

Abstract
Epigenetic ‘clocks’ based on DNA methylation have emerged as the most robust and widely used aging biomarkers, but conventional methods for applying them are expensive and laborious. Here we develop tagmentation-based indexing for methylation sequencing (TIME-seq), a highly multiplexed and scalable method for low-cost epigenetic clocks. Using TIME-seq, we applied multi-tissue and tissue-specific epigenetic clocks in over 1,800 mouse DNA samples from eight tissue and cell types. We show that TIME-seq clocks are accurate and robust, enriched for polycomb repressive complex 2-regulated loci, and benchmark favorably against conventional methods despite being up to 100-fold less expensive. Using dietary treatments and gene therapy, we find that TIME-seq clocks reflect diverse interventions in multiple tissues. Finally, we develop an economical human blood clock (R > 0.96, median error = 3.39 years) in 1,056 demographically representative individuals. These methods will enable more efficient epigenetic clock measurement in larger-scale human and animal studies.
Aging is difficult to study, in part because it is difficult to quantify1. In recent years, researchers attempted to address this problem with aging ‘clocks’, which are machine learning-derived biomarkers trained to predict age or age proxies2. Because clock predictions are not perfect, individuals are often predicted younger or older than their chronological age, and this difference is hypothesized to reflect variation in the biological rate of aging3. Both physiological measurements4,5 and biomolecules3,6,7 have been used to build aging clocks and they are becoming increasingly common readouts to assess longevity interventions. Despite their promise, accurate clocks that are inexpensive and easily applied to large studies are lacking.
The most robust and widely used aging clocks are based on DNA cytosine methylation and are interchangeably referred to as DNA methylation clocks or epigenetic clocks. These clocks include sets of CpGs and corresponding algorithms that use methylation levels to predict age. Epigenetic clocks have been built for humans8–10, mice11–13 and many other mammals14–16; they have been shown to reflect interventions that are associated with longevity17, accelerated aging18 and even cellular rejuvenation19,20. While considerable effort has been devoted to developing more accurate clocks or clocks adjusted according to health outcomes10,21, very little work has been done to make epigenetic clocks more efficient and affordable.
Ideally, epigenetic clocks could be measured for low cost with a scalable technology. However, clocks are predominantly built and assayed using Illumina BeadChip22 microarrays or reduced representation bisulfite sequencing23 (RRBS), which are laborious and cost hundreds of dollars per sample. While these methods are useful for biomarker discovery because they measure hundreds of thousands to millions of CpGs, they are excessive for the accurate measurement of epigenetic clocks that typically only require several hundred loci to be measured. Highly targeted approaches can predict chronological age accurately24–26, such as pyrosequencing or digital PCR. However, these methods are still not optimally cost-effective or scalable and their reliance on low-CpG clocks (for example, 1–15 CpGs) limits the number of aging modules they reflect, making them less relevant to diverse intervention studies. Recently, SyBS, a method that leverages commercial hybridization enrichment library preparation to capture loci syntenic with human methylation BeadChip probes, has been used to construct a dog-specific epigenetic clock27. This method was robust and capable of high-accuracy age predictions from hundreds of loci, but it requires substantial parallel sample processing, expensive commercial enrichment baits and low-plex hybridization reactions28, limiting its scalability and cost-effectiveness. A more ideal approach for economical epigenetic clocks would measure hundreds to thousands of the most highly age-correlated loci while minimizing parallel processing, labor and total cost per sample.
With this in mind, we developed tagmentation-based indexing for methylation sequencing (TIME-seq), an optimized bisulfite sequencing approach to enable low-cost and scalable epigenetic age predictions. To demonstrate its utility, we used TIME-seq to build seven epigenetic clocks for mice and one clock for humans; we applied these clocks to predict age in 2,892 unique samples from nine different tissue and cell types. We benchmarked TIME-seq against gold standard methods and validated our clocks in independent cohorts and interventions that alter the rate of aging. Our methods decrease the costs of epigenetic clock analysis by up to two orders of magnitude, promising to facilitate their use in more large-scale experiments.
Results
Designing TIME-seq for efficient epigenetic age predictions
When designing TIME-seq, we sought (1) scalability (that is, similar effort is required to prepare thousands or dozens of samples) and (2) economy, focusing on minimizing reagents and sequencing costs as well as labor. TIME-seq leverages barcoded and sodium bisulfite-resistant Tn5 transposase adaptors to rapidly index sample DNA for a pooled library preparation (Fig. 1a), which streamlines sample preparation and minimizes the cost of consumables (Supplementary Table 1). After tagmentation and pooling, methylated end repair (5-methyl-deoxycytidine triphosphate (dCTP) replaces dCTP) is performed and pools are prepared for in-solution hybridization enrichment using biotinylated RNA baits (Extended Data Fig. 1a–e). Unlike bisulfite-compatible single-cell indexing approaches29, we designed barcoded TIME-seq adapters to be short (38 nucleotides) for optimal enrichment efficiency because longer adapters are more likely to daisy chain with off-target DNA30 (Extended Data Fig. 1f–h). Baits were produced in-house from single-stranded oligonucleotide libraries (Supplementary Table 2), providing inexpensive enrichments from a regenerable source (Supplementary Information). This enrichment strategy allowed us to achieve deep coverage from thousands of diverse target loci with minimal sequencing costs. After bisulfite conversion of captured DNA and indexed PCR amplification of each pool, Illumina short-read sequencing was performed (Extended Data Fig. 2) and sample reads were demultiplexed based on pool and Tn5 adapter indexes. From mapped reads, a matrix of methylation values for CpGs in each sample was used to train or predict a DNA methylation biomarker.
Fig. 1 |. TIME-seq enables highly efficient epigenetic age predictions.
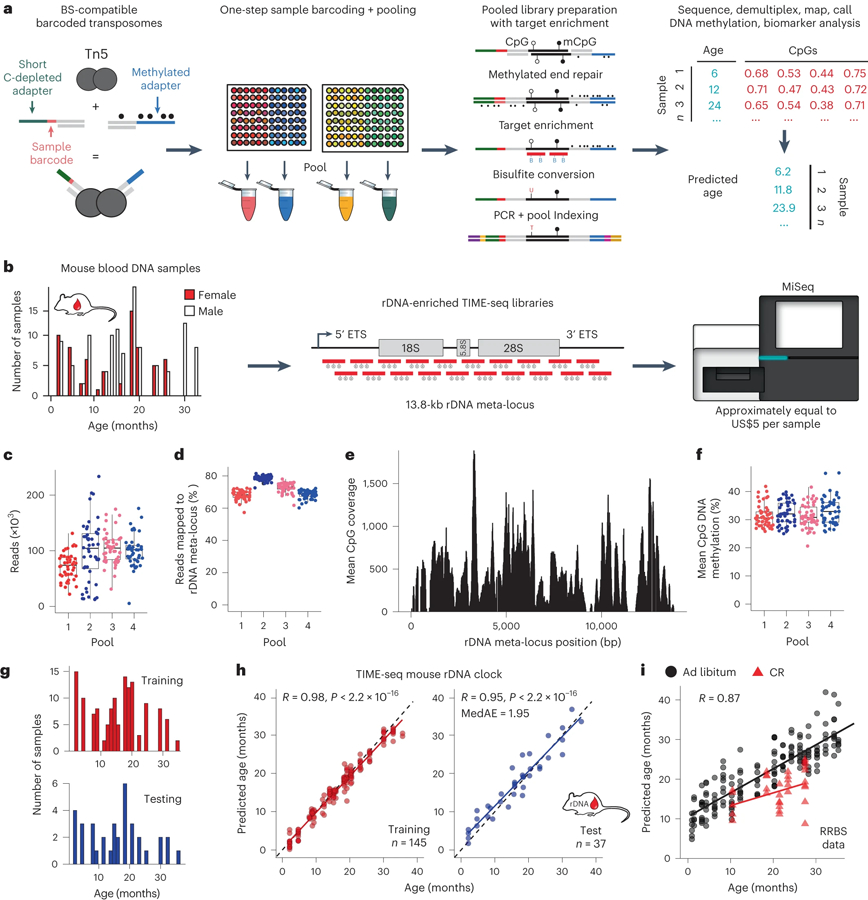
a, Schematic of the TIME-seq library preparation for highly multiplexed targeted methylation sequencing to build and measure DNA methylation-based biomarkers. b, Proof-of-concept rDNA clock experiment schematic; 191 mouse blood DNA samples (histogram) were prepared with TIME-seq enriched for rDNA and sequenced for clock training and testing. c, Reads were demultiplexed from each rDNA clock pool. d, Percentage of demultiplexed reads from each sample that mapped to the rDNA meta-locus. e, Mean coverage at the rDNA meta-locus CpGs in rDNA-enriched TIME-seq libraries. f, Mean CpG methylation from each sample in the four pools. In c,d,f, n = 48 for pools 1–3 and n = 47 for pool 4. The boxplot lengths represent the interquartile range (IQR) with the middle line representing median values and the whiskers 1.5 times the IQR. g, Histogram of training (n = 145; red) and testing (n = 37; blue) samples used to develop the TIME-seq mouse rDNA clock. h, TIME-seq mouse rDNA clock age predictions for the training (red) and testing (blue) datasets. Pearson correlation and MedAE on the test are shown in the top left corner. i, Predictions from a TIME-seq rDNA clock developed using only CpGs with at least 50 reads coverage in the RRBS data used to develop the original mouse rDNA clock. Calorie-restricted (CR) mice are represented as red triangles.
Building a proof-of-concept TIME-seq ribosomal DNA methylation clock
Ribosomal DNA (rDNA) is a highly repetitive locus that shows increased DNA methylation with age in mice, allowing for accurate epigenetic clocks to be constructed20,31. Therefore, we performed a small-scale pilot of TIME-seq in C57BL/6 mouse blood DNA samples using hybridization probes against the described rDNA clock CpGs31 (Extended Data Fig. 3a). Samples were efficiently demultiplexed from each TIME-seq pool; DNA methylation was accurately measured (Extended Data Fig. 3b,c) with a high correlation (R ≥ 0.90) between replicate CpG levels and deep coverage at targeted epigenetic clock loci from fewer than 600,000 reads (Extended Data Fig. 3d–f). Compared to the RRBS libraries of the same samples, TIME-seq libraries had substantially higher overlap with target clock CpGs (Extended Data Fig. 3g). Age prediction using an existing RRBS-based rDNA clock, however, showed only moderate correlation with age in our pilot (R = 0.53, P = 0.0075; Extended Data Fig. 3h), possibly due to the differences in CpG coverage between TIME-seq and RRBS at several clock loci (Extended Data Fig. 3i).
To build a more accurate rDNA clock compatible with TIME-seq, enrichment baits tiling the entire rRNA promoter and coding regions were designed and used to enrich TIME-seq libraries from 191 C57BL/6 mouse blood DNA samples (mice aged 2–35 months) in pools of 47–48 (Fig. 1b). Pools were combined and sequenced on an Illumina MiSeq for a per-sample cost of less than US$5 (see Supplementary Table 3 for the sequencing costs). Most demultiplexed reads (Fig. 1c) from each sample mapped to the rDNA repeat meta-locus (median per pool 68–79%; Fig. 1d), resulted in high coverage for each sample at rDNA CpGs (Fig. 1e) and accurate DNA methylation quantification (Fig. 1f).
To train an epigenetic clock, the 182 samples that passed the quality filters were split approximately 80:20 into training and testing sets (Fig. 1g); elastic net regression (α = 0.05) was applied to the training data. After fine-tuning our model on the training set (R = 0.98), age predictions using the resulting 232 CpG TIME-seq rDNA clock showed a high correlation with age (testing, R = 0.95) and a median absolute error (MedAE) of only 1.95 months in the testing samples (Fig. 1h). To build a clock that could be applied to both TIME-seq and RRBS, we trained a model from TIME-seq data using only CpGs with high coverage in an existing RRBS dataset11. This clock showed a high age correlation (R = 0.89) when applied to RRBS data and reflected the longevity benefit of caloric restriction (Fig. 1i).
Highly efficient TIME-seq multi-tissue and tissue-specific mouse aging clocks
In contrast to rDNA clocks, those built with CpGs from across the genome reflected diverse changes in aging hallmarks and their associated genes3. Therefore, we designed hybridization enrichment probes for 957 distinct CpG islands in gene promoters or other gene regulatory elements previously reported to have high age correlation in mouse blood11 and multi-tissue clocks12,13 (Extended Data Fig. 4a). With these enrichment probes, we prepared TIME-seq libraries using 1,137 DNA samples from mouse blood, liver, skin, kidney, white adipose tissue (WAT) or muscle, and sequenced libraries for an average per-sample cost of U$5.41. From the data, we prepared a high-coverage methylation matrix of 6,370 CpGs for clock training (Fig. 2a). Variation between samples was mainly due to the tissue of origin (Fig. 2b), as expected from quality methylation sequencing datasets from multiple tissues.
Fig. 2 |. Low-cost TIME-seq multi-tissue and tissue-specific clocks applied to 1,137 mouse tissue samples.
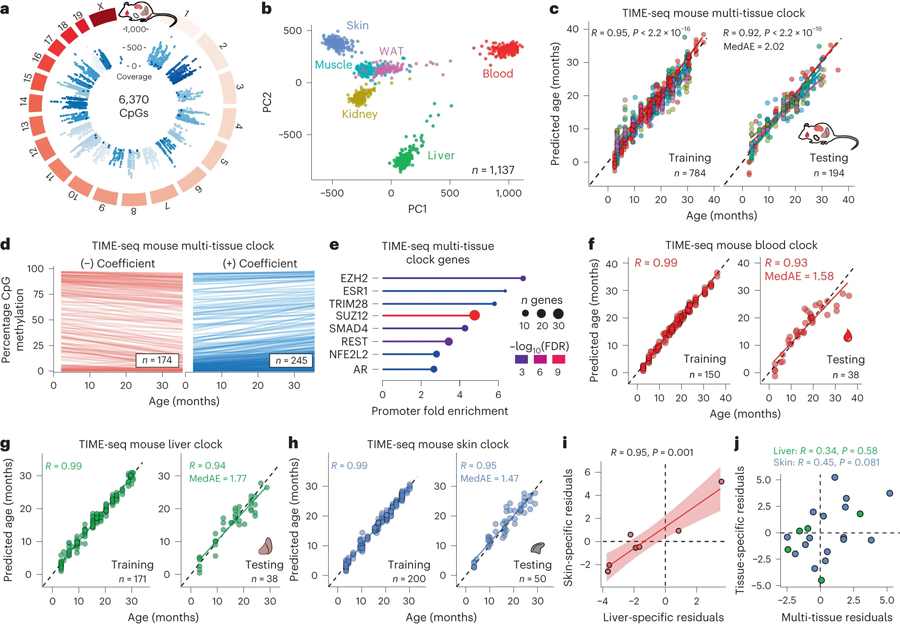
a, Circular genome plot illustrating the position and mean coverage of the 6,370 high-coverage CpGs from the TIME-seq libraries in 1,137 mouse tissue samples. b, Principal component analysis (PCA) for the TIME-seq data colored according to their tissue of origin: liver (green), blood (red), skin (blue), muscle (turquoise), kidney (yellow) and WAT (pink). c, TIME-seq mouse multi-tissue clock training (left) and testing (right) predictions plotted against chronological age. d, Linear models fitted to DNA methylation levels at clock CpGs changing with age in the data used to train the TIME-seq mouse multi-tissue clock. Clock CpGs were split according to their clock coefficient sign, which is represented in the transparency of each line. e, Enrichment of protein binding (based on ENCODE data) at genes associated with the TIME-seq mouse multi-tissue clock. f–h, Tissue-specific TIME-seq clock training (left) and testing (right) datasets for the TIME-seq mouse blood clock (f), liver clock (g) and skin clock (h). For all clocks, the Pearson correlation between predicted and actual age is shown. MedAE is shown for the testing set predictions. i, Age-adjusted residuals for liver and skin clock predictions from the same mice predicted in the testing sets. The shading around the regression line represents the 95% confidence interval (CI). j, Age-adjusted residuals for either skin (blue) or liver (green) clock predictions plotted against the predictions from the multi-tissue clock in the same sample.
Next, we trained and tested the TIME-seq mouse multi-tissue clock (Fig. 2c), which accurately predicted age in mouse blood, liver, skin, kidney and WAT (testing, R = 0.92; MedAE = 2.02 months). As described in previous studies8,20, when using age-correlated CpGs from other tissues, the prediction of age in muscle was less accurate (Extended Data Fig. 4b) and this tissue was excluded from multi-tissue clock training. Our 419-CpG mouse multi-tissue clock contains both positive and negative age-correlated CpGs (Fig. 2d), which are found in genes that are enriched for regulation by polycomb repressive complex 2 (PRC2) components (for example, EZH2 and SUZ12) and the longevity-associated transcription factor REST32 (Fig. 2e). We also trained tissue-specific epigenetic clocks (Fig. 2f–h and Extended Data Fig. 4c,d) for mouse blood (testing, R = 0.93; MedAE = 1.58 months), liver (testing, R = 0.94; MedAE = 1.77 months), skin (testing, R = 0.95; MedAE = 1.47 months), WAT (testing, R = 0.93; MedAE = 1.98 months) and kidney (testing, R = 0.95; MedAE = 2.5 months).
It is still largely unknown what factors influence epigenetic clocks and how different aging clocks relate to each other2. It has been hypothesized that certain clocks exhibit more environmental ‘extrinsic’ influence, whereas other clocks are more intrinsically defined, that is, influenced more by genetic variation3. To understand how our clocks related to each other, we identified mice that contributed tissues to two or more testing datasets in clock development. To control for slight bias in prediction error with age, we calculated the age-adjusted prediction residuals for each clock (Methods) and plotted the pairwise data for each tissue and clock. There was a highly positive correlation (Fig. 2i; R = 0.95, P = 0.001) for the TIME-seq skin and TIME-seq liver clocks with each other, but no significant correlation for either tissue-specific clock with the TIME-seq multi-tissue clock (Fig. 2j). This result suggests there may be some extrinsic factor contributing to synchrony between the TIME-seq liver and skin clocks, whereas our multi-tissue clock is not subject to the same influence and reflects other aging modules.
Validating the robustness of TIME-seq in independent large cohorts of aging mice
To validate the robustness of TIME-seq-based age predictions, we prepared TIME-seq libraries in two separate experiments using blood DNA from an independent cohort of mice (Fig. 3a). A subset of these mice had been tracked longitudinally, assessed using the mouse frailty index33 and had blood composition parameters measured. The TIME-seq blood clock (R = 0.93; Fig. 3b) and the TIME-seq multi-tissue clock (R = 0.9; Fig. 3c) provided accurate predictions that reflected aging longitudinally. For the TIME-seq libraries enriched for rDNA (Extended Data Fig. 5a,b), mice from the same colony as the original training dataset validated clock rDNA clock accuracy ( JAX mice, R = 0.96). As expected, mice from different colonies gave more variable predictions (National Institute of Aging (NIA) mice, R = 0.81) because rDNA copy number varies greatly between different mouse strains, colonies and even individuals within a colony34, and copy number directly relates to aggregated methylation status35.
Fig. 3 |. TIME-seq is a robust and scalable alternative to conventional clock approaches.
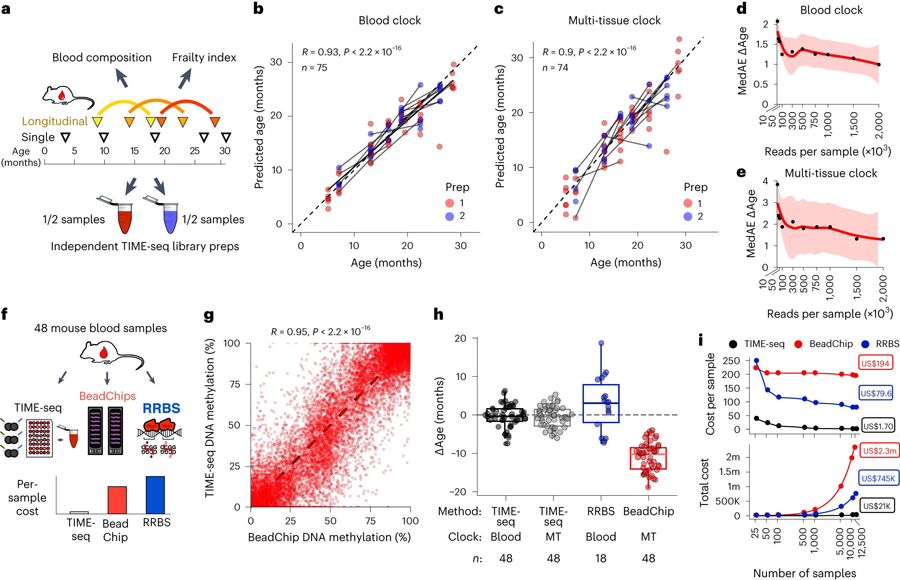
a, Experimental schematic for validation of the TIME-seq age prediction methods in an independent cohort of mice with longitudinal time points, paired frailty index and blood composition data. b,c, TIME-seq age predictions in two independent validation library preparations using the TIME-seq mouse blood clock (n = 75) (b) and the TIME-seq mouse multi-tissue clock (n = 74) (c). The lines connect the same mouse at two different ages. Pearson correlations are shown in the top left corner. d,e, Sequencing saturation simulation to estimate clock accuracy from different read numbers in the validation samples for the TIME-seq mouse blood (d) and multi-tissue (e) clocks. The shading around the locally estimated scatterplot smoothing regression line represents the 95% CI. f, Schematic of the benchmarking experiment to compare TIME-seq to Illumina BeadChip and RRBS. g, Comparison of CpG methylation percentage in TIME-seq and BeadChip. Each dot represents the same CpG from the same sample measured by each technology. h, Comparison of ∆Ages for each method and the associated clocks. The boxplot lengths represent the IQR, with the middle line representing the median values and the whiskers 1.5 times the IQR. i, Comparison of cost per sample (top) and total cost (bottom) for the TIME-seq clocks (black), BeadChip (red) and RRBS (blue) across a range of sample scales. The half-filled circles denote points that are overlapping between TIME-seq clocks. m, million; k, thousand.
To understand if our age prediction methods were synchronous in predicting animals older or younger, we plotted the pairwise age-adjusted prediction residuals for each mouse from each method. While all prediction methods were significantly positively correlated, the TIME-seq multi-tissue clock and blood clock age-adjusted prediction residuals were much more highly correlated with each other and lowly correlated with rDNA clock residuals (Extended Data Fig. 5c), suggesting that rDNA methylation may be capturing a separate aging module only partially reflected in the other clock methods. The high correlation between blood and multi-tissue clock prediction residuals suggests that similar aging modules are reflected by these clocks, in contrast with the multi-tissue clock asynchrony with the skin and liver clocks.
Assessing the influence of blood composition and frailty on epigenetic age with TIME-seq
The difference between predicted age from epigenetic clocks and chronological age (∆Age) has been shown to correlate with a wide variety of age-associated phenotypes3. To test if TIME-seq predictions related to other measures of health or aging, we compared ∆Ages from each approach to the mouse frailty index and blood composition measurements, which have been shown to influence epigenetic clocks2. To control for the raw age correlation of each variable (Extended Data Fig. 5d,e), measurements from each mouse were subtracted from the median value of that variable in similar-aged animals–abbreviated as ∆Medage (blood) and ∆Medage (FI). ∆Medage (blood) values were not correlated with ∆Ages from the deep-sequenced clocks, suggesting that blood cell composition was not driving predictive variance (Extended Data Fig. 5d). Because the frailty index is also highly correlated with age and indicative of age-related decline, we assessed if ∆Ages were correlated with ∆Medage (FI) values (that is, whether mice that are frailer for their age were also predicted older and vice versa). Comparing ∆Medage (FI) to ∆Ages (Extended Data Fig. 5f), we found a low and non-significant correlation. This finding mirrors the previously described low correlation between frailty and human epigenetic age predictions from blood DNA (for example, the Hannum or Horvath clocks36) and suggests that frailty scores are a relatively distinct biomarker of health and functional decline.
To determine the minimum sequencing read number for accurate TIME-seq clock prediction, we simulated a sequencing saturation experiment by extracting reads from each sample at a lower threshold, remapping the subset reads and predicting age with the lower coverage (Fig. 3d,e). For the TIME-seq blood and multi-tissue clocks, prediction accuracy substantially declined with fewer than 500,000 reads per sample.
Benchmarking TIME-seq against conventional methods for epigenetic clock analysis
Next, we benchmarked TIME-seq against the most common technologies for age prediction, Illumina methylation BeadChip and RRBS, in 48 independent mouse blood samples (Fig. 3f). TIME-seq methylation levels from the same CpG and same mouse were highly correlated with both BeadChip (R = 0.95; Fig. 3g) and RRBS (R = 0.92; Extended Data Fig. 5g). Epigenetic age predictions were highly accurate using either the TIME-seq blood or multi-tissue clocks, providing further independent validation (Fig. 3h). In contrast, the recently described BeadChip multi-tissue epigenetic clock37–the only mouse microarray-based clock that is commercially available–uniformly underpredicted the age of the samples, possibly owing to a skewed age and sample distribution in the original study37. Finally, the RRBS-based mouse blood clock11 predicted samples with more accuracy than the BeadChip clock but with more variation than TIME-seq.
To compare the cost of small-scale and large-scale clock analyses, we estimated costs for a range of sample sizes (Fig. 3i and Supplementary Table 4). TIME-seq is increasingly economical at scale, with an estimated per-sample cost of just US$1.70 for 12,500 samples. RRBS is estimated to be more expensive than Illumina BeadChip in small experiments but–also leveraging the efficiency of short-read sequencing–it is increasingly cheaper than BeadChip in large-scale applications. TIME-seq clocks are approximately 50-fold less expensive than RRBS and 100-fold less expensive than Illumina BeadChip-based clocks in the largest simulated experiments, suggesting that TIME-seq clocks may be a more cost-efficient alternative to more conventional methodologies.
Assessing interventions that slow, accelerate and reverse aging in mice with TIME-seq clocks
Our goal for developing TIME-seq was to apply clocks to large-scale intervention experiments such as those obtained in longitudinal mouse aging studies, in vitro screens or large-scale human clinical trials. To understand if TIME-seq clocks could detect differences in aging after interventions, we applied our clocks to controlled treatments associated with age deceleration, acceleration or rejuvenation. We also applied TIME-seq to an in vitro time course to understand if it could be used for the screening experiments.
Two of the most robust interventions that extend mouse healthspan and lifespan are amino acid restriction and caloric restriction38. Conversely, a reduction in healthspan and lifespan is seen when mice are fed a high-fat diet (HFD)39. Using our TIME-seq mouse blood clock, mice that were 40% CR or methionine-restricted (MetR) from age 24 months to 30 months were predicted younger than their controls fed ad libitum (Fig. 4a,b; MetR, P = 0.022; 40% CR, P = 0.031). Likewise, the TIME-seq liver clock predicted that the livers of mice that experienced 30% caloric restriction for 10 months were younger (P = 0.046) than their controls fed ad libitum (Fig. 4c,d). HFD-fed mice were predicted older than mice of the same age on a standard diet using the TIME-seq liver clock (Fig. 4e,f; P = 0.026) and the TIME-seq multi-tissue clock (Extended Data Fig. 6a; P = 0.0064). These data suggest that TIME-seq clocks can detect age deceleration and acceleration from multiple tissues, even when interventions are initiated late in life.
Fig. 4 |. TIME-seq clocks reflect interventions that slow, accelerate and reverse aging and can be used for in vitro studies.
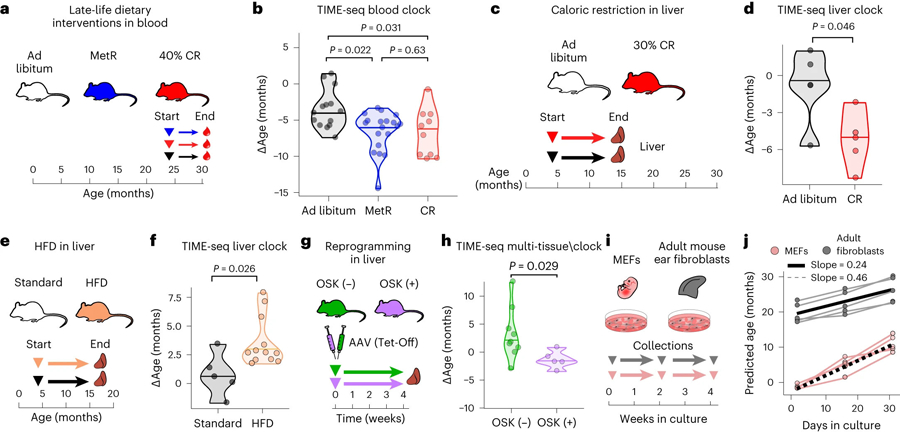
a, Schematic of dietary restriction treatments started in late life. Groups of mice were treated with a 40% CR diet, MetR diet and ad libitum diet for 6 months starting at 24 months of age. b, Comparison of ∆Ages from TIME-seq blood clock predictions from blood samples from MetR (n = 19), CR (n = 8) and ad libitum (n = 13) fed groups of mice. Group comparison was performed with a pairwise two-sided Wilcoxon test with false discovery rate (FDR) correction for multiple comparisons. c, Schematic of the experiment to assess the effect of a CR diet on mouse liver epigenetic age. Mice were treated with 30% CR or control ad libitum diets starting at 4 months of age for 10 months; then livers were collected. d, Comparison of ∆Age from mice fed ad libitum (n = 4) and CR (n = 5) mice using the TIME-seq mouse liver clock. e, Schematic of HFD treatment and liver collection. Mice were treated for 13 months starting at 4 months. f, Comparison of predicted ages for standard (n = 5) and HFD mice (n = 12) using the TIME-seq liver clock. f, Statistical comparison between groups was performed using a two-sided Student’s t-test after assessing normality with a Shapiro–Wilk test. g, Schematic of AAV treatments with OSK-expressing or control (GFP) cassettes. Livers were collected 1 month after AAV injection from two sets of mice (first set: control, n = 4 treatment, n = 2 aged 15–17 months; second set: control, n = 9; treatment, n = 5 aged 24 months). h, Comparison of ∆Ages in the livers of mice with (OSK+) or without (OSK−) OSK expression for 1 month. Statistical comparison was performed with a two-sided Wilcoxon test. i, Schematic of the experiment to assess MEFs and mouse adult ear fibroblasts in a cell culture time course lasting 1 month, with collection every 2 weeks. j, TIME-seq multi-tissue clock predictions from cell culture samples collected across the time course. The slope represents the change in predicted age per day in culture based on linear models fitted to data from each cell type.
Epigenetic reprogramming can rejuvenate aged tissues, driving gene expression and epigenetic changes toward a more youthful and regenerative state19,40,41. To understand if our clocks reflected rejuvenation, we assayed mouse livers treated for 1 month with an adeno-associated virus (AAV) expressing an Oct-4, Sox-2 and Klf-4 (OSK) polycistronic gene cassette (Fig. 4g). With the TIME-seq multi-tissue clock, mice with OSK expression were predicted significantly younger than control AAV-injected mice (Fig. 4h; P = 0.029). TIME-seq liver clock predictions were not significantly different between groups (Extended Data Fig. 6b), perhaps reflecting differences between intrinsic and extrinsic aging modules. These results suggest that the TIME-seq multi-tissue clock reflects epigenetic rejuvenation in vivo.
TIME-seq clocks reflect developmental stage and different rates of epigenetic aging in cultured cells
Epigenetic clocks have been shown to ‘tick’ as cells are grown and passaged in culture18,42,43, providing a promising way to screen for aging interventions in vitro. To test if TIME-seq clocks also work on cultured cells, we grew five independent lines of low-passage mouse embryonic fibroblasts (MEFs) or adult mouse ear fibroblasts for 1 month and collected cells at three different time points (Fig. 4i). Using the TIME-seq mouse multi-tissue clock, MEFs were initially predicted a subzero (embryonic) age; their epigenetic age steadily increased at a rate of approximately 2 weeks for every day in culture (Fig. 4j). Adult fibroblast lines were initially predicted between 17 and 23 months of age; their epigenetic ages increased at approximately half the rate as MEFs. The results were similar using the TIME-seq mouse skin clock, albeit with cells predicted to age at a slower rate (Extended Data 7c). These data indicate that we can use TIME-seq to track aging in cultured cells and provide further evidence for a different rate of aging between adult and embryonic cells18.
Developing a highly accurate and efficient human TIME-seq clock for human blood
Most human clock studies train their age prediction models on publicly available microarray data from dozens of past studies8,10,18 because new, large-scale experiments using microarrays are exorbitantly expensive and laborious. To develop a TIME-seq epigenetic clock that could be used in new large-scale human studies, we obtained 1,056 human blood DNA samples from demographically representative individuals aged 18–103 years (Fig. 5a). Most of the samples were designated for initial clock training and testing (n = 796), whereas a subset was used for independent sample preparation and validation (n = 260). Using enrichment probes against age-correlated loci from 11 described clocks44, TIME-seq libraries were prepared and sequenced for a per-sample cost of U$6.24. We achieved high coverage from 9,379 CpGs across the genome (Fig. 5b), and sample data largely separated by age in the first two principal components (Fig. 5c), validating that age-associated CpGs were enriched in the dataset.
Fig. 5 |. Highly accurate epigenetic age predictions in 1,056 human blood samples using TIME-seq.
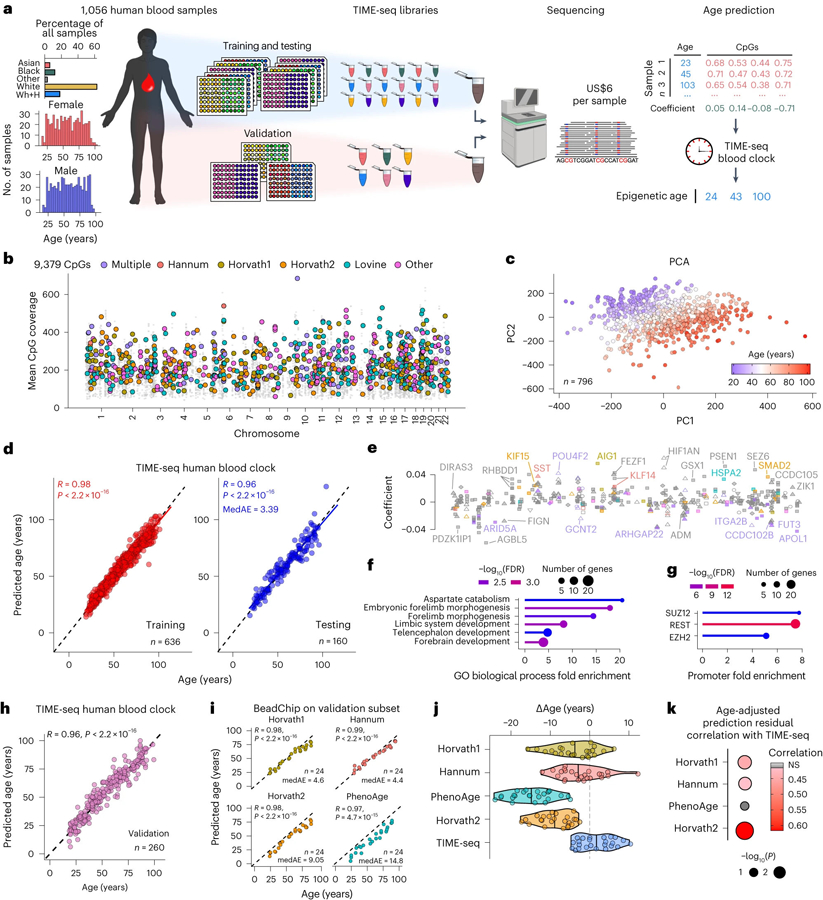
a, Schematic of the experimental design to train, test and validate TIME-seq in 1,056 human blood DNA samples. Wh+H indicates White (Hispanic) ethnicity as denoted in the biobank metadata. b, Coverage of 9,379 CpGs from across the human genome. The colored dots represent CpGs described in the Illumina BeadChip clocks, whereas the smaller gray dots are the other enriched CpGs. c, PCA of the methylation matrix for the training and testing samples, colored according to age from youngest (blue) to oldest (red). d, Predicted ages from the TIME-seq human blood epigenetic clock. Pearson correlation between predicted and actual age is shown for the training (right; R = 0.98, P < 2.2 × 10−16) and testing (left; R = 0.96, P < 2.2 × 10−16) datasets. The MedAE is shown for the testing dataset. e, Annotation of the 405 clock CpGs with the coefficient on the y axis. The x axis (not shown) is the genomic space in the same style as b from left (chromosome 1) to right (chromosome 22). CpGs and gene names are colored with the same color key as in b. Feature annotation is coded according to shape as follows: 5′ UTR (open circle), exon (filled diamond), intergenic (open triangle), intron (filled triangle), noncoding (inverted filled triangle), promoter (filled square) and transcription termination site (inverted open triangle). f,g, Gene Ontology (GO) analysis for the enrichment of biological processes (f) or transcription factor binding sites (g) in genes associated with clock CpGs. h, TIME-seq human blood clock predictions in 260 independently prepared human blood DNA samples. Pearson correlation between predicted and actual age is shown (R = 0.96, P < 2.2 × 10−16). i, Predicted ages from each BeadChip-based clock in a subset of the validation set sample (n = 24). Pearson correlation between predicted and chronological age is shown, as well as MedAE in the subset. For Horvath1, Horvath2 and Hannum, P < 2.2 × 10−16. For PhenoAge, P = 4.7 × 10−15. j, ∆Age values (difference in predicted and chronological age in units of years) for each age prediction method. k, Comparison of Pearson correlations between age-adjusted prediction residuals for each TIME-seq-based prediction and BeadChip-based clocks.
With these data, we trained and tested the TIME-seq human blood clock (Fig. 5d), observing high age correlation in training (R = 0.98) and testing predictions (R = 0.96) with an MedAE of just 3.39 years, comparable to the most widely used human epigenetic clocks8,9. Gene set enrichment analysis of the resulting 405 CpG clock (Fig. 5e) revealed an association with developmental biological processes (Fig. 5f), including genes with promoter enrichment of PRC2-associated proteins and REST similar to our mouse clocks (Fig. 5g).
To validate our clock in a large and independent human experiment, we prepared and sequenced TIME-seq libraries from the remaining human blood DNA samples. With the TIME-seq human blood clock, we observed similar predictive accuracy in the validation dataset (n = 260) compared to the initial testing set (R = 0.96; Fig. 5h). Next, we performed Infinium MethylationEPIC BeadChip analysis on 24 random human samples from this validation cohort to benchmark TIME-seq-based age predictions against conventional microarray human clock predictions. Like the mouse BeadChip analysis, human BeadChip methylation was highly correlated with TIME-seq methylation in the same CpG and sample (R = 0.93, P < 2.2 × 10−16; Extended Data Fig. 7). DNA methylation age predictions using four of the most used BeadChip-based clocks (Horvath1, Hannum, Levine-PhenoAge, Horvath2) were all highly correlated with age (Fig. 5i), although at least two of the BeadChip-based clocks were uniformly predicted under the chronological ages in these samples (PhenoAge and Horvath2). The TIME-seq human blood clock had the lowest median error of any of the age prediction methods in this subset of samples (MedAE = 3.7 years; Fig. 5j). Next, we compared the age-adjusted prediction residuals for each method, finding that the TIME-seq human blood clock predictions had the strongest correlation with the Horvath2 clock trained only on blood and skin (R = 0.61, P = 0.00135; Fig. 5k). The only clock that the TIME-seq human blood clock prediction residuals were not significantly correlated with was PhenoAge, a clock that is designed to reflect age-related mortality risk. These results suggest that the TIME-seq human blood clock is comparable to previously described and widely employed BeadChip-based chronological age epigenetic clocks.
Discussion
Epigenetic clocks are increasingly ubiquitous tools for both clinical and basic aging research. Unfortunately, they have been relatively expensive and laborious to measure, limiting their application to modest-sized or extremely well-funded experiments. In this study, we present TIME-seq, a flexible and scalable targeted sequencing approach that decreases the costs of epigenetic age analysis by up to 100-fold in large experiments. Using TIME-seq, we built and validated epigenetic clocks to predict age in over 2,800 unique samples. This scale was enabled by immediate sample barcoding that facilitated low-cost, pooled library preparation compatible with efficient hybridization-based enrichment and bisulfite conversion. Compared to traditional RRBS library preparations or BeadChip sample preparation, which can take anywhere from 4–9 days45 and cost upwards of US$30–50 per sample, reagent cost for TIME-seq libraries is only US$0.65 per sample; without any automation, a trained technician can easily prepare upwards of 800 samples in only 1.5 days (approximately 12 h of hands-on time). Further, input DNA for TIME-seq (100 ng) is the same as standard RRBS libraries and 3–5 times less than DNA methylation microarray, enabling longitudinal measurement of epigenetic age from low-yield DNA extractions, such as a mouse cheek bleed. Compared to highly targeted techniques (for example, droplet digital PCR) or commercial hybridization methods (for example, SyBS), TIME-seq is more flexible–capable of high enrichment of thousands to tens of thousands of CpGs–while minimizing sample preparation time, costs and parallel sample processing. At larger scales, we estimate that the cost of measuring TIME-seq clocks is substantially lower. Conservatively, we estimate that 12,500 samples could be prepared and sequenced for clock prediction on a single NovaSeq S4 flow cell for just US$1.70 per sample.
A central goal of aging research has been to develop biomarkers capable of detecting differences in the biological rate of aging1. We show that TIME-seq clocks can be used to detect the effect of interventions that accelerate or slow the rate of aging and rejuvenate aged tissue. TIME-seq clocks also reflect the age and developmental stage of cultured primary cells. Designed for low cost and scale, TIME-seq is uniquely capable of biomarker application to large intervention studies, such as screens or longitudinal clinical trials. Whether or not TIME-seq clocks can detect age-altering interventions in every tissue, or if they will associate with other metrics of functional decline, is not known and will require additional experiments. For instance, our analysis of TIME-seq clocks in mouse blood showed the lack of robust correlation between epigenetic age and frailty index in mice. A low correlation between baseline frailty and epigenetic age has also been described for human clocks36, suggesting that DNA methylation biomarkers might be specifically designed to predict frailty or a frailty-adjusted age proxy in mice or humans.
In the current study, we rationally designed hybridization probes to enrich loci that were known to correlate with chronological age, our target metric. However, when such data are not available, more genomic area might be enriched to identify CpGs with high correlation relative to the phenotype of interest (for example, mortality, frailty or cancer status). These libraries would initially be more expensive to sequence, but the number of baits could be reduced once model CpGs were discovered. This separation of more expensive biomarker discovery from low-cost measurement is key to the widespread adoption and routine use of DNA methylation clocks as well as clocks based on other biomolecules7,46.
There are trade-offs to consider when using TIME-seq. First, being designed for minimal cost and maximal efficiency, TIME-seq has the same limitations as other Tn5-based library preparations. For example, samples with improperly normalized DNA tend to drop out because of either over-tagmentation or under-tagmentation. During this study, we developed increasingly automated and reliable approaches for DNA normalization at scale (Supplementary Information), which limited sample dropout. Sample normalization could also be addressed using on-bead tagmentation47 to control for variation in starting DNA content. Likewise, techniques for increasing Tn5-based library efficiency might be used48. Second, we only present epigenetic clocks that are trained to predict chronological age and do not incorporate additional phenotypic information to adjust age before prediction as has been done, for example, DNAme PhenoAge and GrimAge10,21. To train these biological age TIME-seq clocks, we would need to obtain a large cohort of DNA with similarly rich phenotypic data, which may be within the scope of a different study. Nonetheless, we see that our clocks reflect interventions associated with altered aging phenotypes in mice.
Ultimately, further automation of our TIME-seq sample processing pipeline could enable a single researcher to prepare thousands of samples at once, enabling low-cost age predictions from extremely large cohorts such as the UK Biobank. Such a study would be a powerful resource to identify genetic and lifestyle factors that influence aging at the population scale.
Methods
TIME-seq library preparation
Tn5 was purified and prepared using previously described protocols49. See Supplementary Information for more details specific to TIME-seq. For the TIME-seq library preparations, samples were organized into relatively even pools; 10 μl of DNA (10 ng μl−1, 100 ng total) from each sample was distributed into separate wells of strip tubes (or 96-well plates) for tagmentation. Then, 100 ng of unmethylated lambda phage DNA (catalog number D1521, Promega Corporation) was tagmented with each pool. Lambda DNA that came through at a low percentage of demultiplexed reads served to estimate bisulfite conversion efficiency. To tagment samples, 12.5 μl of 2× tagmentation buffer (20 mM Tris-HCl, pH 7.8, 10 mM dimethylformamide, 10 mM MgCl2) was added to each sample. Next, 2.5 μl of uniquely indexed TIME-seq transposase was added and the reaction was immediately mixed by pipetting 20 times. Once transposase was added to each sample in a pool, the samples were placed at 55 °C for 15 min. After incubation, 7 μl of STOP buffer (100 mM MES, pH 5, 4.125 M guanidine thiocyanate, 25% isopropanol, 10 mM EDTA) was added, pools were vortexed and pulse-spun in a centrifuge and the reaction was incubated at 55 °C for an additional 5 min.
After stopping the reactions, samples from each pool were combined into a single tube, typically a 5-ml LoBind tube (catalog number 0030122348, Eppendorf) or 15-ml Falcon tube (catalog number 229410, CELLTREAT); 118 μl per sample of DNA binding buffer (catalog number D4004–1-L, Zymo Research) was added. Pools were then applied to Clean & Concentrator 25 (catalog number D4033, Zymo Research) columns. If the volume of the pool exceeded 5 ml, each pool was passed in equal volume through two separate columns. After two washes, pools were eluted in 41 μl (typically yielding 39 μl after elution) and 1 μl was removed to assess tagmentation fragment size and yield by D5000 ScreenTape (catalog number 5067–5588, Agilent Technologies) on an Agilent TapeStation.
For methylated end repair, eluted pools were combined with 5 μl of New England Biolabs buffer 2, 5 μl of 5 mM deoxynucleotide triphosphate containing 5-methyl-dCTP (catalog number N0356S, New England Biolabs) instead of dCTP, and 2 μl of Klenow Fragment (3′ → 5′ exo-) (catalog number M0212L, New England Biolabs). The reactions were incubated at 37 °C for 30 min and then cleaned up with a Clean and Concentrator 25 column. To elute pools, 30 μl of heated elution buffer was applied to the column and incubated for 1 min before being spun. Eluted DNA was then passed through the column a second time and concentrated for hybridization to 5 μl with a SpeedVac Concentrator (Eppendorf).
For each pool, DNA, RNA and hybridization mixtures were prepared in separate strip tubes (one per pool). On ice, DNA mixtures were prepared by adding 5 μl of concentrated tagmented DNA from each pool, 3.4 μl of 1 μg μl−1 mouse cot-1 (catalog number 18440016, Thermo Fisher Scientific) or human cot-1 (catalog number 15279011, Thermo Fisher Scientific), and 0.6 μl of 100 μM TIME-seq hybridization blocking primers (Integrated DNA Technologies). RNA mixtures were prepared on ice by combining 4.25 μl of nuclease-free H2O with 1 μl of SUPERase•In RNase Inhibitor (catalog number AM2696, Thermo Fisher Scientific), mixing and then adding 0.75 μl (750 ng total) of the biotin-RNA baits. The methods for the design and production of RNA baits can be found in Supplementary Information and the bait sequences are found in Supplementary Table 5. Hybridization mixtures were kept at room temperature and consisted of 25 μl 20× SSPE (catalog number AM9767, Thermo Fisher Scientific), 1 μl of 0.5 M EDTA, 10 μl 50× Denhardt’s buffer (1% w/v Ficoll 400, 1% w/v polyvinylpyrrolidone, 1% w/v BSA) and 13 μl of 1% SDS. Once the mixtures were prepared for each pool, the DNA mixtures were placed in a thermocycler and incubated for 5 min at 95 °C. Next, the thermocycler cooled to 65 °C and the hybridization mix was added to the thermocycler. After 3 min at 65 °C, the RNA mix was added to the thermocycler and incubated for 2 min at 65 °C. Next, the thermocycler lid was opened, and, keeping all tubes in the thermocycler well, 13 μl of heated hybridization buffer was transferred to the RNA bait mixture, followed by 9 μl of the denatured TIME-seq pooled DNA. This step was done quickly to limit temperature change during transfer, typically with a multichannel pipette for multiple pools. The combined mixtures were pipetted to mix 3–5 times and capped and the thermocycler lid was closed. The hybridization reaction was then incubated at 65 °C for 4 h.
To capture biotin-RNA:DNA hybrids, 125 μl of streptavidin magnetic beads was washed three times in 200 μl of binding buffer (1 M NaCl, 10 mM Tris-HCl, pH 7.5, 1 mM EDTA) and resuspended in 200 μl of binding buffer. With the reaction still in the thermocycler, the streptavidin beads were added to the reactions and then quickly removed to room temperature. The reactions were rotated at 40 r.p.m. for 30 min to allow for biotin-streptavidin binding and then placed on a magnetic separation rack (20–400, Sigma-Aldrich) until the solution was clear. Next, the beads were resuspended in 500 μl of hybridization wash buffer 1 (1× SSC, 0.1% SDS) and incubated at room temperature for 15 min. The beads were separated again on the magnetic separation rack and quickly resuspended in 500 μl of preheated 65 °C wash buffer 2 (0.1× SSC, 0.1% SDS), then incubated for 10 min at 65 °C. This step was repeated for a total of three heated washes. On the final wash, beads were magnetically separated, resuspended in 22 μl of 0.1 M NaOH, moved to a new strip tube (to avoid droplets on the original tube mixing with the elution buffer) and incubated for 10 min. After 10 min, beads were separated and 21 μl of the eluted single-stranded DNA from each pool was moved to another new strip tube for the bisulfite conversion reaction.
Bisulfite conversion was done using the EpiTect Fast Bisulfite Conversion Kit (catalog number 59824, QIAGEN), which was chosen due to the inclusion of DNA protect buffer (limiting DNA strand breakage in the bisulfite reaction) and carrier RNA that helps yield from low-concentration reactions. The volume of the eluted DNA was adjusted to 40 μl using nuclease-free H2O; 85 μl of bisulfite solution was added, followed by 15 μl of the DNA protect buffer, and the solution was mixed thoroughly. Bisulfite conversion and cleanup proceeded according to standard kit instructions. DNA was eluted in 23 μl of kit elution buffer. The initial elution was passed through the column a second time.
PCR amplification was done in a 50-μl reaction containing 23 μl of the eluted DNA, 1 μl of 25 μM P7 indexed primer (see Supplementary Table 5 for the primer sequences), 1 μl of 25 μM P5 indexed primer and 25 μl New England Biolabs Q5U 2× Master Mix. Reactions were amplified with the following program: initial denaturation at 98 °C for 30 s; 19 cycles of 98 °C for 30 s, 65 °C for 30 s and 72 °C for 1 min; and a final elongation at 72 °C for 3 min. After the PCR reactions were finished, they were cleaned using 1.8× CleanNGS SPRI Beads (catalog number CNGS005, Bulldog-Bio). Library fragment size and yield were assessed using a D1000 (catalog number 5067–5582, Agilent Technologies) or High Sensitivity D1000 ScreenTape (5067–5584, Agilent Technologies) on an Agilent TapeStation. Pools were combined for sequencing.
Sequencing
TIME-seq library sequencing requires two custom sequencing primers (Supplementary Table 5) for read 2 (Tn5 index) and index read 1 (i7 index), which were spiked into standard primers for all sequencing runs so that standard or control libraries (for example, phiX) could also be read out. A complete list of sequencing platforms used for each experiment is found in Supplementary Table 3.
Sequenced read demultiplexing and processing
TIME-seq pools were demultiplexed using sabre (https://github.com/najoshi/sabre) to identify the internal Tn5 barcode with no allowed mismatches and separate reads into unique FASTQ files for each sample. Cutadapt (v.2.5) was used to trim adapters (paired-end option: -G AGATGTGTATAAGAGANAG -a CTNTCTCTTATACACATCT -A CTNTCTCTTATACACATCT; single-end option: -A CTNTCTCTTATACACATCT). Reads were mapped with bowtie2 (v.2.3.4.3) using Bismark50 (v.0.22.3; options -N 1 --non_directional) to bisulfite-converted genomes (bismark_genome_prepararation) mm10, hg19 or custom rDNA loci (see the bait design methods); reads were subsequently filtered using the Bismark function filter_non_conversion (option --threshold 11). Importantly, the latter step does not (as the Bismark function name suggests) reflect non-conversion from sodium bisulfite treatment, rather it removes a small percentage of reads (0.5–3%) that are artificially fully methylated during the methylated end repair of the reads, which has been described previously51. Next, the Bismark function bismark_methylation_extractor was used to call methylation for each sample with options to avoid overlapping reads (--no-overlap) in paired-end sequencing and to ignore the first 10 bp of each read (if single end: --ignore 10 and --ignore_3prime 10; if paired end: --ignore_r2 10 --ignore_3prime_r2 10 as well), which precludes bias from methylated cytosines added in the Tn5 insertion gap during end repair. Reads that failed to map as pairs but that mapped individually were processed with the same pipeline and joined to the methylation data using bismark2bedGraph. Bisulfite conversion efficiency was assessed from unmethylated lambda DNA mapped to the bisulfite-converted Enterobacteria phage λ genome (iGenomes, Ilumina) and was generally 99% or higher.
Epigenetic clock training, testing and analysis in the validation cohorts
R (v.4.0.2) was used for all data analysis, including data organization, clock training and testing, and applying clocks to the validation data. For clock training, methylation and coverage data were taken from the bismark.cov files (output of bismark_methylation_extractor), which contain CpG location and methylation percentage (0–100). Because high-coverage rDNA loci have been shown to make better age prediction models20, mouse rDNA methylation data were filtered to include only CpGs with high coverage (≥200) in more than 90% of samples at each CpG in the coverage matrix. For other mouse and human clock CpG enrichment datasets, CpGs were filtered to have at least coverage 10 in 90% of the samples.
To build epigenetic clocks from deep-sequenced TIME-seq data, samples were randomly selected from discrete age groups (for example, 25–55 weeks old for mice, 30–40 years old for humans) in an approximately 80:20 training to testing ratio. From the training dataset, penalized regression models were trained to predict age from methylation values with the R package glmnet52 with alpha set to 0.05 or 0.1 (elastic net). To further refine the model, age predictions from the training data were regressed against age and the coefficients from this simple linear model were included to adjust the elastic net model to account for small over-prediction or under-prediction in the youngest and oldest samples and produced predictions with more normal ∆Age distribution across the lifespan. Elastic net models were trained using random training-testing splits; a model with high Pearson correlation and low median error in the testing set was selected. This same process was applied to build the TIME-seq rDNA clock for application to the RRBS data, filtering for CpGs in TIME-seq data that had minimum coverage of 50 reads in the RRBS mouse blood clock dataset11.
Applying clocks to validation and intervention samples
Clocks were applied to the validation and intervention samples by joining bismark.cov files with the corresponding clock CpG coefficients. When a clock CpG was not covered, the missing value was replaced by the average methylation percentage at that CpG from all other samples in the experiment. Samples were excluded if more than 10% of clock CpGs were missing, or samples contained fewer than 100,000 reads. To calculate the weighted methylation () from the elastic net regression coefficients, each clock CpG methylation percentage was multiplied by the corresponding clock coefficient, and these values were summed as follows:
where is the total number of clock CpGs and is a number from 0–100 representing percentage methylation. To calculate the predicted age, the intercept from the elastic net regression is added to and this value is adjusted with the simple linear regression coefficients and , as follows:
Clocks predict age in units of weeks for the TIME-seq mouse rDNA clock, months for all other mouse clocks and years for the TIME-seq human blood clock. See Supplementary Table 6 for all clock positions, coefficients and intercepts used in this study.
Comparison between age prediction methods in the same mouse or sample
While age predictions were highly accurate, we still observed slight bias in the prediction for the oldest and youngest samples, which could influence the correlation between predictions made in the same mouse or sample. To control for this bias and compare predictions, we regressed predicted ages against chronological age for the entire set of data (for example, the testing dataset from clock training or the entire validation set) using the lm() function in R and calculated the residual for each prediction. For brevity, we refer to these values as the age-adjusted prediction residuals in our ‘Results’ section.
Animal assessments
All mouse experiments were conducted according to an animal protocol (reference number IS00000927) that was reviewed and approved by the Institutional Animal Care and Use Committee of the Harvard Medical Area Standing Committee on Animals. Male and female C57BL/6N mice were obtained from the NIA and group-housed (three or four mice per cage) at the Harvard Medical School in ventilated microisolator cages with a 12:12 h light–dark cycle at 71 °F (21.7 °C) with 45–50% humidity. Mouse blood samples (150–300 μl) were collected in anesthetized mice (3% isoflurane) from the submandibular vein into tubes containing approximately 10% by volume of 0.5 M EDTA. Blood was spun at 1,500g for 10 min and plasma was removed. Blood cell pellets were stored frozen at −80 °C. For the validation experiments, a subsample of whole blood was stored on ice and processed within 4 h with the Hemavet 950 (Drew Scientific) to give 20 whole blood count parameters. Frailty was assessed using the mouse clinical frailty index33, a noninvasive assessment of 31 health deficits in mice. Two-hundred mouse (C57BL/6N) ocular vein blood samples were collected by researchers at the Jackson Laboratory’s Nathan Shock Center according to methods described previously53. These samples were used for TIME-seq clock training and testing.
Benchmarking TIME-seq against BeadChip and RRBS
For the mouse benchmarking experiments, 48 mouse blood DNA samples (independent from the validation set) were prepared with TIME-seq using mouse clock enrichment baits; 500 ng of DNA from the same 48 samples was sent to FOXO Technologies for analysis on the Infinium Mouse Methylation BeadChip (catalog number 20041559, Illumina). Eighteen of these DNA samples with at least 100 ng of DNA remaining were prepared and analyzed with RRBS using published library preparation methods12. RRBS samples were pooled and sequenced on an Illumina NovaSeq to a median depth of 38 million read pairs and mapped with Bismark (v.0.22.3); methylation data were extracted with bismark_methylation_extractor with the option --no_overlap.
For the analysis of CpG methylation levels, loci common to each sample from each method (and more than 20 coverage for the sequencing approaches) were identified; the methylation levels from each technology were plotted pairwise as shown in Fig. 4. For the clock analysis, the TIME-seq mouse blood and multi-tissue clocks were applied to the TIME-seq data as described above, whereas the RRBS-based mouse blood clock was applied in a similar fashion using the reported loci, weights and formula12, replacing any missing values with the mean methylation from all other samples. The BeadChip clock was applied to BeadChip data as reported elsewhere54.
For the comparison of costs at various scales, consumable costs were estimated (without consideration of labor) as follows: (1) TIME-seq costs were estimated as the library preparation cost per sample (US$0.65, detailed in Supplementary Table 1) and the cost of sequencing reagents sufficient to sequence each sample to at least 750,000 reads, which is 50% more than the minimum depth we report for accurate age estimation; (2) for BeadChip, costs were estimated at US$25 per sample for DNA preparation below 1,000 samples and US$20, US$17.5 and US$15 per sample for volumes of 4,992, 9,984 and 12,000 samples. The costs of consumables for the BeadChip assay were taken from the list price for Infinium Mouse Methylation BeadChip on Illumina’s website (catalog numbers 20041558, 2004159 and 20041560); (3) for RRBS, sample preparation costs were estimated at US$50 per sample below 1,000 samples and US$30 per sample for volumes of 1,000 and 5,000 samples, and US$20 per sample for volumes of 10,000 and 12,500 samples. The costs of sequencing reagents were estimated to provide median reads per sample of 45 million.
Mouse intervention studies
For the late-life dietary interventions, male and female C57BL/6N mice were obtained from the NIA at 19 months of age and housed at the Harvard T.H. Chan School of Public Health. Mice were group-housed three or four per cage for the duration of the study in static isolator cages at 71 °F (21.7 °C) with 45–50% humidity, on a 12:12 h light–dark (7:00–19:00) cycle. After arrival, mice were fed a control diet (Research Diets) until the start of the study. Mice were then randomized to one of three groups: ad libitum diet, MetR (0.1% methionine) or 40% CR. CR was started in a stepwise fashion decreasing food intake by 10% per week until it reached 40% CR at week 4. CR intake was based on ad libitum intake. Mice were monitored weekly for body weight and food intake. Fasting blood samples (4–6 h) were taken when mice were killed (after 6 months on the diet) using cardiac puncture. Approximately 200 μl of whole blood in 1 μl of 0.5 mM EDTA was collected. The tube was spun and the plasma was removed. The remaining blood pellet was frozen at −80 °C until further analysis. Custom mouse diets were formulated at Research Diets (catalog numbers A17101101 and A19022001).
For our liver calorie restriction studies, male C57BL/6J wild-type (WT) mice bred in-house at the Harvard Medical School were housed singly with ad libitum access to water at the New Research Building. Male C57BL/6J mice were housed singly in the animal facility of the New Research Building at the Harvard Medical School. These mice were first given ad libitum access to water before being switched to house chow (LabDiet 5053), either ad libitum or 30% CR starting at 3 months old. Food intake was gradually reduced 10% per week for CR mice; body weight and food intake were monitored weekly. For both treatment groups of mice, food was placed on the floor of the cage each day at 8:00 ± 1 h. Livers were collected after approximately 10 months of treatment.
For our studies of HFD in the liver, male C57BL/6J WT mice were housed three or four animals per cage with access to water in the New Research Building animal facility at the Harvard Medical School. Approximately 3–4 months before the experiment, mice were switched to house chow (LabDiet 5053). Next, a subset of mice were switched to high-fat AIN-93G diet (modified by adding hydrogenated coconut oil to provide 60% of calories from fat) starting at approximately 4 months of age for the remainder of their lives. Livers were collected from mice after 13 months of treatment.
To assess the effect of partial reprogramming in mouse liver, male C57BL/6 WT mice were ordered from the Charles River Laboratories. After being acclimated in the housing facility for at least 1 week, mice were injected with AAV.PHP.eB viruses via the retro-orbital route to express either green fluorescent protein (GFP) or OSK in the liver. A Tet-Off system was used to control the expression of GFP and OSK. Specifically, AAV encoding TRE-OSK was coinjected with AAV encoding CMV-tTA, and AAV encoding TRE-GFP was coinjected with AAV encoding CMV-tTA to express either OSK or GFP. One month after the AAV injection, mice were killed and the liver tissues were collected for genomic DNA extraction for the TIME-seq experiment.
Cell culture time course
Five independent cell lines of low-passage MEFs derived from C57BL/6 mice were thawed and cultured in low-oxygen conditions (3% v/v) in DMEM with 17% FBS (Seradigm) and 1% penicillin/streptomycin plus 3.8 μl of β-mercaptoethanol per bottle of DMEM (500 ml). Likewise, five cell lines of low-passage adult ear fibroblasts were cultured with DMEM plus 10% FCS, 1% penicillin/streptomycin and 3.6 μl of β-mercaptoethanol per bottle of DMEM in the same low-oxygen conditions. After thawed cell lines became confluent in a 150-mm cell culture dish, cells were split into a 12-well cell culture dish and an aliquot of more than 1 million cells was taken for the initial time point by first washing cells with cold PBS and then freezing at −80 °C. Two more samples of each cell line were collected over the following 28 days in the same fashion, expanding each culture to a 100-mm dish and collecting cell lines simultaneously.
Human whole-blood DNA samples
Human blood DNA samples were selected from the database of the Mass General Brigham Biobank, a biorepository of patient samples at Mass General Brigham (parent organization of the Massachusetts General Hospital and Brigham and Women’s Hospital). Patients who donated samples to this biobank provided written informed consent before their inclusion in this study. To select samples, the biobank database was queried for individuals from the ‘Healthy Populations’ cohorts for whom DNA samples were available. From this pool of individuals, samples were selected for clock development that were demographically representative of the US population from ages 18 to 103 years. DNA samples were obtained from the biobank and deidentified before distribution for TIME-seq. Sample handling, data analysis and study design were approved by the Mass General Brigham Institutional Review Board (protocol number 2021P003059). Deidentified DNA samples were split into an initial cohort for model training and testing and a validation cohort. Cohorts were normalized separately to a starting concentration of 10 ng μl−1, prepared with TIME-seq, and sequenced on an Illumina NovaSeq.
For human BeadChip benchmarking, 300 ng of DNA from a subset of DNA samples was sent to TruDiagnostic for analysis on Infinium MethylationEPIC BeadChip. Data were returned as raw IDAT files, which were processed into beta values using the SeSAMe toolkit (v.1.16.1) in R. Age predictions from BeadChip-based clocks were made using the predictAge() command in SeSAMe.
Statistics and reproducibility
For the intervention experiments, no statistical methods were used to predetermine sample size, but sample sizes were similar to those used in previously described mouse epigenetic clock intervention experiments11–13. The investigators were not blinded to allocation during the experiments and outcome assessment. The statistical analysis listed in the figure legends, including Pearson correlation analysis, two-sample two-sided Student’s t-test, two-sided Wilcoxon test with or without multiple testing correction and Shapiro–Wilk test of normality, were implemented in R. Clock training and testing datasets were selected randomly using the R package dplyr (v.1.0.2) with the function sample_n(). Before benchmarking, data quality cutoffs were selected to filter low-quality data before clock training and testing analysis. For the mouse rDNA clock dataset, samples with a high percentage of flagged fully methylated reads (>3%), indicative of degraded single-stranded DNA, were filtered. For the human and mouse multi-tissue and tissue-specific clock training and testing datasets, low-coverage samples with fewer than 300,000 reads on target (within 1 kb of the target loci) were filtered. In the validation and intervention analysis, no samples were excluded except those with low-quality data. Low quality was predefined as a sample having fewer than 100,000 reads on target or a sample with more than 10% of clock CpGs with low coverage, that is, covered by fewer than ten reads. The Pearson correlation between DNA methylation values of technical replicates of the mouse blood DNA samples was assessed in two separate TIME-seq library preparations (shown in Fig. 1). The reproducibility of the TIME-seq epigenetic clocks was characterized by independently prepared and well-distributed (age and sex) validation sets of mouse and human DNA.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Biotinylated-RNA bait production and initial hybridization enrichment testing.
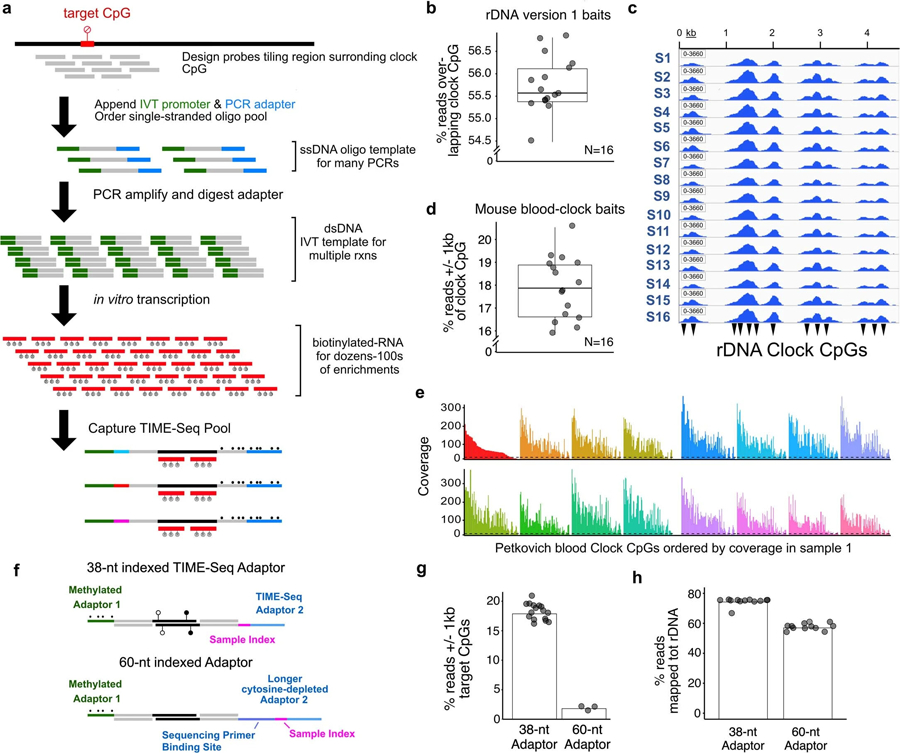
a, Schematic of steps involved in production of biotinylated-RNA baits from single-stranded oligo pools for target enrichment in TIME-Seq libraries. The percent of reads overlapping target RRBS mouse rDNA clock CpGs (b) and an IGV browser screenshot of mapped-read pileups (c) using version 1 rDNA baits for enrichment of a TIME-Seq pool. Reads on-target (d) and mouse RRBS blood clock (Petkovich et al., 2017) CpG coverage (e) using mouse-blood specific baits in a pilot experiment targeting non-repetitive clock loci. Dotted line represents coverage cut-off of 10. Pools in both rDNA and blood clock pilot enrichments were sequenced with approximately 1 million paired end (PE) reads each in pool of 16 samples. (f) Adaptor design schematic for comparison of TIME-Seq adaptors with longer barcoded adaptors. Comparison of on-target reads in short TIME-Seq and long cytosine-depleted adaptor designs for both mouse blood clock (g) and (h) rDNA (version 1) baits enrichments.
Extended Data Fig. 2 |. TIME-Seq library and sequencing schematic.
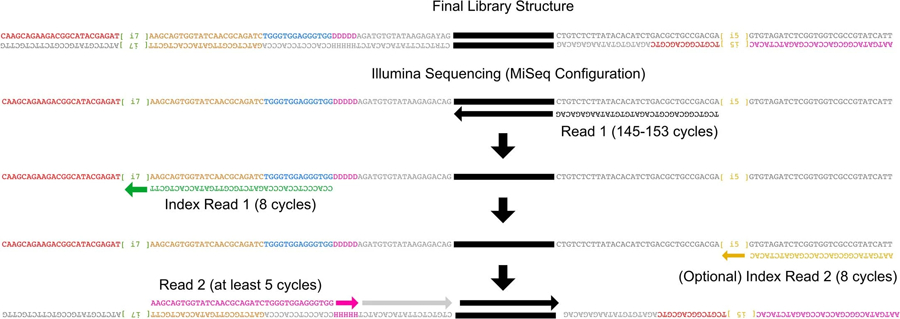
Schematic representation of final library structure (top) and Illumina sequencing (bottom) steps required to sequence TIME-Seq libraries. Index read 1 and read 2 primers are custom primers.
Extended Data Fig. 3 |. Small pilot-experiment sample metrics, correlation of rDNA CpG methylation, and age predictions using a reported RRBS-based rDNA clock.
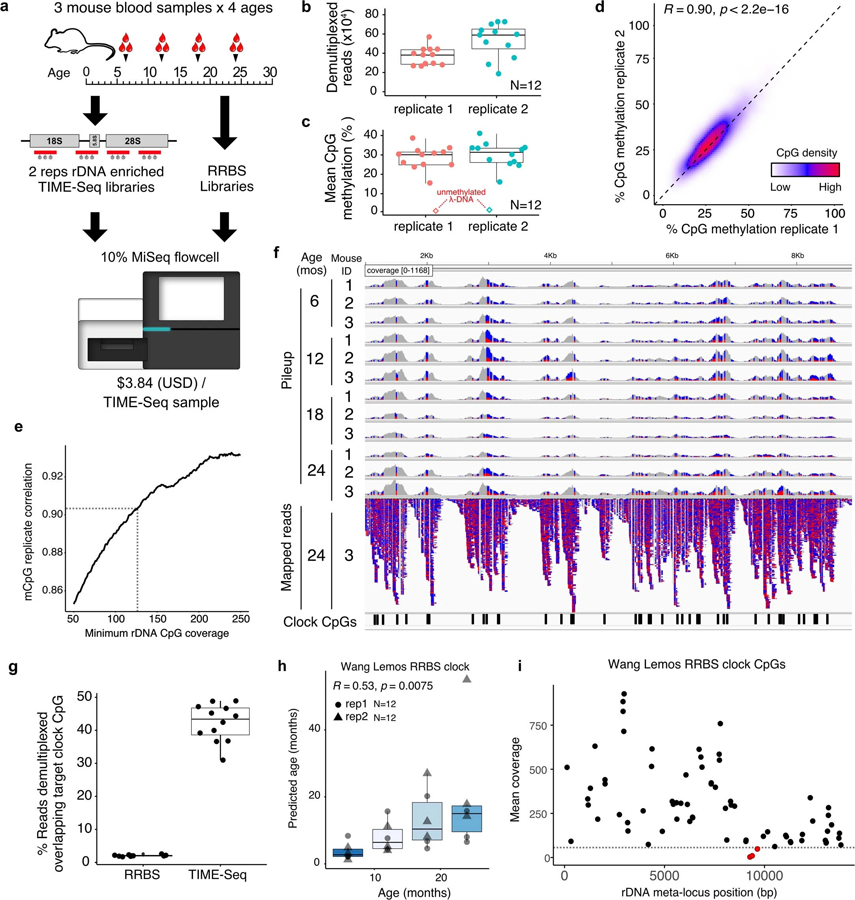
a, TIME-Seq pilot experimental design using mouse blood DNA from 4 age groups and preparing 2 replicates of each sample with rDNA baits (version 1) as well as RRBS libraries to be sequenced as a fraction of an Illumina MiSeq sequencing run. b, Demultiplexed reads from TIME-Seq pools. c, Mean CpG methylation from reads mapped to the mouse ribosomal DNA meta-locus. Unmethylated lambda phage DNA control is represented as a diamond. d, Percent methylation from reads mapped to ribosomal DNA meta-locus in replicate 1 and replicate 2 in CpGs with coverage of at least 125 reads. e, Replicate correlation from different coverage cutoffs in the rDNA. f, Pileup tracks for samples from a TIME-Seq pool (replicate 1) as well as mapped reads from one sample (mouse ID 3, aged 24 months). Reads are colored by mismatch: blue for T (unmethylated) and red for C (methylated). RRBS rDNA clock coordinates are illustrated on the bottom by black rectangles. g, Percent of reads directly overlapping clock CpGs from TIME-Seq libraries (N = 12; mean from 2 replicates) and shallow-sequenced RRBS libraries (N = 10). h, RRBS rDNA clock predictions using TIME-Seq data enriched for clock loci (N = 12, both replicates) i, Coverage of each clock locus in the original RRBS rDNA clock. CpGs shown in red have a mean coverage of less than 50. Boxplot lengths (panels b, c, g, h) represent the interquartile range (IQR) with the middle line representing median values and the whiskers 1.5 times the IQR.
Extended Data Fig. 4 |. Additional data related to mouse multi-tissue and tissue-specific clock training and testing.
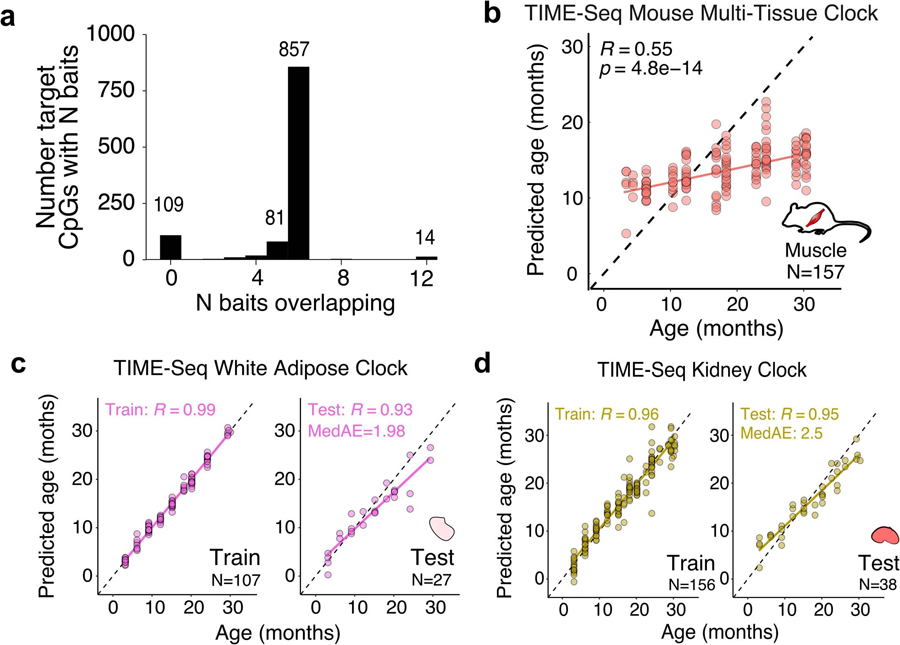
a, Baits overlapping target loci used for mouse clock CpG enrichment. b, Age predictions from the TIME-Seq Mouse Multi-tissue Clock applied to the 157 mouse muscle samples. Pearson correlation between predicted and actual age is shown. c, TIME-Seq White Adipose Clock train (N = 107) and testing set (N = 27) predictions. d, TIME-Seq Kidney Clock train (N = 156) and testing set (N = 38) predictions. For panels c and d, Pearson correlation between predicted and actual age is shown for train and test. The median absolute error is shown for the testing set.
Extended Data Fig. 5 |. Additional Data from validation and benchmarking of TIME-Seq.
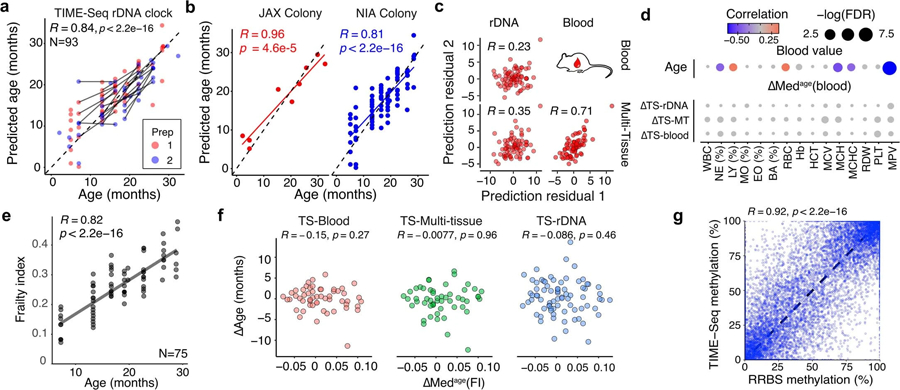
a-b, TIME-Seq Mouse rDNA Clock predictions with samples colored for (a) validation library preparation (prep) and (b) cohort of the mouse. Pearson correlation is shown for each panel. c, Correlation between age-adjusted prediction residuals in the validation sets from the different prediction approaches. d, Correlation and significance matrix between ∆Age from each approach and ∆Medage(blood), that is, the difference in median value from similar aged mice for each blood measurement. The color and size of each circle represent the correlation and p-value significance, respectively. WBC = white blood cell count, NE (%) = percent of neutrophils, LY (%) = percent of lymphocytes, MO (%) = percent of monocytes, EO (%) = percent of eosinophils, BA (%) = percent of basophils, RBC = red blood cell count, Hb = hemoglobin, HCT = hematocrit, MCV = mean corpuscular volume, MCH = mean corpuscular hemoglobin, MCHC = mean corpuscular hemoglobin concentration, RDW = red blood cell distribution width, PLT = platelets, MPV = mean platelet volume. e, Frailty indexes for each of the assayed mice along with Pearson correlation with age. f, Comparison of ∆Age and ∆Medage(FI) for mice in the validation cohort. Pearson correlation is shown without adjusting for multiple comparisons. g, Comparison between TIME-Seq CpG methylation and RRBS methylation in the same sample and CpG. Pearson correlation between CpG methylation levels is shown.
Extended Data Fig. 6 |. Additional Data for TIME-Seq clocks applied to intervention mice and an in vitro time course.
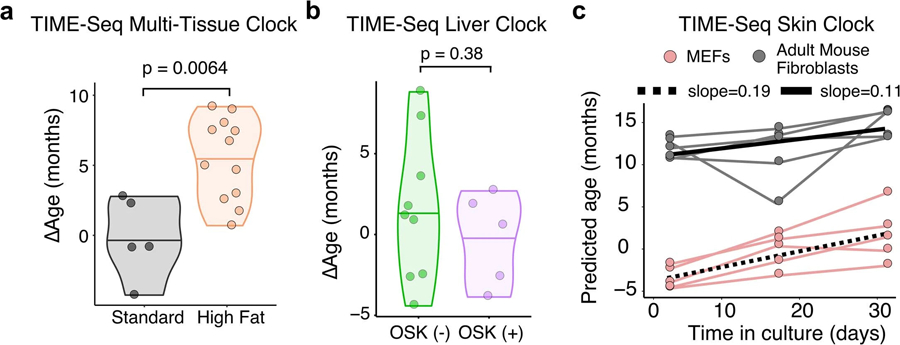
a, Comparison TIME-Seq Multi-Tissue Clock predictions of high-fat diet mouse liver (N = 12) with standard diet controls (N = 5). b, Comparison of TIME-Seq Liver Clock predictions in OSK-expressing, (+) OSK (N = 5), and control, (−) OSK (N = 9), mice. For panels a-b, statistical comparison between groups was performed using a two-sided Student’s t-test after assessing normality with Shapiro-Wilk’s test. c, Predictions of cell culture samples using the TIME-Seq Mouse Skin Clock. The slope from the linear models fit to data points from each cell line is shown.
Extended Data Fig. 7 |. Comparison of methylation levels from TIME-Seq and BeadChip on the same samples.
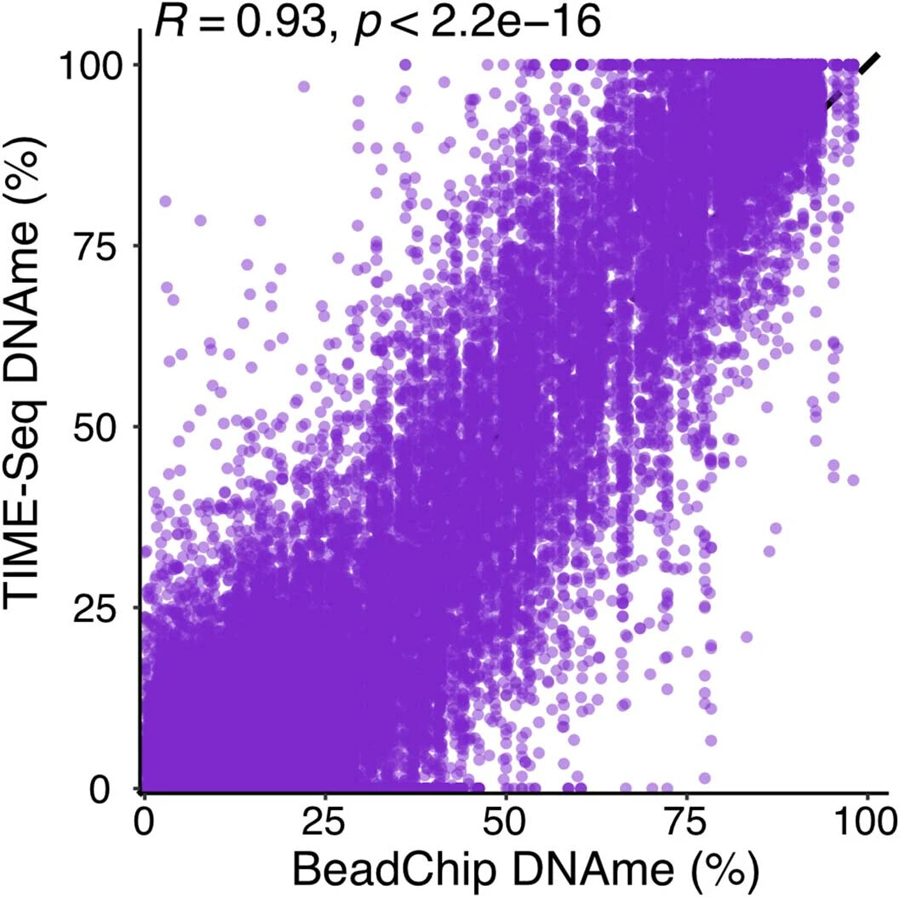
Pearson correlation between BeadChip and TIME-Seq DNAme values for each sample and CpG (R = 0.93, p < 2.2e − 16).
Supplementary Material
Acknowledgements
We thank the following individuals for their contributions to this work: R. Rogers for help editing this paper; A. Nguyen Ba and S. Boswell for providing the Tn5 purification and reaction protocols; C. Ricci-Tam for providing the initial hybridization enrichment protocols; the Bauer Core Facility at Harvard for sequencing and DNA extraction; the Harvard Microbiology Department for allowing us access to their Illumina MiSeq; the Seidman laboratory at the Harvard Medical School for allowing us to use their Agilent TapeStation; and the Jackson Laboratory Nathan Shock Center of Excellence in the Basic Biology of Aging (National Institutes of Health (NIH) number AG038070) for providing 200 mouse blood samples. This work was supported by the following grants, research programs and organizations, and gifts: the National Science Foundation Graduate Research Fellowship (number DGE1745303 to P.T.G.); the NIH/NIA F99/K00 Fellowship (number AG073499 to P.T.G.); the Diamond/AFAR Postdoctoral Transition Award in Aging (number DIAMOND19036 to A.E.K.); the NIH/NIA K99/R00 Fellowship (number AG070102 to A.E.K.); the Intramural Research Program of the NIH/NIA (to N.N.H. and M.K.E.); several NIH/NIA research project grants (numbers R01AG019719 and R21HG011850 to D.A.S.; number P01AG055369 to S.J.M.; and numbers AG065403 and AG047200 to V.N.G.); the Glenn Foundation for Medical Research (to D.A.S.); the Milky Way Research Foundation (to D.A.S.); gifts from M. Chambers, R. Rosenkranz, T. Robbins, P. Diamandis, S. Aoki, D. and S. Hoff, D. Dalio and Dalio Philanthropies, and the Glenn Foundation for Medical Research (to D.A.S.).
Footnotes
Code availability
The code for demultiplexing and read processing, and clock analysis, is provided on GitHub at https://github.com/patricktgriffin/TIME-Seq.
Competing interests
P.T.G. and D.A.S. are named inventors on a patent application related to TIME-seq methods filed by the Harvard Medical School (patent application number PCT/US21/37069) and licensed to Tally Health. D.A.S. is a founder and equity owner of Tally Health. P.T.G. has received minor equity compensation as a consultant to Tally Health. Additional information on D.A.S. affiliations not directly related to this work can be found at https://sinclair.hms.harvard.edu/david-sinclairs-affiliations. The other authors declare no competing interests.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s43587-023-00555-2.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s43587-023-00555-2.
Peer review information Nature Aging thanks Andrew Adey, Trey Ideker, Andrea Maier and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Data availability
Raw and processed sequencing data are available at the NCBI Gene Expression Omnibus under accession number GSE232346. The microarray data used for benchmarking are listed in the SuperSeries with the sequencing data under accession number GSE245630. Source data are provided with this paper.
References
- 1.Sprott RL Biomarkers of aging and disease: introduction and definitions. Exp. Gerontol 45, 2–4 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Bell CG et al. DNA methylation aging clocks: challenges and recommendations. Genome Biol 20, 249 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horvath S & Raj K DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet 19, 371–384 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Schultz MB et al. Age and life expectancy clocks based on machine learning analysis of mouse frailty. Nat. Commun 11, 4618 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bobrov E et al. PhotoAgeClock: deep learning algorithms for development of non-invasive visual biomarkers of aging. Aging 10, 3249–3259 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lehallier B et al. Undulating changes in human plasma proteome profiles across the lifespan. Nat. Med 25, 1843–1850 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fleischer JG et al. Predicting age from the transcriptome of human dermal fibroblasts. Genome Biol 19, 221 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horvath S DNA methylation age of human tissues and cell types. Genome Biol 14, R115 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hannum G et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 49, 359–367 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levine ME et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 10, 573–591 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petkovich DA et al. Using DNA methylation profiling to evaluate biological age and longevity interventions. Cell Metab 25, 954–960 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meer MV, Podolskiy DI, Tyshkovskiy A & Gladyshev VN A whole lifespan mouse multi-tissue DNA methylation clock. eLife 7, e40675 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thompson MJ et al. A multi-tissue full lifespan epigenetic clock for mice. Aging 10, 2832–2854 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levine M et al. A rat epigenetic clock recapitulates phenotypic aging and co-localizes with heterochromatin. eLife 9, e59201 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robeck TR et al. Multi-species and multi-tissue methylation clocks for age estimation in toothed whales and dolphins. Commun. Biol 4, 642 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilkinson GS et al. DNA methylation predicts age and provides insight into exceptional longevity of bats. Nat. Commun 12, 1615 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang T et al. Epigenetic aging signatures in mice livers are slowed by dwarfism, calorie restriction and rapamycin treatment. Genome Biol 18, 57 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horvath S et al. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging 10, 1758–1775 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu Y et al. Reprogramming to recover youthful epigenetic information and restore vision. Nature 588, 124–129 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kerepesi C, Zhang B, Lee S-G, Trapp A & Gladyshev VN Epigenetic clocks reveal a rejuvenation event during embryogenesis followed by aging. Sci. Adv 7, eabg6082 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu AT et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging 11, 303–327 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pidsley R et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol 17, 208 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meissner A et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res 33, 5868–5877 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han Y et al. Epigenetic age-predictor for mice based on three CpG sites. eLife 7, e37462 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han Y et al. New targeted approaches for epigenetic age predictions. BMC Biol 18, 71 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han Y et al. Targeted methods for epigenetic age predictions in mice. Sci. Rep 10, 22439 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang T et al. Quantitative translation of dog-to-human aging by conserved remodeling of the DNA methylome. Cell Syst 11, 176–185 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wendt J, Rosenbaum H, Richmond TA, Jeddeloh JA & Burgess DL Targeted bisulfite sequencing using the SeqCap Epi enrichment system. Methods Mol. Biol 1708, 383–405 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Mulqueen RM et al. Highly scalable generation of DNA methylation profiles in single cells. Nat. Biotechnol 36, 428–431 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rohland N & Reich D Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res 22, 939–946 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang M & Lemos B Ribosomal DNA harbors an evolutionarily conserved clock of biological aging. Genome Res 29, 325–333 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zullo JM et al. Regulation of lifespan by neural excitation and REST. Nature 574, 359–364 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Whitehead JC et al. A clinical frailty index in aging mice: comparisons with frailty index data in humans. J Gerontol. A 69, 621–632 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Watada E et al. Age-dependent ribosomal DNA variations in mice. Mol. Cell. Biol 40, e00368–20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodriguez-Algarra F et al. Genetic variation at mouse and human ribosomal DNA influences associated epigenetic states. Genome Biol 23, 54 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seligman BJ, Berry SD, Lipsitz LA, Travison TG & Kiel DP Epigenetic age acceleration and change in frailty in MOBILIZE Boston. J. Gerontol. A 77, 1760–1765 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou W et al. DNA methylation dynamics and dysregulation delineated by high-throughput profiling in the mouse. Cell Genom 2, 100144 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee MB, Hill CM, Bitto A & Kaeberlein M Antiaging diets: separating fact from fiction. Science 374, eabe7365 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baur JA et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444, 337–342 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ocampo A et al. In vivo amelioration of age-associated hallmarks by partial reprogramming. Cell 167, 1719–1733 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gill D et al. Multi-omic rejuvenation of human cells by maturation phase transient reprogramming. eLife 11, e71624 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matsuyama M, WuWong DJ, Horvath S & Matsuyama S Epigenetic clock analysis of human fibroblasts in vitro: effects of hypoxia, donor age, and expression of hTERT and SV40 largeT. Aging 11, 3012–3022 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matsuyama M et al. Analysis of epigenetic aging in vivo and in vitro: factors controlling the speed and direction. Exp. Biol. Med 245, 1543–1551 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Z et al. Underlying features of epigenetic aging clocks in vivo and in vitro. Aging Cell 19, e13229 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gu H et al. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat. Protoc 6, 468–481 (2011). [DOI] [PubMed] [Google Scholar]
- 46.Lehallier B, Shokhirev MN, Wyss-Coray T & Johnson AA Data mining of human plasma proteins generates a multitude of highly predictive aging clocks that reflect different aspects of aging. Aging Cell 19, e13256 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bruinsma S et al. Bead-linked transposomes enable a normalization-free workflow for NGS library preparation. BMC Genomics 19, 722 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mulqueen RM et al. High-content single-cell combinatorial indexing. Nat. Biotechnol 39, 1574–1580 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nguyen Ba AN et al. Barcoded bulk QTL mapping reveals highly polygenic and epistatic architecture of complex traits in yeast. eLife 11, e73983 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krueger F & Andrews SR Bismark: a flexible aligner and methylation caller for bisulfite-seq applications. Bioinformatics 27, 1571–1572 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suzuki M et al. Whole-genome bisulfite sequencing with improved accuracy and cost. Genome Res 28, 1364–1371 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Friedman J, Hastie T & Tibshirani R Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw 33, 1–22 (2010). [PMC free article] [PubMed] [Google Scholar]
- 53.Ackert-Bicknell CL et al. Aging research using mouse models. Curr. Protoc. Mouse Biol 5, 95–133 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou W et al. DNA methylation dynamics and dysregulation delineated by high-throughput profiling in the mouse. Cell Genom 2, 100144 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed sequencing data are available at the NCBI Gene Expression Omnibus under accession number GSE232346. The microarray data used for benchmarking are listed in the SuperSeries with the sequencing data under accession number GSE245630. Source data are provided with this paper.