf4, Sox4, and Sox9 (Oskarsson et al., 2014; Prager et al., 2019) (Figure 3A). The top upregulated pathways in QCCs across the 3 models were hypoxia-related (e.g., angiogenesis) and glucose metabolism-related (Figures 3B, 3C, S3C, and S3D). To test whether these QCCs were indeed in a hypoxic niche, we visualized pimonidazole (Hypoxyprobe) along with CD3 in mVenus-p27K-expressing tumors. Pimonidazole+ regions partially co-localized with clusters of QCCs and inversely correlated with T cell-rich regions (Figures 3D and 3E). Accordingly, areas of surviving GFP+ cancer cells after Jedi T cell attack also showed augmented pimonidazole signal (Figure 3F). This suggested that the hypoxia-induced factor 1-alpha (HIF1A) signature observed in the RNA-seq data was the result of local hypoxia. HIF1A activation leads to expression of glycolytic genes such as the glucose transporter Slc2a1 or Glut1. QCCs showed a >2-fold increase in uptake of 2-NBDG, demonstrating that these cells were more glycolytic than their proliferative counterparts, a feature that had been described in tumor-initiating cells (Paolicchi et al., 2016; Prager et al., 2019; Semenza, 2017) (Figure 3G). Hence, QCCs in breast cancer reside in hypoxic micro-niches and display an enhanced glycolytic phenotype as reported by others (Fluegen et al., 2017; Hu et al., 2003).
Figure 3. QCCs are hypoxic and glycolytic.

(A) RNA-seq analysis of mCherry+ mVenus-p27KHigh and mCherry+ mVenus-p27KNeg cells from 4T07, EMT6, and D2A1 tumors. Heatmaps show differentially expressed genes involved in stemness and chemotherapy resistance
(B) Pathway enrichment analysis from (A) showing top upregulated pathways by adjusted p value.
(C) Heatmap shows hypoxia-induced differentially expressed genes in (B). Data is color coded to reflect gene expression Z scores.
(D and E) Pimonidazole in 4T07 (D) and EMT6 (E) tumors expressing mVenus-p27K (n = 3 mice/model).
(F) Pimonidazole in GFP:mCherry 4T07 tumors 7 days after treatment with 5M Jedi T cells (n = 5 mice).
(G) Mean ± SEM of mean fluorescent intensity (MFI) of 2-NBD-glucose (n = 3 mice). ***p < 0.001.
The QCC niche displays suppressive fibroblasts and more exhausted T cells
To uncover how QCCs limited T cell function at single-cell resolution, we adapted a protocol for isolation of cells from specific tissue areas for scRNA-seq (Medaglia et al., 2017) to our tumor setting. We used transgenic mice expressing the photo-convertible protein Kaede to grow tdTomato-p27K-expressing tumors and generated consecutive slices of live tumor (Figures 4A and 4B). This approach, which we named photo-conversion of areas to dissect micro-environments (PADME-seq), allowed us to profile infiltrating cells from distinct sub-regions within the same tumor mass (inside and outside clusters of QCCs). We performed two separate experiments in which we FACS-sorted equivalent numbers of live photo-converted cells from each region. After discarding doublets and cells with low mRNA quality, we had 1,743 and 975 cells from inside the areas of QCCs for each experiment respectively, and 1,968 and 2,259 in non-QCC regions. All major tumor infiltrating cell types (macrophages, fibroblasts, T cells, dendritic cells, NK cells, and neutrophils) were present both inside and outside regions of QCCs (Figures 4C and S4A–S4C). Since immunofluorescence analysis had shown that immune infiltrates (Figure 2G) were reduced in QCC regions, we first investigated whether fibroblasts were phenotypically different. Unsupervised GSEA comparing fibroblasts from p27KHigh versus p27KNeg areas revealed that immune-related defense pathways were downregulated in the QCC niche (Figures 4D and 4E). Conversely, collagen-deposition pathways were upregulated (Figures 4D and 4E). Notably, we compared our data with a previously published fibroblast signature identified in ICB-resistant patients (Dominguez et al., 2020) and confirmed a suppressive fibroblast phenotype inside the QCC niche (Figure S4D).
Figure 4. scRNA-seq analysis inside the QCC niche reveals higher T cell exhaustion.

(A) Schematic of PADME-seq.
(B) Representative image after photo-conversion. White dashed line marks photoconverted area.
(C) Uniform manifold approximation and projection (UMAP) visualization of cell clusters identified from PADME-seq.
(D and E) Unsupervised gene-set enrichment analysis performed in the fibroblast population comparing cells from p27KHigh versus p27KNeg areas. (D) GO term pathways. (E) Heatmap with the average expression across populations in set signature genes from (D). Z score normalized data are shown.
(F) UMAP visualization of reclustered T cells from (C).
(G) Unsupervised gene-set enrichment analysis performed in all CD8 T cells comparing p27High versus p27Neg areas.
(H) Distribution of CD8+ T cell sub-populations identified in (F). **adj. p value < 0.01.
(I) Flow cytometry analysis of infiltrating cells in p27KHigh areas versus p27KNeg (n = 4 mice).
(J and K) Exhausted PD1+ TIM3+ CD3+ T cells by CODEX in untreated 4T07 tumors. (J) CD3+ T cell density in p27KHigh areas and p27KNeg. (K) Exhausted (TIM3+) and non-exhausted (TIM3—) CD3+ T cells inside p27KHigh versus p27KNeg areas. Bar marks the median (multiple areas from 4 tumors). Non-parametric Wilcoxson test was used.
(L–N) Quantification of exhausted PD1+ TIM3+ CD3+ T cells by CODEX in Thy1.1:GFP tumors after treatment with 5M Jedi T cells (multiple areas from 3 tumors).
(L) Representative image, arrows point to T cells. (M) Exhausted (TIM3+) and non-exhausted (TIM3—) CD3+ T cells in GFP+ versus Thy1.1+ regions. (N) TIM3+/ CD3+ T cell ratio in GFP+ areas versus Thy1.1+ areas. Bar marks the median. *p < 0.05, **p < 0.01, ***p < 0.001.
Although immune infiltration was lower in areas of QCCs, some T cells were able to enter. To understand why these T cells could not kill QCCs, we analyzed gene expression profiles of T cells inside and outside QCC areas. First, to elucidate the heterogeneity in T cell subsets, we reclustered the Cd3+ T cell cluster (Figures 4F and S4E). This revealed one subset of γδT cells, one of CD4+ T cells, and five of CD8+ T cells. One CD8+ T cell subset displayed high expression of myeloid-specific genes such as Lyz2 and was not included in further analysis (Figure S4E). We performed GSEA of the remaining CD8+ subsets comparing cells inside p27KHigh regions with cells outside. This revealed downregulation of multiple immune functions and signaling pathways in CD8+ T cells inside the QCC niche (Figure 4G). Additionally, we constructed a co-expression network, which showed downregulation of genes related to T cell activation in CD8+ T cells inside QCC areas (Figures S4F and S4G). Taken together, these analyses suggest that CD8+ T cells in the QCC niche are more dysfunctional and less fit for anti-tumor immunity.
Further, we classified the 4 identified sub-populations of CD8+ T cells using previous literature (Beltra et al., 2020; Miller et al., 2019) as: terminally exhausted (Prf1, Havcr2, Gzmb, and Il2rb), intermediate exhausted (Tnfrsf4, Lag3, Ifng, Ccl3, Tnfrsf9, and Csf1), proliferative (mKi67 and Cdk1), and progenitor exhausted (Cd28, Cxcr3, Ifit1, and Stat1) (Figures 4F, S4E, and S4H). The percentage of terminally exhausted CD8+ T cells was higher inside the QCC niche, whereas proliferative CD8+ T cells were more frequent outside the QCC niche (Figure 4H). Terminally exhausted T cells displayed the highest expression of Havcr2 (TIM3+) (Figure S4H), a hallmark of the most dysfunctional T cells (Sakuishi et al., 2010; Singer et al., 2016). Thus, to validate this T cell exhaustion signature inside the QCC niche, we quantified PD1+ TIM3+ CD8+ T cells by flow cytometry using the same photo-labeling system (Figure 4I). Additionally, we used CODEX (Akoya) for multiplexed immunofluorescence to quantify exhausted T cells with respect to p27+ cells (Figures 4J, 4K, and S4I). Both approaches confirmed that less exhausted TIM3– CD8+ T cells were more abundant in proliferative tumor areas (Figures 4I–4K and S4I). CODEX analysis also confirmed a significant reduction in overall immune infiltration inside the QCC nice (Figure S4J). Furthermore, we used CODEX to characterize T cell exhaustion in GFP-expressing tumors upon treatment with Jedi T cells. This confirmed that the small clusters of surviving GFP+ cancer cells after Jedi attack (described in Figure 1) displayed lower T cell infiltration (Figure S4K) and these few T cells were more exhausted (Figures 4L–4N). Thus, all these data demonstrated that the QCC niche was less infiltrated, with more exhausted T cells.
Dendritic cells inside clusters of QCCs express lower levels of key genes for T cell immunity
To uncover the immune populations that could contribute to T cell dysfunction in response to local hypoxia, we quantified a hypoxic signature comparing cells inside and outside QCC regions. All subsets of conventional dendritic cells (cDCs) showed an enrichment in hypoxia-induced genes: DC1 (identified by Clec9a, Xcr1, and Wdfy4), DC2 (Itgax, Sirpa, and Cd209a), and mregDCs (Ccr7, Cd80, Cd200, and Cd247) (Maier et al., 2020) (Figures 5A, S5A, and S5B). Unsupervised GSEA of all cDCs showed a downregulation of innate immunity and immune-response pathways inside the QCC niche (Figure 5B). DC2s displayed a similar result, presenting a highly immune-dysfunctional state (Figures 5C and S5C). Key IFN-induced genes and other important genes for DC function were reduced in DC2 located in the QCC niche (Figure 5D). We then focused on mregDCs, which are DCs that have taken up tumor antigen, being key for anti-tumor immunity (Maier et al., 2020). Since this is a newly identified subset, we analyzed expression of functionally relevant genes based on the literature (Figure 5E). mregDCs in the QCC niche displayed lower expression of MHCI and MHCII-related genes, suggesting a diminished ability to present antigen. They also displayed lower expression of Cd81 and Il12a and Il12b, a key cytokine for T cell responses (Garris et al., 2018). Moreover, mregDCs in the QCC niche had lower expression of Fscn1 (Fascin1), which is expressed in migratory DCs (Miller et al., 2012), suggesting a reduced capacity to migrate to lymph nodes.
Figure 5. Dendritic cells within the QCC niche downregulate key genes for T cell activation.

(A) Difference in expression of hypoxic signature comparing cells from p27KHigh versus p27KNeg regions from Figure 4C. *adj. p value < 0.05.
(B) Gene-set enrichment analysis from all cDCs (DC1, DC2, and mregDC) comparing cells from p27KHigh versus p27KNeg areas.
(C) Gene-set enrichment analysis from DC2, comparing cells from p27KHigh versus p27KNeg areas.
(D) Heatmap of differentially expressed interferon-induced genes comparing DC2 from p27KHigh versus p27KNeg areas.
(E) Heatmap of differentially expressed key functional genes in mregDC from p27KHigh versus p27KNeg areas. Z score normalized data is shown.
(F) Representative immunofluorescence image of MHCII and CD11c staining in mVenus-p27K-expressing tumors (n = 3 tumors). Yellow dashed line marks mVenus-p27KHigh area. Arrows point at CD11c+ MHCIILow cells.
(G) MHCII in CD11c+ cells in D. Bar marks the median (4 images from 3 tumors).
(H) MHCII in DCs from untreated 4T07 tumors (Figures 4J and 4K) from CODEX. DCs were identified as CD45+ CD11c+ F4/80— CD3—. Bar marks the median (n = 4 tumors).
(I) Representative images from (H). Arrows point at F4/80-, CD11c+ cells.
(J) MHCII in DCs by CODEX from GFP:Thy1.1 tumors after treatment with 5M Jedi T cells (Figures 4L–4N). Bar marks the median. *p < 0.05, ****p < 0.0001.
To validate our findings about cDCs from PADME-seq at the protein level, we quantified MHCII in DCs by both conventional immunofluorescence and CODEX. In all settings, DCs displayed lower MHCII when they were located inside areas of surviving GFP+ tumor cells or inside clusters of QCCs in untreated mice (Figures 5F–5J). Therefore, DCs within the QCC niche displayed a phenotype consistent with an impaired ability to recruit and activate T cells. Next, we investigated whether dysfunction in DCs was the direct effect of hypoxia by exposing bone marrow-derived DCs (Agudo et al., 2014) to low oxygen levels. Steady-state DCs did not display any difference in key functional markers during hypoxia (Figure S5D). Upon treatment with lipopolysaccharide (LPS) and polyI:C, all DCs similarly became activated and upregulated MHCII, CD86, and CD40 regardless of oxygen concentrations. Thus, low oxygen had no effect on the ability of DCs to respond to danger signals (Figure S5D).
Specific activation of HIF1a in tumor cells abrogates anti-tumor immunity
We assessed whether the immune-suppressive niche orchestrated by QCCs was the result of HIF1a activation in tumor cells by engineering 4T07 cells to express a constitutively active HIF1a (HIF1aSTBL) (Hu et al., 2007). HIF1aSTBL-expressing cells grew slower in vitro (Figure S6A) but upon in vivo injection, breast carcinomas showed no statistical differences in weight compared with WT (Figure S6B). HIF1aSTBL led to a reduced infiltration of CD8+ T cells, and specifically of less exhausted TIM3– T cells (Figures 6A–6C). Moreover, MHCII was lower in DCs (Figures 6D and 6E), similar to the phenotypes observed inside clusters of QCCs. To investigate whether these alterations could cause resistance to T cell immunity, we generated breast tumors from Cherry+ 4T07 cells mixed with either GFP+ wild-type or GFP+ HIF1aSTBL cells (Figure 6F). Although only a fraction of cancer cells had HIF1aSTBL, DCs showed a trend to lower MHCII levels (Figures S6C and S6D). Moreover, mixed mCherry with GFP-HIF1aSTBL-expressing tumors showed a ~50% reduction in CD8+ T cell infiltration and the infiltrating T cells were mostly excluded from GFP+ regions (Figures 6G and 6H). Upon adoptive transfer of Jedi T cells, WT tumors showed <6% of surviving GFP+ cells, demonstrating efficient killing. Conversely, ~26% of GFP+ HIF1aSTBL cells survived, constituting a 4-fold increase (Figures 6I and 6J). To investigate the therapeutic benefit of HIF1a modulation in cancer cells, we generated Hif1a knockout GFP+ 4T07 cells (Figure S6E). Mice with either WT GFP+ or Hif1a−/− GFP+ tumors were treated with Jedi T cells. This led to a larger reduction in size in Hif1a−/− tumors compared with WT (Figure 6K). Both endogenous CD8+ T cells and transferred CD45.1+ Jedi T cells displayed increased numbers and reduced exhaustion in Hif1a−/− tumors (Figures 6L–6N). These results suggest that HIF1a activation in cancer cells restricts T cell infiltration and increases T cell exhaustion, possibly through forcing DCs into a less activated phenotype (Figure 6O). Our data, together with previous reports correlating HIF1a to chemotherapy resistance (Lu et al., 2017), suggest that HIF1a activation in tumor cells induces a multi-resistant phenotype. Thus, our work has identified a crucial cancer population that orchestrates an immune-suppressive TME and must be eliminated for treatment success.
Figure 6. HIF1a in tumor cells modulates T cell infiltration and exhaustion, controlling killing of tumor cells.

(A) Percentage of CD8+ T cells in WT or Hif1aSTBL 4T07 tumors (n = 5 mice). (B and C) T cell exhaustion in CD8+ T cells from (A).
(B) Representative plot.
(C) Mean ± SEM of PD1+ TIM3— CD8+ T cells (n = 6 mice).
(D) MHCII in tumors in (A) (n = 3 mice).
(E) Mean ± SEM of MFI from (D).
(F) Schematic: mixed WT mCherry with WT GFP or WT mCherry with Hif1aSTBL GFP 4T07 cells were used to grow tumors. Mice were treated with 7M Jedi T cells
(n = 5 mice).
(G) Mean ± SEM of CD8+ T cells per gram of tumor from mice in (F) (n = 5 mice).
(H) CD3 staining in tumors from (F).
(I) Flow cytometry of surviving GFP+ cells in tumors from (F).
(J) Mean ± SEM of GFP:mCherry cells ratio in tumors from (F).
(K) Area under the curve of tumor size in Hif1a_/_GFP and WTGFP tumors upon treatment with 5M Jedi T cells. Arrowindicates time of Jedi injection (n = 4 mice).
(L) Mean ± SEM of percentage of CD8+ T cells in tumors from (K).
(M) Mean ± SEM of percentage of Jedi T cells (CD45.1+) in tumors from (K).
(N) Mean ± SEM of percentage of TIM3_ PD1+ Jedi T cells in tumors from (K).
(O) Effects HIFaSTBL in tumor cells. *p < 0.05, **p < 0.01, ***p < 0.001.
DISCUSSION
The abundance and types of immune infiltration correlate with response to ICB. Understanding how the location and phenotype of infiltrating cells are determined remains an outstanding question in cancer research. Here, we found that QCCs formed micro-territories with lower immune infiltration, uncovering a hidden driver of immune exclusion. Using PADME-seq, we discovered that QCCs survived during adoptive T cell therapy through orchestration of a local hypoxic and immune-suppressive milieu that led to augmented T cell exhaustion.
Since most cancer studies focus on mechanisms controlling tumor growth, quiescence in the tumor mass has been less studied, with some exceptions (Fluegen et al., 2017; Pascual et al., 2017). Of note, we did not identify QCCs in TNBC by studying heterogeneity in cell cycle, but in an unbiased manner by isolating tumor cells that had resisted T cell attack. This intra-tumor quiescent phenotype is different from the process known as cellular tumor dormancy, which occurs in disseminated tumor cells (Ghajar, 2015; Sosa et al., 2014). Notably, QCCs displayed enhanced tumor initiation potential upon isolation and re-injection, demonstrating their potential for disease recurrence. Importantly, reports from the IMpassion130 trial analyzing RNA-seq data from >800 TNBC patients revealed “proliferation” as a feature that correlated with response to ICB. Conversely, “angiogenesis” (a hypoxia-induced pathway that we observed in QCCs) was associated with resistance to ICB (Emens et al., 2021). Thus, a metabolically distinct cancer sub-population of quiescent cells comprises the fittest to survive upon T cell attack, becoming reservoirs of resistance (and probably recurrence) during ICB treatment in TNBC.
One of the strongest mechanisms to escape from T cell killing is by downregulation of antigen presentation (Patel et al., 2017). For example, disseminated pancreatic cancer cells survive during anti-tumor T cell responses by losing MHCI protein (Pommier et al., 2018), while quiescent stem cells decrease MHCI both at the RNA and protein level (Agudo et al., 2018). Nevertheless, the quiescence program in primary breast tumors that we uncovered here did not reduce MHCI, illustrating an orthogonal mechanism to previously published means of T cell immune evasion.
Our PADME-seq method exposed that while terminally exhausted CD8+ T cells were more abundant inside the QCC niche, proliferating CD8+ T cells were more likely found outside these niches. Similarly, the percentage of PD1+ TIM3+ CD3+ T cells was greater inside clusters of QCCs. The underlying mechanism/s inducing this exhausted phenotype along with their location within the tumor mass had not been reported. Here we described the spatial distribution of these dysfunctional TIM3+ CD3+ T cells, identifying them near QCCs. We also found that HIF1a activation in tumor cells was sufficient to increase the numbers of these more exhausted T cells. Remarkably, genetic activation of HIF1a in T cells had been shown to improve their ability to eliminate tumors. Conversely, HIF1a loss in T cells had been demonstrated to impair anti-tumor T cell immunity (Doedens et al., 2013; Palazon et al., 2017). Thus, these published data suggest that abrogated T cell function in regions of QQCs is not the result of HIF1a activation in T cells. Instead, we uncovered a circuit by which cancer metabolism altered T cells in an indirect manner through induction of suppressive fibroblasts and DCs. This finding can explain the incongruency of the previously reported beneficial role of HIF1a activation in T cells from the overall detrimental effect of hypoxia on response to immunotherapy (Jayaprakash et al., 2018; Scharping et al., 2017).
In summary, our work discovered that primary TNBC contained a population of QCCs with the ability to resist T cell attack. These QCCs, through activation of HIF1a, orchestrated a micro-niche with unfit DCs, suppressive fibroblasts, reduced T cell infiltration, and enhanced T cell exhaustion. The development of strategies to overcome immune suppression around QCCs and eradicate these cells will be key to improving responses to immunotherapy in TNBC and preventing recurrence after these treatments.
Limitations of the study
In this study, we identified quiescence as the top feature of tumor cells that escape from T cell killing in primary breast cancer. We showed that these surviving cancer cells were already in a p27+ quiescent state prior to T cell adoptive transfer. However, the transcriptomes of surviving GFP+ cancer cells from Jedi-treated mice and p27KHigh cancer cells from untreated animals did not show a clear overlap, mostly due to an increase in IFN and inflammatory signatures in GFP+ cells in response to strong Jedi cell killing. This makes it difficult to determine the correspondence between both quiescent populations, and we cannot exclude that they may be heterogeneous. Moreover, QCCs were not terminally arrested but instead showed a higher tumor-initiating potential than their proliferative counterparts. We did not uncover whether this enhanced potential was the result of an inherent resistance to cell death, their immune evasion properties, or higher stemness potential. Additional studies will be necessary to deepen into this feature of QCCs.
Furthermore, we observed a clear hypoxia signature in the QCC niche and demonstrated that HIF1a activation in tumor cells was sufficient to recapitulate the immune evasion mechanisms found in the QCCs’ microenvironment. Nevertheless, we did not specifically identify a direct molecular link between HIF1a+ cancer cells and downstream impaired responses in DCs and T cells. Lack of oxygen did not alter DC function, but deprivation of nutrients along with unknown secreted factors by QCCs could be the underlying mechanisms. Uncovering the complex cross-talk between QCCs and the diverse immune and stromal components of the TME will require further investigations.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Requests for resources, reagents and further information regarding this manuscript should be addressed and fulfilled by the lead contact, Judith Agudo (judith_agudo@dfci.harvard.edu).
Materials availability
Plasmids and cell lines newly generated in this work are available upon request to the lead contact.
Data and code availability
Bulk RNAseq and scRNAseq have been deposited at GEO and are publicly available as of the date of publication. Accesion number is listed in the key resources table. Original western blot and microscopy images will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
| aCD11c PE | BioLegend | RRID: AB_313776 Clone:N418 |
| aCD11c PE/Cy7 | BioLegend | RRID: AB_493569 Clone:N418 |
| aCD3 AF647 | BioLegend | RRID: AB_389323 Clone:17A2 |
| aCD3e biotin | BioLegend | RRID: AB_312668 Clone:145–2C11 |
| aCD4 AF647 | BioLegend | RRID: AB_493372 Clone:RM4–5 |
| aCD40 PE/cy7 | BioLegend | RRID: AB_10933422 Clone:3/23 |
| aCD45 AF647 | BioLegend | RRID: AB_493534 Clone:30-F11 |
| aCD45 AF700 | BioLegend | RRID: AB_493714 Clone:30-F11 |
| aCD45.1 AF647 | BioLegend | RRID: AB_492864 Clone:A20 |
| aCD64 APC | BioLegend | RRID: AB_11219205 Clone:X54–5/7.1 |
| aCD8 AF594 | BioLegend | RRID: AB_2563237 Clone:53–6.7 |
| aCD8 Pe/Cy7 | BioLegend | RRID: AB_312760 Clone:53–6.7 |
| aCD86 PE | BioLegend | RRID: AB_313150 Clone: GL-1 |
| aGr1 APC | BioLegend | RRID: AB_313376 Clone:RB6–8C5 |
| aH2Kd AF647 | BioLegend | RRID: AB_493063 Clone:AF6–88.5 |
| aH2Kd APC | BioLegend | RRID: AB_10640118 Clone:SF1–1.1 |
| aHIF-1α XP® Rabbit mAb | Cell signaling | RRID: AB_2799095 Clone:D1S7W |
| aHuman CD3 | Agilent | RRID: AB_2631163 Clone:F7.2.38 |
| aHuman E-Cadherin | CST | RRID: AB_2291471 Clone:24E10 |
| aHuman Ki67 | Biocare | RRID: AB_2721189 Clone:SP6 |
| aHuman p27K | BD | RRID: AB_397636 Clone:57/Kip1/p27 |
| aIFNAR1 biotin | BioLegend | RRID: AB_1134250 Clone:MAR1–5A3 |
| aKi67 mAb | Cell Signaling | RRID: AB_2620142 Clone:D3B5 |
| aMHCII APC | BioLegend | RRID: AB_313328 Clone:M5/114.15.2 |
| aMHCII PB | BioLegend | RRID: AB_493528 Clone:M5/114.15.2 |
| aPD1 PerCP/Cy5.5 | BioLegend | RRID: AB_10550092 Clone:29F.1A12 |
| aTCRb biotin | BioLegend | RRID: AB_313426 Clone: H57–597 |
| aTIM3 APC | BioLegend | RRID: AB_2562997 Clone:B8.2C12 |
| Donkey anti-rabbit IgG AF647 | BioLegend | RRID: AB_2563202 Clone:Poly4064 |
| Goat anti-mouse AF555 | Invitrogen | RRID: AB_2535849 |
| IRDye 800CW Goat anti-Rabbit | LI-COR | RRID: AB_2651127 |
| Rat IgG1, κ Isotype Ctrl | BioLegend | RRID: AB_11150772 Clone: RTK2071 |
| Rat IgG2a, κ Isotype Ctrl | BioLegend | RRID: AB_11147167 Clone: RTK2758 |
| Rat IgG2b, κ Isotype Ctrl | BioLegend | RRID: AB_11149687 Clone: RTK4530 |
| Mouse IgG1, κ Isotype Ctrl | BioLegend | RRID: AB_11148942 Clone: MG1–45 |
| American Hamster IgG Isotype Ctrl | BioLegend | RRID: AB_11203529 Clone: HTK888 |
| Rabbit IgG Isotype Ctrl | Thermofisher scientific | RRID: AB_243593 |
| Anti-mouse CD16/32 | BioLegend | RRID: AB_2561482 Clone: 93 |
| CD45-BX007—Alexa Fluor™ 750-RX007 | Akoya Biosciences | Cat#4450002, Clone: 30-F11 |
| MHCII-BX014-Atto 550-RX014 | Akoya Biosciences | Cat#4250003, Clone: M5/114.15.2 |
| CD3-BX021—Cy5-RX021 | Akoya Biosciences | Cat#4350014, Clone: 17A2 |
| CD11c-BX030-Cy5-RX030 | Akoya Biosciences | Cat#4350013, Clone: N418 |
| Anti-p27 KIP 1 | Abcam | RRID: AB_2811037, Clone: Y236 |
| Anti-GFP | Abcam | RRID: AB_305643 |
| Anti-mouse CD90.1 (Thy-1.1) | BioLegend | RRID: AB_314013 Clone: OX-7 |
| Anti-mouse F4/80 | BD Biosciences | RRID: AB_2739222 Clone: T45–2342 |
| Anti-mouse CD366 (Tim-3) | Biolegend | RRID: AB_1626128 Clone: B8.2C12 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| One Shot™ Stbl3™ Chemically Competent E. coli | Thermofisher Scientific | cat#C737303 |
|
| ||
| Biological samples | ||
|
| ||
| anonymized TNBC patients | Brigham and Women's Hospital Pathology Core | protocols 11–104 and 93–085 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Iscove's Modified Dulbecco's Medium (IMDM) | Thermofisher Scientific | Cat#31980030 |
| USDA FBS | Life Technologies | Cat#10437028 |
| Penicillin-Streptomycin | Life technologies | Cat#15140122 |
| Glutamax | Life Technologies | Cat#35050061 |
| Polybrene Transfection Reagent | Thermofisher Scientific | Cat#TR1003G |
| T4 Ligase | NEB | Cat#M0202L |
| BamHI-HF | NEB | Cat## R3136L |
| NheI-HF | NEB | Cat#R3131S |
| CutSmart® Buffer | NEB | Cat#B7204 |
| AgeI-HF | NEB | Cat#R3552S |
| SalI-HF | NEB | Cat#R3138S |
| BsaBI | NEB | Cat#R0537S |
| Q5® High-Fidelity DNA Polymerase | NEB | Cat#M0491S |
| BclI-HF | NEB | Cat#R3160S |
| NotI-HF | NEB | Cat#R3189S |
| Calcium Chloride, Anhydrous (Pellets, 4–20 mesh, for Desiccators), Fisher Chemical™ | Fisher Scientific | Cat#C614–500 |
| UltraPure 0.5 M EDTA, pH 8.0 | Life Technologies | Cat#15575020 |
| Alt-R® S.p. Cas9 Nuclease V3 | IDT | Cat#1081059 |
| Hyaluronidase | StemCell | Cat#07461 |
| Collagenase IV | StemCell | Cat#17104019 |
| DNaseI | Sigma Aldrich | Cat#10104159001 |
| RBC lysis buffer | Biolegend | Cat#420301 |
| BSA | Cell Signaling Technology | Cat#9998S |
| Dapi | Biolegend | Cat#422801 |
| 10X CODEX Buffer | Akoya Biosciences | Cat#7000001 |
| CODEX Assay Reagent | Akoya Biosciences | Cat#7000002 |
| Nuclear Stain | Akoya Biosciences | Cat#7000003 |
| CODEX Storage Buffer | Akoya Biosciences | Cat#232107 |
| BX002-Atto 550-RX002 | Akoya Biosciences | Cat#5450023 |
| BX006-Cy5-RX006 | Akoya Biosciences | Cat#5450027 |
| BX017-Atto 550-RX017 | Akoya Biosciences | Cat#5250001 |
| BX027-Cy5-RX027 | Akoya Biosciences | Cat#5350004 |
| BX042-Cy5-RX042 | Akoya Biosciences | Cat#5350008 |
| 7-AAD | Biolegend | Cat#420404 |
| Trizol LS | Life Technologies | Cat#10296010 |
| Trizol | Life Technologies | Cat#15596018 |
| D(+)-SUCROSE | Fisher Scientific | Cat#AC177140010 |
| PARAFORMALDEHYDE 32% SOL. EM GRADE | VWR | Cat#100496–496 |
| 16% Paraformaldehyde Aqueous Solution, EM Grade | Fisher Scientific | Cat#50–980-487 |
| FB OCT COMPOUND CLEAR 4OZ | Fisher Scientific | Cat#23730571 |
| Normal Mouse Serum | Jackson ImmunoResearch Laboratories | Cat#015–000-120 |
| Avidin/Biotin Blocking System | BioLegend | Cat#927301 |
| ProLong Diamond Antifade Mountant | Life Technologies | Cat#P36961 |
| Doxycycline hyclate | Biogems | Cat#2431450 |
| Poly-L-lysine solution | Sigma Aldrich | Cat# P8920–500ML |
| 2-NBDG | VWR | Cat#76021–666 |
| Recombinant Murine Flt3-Ligand | VWR | Cat#10780–338 |
| Lipopolysaccharides from Escherichia coli O111:B4 γ-irradiated |
Sigma Aldrich | Cat#L4391–1MG |
| Poly I:C | Invitrogen | Cat#tlrl-pic |
| Pierce RIPA Buffer | VWR | Cat#PI89900 |
| NuPAGE Sample Reducing Agent | Life Technologies | Cat#NP0009 |
| Novex 4–20% Tris-Glycine gels | Life Technologies | Cat#P04205BOX |
| Nitrocellulose Sandwiches | Cell Signaling Technology | Cat#12369P2 |
|
| ||
| Critical commercial assays | ||
|
| ||
| QIAprep Spin Miniprep Kit | Qiagen | Cat#27106 |
| EndoFree Plasmid Maxi Kit | Qiagen | Cat#12362 |
| miRNeasy Micro Kit | Qiagen | Cat#217084 |
| SF Cell Line 4D-NucleofectorTM X Kit S | Lonza | Cat#V4XC-2032 |
| EasySep™ Mouse CD8+ T Cell Isolation Kit | StemCell | Cat#19853 |
| QIAquick PCR Purification Kit | Qiagen | Cat#28104 |
| QIAquick Gel Extraction Kit | Qiagen | Cat#28706 |
| Hypoxyprobe-Biotin Kit | Hypoxyrprobe | Cat#HP10–1000kit |
| EdU-Click 647 | BaseClick | Cat#BCK-EdU647 |
| CODEX Staining Kit | Akoya Biosciences | Cat#7000008 |
| CODEX Conjugation Kit | Akoya Biosciences | Cat#7000009 |
|
| ||
| Deposited data | ||
|
| ||
| Sequencing datasets | This paper | Available at NCBI accession # GSE198715 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| 4T07 | Dr. Robert Weinberg, MIT | N/A |
| D2A1 | Dr. Robert Weinberg, MIT | N/A |
| EMT6 | ATCC | Cat#CRL-2755™ |
| Hek293:Lenti-X™ 293T Cell Line | Takara | Cat#632180 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: Balb/c :BALB/cJ | The Jackson Laboratory | Strain #:000651 |
| Mouse: NSG: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | The Jackson Laboratory | Strain #:005557 |
| Mouse: Jedi: Ptprca TcrbLn1Bdb TcraLn1Bdb H2d/J | Dr. Brian Brown laboratory, Mount Sinai | N/A |
| Mouse:PD1-/-:B6.Cg-Pdcd1tm1.1Shr/J | The Jackson Laboratory | Strain #:028276 |
| Mouse:B6:C57BL/6J | The Jackson Laboratory | Strain #:000664 |
| Mouse:Kaede:Kaede | Dr. Osami Kanagawa, University of Lyon | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Alt-R® CRISPR-Cas9 tracrRNA | IDT | Cat#1072533 |
| Hifla gRNA: AGTGCACCCTAACAAGCCGG | IDT | Alt-R® CRISPR-Cas9 crRNA |
| Ifnarl gRNA: GCTCGCTGTCGTGGGCGCGG | IDT | Alt-R® CRISPR-Cas9 crRNA |
|
| ||
| Recombinant DNA | ||
|
| ||
| 3rd Generation Lentiviral packaging vectors | Dr. Brian Brown, Mount Sinai | N/A |
| PGK:eGFP Lentiviral vector | Dr. Brian Brown, Mount Sinai | N/A |
| mVenus:p27k reporter | Dr. Toshio Kitamura, University of Tokyo | N/A |
| PGK:mCherry Lentiviral vector | Dr. Brian Brown, Mount Sinai | N/A |
| H2B-mCherry | Addgene | Cat#51007 |
| H2B-tdTomato | Addgene | Cat#58101 |
| rtTA | Addgene | Cat#104543 |
| TetON lentiviral expression vector | Addgene | Cat#131687 |
| Hifla STBL: mHif-1α MYC (P402A/P577A/N813A) | Addgene | Cat#44028 |
|
| ||
| Software and algorithms | ||
|
| ||
| FlowJo 10 | BD | https://www.flowjo.com |
| Prism 9 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| FiJi® (v2.0.0-rc-69/1.52i) | ImageJ | https://imagej.net/software/fiji/ |
| Seurat | Satija Lab | https://satijalab.org/seurat/articles/pbmc3k_tutorial.html |
| DESeq2 | Bioconductor | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Akoya CODEX Processor® | Akoya | https://www.akoyabio.com/phenocycler/software/ |
| clusterProfiler | Bioconductor | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| bigSCale2 | Iacono et al. (2019) | https://github.com/iaconogi/bigSCale2 |
| Benchling | Benchling | ttps://www.benchling.com |
| Biorender | Biorender | https://biorender.com/ |
|
| ||
| Other | ||
|
| ||
| Compresstome® | Precisionary | VF-310–0Z |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Two-month-old BALB/cJ females (Stock No. 000651) from Jackson Laboratories were used for all experiments as tumor-bearing mice, unless otherwise stated. NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ a.k.a. NSG mice (Stock No. 005557 from Jackson Labs) were used as non-T cell bearing controls. Just EGFP Death Inducing (Jedi) mice (Ptprca TcrbLn1Bdb TcraLn1Bdb H2d/J) (Agudo et al., 2015) in BALB/cJ background were obtained from Dr. Brian Brown. These Jedi mice were bred with PD1−/− mice (B6.Cg-Pdcd1tm1.1Shr/J, Stock No. 028276, from Jackson Labs) to generate PD1−/− Jedi, ensuring the H2-Kd haplotype and CD45.1 congenital marker were also selected. F1 from BALB/cJ crossed with C57BL/6J mice (Stock No. 000664 from Jackson Labs) were used for tumor implantation in experiments using PD1−/− Jedi T cells adoptive transfer. Kaede mice were previously reported (Tomura et al., 2008) and provided by Dr. Osami Kanagawa at RIKEN, Japan. Kaede mice were crossed with BALB/cJ one generation for injection of 4T07 breast cancer cells for tumor formation. All animal procedures were approved by Dana-Farber Cancer Institute IACUC and the Harvard Medical School IACUC and performed according to DFCI protocol #17–017 and HMS protocol # IS00002540. The study is compliant with all relevant ethical regulations regarding animal research.
Cell lines
4T07 and D2A1 breast cancer cell lines were a gift from Dr. Robert Weinberg. EMT6 breast cancer cells were purchased from ATCC. HEK293T cells were purchased from Takara. All cell lines were cultured in Iscove’s Modified Dulbecco’s Medium (IMDM), 10% heat inactivated FBS (Gibco), 1% Penicillin-Streptomycin (Gibco) and 1% Glutamax (Gibco). 4T07, EMT6 and D2A1 cells were engineered to express the desired fluorescent proteins and markers by stable transduction with lentiviral vectors (LV). All cells were tested for mycoplasma prior to injection.
GFP and mCherry-expressing 4T07 cells used in Figure 1 were subjected to RNA-sequencing analysis to ensure no bias had occurred during generation of the cell lines. This analysis revealed no differences between the cell lines.
Human FFPE samples and Bulk RNAseq data
This study was performed in accordance with the guidelines of ethical regulation for human samples under approved protocols 11–104 and 93–085 at Dana Farber Cancer Institute.
Sections from Formalin-Fixed Paraffin Embedded Primary Triple Negative Breast Cancer from treatment naïve unidentified patients were obtained from the Brigham and Women’s Hospital Pathology core.
Bulk RNAseq data cohort was formed by 29 patients with confirmed mTNBC defined as <1% progesterone receptor, <1% estrogen receptor and HER2 negative by ASCO/CAP guidelines on a metastatic biopsy. Patient regimens: treated with an ICB alone (pembrolizumab, n = 6, NCT02447003; atezolizumab, n = 4, NCT01375842) or as part of a combination regimen with chemotherapy (pembrolizumab + eribulin, n = 8, NCT02513472; atezolizumab + nab-paclitaxel, n = 5, NCT0163970) or a targeted therapy (nivolumab + cabozantinib, n = 6, NCT03316586). Samples were sequenced before ICB treatment. PFS was defined as the date of starting immunotherapy to the date of progression, death, or last follow-up. Durable responders were considered as free of disease progression at time of analysis (5 patients) with a PFS of 26–60 months.
Plasmids
Third generation lentiviral packaging and lentiviral vector (LV) backbone with eGFP expression under PGK promoter were a gift from Dr. Brian Brown (Brown et al., 2006). The mVenus-p27K insert sequence was a gift from Dr. Kitamura (Oki et al., 2014). Fluorescent protein sequences were obtained from the following vectors: mCherry (Dr. Brian Brown gift), H2B-mCherry (Addgene #51007) and H2B-tdTomato (Addgene #58101). The eGFP lentiviral plasmid was used as backbone to generate vectors expressing mCherry, H2B-mCherry, mVenus-p27K, tdTomato-p27K and a shuttle vector for co-expressing two proteins with Internal Ribosome Entry Site (IRES) element by PCR, digestion by BamHI and SalI restriction enzymes and T4 DNA Ligase.
The mVenus fluorescent protein (yellow) was replaced by tdTomato by PCR from H2B-tdTomato and using BamHI and BsaBI restriction enzymes to make a tdTomato-p27k fusion protein in red color. Same strategy was used to create a fusion protein with rtTA-p27K for the genetic circuit in Figure 1O. The rtTA sequence was obtained from Addgene #104543. The doxycycline expression vector was obtained from Addgene #131687 in which H2B-tdTomato fluorescent protein was introduced using BclI and NotI.
The fluorescent proteins mCherry or eGFP were cloned into the same third generation LV backbone with a normoxia-stable form of HIF1α, mHif-1α MYC (P402A/P577A/N813A) (Addgene #44028), here referred as HIF1aSTBL. The resulting vectors had therefore the following structure mCherry-IRES-HIF1aSTBL and GFP-IRES-HIF1aSTBL. DNA was extracted by miniprep or maxiprep kits (QIAGEN) after transformation in Stbl3 E.coli (ThermoFisher). All plasmids were verified by Sanger sequencing.
METHOD DETAILS
Tumor injection
Breast tumors were induced by intra-mammary fat pad injection of indicated breast cancer cell lines (4T07, EMT6 and D2A1). The injections were performed after introducing a 2mm incision to visualize the 4th mammary fat pad under isoflurane anesthesia. A total of 250,000 cells in 50μL sterile PBS were injected in the fat pad of the mammary gland. Local analgesic was used for pain relieve. Tumors were measured with a caliper and monitored twice a week and tumor volume was calculated as (length*width*width)/2.
Lentiviral production and transduction
Third generation lentivirus were produced by Ca3(PO4)2 transfection into HEK293T cells (Brown et al., 2006). Supernatants were collected, passed through a 0.22mm filter, aliquoted and frozen, or directly used to transduce breast cancer cell lines.
For LV transduction, tumor cells were cultured with filtered LV in the presence of 1:2000 Polybrene Transfection Reagent (Millipore) for 16–24 hours, expanded, and subsequently FACS-sorted with Aria III cell sorter (BD Biosciences) using the expressed fluorescent reporter. All transgenes introduced by LV transduction were expressed under the ubiquitous promoter PGK.
Generation of knockout cell lines
Amaxa 4D-Nucleofector X Unit device (Lonza) was used to generate knockout cells lines through CRISPR/Cas9-mediated genome editing. Briefly, 2.5μL 200μM trRNA, 2.5μL 200μM gRNA and 5μL 20μM recombinant CAS9 (Integrated DNA Technologies) were mixed to make Cas9/gRNA ribonucleoprotein (RNP). The RNP was delivered into tumor cell lines using program A549 by SF Cell Line 4D-NucleofectorTM X Kit S (Lonza). 200,000 cells were used per reaction and recovered in 6-well plate for 4 days after nucleofection, and then subjected to a 2nd round of the same process. The efficiency of Ifnar1−/− was validated by flow cytometry analysis. Hif1a−/− cells were validated by western blot using HIF-1a (D1S7W) rabbit antibody from Cell Signaling Technology. The guide RNA sequences were: HIF1a (AGTGCACCCTAACAAGCCGG), Ifnar1 (GCTCGCTGTCGTGGGCGCGG).
Adoptive transfer of CD8+ T cells
Jedi CD8+ T cells as well as PD1−/− Jedi CD8+ T cells were purified from spleens and lymph nodes (LNs) after obtaining a single cell suspension by mechanical disruption and filtering through a 70μm cell strainer. CD8+ T cells were selected with the mouse CD8+ T cells isolation kit (STEMCELL) following manufacturer’s instruction. 5–7×106 T cells (unless stated otherwise) were injected via tail vein per mouse in 200μL sterile PBS.
Flow cytometry and FACS-sorting
Tumors were collected and placed in 500μL of digestion solution (400U/mL CollagenaseIV, 6.8U/mL Hyaluronidase, 20μg/mL DNaseI and 10%FBS in HBSS). After mincing, they were transferred to a 10mL tube with 5mL digestion solution and incubated at 37 °C for 20 minutes on a rotator. Digested samples were then filtered through a 100μm cell strainer followed by red blood cell (RBC) lysis (Biolegend) for 5 minutes at room temperature (RT). Single cell suspensions were blocked with 1:100 dilution of TruStain FcX (#101319 Biolegend) in flow buffer (2mM EDTA 0.1% BSA PBS). Samples were then stained with the appropriate antibodies. DAPI (1:10,000) or 7AAD (1:100) were used as viability markers. Samples were analyzed by CytoFLEX (Beckman). Cells were FACS-sorted by Aria III cell sorter (BD Biosciences) and collected in 15mL tubes with 5mL of media. Data were analyzed using FlowJo v.10 software.
Bulk RNA-sequencing
GFP+ and mCherry+ 4T07 cells in Figure 1 were directly FACS-sorted into TRIzol LS (Invitrogen) and submitted directly for RNA extraction and low-input RNA-sequencing to GENEWIZ. FACS-sorted mVenusp27KHigh and mVenus-p27KNeg 4T07, EMT6 and D2A1 cells from primary tumors were collected in media, centrifuged, and resuspended in 1mL TRIzol (Invitrogen). RNA was extracted with miRNeasy Micro kit (QIAGEN) following the manufacturer’s instructions. RNA samples were submitted to GENEWIZ for bulk RNA-sequencing.
Immunofluorescence
Tumors were harvested and fixed in 20% Sucrose 4% Paraformaldehyde (PFA) solution in PBS overnight at 4°C. They were then embedded in OCT compound (Fisher Health Care) and stored in −80°C. Sections (10μm) were blocked in 10% Fetal Bovine Serum, 2% Normal Mouse Serum, 0.1% Tween-20 TBS for 1 hour at RT and incubated for 3 hours at RT or overnight at 4°C with primary antibodies. Tissue was washed and, when needed, incubated with secondary fluorescently labelled antibodies for 2 hours before nuclear staining with DAPI (ThermoFisher). For Nuclear marker Ki67, samples were permeabilized with 1% TritonX-100 at 37 °C for 30min prior to staining. For biotin antibodies, an extra blocking step was done with Avidin/Biotin Blocking System (Biolegnd) following the instruction of the Kit. Antibodies were used at 1:100 dilution in blocking buffer. The stained tissue was then mounted on ProLong™ Diamond Antifade Mountant (Invitrogen) and covered with a #1.5 coverslip.
For assessment of tumor hypoxia, Pimonidazole (75mg/Kg of body weight) was intraperitoneally injected 30–60min prior to euthanasia. Staining was performed with Hypoxyprobe® following manufacturer instructions.
Microscopy methods
Microscopy methods are reported following the guidance of (Montero Llopis et al., 2021).
Images for Figures 2D, 3E, and S1O were acquired in Yokogawa CSU-W1 single disk (50 μm pinhole size) spinning disk confocal unit attached to a fully motorized Nikon Ti inverted microscope equipped with a Nikon linear-encoded motorized stage with a PI 250 μm range Z piezo insert, an Andor Zyla 4.2 plus (6.5 μm photodiode size) sCMOS camera using a Nikon Plan Apo 20x/0.75 NA DIC air objective. The final digital resolution of the image was 0.325 μm/pixel. Fluorescence from mVenus, Alexa Fluor 555 (or mCherry) and Alexa Fluor 647 was collected by illuminating the sample with solid-state directly modulated 488 nm diode 100 mW (at the fiber tip) laser line, a solid state, directly modulated 561 nm DPSS 100mW (laser tip) laser line or a solid state, directly modulated 640 nm diode 100mW (laser tip) laser line in a Toptica iChrome MLE laser launch, respectively. A hard-coated Semrock Di01-T405/488/568/647 multi-bandpass dichroic mirror was used for all channels. Signal from each channel was acquired sequentially with either a hard-coated Chroma ET525/36 nm, Chroma ET605/52 nm or Chroma ET705/72m emission filters in a filter wheel placed within the scan unit, for mVenus, Alexa Fluor 555 (or mCherry) and Alexa Fluor 647 channels, respectively. Fluorescence from DAPI was collected by illuminating the sample with a Lumencore SOLA 395 LED based light engine using a Chroma 49000 filter cube. Nikon Elements AR 5.02 acquisition software was used to acquire the data. Data was saved as ND2 files.
Images for Figures 1L, 2D, 2F, 5F, S1R, S2C, S2E, S2H, and S2I were acquired using a motorized Nikon Ti inverted widefield microscope equipped with a SOLA SE LED light engine, Prior ProScan II linear encoded motorized stage, a Shutter Instruments Lambda 10–3 motorized excitation filter wheel, a LUDL MAC3000 motorized emission filter wheel located in front of the camera on the side port. A Hamamatsu Orca Flash 4.0 v3, 6.5 μm photodiode size sCMOS camera was used for detection. Images were acquired using a Nikon Plan Apo 20x/075 Ph2 air objective lens. Final digital resolution of the image was 0.325 mm/pixel. Signal from DAPI, FITC, RED, FAR RED was collected by illuminating the sample with a hard-coated Chroma AT350/50x, Chroma ET490/20X, Chroma ET555/25x and Chroma ET 645/30x excitation filter, a hard-coated Chroma ET 69002bs multiband dichroic DAPI/FITC/TRITC/Cy5 and a Chroma ET455/50m, Chroma ET525/50m, Chroma ET605/52M, Chroma ET700/75m emission filter, respectively. Images from each experiment were collected under identical imaging conditions using Nikon Elements 5.2 AR acquisition software and a 12-bit low gain digitizer. Images were saved as ND2 files
Images for Figures 1D, 1J, 2G, 2H, 3D, 3F, 6H, S1C, S1D, and S1I were acquired using a motorized Zeiss Axio Observer 7 inverted microscope equipped with a LED Colibri 7 fluorescence light source, and motorized scanning stage with a Z-piezo insert of 500 μm range. A Photometrics Prime BSI sCMOS 6.5 um photodiode size was used for detection. Images were acquired using a Zeiss 20x Plan Apo 20x/0.8 DICII air or a Zeiss Plan Apo 63x/1.4 DICII oil objective lens with Zeiss 518F immersion oil; the final digital resolution of the image was 0.325 μm/pixel or 0.1 μm/pixel, respectively. Signal from DAPI, GFP/mVenus, Alexa Fluor 555, mCherry, Alexa Fluor 647 and Alexa Fluor 750, was collected by illuminating the sample with a Zeiss 90 HE DAPI/GFP/Cy3/Cy5 multi-band pass filter cube or a Zeiss 110 HE DAPI/GFP/Cy3.5/Cy7 multi-band-pass filter cube. Images were collected using Zen Blue (Zen 2.6 Pro) acquisition software and saved as.czi files.
Evaluation of cell cycle
To evaluate cell proliferation in vivo, animals were injected intraperitoneally with 10mg/Kg of EdU (5-ethynyl-2’-deoxyuridine) daily during the last 5–8 days of tumor growth. Tumors were harvested and processed for immunofluorescence staining as described above. EdU was stained using the immunofluorescence BCK-EdU647 staining kit (BaseClick) following manufacturer’s instructions.
Evaluation of cell cycle state prior to adoptive transfer of Jedi T cells was performed using a genetic circuit as explained in Figure 1O. 4T07 cells carrying both GFP and the genetic circuit were injected in the mammary fat pad. Animals received one intraperitoneal injection of Doxycycline (50mg/kg diluted in sterile PBS) 30h prior to adoptive transfer of Jedi T cells to ensure doxycycline had been eliminated from the system (Bocker et al., 1981; Lucchetti et al., 2019). 5 million Jedi T cells were injected on day 6 of tumor growth. H2B-tdTomato+ tumor cells were quantified by flow cytometry on day 12 of tumor growth.
Tumor initiation assay
To test the tumor initiation potential of Jedi-resistant cancer cells: GFP or mCherry-expressing 4T07 cells were mixed 1:1 ratio and injected into the mammary fat pad of BALB/c mice. 7 days after tumor inoculation 5M of Jedi T cells were adoptively transfer. In parallel, double transduced tdTomato-p27K and GFP-expressing 4T07 were injected into mammary fat pad of NSG mice as controls with no T cell killing. 14 days after tumor inoculation tumors from both groups were processed and surviving GFP+ cells were FACS-sorted. 500 live (DAPI-) were injected into mammary fat pad of new BALB/c mice.
To test the tumor initiation potential of p27KHigh cells: mCherry and mVenus-p27K expressing breast tumors (4T07, D2A1 or EMT6) were processed to obtain single cell suspensions by collagenase digestion. mCherry+ mVenus–p27KNeg and mCherry+ mVenus-p27KHigh cells were FACS-sorted. Tumor initiation potential was assessed by injecting 500 live (DAPI-) cells (for EMT6) and 1,000 cells (4T07 and D2A1) into mammary fat pad of new BALB/c mice.
Glucose uptake
Tumor single cell suspensions were resuspended in 200μM 2NBDG (BioVision) in flow buffer for 20 min at 37 °C, using equivalent number of cells across tumors. Samples were then analyzed by flow cytometry.
Photo-conversion of Areas to Dissect Micro-Environments (PADME-seq)
tdTomato-p27K-expressing 4T07 cells were injected into the mammary fat pad of Kaede x BALB/c F1 females. Tumors were embedded in 2% low melting agarose and cut into 300mm slices by the Compresstome® VF 310–0Z vibrating microtome (Precisionary Instruments). Setting of the machine were 3 for vibration and 1–2 for speed depending on tumor consistency. Tumors were sectioned to obtain 2 consecutive slices together from 2 depths/regions from each tumor mass. Consecutive slices were used to photoconvert cells infiltrating tdTomato-negative areas and then cells in regions with tdTomato-p27KHigh cells. Slices were kept in a 12 well plate with IMDM media on ice. Photoconversion was performed in a Zeiss LSM710 single point-scanning confocal unit with galvanometer mirrors attached to a fully motorized Axio-Observer Z1 equipped with a Zeiss motorized stage using a Zeiss Plan Apochromat 20x/0.8 NA DIC air objective. Photoconversion of Kaede was performed using a 405nm 30mW diode AOTF modulated line set to 0.2 % transmittance laser and a time series of 325 cycles at a 1.58msec/pixel pixel dwell times, using a 405 long-pass dichroic mirror. Kaede green and red (after photoconversion) fluorescence was collected by illuminating the sample with an argon multi-line 488 nm 25 mW Argon AOTF modulated line set to 0.2 % transmittance and a 561 nm 20mW DPSS AOTF modulated line set to 0.2 % transmittance, using a multi-bandpass dichroic mirror with 488/561cut off wavelengths (MBS 488/561). The emission wavelength range for the green and red fluorescence signal was set to 499–560 nm and 571–735 and collected by multi-alkali PMTs. No offset was applied. The microscope was controlled by ZEN Black SP2 acquisition software, scanned unidirectionally with a pixel dwell time of 1.58μsec/pixel, no averaging, a 12-bit digitizer, zoom 1.0x, a pixel size of 0.83 mm and pinhole set to 1 A.U. for 488 nm wavelength. Images were saved with the.czi file format.
Photoconverted slices were digested in collagense IV + DNAse as described above. Samples were digested for 15 min at 37°C and pass through a 100μm cell strainer followed by RBC lysis. Cells were washed and resuspended in 1mL flow buffer with DAPI (1:10,000) for sorting. Green/red double positive live cells were considered as the successful photoconverted ones and FACS-sorted into 1.5mL Eppendorf tubes with 500μL media at AriaIII.
Co-detection by indexing (CODEX) tissue staining
mCherry from the original Figure 1 setting was replaced by Thy1.1 as a tumor marker. The CODEX technology requires three spectrally distinct dyes (Atto 550, Cy5, Alexa Fluor™ 750) for each imaging cycle and Atto550 overlaps with mCherry. Thy1.1 was not immunogenic in BALB/c mice and antibody in the panel was used to detect Thy1.1 (as the non-GFP cancer cells). Wildtype (untrunsduced) or Thy1.1:GFP-expressing mixed 4T07 tumors were harvested and dehydrated in 20% sucrose in PBS overnight at 4°C. Tumors were then embedded in OCT compound and stored in −80°C. Sections (8μm) were obtained onto poly-L-lysine-coated coverslips and blocked for 1h at RT in Akoya CODEX Blocking Buffer® with IgG isotype controls and anti-mouse Fc receptors (93, Biolegend #101330). Then tissues were stained overnight at 4°C with the oligonucleotides-barcoded primary antibody cocktail, which were prepared following Akoya Biosciences CODEX Fresh-Frozen Tissue Staining protocol. The following primary antibodies were purchased from Akoya Biosciences: CD45, MHCII, CD3, CD11c. The rest primary antibodies were conjugated to Akoya barcodes according to Akoya Biosciences CODEX Antibody Conjugation protocol: p27, GFP, Thy1.1, F4/80, TIM3. Secondary fluorophore-tagged oligonucleotides (reporters) and DAPI (Akoya #7000003) were automatically added and washed away for each imaging cycle using Akoya CODEX system to allow multiplexed visualization on a single tissue section. Images (Figures 4L, 5G, and S4F) were acquired using a motorized Zeiss Axio Observer 7 inverted microscope equipped with a LED Colibri 7 fluorescence light source, and motorized scanning stage with a Z-piezo insert of 500 μm range. A Photometrics Prime BSI sCMOS 6.5 um photodiode size was used for detection. Images were acquired using a Zeiss 20x Plan Apo 20x/0.8 DICII air; the final digital resolution of the image was 0.325 μm/pixel. Signal from DAPI, Atto 550, Cy5 and Alexa Fluor™ 750, was collected by illuminating the sample with a Zeiss 90 HE DAPI/GFP/Cy3/Cy5 multi-band pass filter cube or a Zeiss 110 HE DAPI/GFP/Cy3.5/Cy7 multi-band-pass filter cube. Images were collected using Zen Blue (Zen 2.6 Pro) acquisition software and saved as.czi files. Analysis was done using CODEX Multiplex Analysis Viewer CODEXMAV (v1.5.0.8) plugin in Fiji (v2.1.0/1.53c).
Bone marrow derived dendritic cell (BM-DC) generation and culture in different oxygen concentrations
BM-DCs were generated using Flt3l as already described in Agudo et al. (2014). At day 9 BMDC were recovered with flow buffer 10mM EDTA and 90,000 live cells/well were seeded in a 96 well U bottom plate. Cells were conditioned for 2 hours to the required oxygen level (normoxia, 5% or 1%O2). Then either LPS (50ng/ml) or PolyI:C (5μg/ml) was added. Cells were kept with either LPS or PolyI:C for 18h at the corresponding oxygen level (so total 20h). Cells were detached with flow buffer 10mM EDTA and processed for flow cytometry assay.
Western blot
Whole cell lysate (WCL) was collected from 1 million WT or Hif1a−/− cells using RIPA buffer (Cell Signaling Technology, CST) with Pierce Protease Inhibitor (Thermo Fisher Scientific). After incubating for 10 min on ice, WCL was spined for 15 min at 13,000 rpm at 4°C to get the supernatant. 3X loading buffer with DTT (CST) was added and boiled at 100°C for 5 min to denature. Protein samples were separated by Novex 4–20% Tris-Glycine gels (Invitrogen) and transferred to nitrocellulose membranes (Cell Signaling Technology, CST). The membranes were blocked by 5% milk TBST (0.1% Tween 20), then incubated with rabbit anti-HIF1A (D1S7W, 1:1000, CST) and mouse anti-Actin (8H10D10, 1: 5000, CST) overnight at 4°C. Followed by washing use TBST for 3 times, the membranes were incubated with AF555 Goat anti-mouse (1:5000,Invitrogen) and IRDye 800CW Goat anti-Rabbit (1:5000, LI-COR Biosciences) fluorescent secondary antibodies for 2 hours at room temperature. The membranes were imaged using Bio-Rad ChemiDoc MP Imaging System.
QUANTIFICATION AND STATISTICAL ANALYSIS
Computational analysis of immunofluorescent images of mouse tumors
All images were analyzed using FiJi® (v2.0.0-rc-69/1.52i) (Schindelin et al., 2012).
T cell density per area was analyzed in tumors from Figures 1E, 1F, 2E, S1E, S2F, and S2G after CD3 staining. Areas were drawn using mVenus-p27K, GFP or mCherry and blindly to CD3 signal. After drawing the regions of interest (ROIs), CD3 signal was revealed, and T cells were counted manually. Area was calculated by FiJi after scaling images. Density of T cells was calculated as number of T cells/area (μm2).
Colocalization analysis for nuclear markers (Ki67, mVenus-p27K and EdU) was done using DAPI channel for segmentation with the Trainable Weka Segmentation (v3.2.29) plugin. Nuclear particles were converted into a binary mask to analyze the fluorescent intensity in the different channels with the function Analyze Particles. Intensities of the different channels in the same particle were plotted to assess correlation or exclusion patterns.
Quantification of MHCII fluorescent intensity in DCs (Figure 5G) was performed on immunofluorescence images with CD11c and MHCII staining on mVenus-p27K-expressing 4T07 breast tumors. ROIs were drawn using only mVenus-p27K, being blind to both CD11c and MHCII. Afterwards, CD11c channel was used to create a binary mask after thresholding with Otsu method. Fluorescent intensity of the MHCII channel was measured using Analyze Particles function using particles generated by the CD11c mask.
Analysis of immunofluorescent FFPE sections from human TNBC
FFPE sample slides from 10 anonymized TNBC patients were stained by the BWH Pathology Core with E-Cadherin (24E10, CST), p27K (57/Kip1/p27, BD), Ki67 (SP6, Biocare) and CD3 (F7.2.38, Agilent). Samples were imaged with Zeiss Axio Observer 7 inverted microscope as explained in the immunofluorescence method section. For T cell enrichment analysis areas surrounding T cells that were able to infiltrate E-Cadherin+ areas were drawn and then evaluated for Ki67 or p27 positive cells. Total number of positive cells were normalized by area to obtain density of p27+ and Ki67+ cells in the vicinity of T cells.
Mouse Bulk RNAseq analysis
GENEWIZ performed basic computational analysis for quality controls and constructed the gene matrices. Gene matrices were normalized using DESeq2 size factors and filtering for a minimum of 10 read per condition (60–80 rowSums). The resulting genes with adjusted p-Value < 0.05 were considered significant for GO term enrichment analysis at https://david.ncifcrf.gov/. Log2 fold change ≥0.5 was considered as upregulated and ≤ −0.5 as downregulated genes. Volcano plots were generated with the package EnhancedVolcano from Bioconductor. Heatmaps were generated in excel using Z-score across different samples in a given gene after after counts per million mapped reads (CPM) normalization.
CODEX image processing and analysis
Raw images were processed using Akoya CODEX Processor® for background subtraction, deconvolution, extended depth of field and shading correction. CODEXMAV plugin in ImageJ was used for visualization and selection of cell populations. To assess T cell exhaustion, p27 positive versus negative areas or Thy1.1+ versus GFP+ areas in tumor images were drawn and then evaluated for TIM3 expression on T cells (CD45+ CD3+ MHCII-CD11c-F4/80-). To assess DC function, same selected areas were used. DCs were considered as CD45+CD11c+F4/80-CD3-. MHCII intensities were recorded for each DC. T cells were normalized dividing the total number of T cells by the area (μm2) of the ROI. MHCII intensities from DC were normalized and centered using a Z-score to be able to pool the different experiments eliminating the batch effect.
RNA-seq analysis from TNBC patient cohort
RNAseq data was aligned using the STAR software (Dobin et al., 2013). Further transcript per million (TPM) quantification was performed using RSEM (Li and Dewey, 2011). Canonical pathways compiled in MSigDB from pathway databases, namely the C2 CP gene sets, were used for gene set enrichment analysis (GSEA) analysis using 1000 permutations by phenotype (Subramanian et al., 2005).
PADME-seq 10X genomics and analysis
Count matrix generation
FACS-sorted live cells were submitted to the Bigham and Women’s Hospital Single Cell Genomics Core for 10x genomics. Samples from different areas were tagged using hashed antibodies and then pooled to reduce technical variability in the preparation of the 10X library following an approved protocol from the core (Table S1). The BWH Single Cell Genomics Core performed basic computational analysis using Cell Ranger pipeline and provided us the filtered matrix of read counts per gene in each cell.
Filtering, normalization, clustering and UMAP generation
Seurat v4.0.3 package (Hao et al., 2021) for R was used to analyze the scRNA-seq data. Following Seurat-guided clustering tutorial (Source: vignettes/pbmc3k_ tutorial.Rmd), cells were filtered using <15% of mitochondrial genes reads to eliminate dead cells. We distinguish unique hashtags from each cell by excluding the cells that have > 250 read count in both hashtags. We excluded cells outlier to the 200–25000 read count (nCount RNA) to eliminate doublets and debris. Having filtered our data, we normalized, scaled and log-transformed the matrix (NormalizeData function) and then we centered the mean to 0 and the variance to 1 (ScaleData function) without regressing out variables.
Next, we performed a linear dimensional reduction by PCA using the top 2,000 most variable genes (FindVariableFeatures using the vst method and a span of 0.3). Based on an elbow plot (function ElbowPlot) we selected 30 PCs to calculate the shared nearest neighbor graph based on the calculated Euclidean distance (FindNeighbors function) and clustered our data using the Louvain algorithm (Blondel et al., 2008) with a resolution of 0.5, with the Seurat function FindClusters (McDavid et al., 2013, 2017; Trapnell et al., 2014; Love et al., 2014). Clustered data was displayed using the UMAP dimensional reduction (RunUMAP function, McInnes et al., 2020). Clusters were identified using well known markers for immune populations and fibroblasts as described in the Figures 4C, S4A, S4E, S4H, S5A, and S5B.
Cells from the T cell cluster were subjected to another round of unsupervised clustering as described above, except 10 PCs were used, to find further differences among QCC and non-QCC areas.
Differential expression analysis
Differential expression analysis was performed using the Seurat function FindMarkers. Genes were pre-filtered for a minimum log fold change of 0.25 and detectable expression in at least 10% of cells in the cluster, then compared for differential expression using DESeq2 (Love et al., 2014). Genes with an FDR-adjusted p-value less than 0.1 were selected as input for gene-set enrichment analysis using GO biological processes as implemented in the clusterProfiler package from Bioconductor (Wu et al., 2021a), using a set size between 3 and 800 genes and an FDR-adjusted p-value cutoff of 0.05. Relevant genes were curated from the GSEA to generate heatmaps. Average gene expression for each tumor and area was generated using AverageExpression.
Heatmaps from Figures S4E and S5A were generated with the individual cell values and the top 10 differentially expressed genes per cluster. Heatmaps with specific genes selected from the differential analysis between QCC and non-QCC areas and selected signatures among the same cluster were generated using the average value of cells coming from the same tumor and area with the function AverageExpression.
Cell abundance comparisons
We performed a Fisher’s exact test for each cell type between the two different groups followed by Benjamini-Hochberg (BH) correction to obtain adjusted p value.
Co-expression gene network construction and analysis in CD8 T cells
To understand differences in the regulation of CD8+ T cells, we generated a co-expression gene network. Because of the zero-inflated and highly noisy nature of single-cell transcriptome data, we inferred co-expression of gene pairs using BigSCale2 (Iacono et al., 2019), which pools cells with similar expression profile. We considered the set of genes from the co-expression network that were direct neighbors of the 124 genes that had been identified as downregulated in the DEG analysis, and we refer to this as the DEG module. The DEG module was visualized by cytoscape (ver 3.7.1) (Shannon et al., 2003). We obtained the enriched pathways for the DEG module by GO enrichment analysis (Mi et al., 2019) implemented in Gene Ontology website (http://geneontology.org/).
Hypoxic score
Hypoxic signature scores were calculated using the Seurat function AddModuleScore that calculates the average expression of a given signature per cell and subtracts it from the average expression of randomly selected control features. AddModuleScore (Tirosh et al., 2016) was run using 100 control features. Scores were averaged within all cells from the same cluster and tumor region (p27K+ or p27K-areas). To compare the hypoxic signature across populations we generated a delta score subtracting the average score of p27K-areas to that of p27K+ areas for each cluster of cells. Hypoxic signature was formed by: Hif1a, Slc2a1, Vegfa, Hmox1, Bnip3, Nos2, Mmp2, Sod3, Cited2, Ldha. This list is constituted by genes from the GO:0001666 “Response to hypoxia” that were upregulated in mVenus-p27K+ QCCs. Statistical analysis was performed by two-sided Wilcoxon rank sum test followed by BH correction.
Statistical analysis
All analysis was performed using data from at least three independent biological replicates (exact number of replicates are stated in the figure legend). Unless stated otherwise, all statistical analyses were performed in PRISM6 software using paired or unpaired Student’s t test.
Supplementary Material
Highlights.
Antigen-expressing tumor cells that escape from T cell attack are quiescent
Quiescent cancer cells (QCCs) form clusters that exclude immune infiltrates
PADME-seq reveals an increase in terminally exhausted T cells in the QCC niche
HIF1a activation in cancer cells augments T cell exhaustion and reduces T cell killing
ACKNOWLEDGMENTS
We thank G. Freeman, K. Polyak, S.K. Dougan (DFCI), M. Haigis (HMS), C. Kim (BCH), and S. McAllister (BWH) for helpful discussions and review of the manuscript. We thank R. Weinberg (MIT) for providing 4T07 and D2A1 cells. We thank P.L. Sulkowski for helping us with hypoxic chamber experiments. We thank the Micron Microscopy Core (HMS) and the Molecular Imaging Core (DFCI) for image acquisition and the Brigham and Women’s Hospital Single Cell Core for generation of the scRNA-seq data. P.B. was supported by La Caixa pre-doctoral fellowship (LCF/BQ/AA18/11680040) funded by “La Caixa” Foundation (ID 100010434). S.A.S. acknowledges support from the NCI (R50RCA211482). S.M. is funded by the T32 NIH grant (T32CA207021). A.W. is funded by the Claudia Adams Barr program for Innovative Cancer Research. J.-W.C. and M.H. were supported by the Helmsley foundation (126320) and startup funds from the Evergrande Center. J.A. was supported by the Mary Kay Foundation, the Susan G. Komen Career Catalyst award, the Smith Family Awards Program for Excellence in Biomedical Research, the Ludwig Center at Harvard, the Ira Schneider Foundation, and the DF/HCC Breast SPORE: grant 1P50CA168504. Schemes were created with BioRender.com
S.A.S.: non-financial support (Bristol-Myers Squibb), equity (Agenus Inc., Agios Pharmaceuticals, Breakbio Corp., Bristol-Myers Squibb, and Lumos Pharma). E.A.M.: SAB compensation (Merck, Genentech, and Genomic Health), uncompensated steering committees (Bristol-Myers Squibb, Lilly, and Genentech). K.W.W.: scientific advisory board (TCR2 Therapeutics, T-Scan Therapeutics, SQZ Biotech, and Nextechinvest), sponsored research funding (Novartis), co-founder (Immunitas Therapeutics). S.M.T.: institutional research funding (AstraZeneca, Lilly, Merck, Nektar, Novartis, Pfizer, Genentech/Roche, Immunomedics, Exelixis, Bristol-Myers Squibb, Eisai, Nanostring, Cyclacel, Odonate, and Seattle Genetics); adviser/consultant (AstraZeneca, Lilly, Merck, Nektar, Novartis, Pfizer, Genentech/Roche, Immunomedics, Bristol-Myers Squibb, Eisai, Nanostring, Puma, Sanofi, Celldex, Paxman, Puma, Silverback Therapeutics, G1 Therapeutics, Gilead, AbbVie, Anthenex, OncoPep, Outcomes4Me, Kyowa Kirin Pharmaceuticals, Daiichi-Sankyo, Ellipsis, Infinity, and Samsung Bioepsis Inc). R.B.-S.: consulting (AstraZeneca, Elli Lilly, Libbs, Merck, Roche, and Zodiac), non-CME services received directly from commercial interest or their Agents (Bard Access, BMS, Eli Lilly, Libbs, Merck, Novartis, and Pfizer, Roche), contracted research funding (Roche and BMS), travel/accommodation/expenses (Eli Lilly, Roche, Daichi Sankyo, and Merck). M.H.: co-founder and adviser (Neomer Diagnostics).
Footnotes
DECLARATION OF INTERESTS
P.B., A.B.-M., Y.H., A.W., J.-W.C., G.E., I.A., S.M., J.S., X.Z., X.S., Z.K., A.R., E.T.R., R.P., and J.A. have no conflicts of interest to report.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science.
One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2022.03.033.
REFERENCES
- Agudo J, Park ES, Rose SA, Alibo E, Sweeney R, Dhainaut M, Kobayashi KS, Sachidanandam R, Baccarini A, Merad M, and Brown BD (2018). Quiescent tissue stem cells evade immune surveillance. Immunity 48, 271–285.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agudo J, Ruzo A, Park ES, Sweeney R, Kana V, Wu M, Zhao Y, Egli D, Merad M, and Brown BD (2015). GFP-specific CD8 T cells enable targeted cell depletion and visualization of T-cell interactions. Nat. Biotechnol. 33, 1287–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agudo J, Ruzo A, Tung N, Salmon H, Leboeuf M, Hashimoto D, Becker C, Garrett-Sinha L-A, Baccarini A, Merad M, and Brown BD (2014). The miR-126–VEGFR2 axis controls the innate response to pathogen-associated nucleic acids. Nat. Immunol. 15, 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, Casella V, Ngiow SF, Khan O, Huang YJ, et al. (2020). Developmental relationships of four exhausted CD8+ T cell subsets reveals underlying transcriptional and epigenetic landscape control mechanisms. Immunity 52, 825–841.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, et al. (2018). Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 24, 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondel VD, Guillaume JL, Lambiotte R, and Lefebvre E (2008). Fast unfolding of communities in large networks. arXiv. 10.48550/arXiv.0803.0476. [DOI] [Google Scholar]
- Böcker R, Estler CJ, Maywald M, and Weber D (1981). Comparison of distribution of doxycycline in mice after oral and intravenous application measured by a high-performance liquid chromatographic method. Arzneimittelforschung. 31, 2116–2117. [PubMed] [Google Scholar]
- Brown BD, Venneri MA, Zingale A, Sergi LS, and Naldini L (2006). Endogenous microRNA regulation suppresses transgene expression in hematopoietic lineages and enables stable gene transfer. Nat. Med. 12, 585–591. [DOI] [PubMed] [Google Scholar]
- Correia AL, Guimaraes JC, Auf der Maur P, De Silva D, Trefny MP, Okamoto R, Bruno S, Schmidt A, Mertz K, Volkmann K, et al. (2021). Hepatic stellate cells suppress NK cell-sustained breast cancer dormancy. Nature 594, 566–571. [DOI] [PubMed] [Google Scholar]
- Cortes J, Cescon DW, Rugo HS, Nowecki Z, Im SA, Yusof MM, Gallardo C, Lipatov O, Barrios CH, Holgado E, et al. (2020). Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): a randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 396, 1817–1828. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, Johnson RS, and Goldrath AW (2013). Hypoxia-inducible factors enhance the effector responses of CD8+ T cells to persistent antigen. Nat. Immunol. 14, 1173–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez CX, Müller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, Breart B, Foreman O, Bainbridge TW, Castiglioni A, et al. (2020). Single-cell RNA sequencing reveals stromal evolution into LRRC15+ myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discov. 10, 232–253. [DOI] [PubMed] [Google Scholar]
- Emens LA, Goldstein LD, Schmid P, Rugo HS, Adams S, Barrios CH, Schneeweiss A, Dieras V, Iwata H, Chang C-W, et al. (2021). The tumor microenvironment (TME) and atezolizumab + nab-paclitaxel (A+nP) activity in metastatic triple-negative breast cancer (mTNBC): IMpassion130. J. Clin. Oncol. 39, 1006. [Google Scholar]
- Fluegen G, Avivar-Valderas A, Wang Y, Padgen MR, Williams JK, Nobre AR, Calvo V, Cheung JF, Bravo-Cordero JJ, Entenberg D, et al. (2017). Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat. Cell Biol. 19, 120–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garris CS, Arlauckas SP, Kohler RH, Trefny MP, Garren S, Piot C, Engblom C, Pfirschke C, Siwicki M, Gungabeesoon J, et al. (2018). Successful anti-PD-1 cancer immunotherapy requires T cell-dendritic cell cross-talk involving the cytokines IFN-γ and IL-12. Immunity 49, 1148–1161.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghajar CM (2015). Metastasis prevention by targeting the dormant niche. Nat. Publ. Gr. 15, 238–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinohara K, Wu HJ, Vigneau S, McDonald TO, Igarashi KJ, Yamamoto KN, Madsen T, Fassl A, Egri SB, Papanastasiou M, et al. (2018). KDM5 histone demethylase activity links cellular transcriptomic heterogeneity to therapeutic resistance. Cancer Cell 34, 939–953.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu CJ, Sataur A, Wang L, Chen H, and Simon MC (2007). The N-terminal transactivation domain confers target gene specificity of hypoxia-inducible factors HIF-1α and HIF-2α. Mol. Biol. Cell 18, 4528–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C-J, Wang L-Y, Chodosh LA, Keith B, and Simon MC (2003). Differential roles of hypoxia-inducible factor 1μ (HIF-1μ) and HIF-2μ in hypoxic gene regulation. Mol. Cell. Biol. 23, 9361–9374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacono G, Massoni-Badosa R, and Heyn H (2019). Single-cell transcriptomics unveils gene regulatory network plasticity. Genome Biol. 20, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaprakash P, Ai M, Liu A, Budhani P, Bartkowiak T, Sheng J, Ager CR, Nicholas C, Jaiswal AR, Sun Y, et al. (2018). Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J. Clin. Invest. 128, 5137–5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keir ME, Freeman GJ, and Sharpe AH (2007). PD-1 regulates self-reactive CD8 + T cell responses to antigen in lymph nodes and tissues. J. Immunol. 179, 5064–5070. [DOI] [PubMed] [Google Scholar]
- Lee K-M, Giltnane JM, Balko JM, Schwarz LJ, Guerrero-Zotano AL, Hutchinson KE, Nixon MJ, Estrada MV, Sánchez V, Sanders ME, et al. (2017). MYC and MCL1 cooperatively promote chemotherapy-resistant breast cancer stem cells via regulation of mitochondrial oxidative phosphorylation. Cell Metab. 26, 633–647.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Chen I, Shimoda LA, Park Y, Zhang C, Tran L, Zhang H, and Semenza GL (2017). Chemotherapy-induced Ca2+ release stimulates breast cancer stem cell enrichment. Cell Rep. 18, 1946–1957. [DOI] [PubMed] [Google Scholar]
- Lucchetti J, Fracasso C, Balducci C, Passoni A, Forloni G, Salmona M, and Gobbi M (2019). Plasma and brain concentrations of doxycycline after single and repeated doses in wild-type and APP23 mice. J. Pharmacol. Exp. Ther. 368, 32–40. [DOI] [PubMed] [Google Scholar]
- Maier B, Leader AM, Chen ST, Tung N, Chang C, LeBerichel J, Chudnovskiy A, Maskey S, Walker L, Finnigan JP, et al. (2020). A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 580, 257–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDavid A, Finak G, Chattopadyay PK, Dominguez M, Lamoreaux L, Ma SS, Roederer M, and Gottardo R (2013). Data exploration, quality control and testing in single-cell qPCR-based gene expression experiments. Bioinformatics 29, 461–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDavid A, Finak G, and Yajima M (2017). MAST: model-based analysis of single cell transcriptomics. R package version 1.2.1. https://github.com/RGLab/MAST/. [Google Scholar]
- McInnes L, Healy J, and Melville J (2020). UMAP: uniform manifold aproximation and projection of dimension reduction. arXiv. 10.48550/arXiv.1802.03426. [DOI] [Google Scholar]
- Medaglia C, Giladi A, Stoler-Barak L, De Giovanni M, Salame TM, Biram A, David E, Li H, Iannacone M, Shulman Z, and Amit I (2017). Spatial reconstruction of immune niches by combining photoactivatable reporters and scRNA-seq. Science 358, 1622–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Ebert D, Huang X, and Thomas PD (2019). PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47, D419–D426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, et al. (2019). Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 20, 326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JC, Brown BD, Shay T, Gautier EL, Jojic V, Cohain A, Pandey G, Leboeuf M, Elpek KG, Helft J, et al. (2012). Deciphering the transcriptional network of the dendritic cell lineage. Nat. Immunol. 13, 888–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero Llopis P, Senft RA, Ross-Elliott TJ, Stephansky R, Keeley DP, Koshar P, Marqués G, Gao YS, Carlson BR, Pengo T, et al. (2021). Best practices and tools for reporting reproducible fluorescence microscopy methods. Nat. Methods 18, 1463–1476. [DOI] [PubMed] [Google Scholar]
- Oki T, Nishimura K, Kitaura J, Togami K, Maehara A, Izawa K, Sakaue-Sawano A, Niida A, Miyano S, Aburatani H, et al. (2014). A novel cell-cycle-indicator, mVenus-p27K -, identifies quiescent cells and visualizes G0-G1 transition. Sci. Rep. 4, 4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskarsson T, Batlle E, and Massagué J (2014). Metastatic stem cells: sources, niches, and vital pathways. Cell Stem Cell 14, 306–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazon A, Tyrakis PA, Macias D, Veliç a P, Rundqvist H, Fitzpatrick S, Vojnovic N, Phan AT, Loman N, Hedenfalk I, et al. (2017). An HIF-1α/VEGF-A axis in cytotoxic T cells regulates tumor progression. Cancer Cell 32, 669–683.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D, Kobayashi A, Jiang P, De Andrade LF, Tay RE, Luoma AM, Tsoucas D, Qiu X, Lim K, Rao P, et al. (2018). A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359, 770–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicchi E, Gemignani F, Krstic-Demonacos M, Dedhar S, Mutti L, and Landi S (2016). Targeting hypoxic response for cancer therapy. Oncotarget 7, 13464–13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual G, Avgustinova A, Mejetta S, Martíń M, Castellanos A, Attolini CSO, Berenguer A, Prats N, Toll A, Hueto JA, et al. (2017). Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 541, 41–45. [DOI] [PubMed] [Google Scholar]
- Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, Gartner JJ, Jia L, Steinberg SM, Yamamoto TN, et al. (2017). Identification of essential genes for cancer immunotherapy. Nature 548, 537–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier A, Anaparthy N, Memos N, Kelley ZL, Gouronnec A, Yan R, Auffray C, Albrengues J, Egeblad M, Iacobuzio-Donahue CA, et al. (2018). Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science 360, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prager BC, Xie Q, Bao S, and Rich JN (2019). Cancer stem cells: the architects of the tumor ecosystem. Cell Stem Cell 24, 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, and Anderson AC (2010). Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 207, 2187–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharping NE, Menk AV, Whetstone RD, Zeng X, and Delgoffe GM (2017). Efficacy of PD-1 blockade is potentiated by metformin-induced reduction of tumor hypoxia. Cancer Immunol. Res. 5, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Diéras V, Hegg R, Im SA, Shaw Wright G, et al. (2018). Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N. Engl. J. Med. 379, 2108–2121. [DOI] [PubMed] [Google Scholar]
- Semenza GL (2017). Hypoxia-inducible factors: coupling glucose metabolism and redox regulation with induction of the breast cancer stem cell phenotype. EMBO J. 36, 252–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer M, Wang C, Cong L, Marjanovic ND, Kowalczyk MS, Zhang H, Nyman J, Sakuishi K, Kurtulus S, Gennert D, et al. (2016). A distinct gene module for dysfunction uncoupled from activation in tumor-infiltrating T cells. Cell 166, 1500–1511.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa MS, Bragado P, and Aguirre-Ghiso JA (2014). Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat. Rev. Cancer 14, 611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomura M, Yoshida N, Tanaka J, Karasawa S, Miwa Y, Miyawaki A, and Kanagawa O (2008). Monitoring cellular movement in vivo with photo-convertible fluorescence protein “Kaede” transgenic mice. Proc. Natl. Acad. Sci. USA 105, 10871–10876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellenstein MD, and Visser KE de (2018). Cancer-cell-intrinsic mechanisms shaping the tumor immune landscape. Immunity 48, 399–416. [DOI] [PubMed] [Google Scholar]
- Wolf Y, Bartok O, Patkar S, Eli GB, Cohen S, Litchfield K, Levy R, Jiménez-Sánchez A, Trabish S, Lee JS, et al. (2019). UVB-induced tumor heterogeneity diminishes immune response in melanoma. Cell 179, 219–235.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, et al. (2021a). clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (N Y) 2, 100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R, Li K, Yuan M, and Luo KQ (2021b). Nerve growth factor receptor increases the tumor growth and metastatic potential of triple-negative breast cancer cells. Oncogene 40, 2165–2181. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bulk RNAseq and scRNAseq have been deposited at GEO and are publicly available as of the date of publication. Accesion number is listed in the key resources table. Original western blot and microscopy images will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
| aCD11c PE | BioLegend | RRID: AB_313776 Clone:N418 |
| aCD11c PE/Cy7 | BioLegend | RRID: AB_493569 Clone:N418 |
| aCD3 AF647 | BioLegend | RRID: AB_389323 Clone:17A2 |
| aCD3e biotin | BioLegend | RRID: AB_312668 Clone:145–2C11 |
| aCD4 AF647 | BioLegend | RRID: AB_493372 Clone:RM4–5 |
| aCD40 PE/cy7 | BioLegend | RRID: AB_10933422 Clone:3/23 |
| aCD45 AF647 | BioLegend | RRID: AB_493534 Clone:30-F11 |
| aCD45 AF700 | BioLegend | RRID: AB_493714 Clone:30-F11 |
| aCD45.1 AF647 | BioLegend | RRID: AB_492864 Clone:A20 |
| aCD64 APC | BioLegend | RRID: AB_11219205 Clone:X54–5/7.1 |
| aCD8 AF594 | BioLegend | RRID: AB_2563237 Clone:53–6.7 |
| aCD8 Pe/Cy7 | BioLegend | RRID: AB_312760 Clone:53–6.7 |
| aCD86 PE | BioLegend | RRID: AB_313150 Clone: GL-1 |
| aGr1 APC | BioLegend | RRID: AB_313376 Clone:RB6–8C5 |
| aH2Kd AF647 | BioLegend | RRID: AB_493063 Clone:AF6–88.5 |
| aH2Kd APC | BioLegend | RRID: AB_10640118 Clone:SF1–1.1 |
| aHIF-1α XP® Rabbit mAb | Cell signaling | RRID: AB_2799095 Clone:D1S7W |
| aHuman CD3 | Agilent | RRID: AB_2631163 Clone:F7.2.38 |
| aHuman E-Cadherin | CST | RRID: AB_2291471 Clone:24E10 |
| aHuman Ki67 | Biocare | RRID: AB_2721189 Clone:SP6 |
| aHuman p27K | BD | RRID: AB_397636 Clone:57/Kip1/p27 |
| aIFNAR1 biotin | BioLegend | RRID: AB_1134250 Clone:MAR1–5A3 |
| aKi67 mAb | Cell Signaling | RRID: AB_2620142 Clone:D3B5 |
| aMHCII APC | BioLegend | RRID: AB_313328 Clone:M5/114.15.2 |
| aMHCII PB | BioLegend | RRID: AB_493528 Clone:M5/114.15.2 |
| aPD1 PerCP/Cy5.5 | BioLegend | RRID: AB_10550092 Clone:29F.1A12 |
| aTCRb biotin | BioLegend | RRID: AB_313426 Clone: H57–597 |
| aTIM3 APC | BioLegend | RRID: AB_2562997 Clone:B8.2C12 |
| Donkey anti-rabbit IgG AF647 | BioLegend | RRID: AB_2563202 Clone:Poly4064 |
| Goat anti-mouse AF555 | Invitrogen | RRID: AB_2535849 |
| IRDye 800CW Goat anti-Rabbit | LI-COR | RRID: AB_2651127 |
| Rat IgG1, κ Isotype Ctrl | BioLegend | RRID: AB_11150772 Clone: RTK2071 |
| Rat IgG2a, κ Isotype Ctrl | BioLegend | RRID: AB_11147167 Clone: RTK2758 |
| Rat IgG2b, κ Isotype Ctrl | BioLegend | RRID: AB_11149687 Clone: RTK4530 |
| Mouse IgG1, κ Isotype Ctrl | BioLegend | RRID: AB_11148942 Clone: MG1–45 |
| American Hamster IgG Isotype Ctrl | BioLegend | RRID: AB_11203529 Clone: HTK888 |
| Rabbit IgG Isotype Ctrl | Thermofisher scientific | RRID: AB_243593 |
| Anti-mouse CD16/32 | BioLegend | RRID: AB_2561482 Clone: 93 |
| CD45-BX007—Alexa Fluor™ 750-RX007 | Akoya Biosciences | Cat#4450002, Clone: 30-F11 |
| MHCII-BX014-Atto 550-RX014 | Akoya Biosciences | Cat#4250003, Clone: M5/114.15.2 |
| CD3-BX021—Cy5-RX021 | Akoya Biosciences | Cat#4350014, Clone: 17A2 |
| CD11c-BX030-Cy5-RX030 | Akoya Biosciences | Cat#4350013, Clone: N418 |
| Anti-p27 KIP 1 | Abcam | RRID: AB_2811037, Clone: Y236 |
| Anti-GFP | Abcam | RRID: AB_305643 |
| Anti-mouse CD90.1 (Thy-1.1) | BioLegend | RRID: AB_314013 Clone: OX-7 |
| Anti-mouse F4/80 | BD Biosciences | RRID: AB_2739222 Clone: T45–2342 |
| Anti-mouse CD366 (Tim-3) | Biolegend | RRID: AB_1626128 Clone: B8.2C12 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| One Shot™ Stbl3™ Chemically Competent E. coli | Thermofisher Scientific | cat#C737303 |
|
| ||
| Biological samples | ||
|
| ||
| anonymized TNBC patients | Brigham and Women's Hospital Pathology Core | protocols 11–104 and 93–085 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Iscove's Modified Dulbecco's Medium (IMDM) | Thermofisher Scientific | Cat#31980030 |
| USDA FBS | Life Technologies | Cat#10437028 |
| Penicillin-Streptomycin | Life technologies | Cat#15140122 |
| Glutamax | Life Technologies | Cat#35050061 |
| Polybrene Transfection Reagent | Thermofisher Scientific | Cat#TR1003G |
| T4 Ligase | NEB | Cat#M0202L |
| BamHI-HF | NEB | Cat## R3136L |
| NheI-HF | NEB | Cat#R3131S |
| CutSmart® Buffer | NEB | Cat#B7204 |
| AgeI-HF | NEB | Cat#R3552S |
| SalI-HF | NEB | Cat#R3138S |
| BsaBI | NEB | Cat#R0537S |
| Q5® High-Fidelity DNA Polymerase | NEB | Cat#M0491S |
| BclI-HF | NEB | Cat#R3160S |
| NotI-HF | NEB | Cat#R3189S |
| Calcium Chloride, Anhydrous (Pellets, 4–20 mesh, for Desiccators), Fisher Chemical™ | Fisher Scientific | Cat#C614–500 |
| UltraPure 0.5 M EDTA, pH 8.0 | Life Technologies | Cat#15575020 |
| Alt-R® S.p. Cas9 Nuclease V3 | IDT | Cat#1081059 |
| Hyaluronidase | StemCell | Cat#07461 |
| Collagenase IV | StemCell | Cat#17104019 |
| DNaseI | Sigma Aldrich | Cat#10104159001 |
| RBC lysis buffer | Biolegend | Cat#420301 |
| BSA | Cell Signaling Technology | Cat#9998S |
| Dapi | Biolegend | Cat#422801 |
| 10X CODEX Buffer | Akoya Biosciences | Cat#7000001 |
| CODEX Assay Reagent | Akoya Biosciences | Cat#7000002 |
| Nuclear Stain | Akoya Biosciences | Cat#7000003 |
| CODEX Storage Buffer | Akoya Biosciences | Cat#232107 |
| BX002-Atto 550-RX002 | Akoya Biosciences | Cat#5450023 |
| BX006-Cy5-RX006 | Akoya Biosciences | Cat#5450027 |
| BX017-Atto 550-RX017 | Akoya Biosciences | Cat#5250001 |
| BX027-Cy5-RX027 | Akoya Biosciences | Cat#5350004 |
| BX042-Cy5-RX042 | Akoya Biosciences | Cat#5350008 |
| 7-AAD | Biolegend | Cat#420404 |
| Trizol LS | Life Technologies | Cat#10296010 |
| Trizol | Life Technologies | Cat#15596018 |
| D(+)-SUCROSE | Fisher Scientific | Cat#AC177140010 |
| PARAFORMALDEHYDE 32% SOL. EM GRADE | VWR | Cat#100496–496 |
| 16% Paraformaldehyde Aqueous Solution, EM Grade | Fisher Scientific | Cat#50–980-487 |
| FB OCT COMPOUND CLEAR 4OZ | Fisher Scientific | Cat#23730571 |
| Normal Mouse Serum | Jackson ImmunoResearch Laboratories | Cat#015–000-120 |
| Avidin/Biotin Blocking System | BioLegend | Cat#927301 |
| ProLong Diamond Antifade Mountant | Life Technologies | Cat#P36961 |
| Doxycycline hyclate | Biogems | Cat#2431450 |
| Poly-L-lysine solution | Sigma Aldrich | Cat# P8920–500ML |
| 2-NBDG | VWR | Cat#76021–666 |
| Recombinant Murine Flt3-Ligand | VWR | Cat#10780–338 |
| Lipopolysaccharides from Escherichia coli O111:B4 γ-irradiated |
Sigma Aldrich | Cat#L4391–1MG |
| Poly I:C | Invitrogen | Cat#tlrl-pic |
| Pierce RIPA Buffer | VWR | Cat#PI89900 |
| NuPAGE Sample Reducing Agent | Life Technologies | Cat#NP0009 |
| Novex 4–20% Tris-Glycine gels | Life Technologies | Cat#P04205BOX |
| Nitrocellulose Sandwiches | Cell Signaling Technology | Cat#12369P2 |
|
| ||
| Critical commercial assays | ||
|
| ||
| QIAprep Spin Miniprep Kit | Qiagen | Cat#27106 |
| EndoFree Plasmid Maxi Kit | Qiagen | Cat#12362 |
| miRNeasy Micro Kit | Qiagen | Cat#217084 |
| SF Cell Line 4D-NucleofectorTM X Kit S | Lonza | Cat#V4XC-2032 |
| EasySep™ Mouse CD8+ T Cell Isolation Kit | StemCell | Cat#19853 |
| QIAquick PCR Purification Kit | Qiagen | Cat#28104 |
| QIAquick Gel Extraction Kit | Qiagen | Cat#28706 |
| Hypoxyprobe-Biotin Kit | Hypoxyrprobe | Cat#HP10–1000kit |
| EdU-Click 647 | BaseClick | Cat#BCK-EdU647 |
| CODEX Staining Kit | Akoya Biosciences | Cat#7000008 |
| CODEX Conjugation Kit | Akoya Biosciences | Cat#7000009 |
|
| ||
| Deposited data | ||
|
| ||
| Sequencing datasets | This paper | Available at NCBI accession # GSE198715 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| 4T07 | Dr. Robert Weinberg, MIT | N/A |
| D2A1 | Dr. Robert Weinberg, MIT | N/A |
| EMT6 | ATCC | Cat#CRL-2755™ |
| Hek293:Lenti-X™ 293T Cell Line | Takara | Cat#632180 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: Balb/c :BALB/cJ | The Jackson Laboratory | Strain #:000651 |
| Mouse: NSG: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | The Jackson Laboratory | Strain #:005557 |
| Mouse: Jedi: Ptprca TcrbLn1Bdb TcraLn1Bdb H2d/J | Dr. Brian Brown laboratory, Mount Sinai | N/A |
| Mouse:PD1-/-:B6.Cg-Pdcd1tm1.1Shr/J | The Jackson Laboratory | Strain #:028276 |
| Mouse:B6:C57BL/6J | The Jackson Laboratory | Strain #:000664 |
| Mouse:Kaede:Kaede | Dr. Osami Kanagawa, University of Lyon | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Alt-R® CRISPR-Cas9 tracrRNA | IDT | Cat#1072533 |
| Hifla gRNA: AGTGCACCCTAACAAGCCGG | IDT | Alt-R® CRISPR-Cas9 crRNA |
| Ifnarl gRNA: GCTCGCTGTCGTGGGCGCGG | IDT | Alt-R® CRISPR-Cas9 crRNA |
|
| ||
| Recombinant DNA | ||
|
| ||
| 3rd Generation Lentiviral packaging vectors | Dr. Brian Brown, Mount Sinai | N/A |
| PGK:eGFP Lentiviral vector | Dr. Brian Brown, Mount Sinai | N/A |
| mVenus:p27k reporter | Dr. Toshio Kitamura, University of Tokyo | N/A |
| PGK:mCherry Lentiviral vector | Dr. Brian Brown, Mount Sinai | N/A |
| H2B-mCherry | Addgene | Cat#51007 |
| H2B-tdTomato | Addgene | Cat#58101 |
| rtTA | Addgene | Cat#104543 |
| TetON lentiviral expression vector | Addgene | Cat#131687 |
| Hifla STBL: mHif-1α MYC (P402A/P577A/N813A) | Addgene | Cat#44028 |
|
| ||
| Software and algorithms | ||
|
| ||
| FlowJo 10 | BD | https://www.flowjo.com |
| Prism 9 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| FiJi® (v2.0.0-rc-69/1.52i) | ImageJ | https://imagej.net/software/fiji/ |
| Seurat | Satija Lab | https://satijalab.org/seurat/articles/pbmc3k_tutorial.html |
| DESeq2 | Bioconductor | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Akoya CODEX Processor® | Akoya | https://www.akoyabio.com/phenocycler/software/ |
| clusterProfiler | Bioconductor | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| bigSCale2 | Iacono et al. (2019) | https://github.com/iaconogi/bigSCale2 |
| Benchling | Benchling | ttps://www.benchling.com |
| Biorender | Biorender | https://biorender.com/ |
|
| ||
| Other | ||
|
| ||
| Compresstome® | Precisionary | VF-310–0Z |