

Abstract
DNA ligases are key enzymes involved in the repair and replication of DNA. Prokaryotic DNA ligases uniquely use NAD+ as the adenylate donor during catalysis, whereas eukaryotic enzymes use ATP. This difference in substrate specificity makes the bacterial enzymes potential targets for therapeutic intervention. We have developed a homogeneous chemiluminescence-based hybridization protection assay for Staphylococcus aureus DNA ligase that uses novel acridinium ester technology and demonstrate that it is an alternative to the commonly used radiometric assays for ligases. The assay has been used to determine a number of kinetic constants for S. aureus DNA ligase catalysis. These included the Km values for NAD+ (2.75±0.1 μM) and the acridinium-ester-labelled DNA substrate (2.5±0.2 nM). A study of the pH-dependencies of kcat, Km and kcat/Km has revealed values of kinetically influential ionizations within the enzyme–substrate complexes (kcat) and free enzyme (kcat/Km). In each case, the curves were shown to be composed of one kinetically influential ionization, for kcat, pKa=6.6±0.1 and kcat/Km, pKa=7.1±0.1. Inhibition characteristics of the enzyme against two Escherichia coli DNA ligase inhibitors have also been determined with IC50 values for these being 3.30±0.86 μM for doxorubicin and 1.40±0.07 μM for chloroquine diphosphate. The assay has also been successfully miniaturized to a sufficiently low volume to allow it to be utilized in a high-throughput screen (384-well format; 20 μl reaction volume), enabling the assay to be used in screening campaigns against libraries of compounds to discover leads for further drug development.
Keywords: acridinium ester label, chemiluminescent hybridization protection assay, high-throughput screening, kinetic characterization, lead discovery, Staphylococcus aureus DNA ligase
Abbreviations: AE, acridinium ester; DTT, dithiothreitol; HPA, hybridization protection assay; HTS, high-throughput screening; NHS, N-hydroxysuccinamide; RLU, relative light units
INTRODUCTION
DNA ligases are ubiquitous enzymes that catalyse the formation of a phosphodiester bond between a 5′-phosphate and a 3′-hydroxy group in nicked DNA [1]. These nicks can form during DNA replication, recombination and damage (see [2–5] and references cited therein). Ligases can be classified depending upon their utilization of one of two cofactors during catalysis, NAD+ or ATP. NAD+-dependent ligases are found exclusively in bacteria [6–9], making them a potential target for antibacterial compounds. ATP-dependent ligases have been found in bacteriophages [10], eubacteria [11], archaea [12,13] and viruses [14,15]. Although there is a difference in cofactor requirement, they both use the same catalytic mechanism [16]. The hydrolysis of the cofactor yields an enzyme–adenylate intermediate in which AMP is attached covalently to the α-amino group of a lysine residue within the active site, with the loss of either pyrophosphate or NMN. The AMP moiety is then transferred from the active site lysine to the free 5′-phosphoryl group at the position of the nick in the DNA strand. This process ends with phosphodiester bond formation and subsequent release of AMP from the adenylated DNA intermediate (Scheme 1). Despite this similarity in mechanism, ATP- and NAD+-dependent DNA ligases are structurally very different from each other, apart from the conserved sequence at the active site of both enzymes known as the KXDG (Lys-Xaa-Asp-Gly) motif [17,18], with the lysine residue in the KXDG sequence being the site of adenylation. High-resolution crystal structures of both types of ligase have been solved (bacteriophage T7 in complex with ATP [19]; the adenylation domain of the NAD+-dependent DNA ligase from Bacillus stearothermophilus [20]; the NAD+-dependent DNA ligase from Thermus filiformis [21]; and the Chlorella virus DNA ligase [22]). Both types of ligases are composed of two domains, with the N-terminal domain containing the active-site lysine where adenylation occurs and the C-terminal domain containing the site of DNA binding.
Scheme 1. Schematic representation of the reaction mechanism of NAD+-dependent ligases.

(a) Formation of an enzyme–adenylate complex and NMN. (b) Activation of 5′-phosphate at the site of a nick in the DNA substrate. (c) Nick closure in which a covalent bond is formed between an adjacent 3′-hydroxy group and activated 5′-phosphate in duplex DNA structures with the release of AMP.
The most commonly used method to measure the kinetics of catalysis of DNA ligases uses radiolabelled NAD+ or ATP. Subsequent physical separation of the components of the reaction by gel electrophoresis and visualization of radioactivity by autoradiography can be used to determine the rate of catalysis (e.g. [6,10,18,22,23]). This method for assay of enzyme activity, although very sensitive, has disadvantages in that it is time-consuming and is not amenable for assaying large numbers of samples. A novel assay has been developed that overcomes the drawbacks with the radiometric assay. The assay is a homogeneous chemiluminescence-based HPA (hybridization protection assay) ([24,25] and reviewed in [26]). This type of assay has been applied successfully to measure the activities of enzymes, including human telomerase prepared from cell-free extracts [27,28] and glucose oxidase [29]. The assay developed for Staphylococcus aureus DNA ligase utilizes an AE (acridinium ester)-labelled nicked DNA substrate. The covalent attachment of the AE label to the DNA substrate can be achieved by using a well-known technique such as NHS (N-hydroxysuccinamide) ester labelling [30,31]. The AE label attached to oligonucleotides has been shown to exhibit the same chemiluminescence characteristics as free label [30]. Upon labelling of the oligonucleotide, it is allowed to anneal to two complementary strands forming a nicked substrate. In this substrate form, the AE label can be removed without the generation of chemiluminescence under mild alkali conditions at 40 °C (where the substrate would undergo melting). In contrast, ligation stabilizes the duplex and protects the AE label from this mild alkaline hydrolysis, but the AE label in both substrate and ligated product are susceptible to the alkaline peroxide conditions that generate chemiluminescence. These properties permit a two-step discrimination between substrate and product (Scheme 2), and have general applicability for this and other nucleotide-metabolizing enzymes. Quantification of the protected AE label (in ligated DNA) allows the determination of the amount of product formed without the need for the physical separation of residual unligated DNA substrate or enzyme. The use of this technology has provided an insight into the kinetics and mechanism of action of the S. aureus NAD+-dependent ligase.
Scheme 2. Schematic representation of the HPA for S. aureus DNA ligase.

After ligation of the AE-labelled DNA substrate, the AE label is rendered less susceptible to alkaline hydrolysis. The label from non-ligated DNA is preferentially hydrolysed by mild alkaline stop/selection reagent [360 mM Na2B4O7 containing 240 mM HCl and 0.07% (v/v) Triton X-102, pH 8] with incubation at 40 °C in a water bath or heating block. Addition of 0.5 M NaOH containing 0.3% (v/v) H2O2 results in the decomposition of the AE label from ligated DNA and release of light at 430 nm (chemiluminescence).
We have also extended the scope of the assay by its optimization for use in HTS (high-throughput screening). This required significant miniaturization of the assay to conserve on reagents without compromising sensitivity. An assay format in 384-well microtitre plates has been achieved with a reaction volume of 20 μl. Miniaturization of the assay was an essential prerequisite to the screening of libraries of compounds to find molecules that have the potential to be developed into drugs and has produced an ultrasensitive low-volume HTS-compatible assay for a DNA ligase.
MATERIALS AND METHODS
Materials
ATP, DTT (dithiothreitol), EDTA, H2O2 (30%, v/v), KCl, MgCl2, NAD+, NaOH and all buffer reagents were purchased from Sigma Chemical Co. (Poole, Dorset, U.K.), Ni-chelate chromatography media and a 15Q ion-exchange column was from Amersham Biosciences. The 36-base AE-labelled DNA oligonucleotide was purchased from Molecular Light Technologies (Cardiff, U.K.) and its 18-base complements were from Oswel Research Products (Southampton, U.K.). SDS/PAGE gels and reagents were purchased from Invitrogen. Assay buffers used were at a concentration of 10 mM [sodium acetate (pH 5.2 and 5.6), Hepes (pH 6.0, 6.5, 7.1 and 7.4) and Tris/HCl (pH 8.0), all containing 4 mM MgCl2, 25 μM NAD+, 10 μM lithium dodecyl sulphate and 5 μg/ml BSA]. The mild alkaline stop/selection reagent was 360 mM Na2B4O7 containing 240 mM HCl and 0.07% (v/v) Triton X-102, pH 8. The chemiluminescence-generating flash reagent contained 0.5 M NaOH containing 0.3% (v/v) H2O2, and was prepared fresh weekly. White clear-bottom 384-well microtitre plates were purchased from Nunc (Roskilde, Denmark). The detector used was a Galaxy Lumistar chemiluminescence plate reader (BMG LabTechnologies, Aylesbury, Bucks., U.K.).
Oligomeric AE-labelled DNA substrate
The oligomeric AE-labelled DNA substrate for NAD+-dependent S. aureus DNA ligase comprised two 18-base oligonucleotides annealed to a complementary 36-base oligonucleotide. The latter contained the site of AE-labelling. Annealing of the AE-labelled 36-base oligonucleotide (5′-CGTCCCACACCAAAGCCCTCCATTATAT*TCCTTCTA-3′, where * is the site of the AE label) and two complements each being 18-base oligonucleotides (3′-GCAGGGTGTGGTTTCGGG-5′ and 3′AIITAATATAAIIAAIAT-5′) was performed as follows: the AE-labelled oligonucleotide [200 pmol (20 μl of 10 μM) in storage buffer: 10 mM succinic acid containing 10 mM LiOH and 3.7 mM lithium dodecyl sulphate, pH 4.8] was mixed with the complements [200 pmol of each (2 μl of 100 μM) in 0.1 M Tris/HCl buffer, pH 8.0, containing 10 mM EDTA]. The mixture was heated to 60 °C for 5 min, followed by slow cooling to room temperature (25 °C). The final AE-labelled DNA substrate stock was stored at −20 °C at a concentration of 8.3 μM.
S. aureus DNA ligase
The S. aureus ligA gene was identified by BLAST homology to the Escherichia coli NAD+ dependent ligase P15042 (esco13942). The ligA gene was isolated from S. aureus RN4220 with the following oligonucleotides: 5′-GCTAGCGGGATGGCTGATTTATCGTCTC-3′ and 5′-CTCGAGCTAACTATTTAATTCATTTTGCTTATCTA-3′ which contain NheI and XhoI sites respectively at their 5′ ends (underlined). The subsequent PCR product was inserted into a holding vector, pCR-Blunt (Invitrogen). This was confirmed by sequencing the plasmid. The S. aureus ligA gene has an internal NdeI site, so a linker, which has a 5′ NdeI overhang and blunt 3′ end, was used to cover the 5′ end of the gene. The remainder of the gene was PCR-amplified from the holding vector using an internal primer at the 5′ end and a HindIII-containing primer at the 3′ end. A 3-piece ligation was performed with pET28a(+) cut with NdeI/HindIII to encode an N-terminal His6-tagged protein. The S. aureus ligA construct has been shown by sequencing to have one codon change (Asn→Lys, a noted polymorphism among S. aureus strains) and two silent mutations. The changes are T225→A (Asn→Lys), G1308→A and C1446→T, counting from the start ATG codon. S. aureus DNA ligase was produced in E. coli strain BL21(DE3) harbouring the tRNA vector pRR692. Transformed cells were grown at 37 °C in LB (Luria–Bertani) medium supplemented with 50 μg/ml kanamycin to a D600 of 0.7. The culture was induced by addition of 0.5 mM IPTG (isopropyl β-D-thiogalactoside) and incubated for 3 h at 37 °C. Cells were harvested by centrifugation at 5000 g for 20 min at 4 °C, suspended in 50 mM phosphate buffer (pH 7.5), containing 0.5 M NaCl and 5 mM 2-mercaptoethanol, and lysed by sonication. Insoluble material was removed by centrifugation at 20000 g for 30 min at 4 °C, followed by filtration through a 0.45-μm-pore-size syringe filter. The clarified solution was applied to a Ni-chelate column, unbound material being washed by application of the loading buffer. Bound protein was eluted by application of a linear gradient of loading buffer to 500 mM imidazole. Fractions containing S. aureus DNA ligase predominantly were pooled, dialysed against 10 mM Tris/HCl buffer, pH 7.4, containing 50 mM KCl, 0.1 mM EDTA and 1 mM DTT. The protein was concentrated to 2.9 mg/ml, diluted with an equal volume of glycerol and stored at −80 °C. A sample of the stored protein (0.5 ml at 2.9 mg/ml in storage buffer) was dialysed against 50 mM Tris/HCl buffer, pH 7.4, containing 5 mM 2-mercaptoethanol and 20% (v/v) glycerol, and purified using a 1 ml 15Q ion-exchange column equilibrated with 50 mM Tris/HCl buffer, pH 7.4, containing 5 mM 2-mercaptoethanol and 20% (v/v) glycerol. Protein was eluted by application of a linear gradient of loading buffer to 1 M NaCl. Pure fractions were pooled and dialysed against 10 mM Tris/HCl buffer, pH 7.4, containing 50 mM KCl, 0.1 mM EDTA and 1 mM DTT. The protein was finally concentrated to 3.6 mg/ml, diluted with an equal volume of glycerol and stored at −80 °C. Purity of protein was judged by SDS/PAGE.
Detection of chemiluminescence released upon hydrolysis of the AE label on the DNA substrate
Chemiluminescence assays were carried out in white clear-bottom 384-well microtitre plates. The chemiluminescence plate reader was designed so that reagents could be injected internally while simultaneously measuring chemiluminescence from below the plate. The sensitivity and dynamic range of the plate reader was established by measuring the amount of light emitted at 430 nm from the reactions of solutions of AE-labelled DNA substrate between 0.5 and 10 nM [in volumes of 20 μl in Hepes (pH 7.4) assay buffer (see above)] with an equal volume of a solution of chemiluminescence-generating flash reagent. The lifetimes of chemiluminescence generated as a result of these reactions were determined by collection of light over a 2 s period at intervals of 0.1 s. These experiments were also performed in the presence of S. aureus DNA ligase at concentrations used in the kinetic studies (50 pM).
The kinetics of the mild alkaline, non-chemiluminescent hydrolysis of AE label on substrate and product were investigated independently using 10 nM solutions of each. The solutions of substrate or product (200 μl of 10 nM) were incubated in stop/selection reagent (200 μl; see above) with incubation at 40 °C in a water bath. At approx. 10 min intervals, 20 μl samples were removed and the residual content of AE label was determined by measuring the chemiluminescence emitted upon the addition of flash reagent.
Kinetic assays
Initially, time-course experiments were carried out at room temperature in Hepes (pH 7.4) assay buffer using 0.5 nM enzyme and 10 nM substrate. The pH-dependence of kcat, Km and kcat/Km for catalysis of the AE-labelled DNA substrate by the NAD+-dependent S. aureus DNA ligase were determined from kinetic studies carried out in the following buffers at room temperature and a concentration of 10 mM [sodium acetate (pH 5.2 and 5.6), Hepes (pH 6.0, 6.5, 7.1 and 7.4) and Tris/HCl (pH 8.0); see above]. Reactions were initiated by the addition of enzyme (50 pM final enzyme concentration) to solutions of buffer containing substrate (2–10 nM). Assays were terminated by the addition of an equal volume of a solution of stop/selection reagent followed by incubation at 40 °C in a water bath or heating block for 60 min. AE label on catalytically generated product that contained protected AE label was quantified by measuring the amount of chemiluminescence released upon reaction of the stopped assay mix with flash reagent.
The Km value for S. aureus DNA ligase and NAD+ was determined, as well as IC50 values for two inhibitors of NAD+-dependent DNA ligase from E. coli [32], namely doxorubicin (1) and chloroquine diphosphate (2) in Hepes (pH 7.4) assay buffer (see Figure 7). Kinetic data were analysed using Grafit software (version 4.0.13). Values for Km were determined using the Michaelis–Menten fit (eqn 1). For determination of kcat, eqn (2) was used ([E]T being total protein concentration) and for determination of values for IC50, eqn (3) was used (v0 being the rate in the absence of inhibitor). All experiments were carried out in duplicate. Characterization of the pH-dependent data was by regression of 1/kcat and 1/kcat/Km against [H+], since both these profiles were composed of a single ionization curve [33].
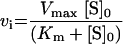 |
(1) |
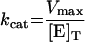 |
(2) |
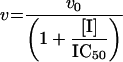 |
(3) |
Figure 7. Structures of (1) doxorubicin and (2) chloroquine diphosphate, two inhibitors of S. aureus DNA ligase.

The IC50 values for inhibition of S. aureus DNA ligase have been determined against these inhibitors using the HPA. For (1), IC50=3.30±0.86 μM; for (2), IC50=1.40±0.07 μM.
HTS assay development
For the HTS-compatible assay, the development buffer used was Hepes (pH 7.4) containing 1% (v/v) DMSO. In these assays, the concentration of enzyme used was 50 pM and substrate concentration was 2 nM (approx. Km). The ratio of signal/background was determined from assays using 96 wells of a 384-well microtitre plate containing no inhibitor (high control) and another 96 wells containing chloroquine diphosphate at 50 μM (low control, [chloroquine diphosphate]> >IC50). The quality and robustness of the assay, represented as Z′ [34] was calculated using eqn (4):
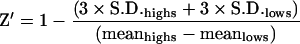 |
(4) |
RESULTS
Cloning, expression and purification of S. aureus DNA ligase
The expression system used in the cloning and production of S. aureus DNA ligase in E. coli yielded approx. 25 mg of S. aureus DNA ligase protein from 500 ml of cell culture. Upon Ni-chelate and ion-exchange chromatography, high-purity DNA ligase was obtained as judged by SDS/PAGE (Figure 1).
Figure 1. Coomassie-Blue-stained SDS/PAGE gel of the final purified S. aureus DNA ligase.

Protein was expressed and purified as described in the Materials and methods section. The positions and sizes (in kDa) of the of marker proteins are shown on the left. Lane 1, protein size markers; lane 2, 2 μg of total protein. The band at approx. 70 kDa is that of S. aureus DNA ligase.
Design and characterization of the AE-labelled oligomeric DNA substrate
The development of an assay for measuring S. aureus DNA ligase activity that would allow its biochemical characterization and also be suitable for HTS has been undertaken and is reported in the present paper. The chemiluminescence-based HPA that has been developed also has general applicability to enzymes of this class. The AE label can be attached covalently to a number of sites in target molecules using well known labelling chemistry, e.g. NHS ester labelling [30,31], and due to its small size, there is minimal effect on the overall structure of the DNA substrate used in the present study. The substrate is stable in its storage buffer at room temperature for an adequate length of time, making it suitable for use in typical HTS campaigns where reagent stability is of significant advantage as it would allow a single batch of substrate to be prepared and used for the entire HTS campaign.
The AE-labelled DNA substrate was characterized before its use in any enzyme assays. A standard curve relating the concentration of AE label (and therefore substrate or product) and chemiluminescence was obtained. A linear relationship between chemiluminescence and concentration of AE-labelled DNA substrate up to 10 nM was shown to exist (Figure 2a). This relationship was found to hold when similar experiments were performed in the presence of S. aureus DNA ligase at concentrations used in subsequent kinetic studies (50 pM). In all assays, the concentration of substrate consumed was below 10 nM where chemiluminescence could be directly correlated to AE label concentration and hence that of substrate or product. The lifetime of the emitted light released upon addition of flash reagent to a solution of the AE-labelled substrate was determined by collecting light over a period of 2 s in 0.2 s intervals over a range of substrate concentrations (0.1–10 nM). This allowed the determination of an appropriate read-time per well to ensure capture of all emitted light (Figure 2b). A 2 s collection time was deemed sufficient to capture all emitted light from the hydrolysis of AE label in both substrate and product up to 10 nM, and was used in all assays that involved the kinetic characterization of S. aureus DNA ligase.
Figure 2. (a) Demonstration of the linear dependence of chemiluminescence (RLU) on AE-labelled DNA substrate concentration in the absence of S. aureus DNA ligase (•) and presence of 50 pM S. aureus DNA ligase (^) and (b) kinetics of chemiluminescence (RLU) generation.

(a) A linear relationship was shown to exist for the concentration of AE-labelled DNA substrate up to 10 nM and approx. 2×105 RLU in the presence (50 pM) and absence of S. aureus DNA ligase. Reactions were carried out in volumes of 20 μl in Hepes (pH 7.4) assay buffer. Chemiluminescence generated upon addition of 20 μl of a solution of flash reagent was detected over a 2 s period. The calibration curve constructed using the closed circles in (a) was used in converting RLU into AE label concentration and hence that of substrate/product. (b) Reaction of AE-labelled DNA substrate in Hepes (pH 7.4) assay buffer with chemiluminescence-generating flash reagent results in the release of light at 430 nm. The concentration of AE-labelled substrate investigated was in the 0.1–10 nM range. In all cases, the lifetime of the emitted light was under 2 s.
Upon sealing of the nick in the substrate catalysed by S. aureus DNA ligase, a duplex is formed, rendering the AE label less susceptible to mild alkaline hydrolysis. In a typical catalytic assay where not all of the substrate underwent ligation, quantification of the amount of product formed required the removal of AE label from the substrate without affecting the label on product. A ‘selection’ step was therefore incorporated into the assay protocol. Under mild alkaline conditions (in the presence of mild alkali stop/selection reagent) the AE label on substrate was preferentially hydrolysed, whereas that on product was protected. To ensure specificity of the selection step, the substrate was designed to have a low melting temperature (<40 °C), but when ligated to form product, to have a significantly higher melting temperature (>80 °C). Thus, if the selection step was carried out at 40 °C, this would allow rapid removal of the AE label from substrate as it would undergo melting, enabling the mild alkali stop/selection reagent to hydrolyse the AE label more rapidly, but unable to do so in product. This difference in melting temperature was made possible by the inclusion of the base inosine into the substrate. To demonstrate the selectivity of the HPA approach, the length of time required to discriminate effectively between AE label protected in product from unprotected AE label in substrate was determined. This was performed by comparing the kinetics of the hydrolysis of the AE label on 2 nM substrate with the hydrolysis of AE label on 2 nM product (that had been generated catalytically). Solutions of substrate or product were incubated in mild alkali stop/selection reagent and aliquots removed at regular time intervals. These were analysed for AE label content by reaction with the light-generating alkaline peroxide treatment using the flash reagent. The dark decay of AE label from substrate was shown to be exponential in nature, with complete removal of the AE label on substrate within 60 min. However, under the same conditions, AE label present in product was essentially completely protected from hydrolysis (Figure 3a). The considerable difference in the kinetics of hydrolysis of the AE label on product and substrate permits the selective removal of the AE label from substrate in the presence of product. This is of benefit, as it alleviates the need for the physical separation of product from substrate in the assay mix before its quantification. The quantification of AE label has been shown to be possible in the presence of other cellular proteins, e.g. as in cell-free extracts of telomerase [27,28]. The exponential decay of AE label in substrate and its essentially full protection in product, allows for an increase in the signal/background ratio with time and a steady value of 200:1 is achieved after a 60 min incubation with the stop/selection reagent (Figure 3b).
Figure 3. (a) Time-course of the hydrolysis of the AE label on unligated (•) and ligated (▪) DNA substrate and (b) the dependence of signal/background of the chemiluminescent HPA for NAD+-dependent S. aureus ligase.

(a) Substrate or product (10 nM) in Hepes (pH 7.4) assay buffer were incubated in mild alkaline stop/selection reagent at 40 °C in a water bath or heating block. The AE label content of samples at various times was quantified by measuring the amount of chemiluminescence (RLU) generated upon reaction with chemiluminescence-generating flash reagent. Hydrolysis of the AE label in substrate by the stop/selection reagent is exponential in nature, but is essentially fully protected in product. (b) Values for signal/background ratio at regular intervals were calculated from the chemiluminescence (RLU) of unligated (•) and ligated (▪) AE-labelled DNA substrate from (a). There is an increase in the signal/background ratio over time, and a steady value of 200:1 after 60 min of incubation with stop/selection reagent was achieved.
Kinetic characterization of S. aureus DNA ligase using the oligomeric AE-labelled DNA substrate
The HPA has been used to characterize the kinetics of catalysis of the AE-labelled DNA substrate by S. aureus DNA ligase. Initially, a time-course experiment at fixed enzyme and substrate concentrations was performed to determine the sensitivity of the assay. Figure 4 shows a typical time course for the S. aureus DNA-ligase-catalysed ligation of the AE-labelled DNA substrate in Hepes (pH 7.4) assay buffer that used 0.5 nM enzyme and 10 nM substrate. A plateau in the amount of chemiluminescence [approx. 200000 RLU (relative light units)] is reached with approx. 45 min of incubation of enzyme and substrate, and is indicative of conversion of essentially all substrate into product. To confirm complete ligation in this case, the predicted yield of chemiluminescence from the hydrolysis of a 10 nM solution of AE-labelled DNA (see calibration graph, Figure 2a) was similar to that obtained (approx. 200000 RLU). This experiment also established the sensitivity range of the assay. In an assay format with a 20 μl assay volume, approx. 1000 RLU can be readily detected, which equated to the detection of approx. 1 fmol of product and is comparable in sensitivity with the usual radiometric assays used for measuring DNA ligase catalytic activities (e.g. [6,10,18,22,23]).
Figure 4. Time course of S. aureus DNA ligase catalysis of the AE-labelled DNA substrate using the HPA.

The assay was carried out using 0.5 nM enzyme and 10 nM AE-labelled DNA substrate in Hepes (pH 7.4) assay buffer. Complete ligation of the AE-labelled substrate is obtained after approx. 60 min of incubation. Upon full ligation of 10 nM AE-labelled substrate, approx. 2×105 RLU were obtained upon analysis, confirming complete ligation.
Having established the sensitivity of the assay, additional studies were carried out that involved the determination of the Km values for S. aureus DNA ligase for both NAD+ and the AE-labelled DNA substrate. These studies showed the existence of hyperbolic dependencies of rate of product formation with an increase in NAD+ and AE-labelled DNA substrate concentration (Figure 5). The value for Km for NAD+ was determined to be 2.75±0.1 μM and Vmax was (6.20±0.05)×10−13 M/s. For the AE-labelled DNA substrate, the Km was determined to be 2.5±0.2 nM and Vmax was (6.15±0.05)×10−13 M/s. In addition, the pH-dependence of kcat and kcat/Km for ligation have been determined (Figure 6). These studies can be used to reveal the pKa value of group(s) that are kinetically influential within the free enzyme and substrate (kcat/Km) and enzyme–substrate complexes (kcat) [35]. The pH range that was chosen for study was limited to a lower value of pH 5 (due to the loss of catalytic competence of the enzyme) and an upper limit of pH 8 (due to the instability of the substrate, the AE label being susceptible to alkaline hydrolysis). There is an increase in the catalytic competence of the enzyme, reflected by an increase in kcat/Km with increase in pH. The curve has been interpreted as being part of a simple sigmoid and thus a single kinetically influential ionization, the characterizing pKa determined as 7.1±0.1 (Figure 6a). The pH-dependence of kcat also has a similar shape to that for kcat/Km and has been analysed as a single sigmoid too. The characterizing pKa for this curve has shifted slightly downwards in pH to 6.6±0.1 (Figure 6b). The pH-dependence of Km for the enzyme and the AE-labelled DNA substrate remains in the ∼3 nM region across the pH range over which the kinetic experiments were performed. The IC50 values have also been determined for S. aureus DNA ligase using two reported inhibitors of E. coli DNA ligase [32] using the HPA. These IC50 values were determined as 3.30±0.86 μM for doxorubicin (1) and 1.40±0.07 μM for chloroquine diphosphate (2) (Figure 7).
Figure 5. Demonstration of the adherence to the Michaelis–Menten equation of (a) catalysis of NAD+ and (b) ligation of the AE-labelled DNA substrate by S. aureus DNA ligase.

(a) Assays were carried out in Hepes (pH 7.4) assay buffer containing 10 nM AE-labelled DNA substrate and 50 pM enzyme, with variation in NAD+ concentration between 2 and 45 μM. The points are experimental and the continuous lines correspond to vi=Vmax[S]0/(Km+[S]0) with Km=2.75±0.1 μM and Vmax=(6.20±0.05)×10−13 M/s. (b) Assays were carried out in Hepes (pH 7.4) assay buffer containing 25 μM NAD+, 50 pM enzyme, with variation in the concentration of AE-labelled DNA between 1 and 10 nM. The points are experimental and the continuous lines correspond to vi=Vmax[S]0/(Km+[S]0) with Km=2.5±0.2 nM and Vmax=(6.15±0.05)×10−13 M/s.
Figure 6. pH-dependence of (a) kcat/Km and (b) kcat for S. aureus DNA-ligase-catalysed ligation of the AE-labelled DNA substrate.

These studies were carried out in the following assay buffers at room temperature: sodium acetate (pH 5.2 and 5.6), Hepes (pH 6.0, 6.5, 7.1 and 7.4) and Tris/HCl (pH 8.0). Reactions were initiated by the addition of enzyme (50 pM final enzyme concentration) to solutions of buffer containing substrate (2–10 nM). The points are experimental and the lines are theoretical for the single ionization equation k=k~/(1+[H+]/Ka), where k=kcat or k=kcat/Km and k~=k~cat or k~cat/K~m (pH-independent kinetic parameters) (a) pKa=7.1±0.1, k~cat/K~m=(4.1±0.1)× 105 M−1·s−1 and (b) pKa=6.6±0.1, k~cat=0.009±0.001 s−1.
Development of an HTS-compatible assay for S. aureus DNA ligase
As well as utilizing the S. aureus DNA ligase HPA in mechanistic studies (see above), the assay has also been optimized to be HTS-compatible. For HTS development, the HPA was miniaturized to a final reaction volume of 20 μl in 384-well microtitre plates. Having established the requirement for a 2 s read-time per well, the total time required to read a 384-well microtitre plate would amount to approx. 13 min. This type of throughput would enable >25000 assays to be performed per day on an automated screening platform. The overall quality and robustness of the assay was determined by performing assays in a significant number of wells (96 wells) that were known to give no inhibition (highs) in Hepes (pH 7.4) assay buffer containing 1% (v/v) DMSO (since in HTS campaigns, the compounds used are often dissolved in DMSO with its final concentration typically being 1%, v/v). An identical number of assays were carried out containing inhibitor at a sufficiently high concentration (chloroquine diphosphate at 50 μM) that would be expected to give full inhibition (lows). The index that is used to measure the robustness of the assay quantitatively, Z′, can be calculated using eqn (4). The closer the value for Z′ to 1, the more robust the assay [34]. In the current assay format, a value for Z′ of 0.75 was calculated.
DISCUSSION
The chemiluminescence-based assay that has been developed has successfully been utilized in the kinetic characterization of S. aureus DNA ligase and has also been shown to possess excellent robustness characteristics that is required for an assay to have HTS-compatibility. It has many advantages over the more commonly used radiometric assays that have been used to date for measuring DNA ligase activities (e.g. [6,10,18,22,23]).
Owing to the broad substrate specificity of DNA ligases, there was considerable flexibility available in designing the sequence of the oligonucleotide substrate. In particular, the substrate was designed to have a relatively low melting temperature (<40 °C), whereas ligated product had a significantly higher melting temperature (>80 °C). As a result of this, incubation of the assay contents at 40 °C resulted in any unligated substrate to undergo melting, but leaving product duplexed. In the presence of the mild alkaline selection reagent, removal of the AE label on substrate was accelerated, leaving that on product essentially intact. Thus, in a typical assay where only a small proportion of the substrate was converted into product, it was possible to selectively remove the AE label from unligated substrate during the selection step leaving AE label on product essentially fully intact. This important facet of the current assay that has been developed allows the quantification of ligated product without any separation steps; in particular, the removal of residual unligated substrate is unnecessary. This increases throughput significantly, and has proved to be instrumental in the successful application of this assay in an HTS environment. In addition, the presence of the S. aureus DNA ligase at concentrations used in typical kinetic studies was demonstrated experimentally not to interfere with the kinetics or yield of chemiluminescence. From the analysis of samples of AE-labelled substrate and product, as well as the time-course experiments, the sensitivity of the assay was established, showing that femtomole quantities of product can be detected, which puts the chemiluminescence assay on a par with alternative radiometric assays (e.g. [6,10,18,22,23]).
A study of the pH-dependence of kcat/Km can be used to reveal kinetically influential ionizations in free enzyme and substrate and for that of kcat kinetically influential ionizations in enzyme–substrate complexes [35]. The pH-dependent kinetic data obtained for DNA ligase catalysis with respect to the AE-labelled DNA substrate was only obtained between pH values of 5–8. The lower limit being loss of catalytic competence of the enzyme and the upper limit due to substrate instability. There is an increase in activity of the enzyme with increase in pH. This has been interpreted as being part of a single sigmoid characterized by a group with a pKa of 7.1±0.1 (Figure 6a). The likely group for this ionization is the active-site lysine residue that is known to be involved in catalysis in this class of enzyme. Although this pKa is significantly lower than the typical pKa of the ε-amino group of lysine (pKa∼10), a similar finding has been reported for the T4 bacteriophage DNA ligase in which its active-site lysine group was shown to be associated with a pKa of 8.4 [36]. This abnormally low value was shown to be due to electrostatic effects of nearby positively charged amino acid side chains within the active site. Similar findings have been shown to exist in other enzymes, e.g. cysteine proteinases of the papain family that have been shown to have active-site thiol groups with pKa values in the 3–4 range, although the typical free thiol group has a pKa of approx. 9 [37,38]. Validation of this in the case of S. aureus DNA ligase would require confirmation by a series of chemical modification studies or by generating site-directed mutants of the enzyme of key residues implicated by the results shown in the present paper.
In the case of the pH-dependence of kcat, which reveals kinetically influential ionizations in the enzyme–substrate complex and the nature of the environment of the group involved in catalysis, the curve is similar to that obtained for kcat/Km (Figure 6a) over the same pH range in that it has been defined as a single sigmoid, but with a pKa of 6.6±0.1 (Figure 6b). As there is no significant change in the shape of the pH profiles, this suggests that the environment of the group is not too dissimilar in the free enzyme and enzyme–substrate complex state. The kcat for the overall rate of ligation of the AE-labelled DNA substrate by S. aureus DNA ligase (pH-independent kcat=0.009 s−1) is essentially the same as that for the ATP-dependent DNA ligase from Neisseria meningitidis (0.008 s−1) [39]. Values for kcat are known to vary to some extent for DNA ligases from other sources (0.02 s−1 for NAD+-dependent DNA ligases from E. coli [1,8]; 0.0025 s−1 for ATP-dependent T4 ligase [40]). These values of kcat are, however, considerably lower than those found for biological catalysts (enzymes [41] and catalytic antibodies generated by active immunization [42]). Also, the fact that kcat and kcat/Km show similar pH profiles over the pH range investigated, the value of Km for S. aureus DNA ligase and AE-labelled DNA substrate used in the present study does not change, remaining at approx. 3 nM. A value for Km for DNA substrates in the low nanomolar region has been shown to exist for a number of DNA ligase systems from a variety of biological sources (NAD+-dependent DNA ligases: 76 nM for Aquifex aeolicus [7], 30 –60 nM for E. coli [43] and 87 nM for Thermus thermophilus [44]; ATP-dependent DNA ligases: 9 nM for vaccinia virus [15] and 30 nM for N. meningitidis [39]).
Further validation of the AE-labelling technology was performed by determining the values of IC50 for the inhibition of S. aureus DNA ligase by two inhibitors of DNA ligase from E. coli [32], namely doxorubicin (1) and chloroquine diphosphate (2) (Figure 7). The IC50 for doxorubicin inhibition of S. aureus DNA ligase (3.30±0.86 μM) is similar to that reported for the NAD+-dependent DNA ligase from E. coli [32] (1.30±0.30 μM). This inhibitor is not selective against the ATP-dependent DNA ligases from T4 bacteriophage (2.0±0.4 μM) and human type I ligase (1.8±0.4 μM) [32], and therefore might not be an appropriate molecule to pursue for selective NAD+-dependent DNA ligase inhibitors. However, the present study shows that chloroquine diphosphate is a significantly more selective inhibitor of the NAD+-dependent ligase from S. aureus (1.40±0.07 μM) than of that of E. coli (53±3 μM), and also exhibits excellent selectivity against the ATP-dependent DNA ligases from T4 bacteriophage (>1500 μM) and human type I ligase (720±80 μM). More recently, pyridochromanones have been identified as specific and potent inhibitors of NAD+-dependent DNA ligase from E. coli [45].
Typically an HTS would be performed using a large number of microtitre (384-well) or nanotitre (1536-well) plates to minimize the requirements for reagents. One of the main objectives of a HTS campaign carried out on a target of interest is in discovering compounds that have the potential for further development in the drug-discovery process. In order to make an HTS an effective exercise, a number of key requirements need to be fulfilled. The cost of performing such a large number of assays is of paramount importance, in particular if the reagents are expensive, as a large number of assays would be carried out (typically 500000). Thus miniaturization of the assay is an integral part of development of HTS-compatible assays (typically to <50 μl). An important aspect of HTS assay development is to optimize the assay conditions in order to obtain a high signal/background ratio. This would enable compounds that are genuine inhibitors of the target to be identified with confidence from noise within the signal window. The current assay format offers a signal/background ratio of 200:1. This is made possible because of the essentially full removal of AE label from residual unligated DNA substrate, with protection of the AE label in product during the 60 min selection step (Figure 3). The quantitative measurement for assay robustness, Z′ (eqn 4), is a coefficient that is reflective of both the assay signal dynamic range and the data variation associated with signal measurements. It is a dimensionless, simple statistical characteristic for an HTS assay, and in the case of the S. aureus DNA ligase assay, has a value of 0.75, indicative of excellent robustness, thus ensuring that compounds that are identified in the current assay would be genuine inhibitors of catalytic activity.
The chemiluminescence-based technology has been successfully exploited in developing an HPA for S. aureus DNA ligase assay exhibits similar sensitivity to the commonly used radiometric assays. The assay has been used to characterize the kinetics of catalysis of the enzyme and has also been demonstrated to be HTS-compatible. This required the assay to be miniaturized to a sufficiently small volume to conserve reagents without compromising sensitivity and quality of the data generated. The development of the HTS assay allows the rapid turnover that is typically required for screening against a large number of compounds. A typical HTS against a library of compounds in the region of 500000 would only require milligram quantities of enzyme with the assay format described in the present study. Developing assays that are amenable to HTS is necessary to facilitate the discovery of novel molecular scaffolds that have the desired properties to be the starting point of potential drug treatments.
Acknowledgments
We thank Dr David Tew, Dr Ian Weeks and Dr Robert Copeland for helpful discussions.
References
- 1.Lehman I. R. DNA ligase: structure, mechanism, and function. Science. 1974;186:790–797. doi: 10.1126/science.186.4166.790. [DOI] [PubMed] [Google Scholar]
- 2.Cherepanov A. V., de Vries S. Dynamic mechanism of nick recognition by DNA ligase. Eur. J. Biochem. 2002;269:5993–5999. doi: 10.1046/j.1432-1033.2002.03309.x. [DOI] [PubMed] [Google Scholar]
- 3.Wilkinson A., Day J., Bowater R. Bacterial DNA ligases. Mol. Microbiol. 2001;40:1241–1248. doi: 10.1046/j.1365-2958.2001.02479.x. [DOI] [PubMed] [Google Scholar]
- 4.Timson D. J., Singleton M. R., Wigley D. B. DNA ligases in the repair and replication of DNA. Mutat. Res. 2000;460:301–318. doi: 10.1016/s0921-8777(00)00033-1. [DOI] [PubMed] [Google Scholar]
- 5.Engler M. J., Richardson C. C. The Enzymes. New York: Academic Press; 1982. pp. 3–29. [Google Scholar]
- 6.Timson D. J., Wigley D. B. Functional domains of an NAD+-dependent DNA ligase. J. Mol. Biol. 1999;285:73–83. doi: 10.1006/jmbi.1998.2302. [DOI] [PubMed] [Google Scholar]
- 7.Tong J., Barany F., Cao W. Ligation reaction specificities of an NAD+-dependent DNA ligase from the hyperthermophile Aquifex aeolicus. Nucleic Acids Res. 2000;28:1447–1454. doi: 10.1093/nar/28.6.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sriskanda V., Shuman S. A second NAD+-dependent DNA ligase (LigB) in Escherichia coli. Nucleic Acids Res. 2001;29:4930–4934. doi: 10.1093/nar/29.24.4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilkinson A., Sayer H., Bullard D., Smith A., Day J., Kieser T., Bowater R. NAD+-dependent DNA ligases of Mycobacterium tuberculosis and Streptomyces coelicolor. Proteins. 2003;51:321–326. doi: 10.1002/prot.10361. [DOI] [PubMed] [Google Scholar]
- 10.Doherty A. J., Wigley D. B. Functional domains of an ATP-dependent DNA ligase. J. Mol. Biol. 1999;285:63–71. doi: 10.1006/jmbi.1998.2301. [DOI] [PubMed] [Google Scholar]
- 11.Cheng C., Shuman S. Characterization of an ATP-dependent DNA ligase encoded by Haemophilus influenzae. Nucleic Acids Res. 1997;25:1369–1374. doi: 10.1093/nar/25.7.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sriskanda V., Kelman Z., Hurwitz J., Shuman S. Characterization of an ATP-dependent DNA ligase from the thermophilic archaeon Methanobacterium thermoautotrophicum. Nucleic Acids Res. 2000;28:2221–2228. doi: 10.1093/nar/28.11.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lai X., Shao H., Hao F., Huang L. Biochemical characterization of an ATP-dependent DNA ligase from the hyperthermophilic crenarchaeon Sulfolobus shibatae. Extremophiles. 2002;6:469–477. doi: 10.1007/s00792-002-0284-5. [DOI] [PubMed] [Google Scholar]
- 14.Ho C. K., Van Etten J. L., Shuman S. Characterization of an ATP-dependent DNA ligase encoded by Chlorella virus PBCV-1. J. Virol. 1997;71:1931–1937. doi: 10.1128/jvi.71.3.1931-1937.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sekiguchi J., Shuman S. Domain structure of vaccinia DNA ligase. Nucleic Acids Res. 1997;25:727–734. doi: 10.1093/nar/25.4.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doherty A. J., Suh S. W. Structural and mechanistic conservation in DNA ligases. Nucleic Acids Res. 2000;28:4051–4058. doi: 10.1093/nar/28.21.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sriskanda V., Shuman S. Conserved residues in domain Ia are required for the reaction of Escherichia coli DNA ligase with NAD+ J. Biol. Chem. 2002;277:9695–9700. doi: 10.1074/jbc.M111164200. [DOI] [PubMed] [Google Scholar]
- 18.Luo J., Barany F. Identification of essential residues in Thermus thermophilus DNA ligase. Nucleic Acids Res. 1996;24:3079–3085. doi: 10.1093/nar/24.15.3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Subramanya H. S., Doherty A. J., Ashford S. R., Wigley D. B. Crystal structure of an ATP-dependent DNA ligase from bacteriophage T7. Cell. 1996;85:607–615. doi: 10.1016/s0092-8674(00)81260-x. [DOI] [PubMed] [Google Scholar]
- 20.Singleton M. R., Hakansson K., Timson D. J., Wigley D. B. Structure of the adenylation domain of an NAD+-dependent DNA ligase. Structure Fold. Des. 1999;7:35–42. doi: 10.1016/s0969-2126(99)80007-0. [DOI] [PubMed] [Google Scholar]
- 21.Lee J. Y., Chang C., Song H. K., Moon J., Yang J. K., Kim H. K., Kwon S. T., Suh S. W. Crystal structure of NAD+-dependent DNA ligase: modular architecture and functional implications. EMBO J. 2000;19:1119–1129. doi: 10.1093/emboj/19.5.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Odell M., Sriskanda V., Shuman S., Nikolov D. B. Crystal structure of eukaryotic DNA ligase–adenylate illuminates the mechanism of nick sensing and strand joining. Mol. Cell. 2000;6:1183–1193. doi: 10.1016/s1097-2765(00)00115-5. [DOI] [PubMed] [Google Scholar]
- 23.Kaczmarek F. S., Zaniewski R. P., Gootz T. D., Danley D. E., Mansour M. N., Griffor M., Kamath A. V., Cronan M., Mueller J., Sun D., et al. Cloning and functional characterization of an NAD+-dependent DNA ligase from Staphylococcus aureus. J. Bacteriol. 2001;183:3016–3024. doi: 10.1128/JB.183.10.3016-3024.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arnold L. J., Jr, Hammond P. W., Wiese W. A., Nelson N. C. Assay formats involving acridinium-ester-labeled DNA probes. Clin. Chem. 1989;35:1588–1594. [PubMed] [Google Scholar]
- 25.Nelson N. C., Kacian D. L. Chemiluminescent DNA probes: a comparison of the acridinium ester and dioxetane detection systems and their use in clinical diagnostic assays. Clin. Chim. Acta. 1990;17:73–90. doi: 10.1016/0009-8981(90)90304-b. [DOI] [PubMed] [Google Scholar]
- 26.Nelson N. C., Reynolds M. A., Arnold L. J. Detection of acridinium esters by chemiluminescence. In: Kricka L. J., editor. Nonisotopic Probing and Blotting. New York: Academic Press; 1992. pp. 275–310. [Google Scholar]
- 27.Lackey D. B. A homogeneous chemiluminescent assay for telomerase. Anal. Biochem. 1998;263:57–61. doi: 10.1006/abio.1998.2807. [DOI] [PubMed] [Google Scholar]
- 28.Hirose M., Abe-Hashimoto J., Tahara H., Ide T., Yoshimura T. New method to measure telomerase activity by transcription-mediated amplification and hybridization protection assay. Clin. Chem. 1998;44:2446–2452. [PubMed] [Google Scholar]
- 29.Brown R. C., Weeks I., Fisher M., Harbron S., Taylorson C. J., Woodhead J. S. Employment of a phenoxy-substituted acridinium ester as a long-lived chemiluminescent indicator of glucose oxidase activity and its application in an alkaline phosphatase amplification cascade immunoassay. Anal. Biochem. 1998;259:142–151. doi: 10.1006/abio.1998.2604. [DOI] [PubMed] [Google Scholar]
- 30.Weeks I., Beheshti I., McCapra F., Campbell A. K., Woodhead J. S. Acridinium esters as high-specific-activity labels in immunoassay. Clin. Chem. 1983;29:1474–1479. [PubMed] [Google Scholar]
- 31.Nelson N. C., Cheikh A. B., Matsuda E., Becker M. M. Simultaneous detection of multiple nucleic acid targets in a homogeneous format. Biochemistry. 1996;35:8429–8438. doi: 10.1021/bi960085+. [DOI] [PubMed] [Google Scholar]
- 32.Ciarrocchi G., MacPhee D. G., Deady L. W., Tilley L. Specific inhibition of the eubacterial DNA ligase by arylamino compounds. Antimicrob. Agents Chemother. 1999;43:2766–2772. doi: 10.1128/aac.43.11.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brocklehurst K. Physical factors affecting enzyme activity: A. pH-dependent kinetics. In: Engel P. C., editor. Enzymology Labfax. San Diego: Bios Scientific Publishers, Oxford/Academic Press; 1996. pp. 175–198. [Google Scholar]
- 34.Zhang J. H., Chung T. D., Oldenburg K. R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 35.Gul S., Sreedharan S. K., Brocklehurst K. Chichester: John Wiley & Sons; 1998. Enzyme Assays; pp. 14–16. [Google Scholar]
- 36.Arabshahi A., Frey P. A. Standard free energy for the hydrolysis of adenylylated T4 DNA ligase and the apparent pKa of lysine 159. J. Biol. Chem. 1999;274:8586–8688. doi: 10.1074/jbc.274.13.8586. [DOI] [PubMed] [Google Scholar]
- 37.Pinitglang S., Watts A. B., Patel M., Reid J. D., Noble M. A., Gul S., Bokth A., Naeem A., Patel H., Thomas E. W., et al. A classical enzyme active center motif lacks catalytic competence until modulated electrostatically. Biochemistry. 1997;36:9968–9982. doi: 10.1021/bi9705974. [DOI] [PubMed] [Google Scholar]
- 38.Noble M. A., Gul S., Verma C. S., Brocklehurst K. Ionization characteristics and chemical influences of aspartic acid residue 158 of papain and caricain determined by structure-related kinetic and computational techniques: multiple electrostatic modulators of active-centre chemistry. Biochem. J. 2000;351:723–733. [PMC free article] [PubMed] [Google Scholar]
- 39.Magnet S., Blanchard J. S. Mechanistic and kinetic study of the ATP-dependent DNA ligase of Neisseria meningitidis. Biochemistry. 2004;43:710–717. doi: 10.1021/bi0355387. [DOI] [PubMed] [Google Scholar]
- 40.Hall Z. W., Lehman I. R. Enzymatic joining of polynucleotides: VI. Activity of a synthetic adenylylated polydeoxynucleotide in the reaction. J. Biol. Chem. 1969;244:43–47. [PubMed] [Google Scholar]
- 41.Fersht A. Enzyme Structure and Mechanism. New York: W. H. Freeman and Company; 1984. Measurement and magnitude of enzymatic rate constants; p. 152. [Google Scholar]
- 42.Gul S., Sonkaria S., Pinitglang S., Florez-Alvarez J., Hussain S., Thomas E. W., Ostler E. L., Gallacher G., Resmini M., Brocklehurst K. Improvement in hydrolytic antibody activity by change in haptenic structure from phosphate to phosphonate with retention of a common leaving-group determinant: evidence for the ‘flexibility’ hypothesis. Biochem. J. 2003;376:813–821. doi: 10.1042/BJ20030716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Modrich P., Lehman I. R. A steady state kinetic analysis of the reaction catalyzed by the enzyme from Escherichia coli. J. Biol. Chem. 1973;248:7502–7511. [PubMed] [Google Scholar]
- 44.Tong J., Cao W., Barany F. Biochemical properties of a high fidelity DNA ligase from Thermus species AK16D. Nucleic Acids Res. 1999;27:788–794. doi: 10.1093/nar/27.3.788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brotz-Oesterhelt H., Knezevic I., Bartel S., Lampe T., Warnecke-Eberz U., Ziegelbauer K., Habich D., Labischinski H. Specific and potent inhibition of NAD+-dependent DNA ligase by pyridochromanones. J. Biol. Chem. 2003;278:39435–39442. doi: 10.1074/jbc.M306479200. [DOI] [PubMed] [Google Scholar]