

ABSTRACT
Infections caused by antimicrobial-resistant Escherichia coli are the leading cause of death attributed to antimicrobial resistance (AMR) worldwide, and the known AMR mechanisms involve a range of functional proteins. Here, we employed a pan-genome wide association study (GWAS) approach on over 1,000 E. coli isolates from sick dogs collected across the US and Canada and identified a strong statistical association (empirical P < 0.01) of AMR, involving a range of antibiotics to a group 1 capsular (CPS) gene cluster. This cluster included genes under relaxed selection pressure, had several loci missing, and had pseudogenes for other key loci. Furthermore, this cluster is widespread in E. coli and Klebsiella clinical isolates across multiple host species. Earlier studies demonstrated that the octameric CPS polysaccharide export protein Wza can transmit macrolide antibiotics into the E. coli periplasm. We suggest that the CPS in question, and its highly divergent Wza, functions as an antibiotic trap, preventing antimicrobial penetration. We also highlight the high diversity of lineages circulating in dogs across all regions studied, the overlap with human lineages, and regional prevalence of resistance to multiple antimicrobial classes.
IMPORTANCE
Much of the human genomic epidemiology data available for E. coli mechanism discovery studies has been heavily biased toward shiga-toxin producing strains from humans and livestock. E. coli occupies many niches and produces a wide variety of other significant pathotypes, including some implicated in chronic disease. We hypothesized that since dogs tend to share similar strains with their owners and are treated with similar antibiotics, their pathogenic isolates will harbor unexplored AMR mechanisms of importance to humans as well as animals. By comparing over 1,000 genomes with in vitro antimicrobial susceptibility data from sick dogs across the US and Canada, we identified a strong multidrug resistance association with an operon that appears to have once conferred a type 1 capsule production system.
KEYWORDS: antibiotic resistance
INTRODUCTION
Resistance has now been documented for nearly every antimicrobial that has ever been developed (1), and many genes and mutations from numerous pathogens have been identified that confer such resistance. There are still, however, many unidentified genes and variants present in all or most bacterial pathogens. One explanation for this gap is that the databases of isolate genotypes and phenotypes used to validate antimicrobial resistance (AMR) prediction algorithms are heavily biased toward bacterial isolates recovered from food production animals and human bloodstream infections.
Companion animals may be exposed to antimicrobial-resistant bacteria from their owners, hospitals, food, or environment, allowing sentinel surveillance of zoonotic infections that are not being captured in human- and livestock-focused approaches. Now, after 6 years of surveillance conducted by the Veterinary Laboratory Investigation and Response Network (Vet-LIRN) (2), an unprecedented set of high-quality complete genomes and AMR phenotypes of pathogenic Escherichia coli in dogs is available. Recent studies of extraintestinal pathogenic E. coli (ExPEC) in dogs from North America (3) and Australia (4) found that dogs and humans are both colonized by a broad range of the same genotypes, which the authors suggest is likely a consequence of shared gut carriage of bacteria strains between dogs and humans because of close proximity and possibly shared diets. Elankumaran et al. identified certain genotypes as likely “spill-back” zoonotic infections. Harrison et al. found that the abundance of AMR determinants in companion animal strains was higher than in those from food production animals. However, neither study compared the genomes and phenotypes for the discovery of novel AMR determinants.
GWAS methods for bacteria have been developed that use, as the variable for genetic correlation, the gene presence/absence characteristics typical of the bacterial accessory genome (pan-GWAS) (5). The size of the accessory genome of different species of bacteria varies a great deal, with E. coli having one of the largest (6, 7). Pan-GWAS of E. coli has been used to identify genetic associations for antibiotic resistance and bloodstream infections (8, 9) and compare E. coli gene content between dog and human isolates (10). Although AMR has been assessed and analyzed in dogs for a range of bacterial species and in a variety of settings and geographic regions [e.g., (11–14)], we are not aware of a publication that employs pan-GWAS to assess the association of genes with resistance phenotypes in E. coli from dogs. The collection analyzed herein involves the periods 2017–2020 and includes in vitro susceptibility data for 34 different antibiotics and over 1,000 isolates from clinical cases or necropsies. Here, we employ a pan-GWAS perspective on E. coli isolates from sick dogs collected as part of the US National Action Plan and its surveillance for resistant bacteria in animals (2, 15). We then go on to explore with evolutionary genetic and protein structural modeling approaches the molecular character of one of the top ranked candidate AMR loci arising from the pan-GWAS analysis that result in working hypotheses regarding how it might function as an AMR mechanism in the cell.
MATERIALS AND METHODS
Samples and genome sequencing
No research animals were used for this study. Pathogenic E. coli isolates from dogs were collected as part of a previously described surveillance program (2). Briefly, 30 animal diagnostics laboratories participating in the Vet-LIRN AMR monitoring program each saved the first four clinical (from a sick or dead animal) E. coli isolates from unique canine cases submitted for diagnostic testing by veterinarians each month. Deidentified isolates were cryopreserved in glycerol and shipped to one of the six assigned sequencing reference laboratories. Whole genome shotgun sequencing of pure cultures was performed using Illumina chemistry (DNA Prep or Nextera XT library preparation), and sequencing was performed on the MiSeq platform (v2 or v3 chemistry with 2 × 250 bp reads). Genomes meeting standardized benchmarks (16) were then uploaded to NCBI, from which they were retrieved for this study. Sample sources included urine, wound, tracheal wash, ear swab, and a variety of tissues. Isolates included in this study were collected from 2017 to 2020 and covered 11 different geographical regions of Canada and USA. All genomes are available in the following NCBI BioProjects: PRJNA318591, PRJNA324565, PRJNA324573, PRJNA481346, PRJNA503851, and PRJNA318589, all under the umbrella of BioProject PRJNA314609.
Genome assembly, capsular polysaccharide gene cluster diagrams, phylogenetic reconstruction, population genetics, and serotyping
All BioProjects were registered with the NCBI Pathogen Detection system (https://www.ncbi.nlm.nih.gov/pathogens/), which performs assemblies from raw Illumina data uploaded to Sequence Read Archive (SRA) and deposits those into GenBank. Any missing assembly was assembled using SKESA (17). Any assembly with >200 contiguous sequences (contigs) was excluded from the study. All genomes were verified to have a canine host in the metadata and identified as E. coli. The final compiled and curated set of dog E. coli genome sequences was 1,778. All genome assemblies were annotated with Prokka v1.14.5 (18). Core genome alignments were generated within Panaroo v. 1.2.9 (19) using the MAFFT algorithm (20). Gaps were then eliminated using Gblocks v. 0.91b (21, 22), and Panaroo was used to generate gene presence/absence matrices for downstream analyses. Trees were built using IQ-TREE (23) v. 2.0.3 and viewed in iTOL (24). Multilocus sequence typing (MLST) was performed according to the Achtman scheme using the MLST tool (25), which makes use of the PubMLST database (26). Serotype predictions were performed with ECTyper (27) for O and H antigens and Kleborate (28) for K antigens. Python package pyGenomeViz (https://github.com/moshi4/pyGenomeViz) was used to plot the aligned capsular polysaccharide (CPS) clusters. The R package RhierBAPS v1.0.1 (29) was used for hierBAPS clustering of the genomes, performed on the same multiple sequence alignment used for the construction of the phylogenetic tree.
Pangenomics, plasmid identification, and Scoary
In vitro antimicrobial susceptibility data (either by a minimum inhibitory concentration or disk diffusion) and interpretations were collected from the veterinary diagnostic laboratories participating in the study, compiled and coded into binary data for each antimicrobial reported (0 = susceptible; 1 = resistant). The laboratories provided susceptibility interpretations according to their standard procedures for clinical testing, and this interpretation was used for analysis. If no interpretation or an intermediate susceptibility was provided, those results were omitted. Correlation of resistance phenotypes with accessory gene presence/absence was performed with Scoary v. 1.6.16 (5). Short lists of candidate loci correlated with resistance were compiled from genes passing both empirical P and Bonferroni correction, at P < 0.01. We were interested in determining putative novel antimicrobial resistance genes, as well as evaluating which of those genes were chromosomal; hence, we first identified those that were known AMR genes, as well as genes carried on plasmids. Significant Scoary genes were compared with known AMR determinants using the AMRFinderPlus (30) database, and genes likely carried on plasmids were identified using Deeplasmid (31). Deeplasmid applies a deep learning model to classify and separate plasmids from bacterial chromosomes and was used to predict plasmids from all assembled genome contigs. Panaroo gene clusters were designated as plasmid-borne only if >10% of the cluster members were located on the predicted plasmids. This resulted in our grouping of the Scoary significant genes into the following four categories: (i) known AMR locus carried on a plasmid, (ii) on a plasmid but not a known AMR gene, (iii) known AMR gene not on a plasmid, and (iv) not a known AMR gene and not on a plasmid. Our efforts to identify putative novel antimicrobial resistance genes focused on genes estimated to be chromosomally encoded because significant Scoary genes judged to be on plasmids may simply be correlated with a resistance phenotype due to linkage on a plasmid carrying a known AMR locus. Plasmids were identified from assemblies using ABRicate (Galaxy Version 1.0.1) with the plasmidfinder database version 2020-Apr-19.
Natural selection
We tested for gene-wide signatures of selection intensity differences across two genes arising from the pan-GWAS Scoary analysis that were correlated with resistance and were components of a group 1 operon for CPS that lacked several critical genes. Our hypothesis was that the absence of critical genes could render this CPS locus non-functional, resulting in a signal of relaxed selection in these two individual CPS genes, and that this in turn could be related to their role in AMR. The two genes in question were allelic variants of wza (polysaccharide export protein) and wzi (capsule assembly protein). The relaxation of selective strength can drive evolutionary innovation and/or foreshadow loss of function; newly developed evolutionary genetic statistical approaches provide the means to identify genes evolving under such relaxed selection pressure (32).
We generated codon-aware alignments following the procedure available at the Github repository (github.com/veg/hyphy-analyses/tree/master/codon-msa). Briefly, in-frame nucleotide sequences were translated and then aligned using MAFFT v7.471 (20). Aligned protein sequences were then mapped back to the nucleotide sequence, and a single copy of each unique sequence was retained. Alignments were screened for the presence of genetic recombination using the genetic algorithm for recombination detection (GARD) method (33). If GARD identified supported recombinant breakpoints, then alignments were partitioned accordingly and a maximum likelihood phylogeny was inferred for each partition using RaxML-NG v0.9.0git (34) under the GTR+Γ nucleotide substitution model. Where necessary, we used phylotree.js (35) to label phylogenetic branches (based on the gene they coded for) for downstream selection analyses. The Hypothesis testing using Phylogenies (HyPhy) v.2.5.43 software package (36) was used to infer signals of selection and gene-wide signals of selection intensity were compared using RELAX (32).
Structural comparison
To generate predicted structural models of Wza2 proteins (ElI3491725.1, ElI6415193.1, EFO5660681.1, and QDM04109.1), GfcE (P0A932), and Wza1 from Klebsiella michiganensis (WP_224378673.1), we used AlphaFold 2.0 (37) accessed with ColabFold (38). The models were generated as monomers, without structural templates and with the mmseqs2_uniref_env multiple sequence alignment (MSA) mode. Predicted structures were then visualized using UCSF ChimeraX (39). AlphaFold 2 predicted structures for Wza2, gfcE, and Wza1, which presented overall similar domain architecture and folding compared with the X-ray crystal structure of Wza1 (PDB 2j58), and an overall high predicted local distance difference test (pLDDT) confidence score (>84) was chosen to map conservative and non-conservative amino acid substitutions. This mapping was performed by identifying substituted positions from protein alignments (20) and comparing the sequence derived from Wza1 reference structure (PDB 2j58) with the corresponding sequences of the AlphaFold 2 predicted structures. Visualizations and highlighting of substituted residues were performed using UCSF ChimeraX. The first 20 residues of Wza1 (PDB 2j58) are absent in the mature protein structure as they constitute the protein’s signal peptide (40). As such, the corresponding residues in the AlphaFold 2 predicted structures were not displayed. The MatchMaker structural alignment tool in UCSF ChimeraX was used to generate octameric representations of the AlphaFold 2 predicted protein monomers, with the Wza1 reference structure (PDB 2j58) used as an octameric template. The octameric representation allowed us to identify residues in domain 2 of each monomer facing the inner pore cavity.
We computed a pairwise TM-score with the tmscoring method (41, 42) between 5 Wza1, 38 Wza2, and four gfcE predicted monomeric structures. All structures were predicted using AlphaFold 2 as described above, and a heatmap for the matrix was constructed to identify structural relationships between pairs.
Phenotypic characterization of strains harboring wza2
In vitro susceptibility of 36 strains identified as harboring wza2 was repeated by antibiotic susceptibility testing (AST) on the automated Sensititre platform (Thermo Fisher, COMPAN1F panel) and interpreted using the CLSI VET01S standard (43). Susceptibility for cefovecin was also measured by disc diffusion on Mueller Hinton agar using commercial 30 µg discs (Thermo Fisher).
Transcriptomics on three of the strains was performed in LB medium with 0.5 µg/mL reconstituted cefovecin (Zoetis) or an equivalent amount of sterile water (same diluent as the antibiotic). Three fresh colonies from each strain were separately inoculated into 1 mL of the broth and incubated at 37°C with shaking for 5 h. Cultures were then immediately pelleted, the supernatant was removed, and the pellet was resuspended in an RNA stabilizer (DNA/RNA shield, Zymo Research). These suspensions were stored at room temperature until processed by the Cornell Transcriptional Regulation and Expression Facility, where RNA extraction and library preparation were performed using Trizol and the NEB directional library preparation kit. Sequencing was performed with PE 2 × 150 bp read length on the NovaSeq 6000 instrument with 20M reads per sample. Reads were trimmed for low quality and adaptor sequences with TrimGalore v0.6.0 (44), a wrapper for cutadapt (45) and fastQC (46). Reads were mapped to the specific reference genome for that strain using STAR v2.7.0e (47). SARTools and DESeq2 v1.26.0 were used to generate normalized counts and statistical analysis of differential gene expression (48, 49). All RNA-seq data have been deposited to NCBI SRA under the BioProject accession PRJNA1053349.
RESULTS
Pan-genome, core-genome phylogeny, and population genetics
The pangenome content for 1,778 canine E. coli isolate genomes meeting all quality thresholds was open, with a total of 23,956 genes (Fig. 1A) and a core genome size nearing plateau at 3,179 genes (Fig. 1B). Additional break-down of components into core (3,191 clusters), shell (16502 clusters), soft-core (235 clusters), and cloud (4028 clusters) is represented in Fig. 1C. Core and soft-core are genes present in 99% and 95%–99% of genomes. Cloud genes are defined as those clusters which contain a single gene (singletons), plus those which have more than one gene, but its organisms are probably clonal due to identical general gene content (colloquially defined as strain-specific genes). Shell genes are defined as those clusters that neither belong to the core genome nor to the cloud genome.
Fig 1.

Canine E. coli pangenome (N = 1,778). (A) Roary (50) matrix containing identified gene clusters. (B) Pangenome curve consisting of all genes in purple and core genes in green. (C) Distribution of the pangenome into cloud, shell, and core components.
A core genome phylogeny with AMR data, phylogroup, and geographic region mapped to it appears in Fig. 2. Eight different phylogroups were represented in that phylogeny, with the majority of isolates falling within B1 and B2, including most of the multidrug-resistant (MDR) clades, with the exception of one major MDR group represented in each of phylogroups A, C, and F. Based on this core-genome phylogeny, there was no evidence for clustering of separate E. coli genotypes in different geographic regions, and there was also no clustering by sample type (Fig. S1) or year (not shown). An examination of accessory gene content did support distinct clusters, not correlated with geographic variation or sample type (urine versus non-urine) but was correlated with phylogroup. HierBAPS analysis identified 20 distinct genetic clusters at hierarchical level two clustering, with a minimum cluster size of N > 16 (Fig. S2). The genetic clusters showed no correlation with geographic regions or year and were highly mixed with isolates from different regions of USA and Canada.
Fig 2.

Core genome phylogeny of E. coli sampled from dog hosts across US and Canadian regions participating in the surveillance program, with geographic regions, phylogroups, and antibiotic resistance phenotypes annotated on that phylogeny. The map was created using the ne_states function within the rnaturalearth R package, the geom_sf function within the sf R package, and ggplot2.
The MLST data depicted a similar mixed picture (Fig. S3) and indicated that ST372 was the predominant ST in all regions of the USA and Canada studied. Sequence type singletons (N = 152) appeared in each region as well. The predominant predicted serovar was O83:H31, followed by O4:H5 and O6:H31 (Fig. S4). No O antigen type 157 was predicted, but several H7 combinations with different O types were found. Sixteen of the canine ST372 strains carried a pUTI89 plasmid (IncFII (29)_1_pUTI89) that has been described as a marker of human infection (51). These were distributed across different serotypes including O83:H31, O4:H31, and O15:H31, which correspond to the ST372 M, G, and L clusters identified by the pan-GWAS analysis performed Elankumuran et al. comparing globally distributed human and dog isolates (10). One strain from our study (ECOL-18-VL-OH-ON-0048) was closely related (SNP distance of 36) to human strain MVAST4963 characterized by Elankumuran et al. as belonging to cluster K. A majority of the dog-associated propanediol operon components highlighted by Elankumuran et al. in clusters G and M1/2 were annotated in the accessory content of 331 of our dog strains, which harbored pduA, pduB, pduC, pduE, pduF,pduL,pduN pduO, pduP, and pduU. The complete gene presence/absence matrix from our set is available in the data repository (https://doi.org/10.7298/pdzk-rq02.2).
Pandemic ExPEC lineages (as reviewed by LW Riley) (52) ST131 (53), ST127 (54), ST73, ST69, and ST95 were also prominent across multiple regions. Canine-derived ST131 strains identified included both O25:H4 and O16:H5 predicted serotypes. Thirty of 55 had two parC point mutations in common with Clade C reference strain EC958 (GCA_000285655.3), and 7 of those also harbored a blaCTX-M beta-lactamase gene. All canine ST131 strains had a pmrB E123D point mutation predicting Colistin resistance, although this phenotype was not assessed. The NCBI Pathogen detection browser identified 11 matched clusters from this set, and those were primarily comprised of clinical strains presumed to be from humans. The canine strains were from urine, wounds, and one lung. The K1 capsule gene neuC, a hallmark of neonatal meningitis causing E. coli (NMEC) (55), was detected in 70 isolates, and kpsF was detected in 598 isolates.
In vitro antimicrobial resistance characterization
In vitro AST data for 50 antimicrobials (or combinations) were obtained from the participating veterinary diagnostic laboratories. Of these, 12 antimicrobials (Bacitracin, Danofloxacin, Florfenicol, Moxiflacin, Oxytetracycline, Rifampin, Spectinomycin, Sulphamethoxine, Tiamulin, Tilmicosin, Tulathromycin, and Tylosin) did not include interpretations and were excluded from further downstream analysis. Four additional antimicrobials (Ciprofloxacin, Levofloxacin, Ofloxacin, and Oxacillin) had only one value and were also excluded. Thirty-four antimicrobials with sufficient interpretation data were used for pan-GWAS analysis on the corresponding 1,133 genomes.
In vitro MDR was relatively common and varied depending on region (Fig. 2; Fig. S5). Resistance to >5 classes of antimicrobials was seen at low levels (<5%) in 3/4 Canadian regions and 4/7 US regions. Resistance to 3–5 antimicrobial classes occurred in every region and was highest in central Canada (24%), followed by west south-central US (17%). In contrast, the proportion of isolates harboring known antimicrobial resistance genes for multiple classes (based on AMR Finder annotation) was similar for either 3–5 or >5 classes, with the exception of two Canadian regions, which had a higher proportion of strains with >5 AMR gene classes.
AMR gene correlation with in vitro susceptibility
Accessory gene content was correlated with resistant/susceptible coding of the AMR data using Scoary, and the genes judged to be significantly correlated (P < 0.01 for both empirical P and Bonferroni) to resistance were divided into our four genomic categories for each antibiotic (Fig. 3).
Fig 3.

Number of candidate genes from Scoary analysis in four genomic categories with significant correlation to resistance (P < 0.01 with both empirical P and Bonferroni correction) by the antibiotic tested.
The total number of significantly correlated genes varied widely depending on the antibiotic, with as few as two (ticarcillin) and as many as 288 (tetracycline). The majority of Scoary-significant genes fell within the category of plasmid-borne but not known AMR—in other words, plasmid cargo genes—and this ranged from 44%–91%. The smallest category was “known AMR not on a plasmid,” so likely chromosomal. The category we focused particular attention on, with regard to identifying putatively novel AMR loci, was the “putative chromosomal AMR gene.” Due to the large number of genes identified, we focused on those associated with MDR. Protein annotations for the top six genes within this category that were Scoary-significant for resistance to multiple antibiotics were (number of antibiotics resistant in brackets) the following: Wzi, capsule assembly protein (10); transcription regulator (9); hypothetical relaxase (8); Wza, polysaccharide export protein (8); transcription regulator (8); and IucC, aerobactin synthase (8). (Table S1). Due to two of these being adjacent to each other on the chromosome, and the strong association with susceptibility of the colanic acid wza, we chose to focus on those for this study and not discuss the other findings.
Wza and Wzi and their associated group 1 CPS
Two group 1 CPS genes—wza and wzi—were correlated with resistance to cefovecin, other cephalosporins, and several other antibiotics (Table S1). Wza is a polysaccharide export protein, and Wzi is a capsule assembly protein involved in group 1 capsules in both E. coli and Klebsiella (56). Wzi is only present in group 1 CPS (57). The wza sequences in our analysis (Excel Table S2; Fig. 4) that were correlated with MDR are wza group 1 allelic variants with protein sequence divergence ranging from 0%–10%. A similar level of divergence of 0%–13% was apparent when our set of 38 Wza was compared with other Wza group 1 sequences randomly chosen from NCBI to reflect a range of sequence divergence. For the purposes of discussion in this manuscript, hereinafter, all Wza proteins from the group 1 CPS arising from these canine E. coli genomes are referred to as Wza2 and other Wza group 1 chosen from GenBank for comparison are referred to as Wza1. Three distinct types of wza genes are present in E. coli: those within group 1 capsules, colanic acid operon, and the group four capsule operon (referred to as gfcE) (57). All three form separate monophyletic clusters (Fig. S6). The most abundant form in our E. coli isolates was wza-colanic acid (hereinafter referred to as wzaCA) (n = 1089), followed by gfcE (group four wza) (n = 289) and then wza2 (n = 38). wzaCA is a soft-core gene in our analysis, which is to be expected, because colanic acid production in E. coli is common (56). No E. coli genomes with wza2 had wzaCA. Those genomes with wzi always also had wza2 and gfcE (38/38); six genomes had gfcE only; 245 genomes had wzaCA and gfcE and 884 genomes had wzaCA only. wzaCA was associated with susceptibility to a variety of antibiotics, including cefovecin and other cephalosporins (Table S1). Phylogenies of solely wza2 and wzi from canine strains had a similar pattern consisting of two distinct clades, one of high divergence and the other a group of 18 identical, plus three nearly identical, sequences. There were 11 different MLST sequence types represented in these wza2/wzi carrying isolates, with 18 isolates belonging to ST162 (Fig. 4). Phylogenies of our group 1 Wza2 with the inclusion of homologous sequences of Wza1 from GenBank did not support the monophyly of the dog derived sequences (Fig. S7). The closest relatives of our Wza2 and Wzi arose from a mixture of E. coli and Klebsiella pneumoniae (Fig. S8). Resistant and susceptible isolates to cefovecin and the other seven (in the case of Wza2) or nine (Wzi) antibiotics came from both the divergent and non-divergent clades of these gene-specific trees. Upon searching the NCBI database for the wza2 operon, strains from food production animals, as well as humans, were close matches (Fig. S8). Active transcription of the wza2 operon was verified for 3 of the dog strains (Fig. S9), with and without cefovecin added at the MIC50 level established in the original publication (58). The levels of normalized transcripts with and without the treatment were nearly identical.
Fig 4.

Group 1 CPS gene content from the 38 isolates carrying wza2 and wzi genes that were correlated with resistance to multiple antibiotics. The phylogeny is based on wza2 with the respective genome IDs as labels. ST of each genome is listed on the right. Color-coding of the isolate genome accessions denotes resistant (red) and susceptible (blue). Group 1 capsules that do not extend to gnd are because of truncated contigs.
A typical E. coli group 1 CPS operon consists of about 12–14 genes between galF and gnd on the E. coli chromosome (Fig. 4), comprising two separate groups. The 5’ part of the locus contains four genes (wzi, wza, wzb, and wzc), always present, and the 3’ region is serotype-specific, variable in gene content and includes enzymes for producing sugar nucleotide precursors, glycosyltransferases (GTs), and two integral inner membrane proteins (Wzy and Wzx) (57). All but two of the CPS depicted in Fig. 4 do not possess the essential gene content for group 1 capsule production. Two essential loci are wzx and wzy, which typically exist together in the same operon. The two sequences that do appear to have intact group 1 capsule gene content are GCA_007012305.1 and GCA_013094695.1. Much of the evolutionary history behind wza2 and its group 1 accompanying wzi is likely linked to lateral gene transfer (LGT) events mediated by the transposons that are prevalent in the CPS of these particular genomes (Fig. 4). One prominent transposon evident in our group 1 CPS sequences is “DDE-type integrase/transposase/recombinase” and is present in the majority of these 38 genomes, residing one or two genes upstream of galF. This same transposon is found in the majority of wzaCA containing soft-core genomes in our data set, immediately upstream of wzaCA, suggesting a common LGT history at some point between these different wza types. Other transposons are also present, including in some instances, as many as four immediately adjacent to one another, residing between galF and wzi (Fig. 4). Two common transposons were Insertion Sequences (IS) IS3 and IS30 elements; IS30 occurring in 8 of 38 genomes and in all but one occasion flanked by IS3, suggesting these paired IS elements represent a possible composite transposon. Both these IS elements share a high sequence identity with other E. coli transposons of the same type.
Relaxed selection
Discerning a signal of selection intensity in targeted sequences (test) requires a comparative group (reference)—in our case, the wzi reference group was comprised of other homologous wzi sequences obtained from NCBI, from outside our study collection. Because E. coli carries different homologous versions of wza, our reference groups for wza2 included E. coli wza from wild-type group 1 capsules (wza1), the colanic acid operon (wzaCA), and the group 4 capsule operon (gfcE). These different forms of wza are, respectively, monophyletic (Fig. S7). In analyses of selection pressure intensity (Table 1), both wza2 and wzi were judged to be evolving under relaxed selection—the entire wza2 gene relative to wzaCA and in the case of wzi, GARD fragment 6 relative to reference wzi sequences. wza2 comparisons to wza1 were significant for relaxed selection for a 5’ region that included primarily the sequence encoding the signal peptide (nucleotides 1–135; GARD fragment 1: k = 0.24, P-value = 0.006). To get an additional comparative perspective on how wza2 might differ from wza1, we compared wza1 with the same outgroup set of wzaCA, and in this case, wza1 was significant for relaxed selection for a 3’ ~57% of the gene (contained within GARD fragment 2: k = 0.67, P-value = 0.002), but not for the 5’ ~43%. Comparisons of both wza2 and wza1 to gfcE were not significant for relaxed selection. gfcE compared with wzaCA was significant for relaxed selection in the 3’ ~55% of the gene. Taken collectively, this suggests that there is a history of relaxed selection in wza2, and what distinguishes it from wza1 in this regard is the 5’ ~12% of the gene, which is only under relaxed selection in wza2. Test results for each of the relevant comparisons can be found in Table 1.
TABLE 1.
RELAX results of wzi, wzaCA, wza1, wza2, and gfcEa
| Gene(s) analyzed | GARD fragment | RELAX | |
|---|---|---|---|
| k | P value | ||
| wzi vs. NCBI wzi sequences (test: wzi, reference: NCBI wzi sequences) | 1 | xx | xx |
| 2 | xx | xx | |
| 3 | xx | xx | |
| 4 | xx | xx | |
| 5 | xx | xx | |
| 6 | 0.24 | 0.006 | |
| 7 | xx | xx | |
|
wza2 vs. wzaCA (test: wza2, reference: wzaCA) |
1 | 0.8 | 0.048 |
| 2 | 0.5 | 0 | |
|
wza1 vs. wzaCA (test: wza1, reference: wzaCA) |
1 | xx | xx |
| 2 | 0.67 | 0.002 | |
|
gfcE vs. wzaCA (test: gfcE,reference: wzaCA) |
1 | xx | xx |
| 2 | 0.2 | 0.014 | |
|
wza2 vs. wza1 (test: wza2, reference: wza1) |
1 | 0.54 | 0.011 |
| 2 | xx | xx | |
| 3 | xx | xx | |
| 4 | xx | xx | |
| 5 | xx | xx | |
The k parameter measures the selection intensity. Intensified selection is inferred when k > 1 and relaxed when k < 1 along test branches.
Wza protein structure comparisons
Previous studies have suggested a possible role of Wza group 1 from E. coli K30 isolates in translocating antibiotic into the bacterial cell, via the central pore that forms in the octameric structure of the protein [summarized by Sun et al. (59)]. This prompted us to investigate structural comparisons between resistant and susceptible isolates carrying Wza2 as well as between Wza2 and Wza1 and WzaCA and GfcE. Structural models of octamers constructed using AlphaFold 2 and compared using Tm-scores (Fig. 5 and 6) suggest that Wza2 has a similar overall structure to Wza1. Comparisons to WzaCA and group 4 GfcE suggest that group 1 Wza (Wza2 and Wza1) was more similar to GfcE than WzaCA. Structural differences between Wza2 from cefovecin-resistant and susceptible isolates were overall not apparent.
Fig 5.

Heatmap generated from computed pairwise TM scores between WzaCA, Wza1, Wza2, and GfcE protein structures. TM scores are between 0 and 1, the closer to 1 (red), the more similar the structures being compared; the further from 1 (blue), the less similar. All sequences used in this study (WzaCA, Wza2, and GfcE) are also annotated based on either their resistance (RES) or susceptibility (SUS) status for cefovecin.
Fig 6.
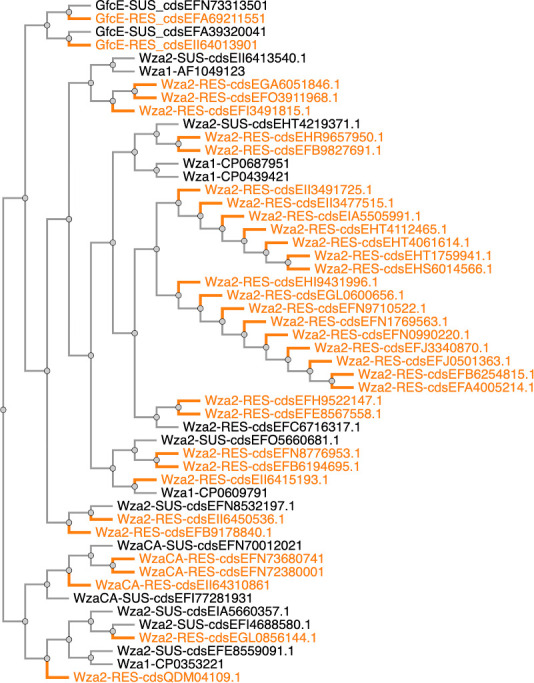
Inferred dendrogram of WzaCA, Wza1, Wza2, and GfcE protein pairwise TM scores. The scores were hierarchically clustered with the unweighted pair group method with arithmetic mean (UPGMA). All sequences used in this study (WzaCA, Wza2, and GfcE) are also annotated based on either their resistance (RES) (highlighted in orange) or susceptibility (SUS) status.
We experimented with the computational docking of cefovecin with Wza2, Wza, and GfcE using SeamDock (60). These data are located in the supplemental data repository. If one considers the entire search space of the protein, cefovecin docked with all three of these proteins but tended to be in different places and could be inside or outside the inner cavity. For Wza2, cefovecin docking was outside either ring 2 or 3. However, if one considers just the internal cavity as a search space (a logical premise for the transmission of an extracellular antibiotic), for both Wza1 and Wza2, cefovecin docks in the D2 domain of the protein in the region where there were a number of non-conservative substitutions. Wza2 was predicted to bind with a slightly better affinity to the antimicrobial (−6.5 kcal/mol vs −5.7 kcal/mol) and included ionic interaction, hydrophobic contact, hydrogen bonds, and weak hydrogen bonds; Wza1 (2j58) did not include any ionic interaction or weak hydrogen bonds. Overall, we consider that these docking results should be regarded with a degree of circumspection since the results are quite sensitive to the initial parameters. Therefore, we consider that although it is likely that cefovecin could indeed dock within the internal pore of Wza1 and Wza2, we cannot confidently conclude that a greater binding affinity of the antibiotic within the internal pore would be expected with Wza2.
An ESPript (61) alignment comparison of a resistant representative of Wza2 and the original 2j58 crystal structure (40) protein sequence identified several non-conservative amino acid changes in Wza2 that mapped to domain D2 in the monomer and the R2 ring in the octamer and tended to cluster along the inner pore of the protein (Fig. 7A). Mapping of non-conservative changes involving a susceptible Wza2 with a seemingly intact group 1 capsule (GCA_013094695.1) was overall similar, but with two notable differences, and these were N181 an K184 compared with K181 and Q184 in the resistant representative. These changes at 181 and 184 were, however, not consistent across other comparisons of resistant and susceptible isolates.
Fig 7.

Changes between Wza1 (crystal structure 2j58) and Wza2 mapped on a model of the monomer and the octamer of a representative resistant (panel A) and susceptible (panel B) Wza2, distinguished by conservative (gray) vs non-conservative (red) mutations. A conservative mutation is considered one that does not dramatically change the biochemical properties of the residue (e.g., L to I or R to K) and the opposite for non-conservative (e.g., D to R). Within the R2 ring (D2 domain of the monomer), there are six non-conservative mutations at positions facing the inner pore cavity, and on the outer “lip” within the R4 ring, there are two mutations in the D4 domain from basic residues to asparagines (N).
DISCUSSION
We found wide variation for both in vitro and predicted resistance profiles in this set of >1,000 E. coli isolates from sick dogs, supporting that dogs may be a reservoir of pathogenic and MDR E. coli with unexplored mechanisms of importance to humans. This could be due to shared exposures to strains from food (62), humans (63), or hospital environments (64, 65). Our population genetics findings indicate recurring sequence types, and predicted serotypes, across every region studied. The most predominant serotype predicted (n = 110) was O83:H31, which has been highlighted (10, 66) as a subset of ST372 shared between human and canine ExPEC strains worldwide but is also highly similar to a strain considered to be a probiotic (67). The next most common serotype, O4:H5 (n = 93) is considered to be highly uropathogenic in humans and several animal hosts (68). The high number of strains harboring ExPEC-associated factors may originate from enteric strains of animal owners that could easily be passed back and forth in households. Food is also an important consideration, as Liu et al. (69) attributed 8.4% of extraintestinal E. coli infections in humans to strains isolated from commercial meat, based on accessory gene content. Our findings are consistent with Elankumuran et al. in the prevalence of pdu propanediol metabolism genes in canine strains, which they hypothesize could be a host adaptation due to diet (10). This is a promising direction for future analyses.
The E. coli capsule is a cell surface structure composed of long-chain polysaccharides involved in protecting the cell, as well as acting as a major virulence component, playing an important role in the bacteria evading or counteracting the host immune system. It has also been suggested that E. coli capsules may act as a physical barrier to impede antibiotics entering the cell (70), resulting in antibiotic resistance. Our goal was to try and identify novel chromosomal loci correlated with AMR. Our Scoary analysis supported two chromosomal genes, wza and wzi, from group 1 cluster as strongly correlated with resistance to cefovecin, other cephalosporins, and a variety of other antibiotics. Wza is an integral outer membrane protein, essential for capsule export and exists as an octamer, with a large central cavity, or pore, 100 Å long and 30 Å wide (40), through which polysaccharides are translocated across the periplasm and outer membrane (71). Earlier studies of E. coli wza1 deletion mutants indicated that wza1 deletion strains were resistant to a number of macrolides (72) and that complementation of wza1 in a strain bearing a deletion of this gene could restore the wild-type susceptibility to erythromycin (73). Su et al. measured the diameter of the Wza1 octamer protein pore as approximately 17 Å and estimated the size of erythromycin as 12 Å, suggesting that the pore may be large enough to allow transmission of the antibiotic into the periplasm [summarized by (59)]. The strong correlation of the wza2/wzi from the group 1 cluster with an array of antibiotics suggests that an opposite effect of inhibition of antibiotic transmission occurs in these widely dispersed strains. This in turn led us to hypothesize that structural differences might be apparent between Wza2-resistant and susceptible strains and possibly this could be related to an inhibition of antibiotic transmission by Wza2.
Based on AlphaFold 2 models compared using Template Modeling score (Tm-score), there were no discernable differences between Wza2 from cefovecin-resistant and susceptible isolates, and there were no differences in comparisons of the protein structure of Wza2 compared with Wza1. Differences were apparent between the two group 1 Wza (Wza2 and Wza1) and WzaCA and GfcE, although the Tm scores were still relatively high, indicating very similar folds (42). The colanic acid operon and the associated wzaCA gene from that operon are absent from the wza2-carrying isolates, but gfcE was present. In isolates where WzaCA is present (which is the vast majority of isolates), it is significantly associated with susceptibility to cephalosporins, as well as a number of other antibiotics, and the other genes in that colanic acid operon share the same pattern. It is possible that WzaCA is a portal for antibiotic transmission, but to our knowledge, this is an untested idea. It is the case that wza1, wzb, and wzc from colanic acid have been reciprocally interchanged with those same three homologous genes from group 1 and function normally (74) in both operons. In comparisons between Wza1 (2j58 PDB crystal structure sequence) and Wza2 sequences, there were a number of non-conservative amino acid substitutions (changing either the polarity or the charge and two instances of A to the cyclic amino acid P) lining the inner pore of Wza2 (Fig. 7). Water interaction with the polarity of the inner pore of Wza1 has been suggested as important in facilitating the export of polysaccharides (40). It is possible that these Wza2 substitutions could involve electrostatic interactions with antibiotic molecules that might affect antibiotic transmission; however, there was no consistent correlation between resistant and susceptible isolates carrying Wza2 regarding the specific character of such substitutions. In our relaxed selection analysis, wza2 was under relaxed selection relative to wzaCA across the entire gene. wza1 was also under relaxed selection relative to wzaCA, but only in the 3’ ~57% of the gene (GARD fragment 2); it was not under relaxed selection in the 5’ ~43% of the gene (GARD fragment 1). The two GARD fragments are judged to have distinct evolutionary histories, with a putative recombination break-point separating the two halves. GARD fragment 1 in wza2 is the region where these non-conservative changes reside. This suggests that relaxed selection pressure on this ~12% of the gene resulted in random nonsynonymous mutations yielding the non-conservative amino acid changes that happen to line the inner pore of the octamer. We suggest that this signal of relaxed selection is a consequence of missing essential gene content in these group 1 capsule sequences, rendering the group 1 CPS non-functional.
It is important to consider that Wza is only one protein in the group 1 CPS assembly pathway. Of particular importance is the interaction that occurs between Wza and Wzc. This second protein is thought to regulate the opening of the PES (polysaccharide export sequence) domain of Wza within the periplasm, which is in a closed conformation in the absence of Wzc interaction. Collins et al. proposed that the polysaccharide enters the central pore of Wza through the opened PES domain and, then moving along the central cavity, exits to the extracellular space through the alpha-helical domain of Wza (75). The interaction with Wzc is thought to trigger the active or open conformation of Wza. Thus, if this association of Wza and Wzc is disrupted because of sequence changes arising from a relaxation of selective constraints on Wza2, then an antibiotic entering the pore at the alpha-helical domain of Wza may not be able to exit into the periplasm or cytoplasm because of a closed conformation of Wza, essentially functioning as an antibiotic trap.
Additional genes within the Wza2 CPS cluster sequence were also correlated with AMR (Table S1). The clearest example of this was wzi; however, other genes do show a bias toward resistance but do not pass our strict statistical criteria because of the reduced sample number for those particular genes, due to their partitioning into different variants. As an example, our data set has two variants of wzc; hence, the Scoary analysis treats them as separate loci, and one of them is significant for empirical P (P < 0.01) but does not pass the Bonferroni correction. Similarly, wbaP (UDP-Gal:phosphoryl-polyprenol Gal-1-phosphate transferase), involved in the first step in biosynthesis of group 1 CPS, is present in several of our CPS, is significant for empirical P (P < 0.01), but does not pass Bonferroni correction. The interactive necessities of the Wzx/Wzy pathway in capsule production suggest that focusing on a single locus as the potential AMR mechanism may be misguided. Failure of the pathway could affect the transmission of antibiotics through the Wza2 pore, ultimately into the periplasm and the cytoplasm, thus rendering the isolates resistant.
These Wza2 capsule clusters carry many of the genes necessary and typical of a functional capsule one assembly locus; however, the gene content is variable between isolates, except in the members of the low divergence Wza2 clade (ST162), and in the majority of instances, they are missing some key loci. Although different serotypes carry gene-specific loci, genes encoding Wzx and Wzy, defining the Wzx/Wzy pathway, are always present in a functional group 1 CPS locus (57). Wzx translocates the O-antigens across the inner membrane, where the O units are polymerized by Wzy. This latter protein is known to exhibit low sequence conservation both between and within bacterial species (76). Our Wza2 genomes carry examples of five different putative Wzy proteins, with low sequence homology. Some form of Wzy, and its representation within this CPS cluster sequence, is essential for group 1 capsule production in E. coli. Certain Salmonella serotypes employ a Wzy polymerase that is located elsewhere in the genome, outside the CPS cluster sequence (77), but this is not reported for E. coli. Wzx, the O-antigen flippase protein, is also missing from the majority of these CPS sequences. Only two of these genomes appear to have both Wzx and Wzy in the CPS sequence, although it is possible this number could be increased by a few, since several contigs are prematurely truncated upstream of gnd (Fig. 5). Thus, key components of the Wzx/Wzy pathway are missing from these putative group 1 CPS cluster sequences. That fact, combined with the relaxed selection evident for wza2 and wzi, suggests this CPS locus is no longer functional as a group 1 capsule assembly system. More specifically, we suggest that a recombinant history, at least partially mediated by the transposons associated with these capsule sequences, resulted in shuffling the gene content in these CPS, rendering it non-functional for capsule production, and this in turn is reflected in the relaxed selection for wza and wzi. Although our examination of Wza protein structure proved inconclusive with regard to distinct patterns associated with resistance, our proposed model would still be that the antibiotic may get trapped in the pore of Wza2, but this could be due to a host of interactive anomalies either involving the core components of Wzi, Wza2, Wzb, and Wzc or involving other missing components in the biosynthetic region of this operon.
All 38 of our isolates carrying the CPS locus depicted in Fig. 5 carry the group 4 cluster sequence, with its wza homolog, gfcE. Our working hypothesis is that these isolates originally carried the group 4 CPS capability and then independently acquired the group 1 CPS via LGT from various lineages. The surrounding gene content of the group 4 CPS is conserved in Wza2 isolates, with the seemingly requisite operon composition for a functioning group 4 capsule (78, 79) and is similarly conserved in non-Wza2 isolates from our collection, suggesting an ancient syntenic inheritance. In contrast, independent LGT events are suggested by the gene content variability of the group 1 CPS between Wza2 isolates, and the wide diversity in STs of this group. Homologous recombination events are undoubtedly part of the history of this group 1 CPS locus, and some of this recombination likely involved the colanic acid operon. All E. coli group 1-producing isolates are unable to produce colanic acid, and it has been suggested that this locus may have been lost during chromosomal rearrangements involving group 1 CPS loci (57).
In our case, the same transposon is present just upstream of galF in our group 1 sequences (Fig. 4) and is just upstream of wza in the vast majority of colanic acid operons in our set of sequences from dogs. IS elements have been implicated previously in the history of group 1 CPS and suggested as a likely factor continuing to mediate recombination between strains (80). Colanic acid and group 1 capsules have similar chemical structure (57) and share similarities in the operon structure, and the wza, wzb, and wzc genes can be interchanged (74). Our wza2 gene tree phylogeny identifies an identical sequence clade of wza2 sequences, all from ST162, that include in their respective cluster sequence, at least one gene locus common in colanic acid operons, gmd, involved in GDP-fucose biosynthesis from mannose-6-phosphate (Man6P). All of this points to an extensive, and likely ongoing, history of LGT between colanic acid and group 1. ST162 is a globally distributed MDR pandemic lineage that has been isolated from clinical, environmental, domestic, and wild animal samples (81), including an endangered species, the Andean condor (82), and, most recently, a pygmy sperm whale (83). Our results suggest the possibility that wza2 and/or the group 1 CPS from ST162 could be a contributing factor to its MDR phenotype.
Our hypothesis is that the majority of our Wza2 group 1 CPS have lost their previous role in capsule production, due to extensive recombination and the consequent loss of certain critical genes. This loss of function is further supported by the signal of relaxed selection we see in genes from this CPS locus that were correlated with resistance. Ultimately, we suggest this has had the unexpected effect of these G1C serendipitously developing into an MDR AMR mechanism. The majority of known AMR mechanisms involve functional loci, and mutations therein, resulting in (i) changes in membrane permeability, which limit the uptake, (ii) modification of a target, (iii) enzymatic inactivation, and (iv) active efflux. There are, however, other examples in Gram-negative bacteria where an analogous loss of function to our proposal for the Wza2 operon results in AMR. For example, colistin resistance in Acinetobacter baumannii is due to mutations within genes essential for lipid A biosynthesis (lpx operon), eliminating the ability to produce lipid A and therefore lipopolysaccharide (LPS) (84). An important difference in this A. baumannii study compared with our proposal is that colistin resistance due to loss of LPS has the corollary effect of increased sensitivity to other clinically relevant antibiotics; we see no such evidence of that in the wza2/group 1 isolates examined here.
The Wza2 cluster sequences are very widely distributed, nearly intact, across a number of bacteria species (Fig. S8), particularly, E. coli and Klebsiella pneumoniae, isolated from different host species (including other mammals, birds, plants, and fly). Whether one considers Wza2 as the primary resistance mechanism, or the probable inactive group 1 CPS cluster, the wide dispersal range of these sequences suggests that this resistant CPS may be a much more important source of AMR than just in this set of dog isolates. Although Wzi and Wza2 were two of only a small handful of putatively novel chromosomal loci correlated with resistance to multiple antimicrobials, it is also true that not all of these 38 genomes carrying these two genes were devoid of any possible confounding influence of known resistance mechanisms, of either plasmid or chromosomal origin. We focused exclusively on gene presence/absence here, and our study did not include mutational resistance. Given the large number of known AMR mechanisms in E. coli, and in our specific data set (Fig. S5), this is to be expected. However, in further attempts to specifically associate a cause-effect relationship of these loci to AMR, it will be important to eliminate the confounding influence of known AMR mechanisms on the role of these putative novel antimicrobial resistance genes. Ongoing and future work in our laboratories regarding Wzi/Wza2 will involve knockouts of these genes and others in the Wza2 CPS cluster, within several strains of the germane genetic background that are devoid of known resistance mechanisms. Another future direction needed is to characterize any capsules that may be formed by these strains.
As genomic surveillance expands to additional hosts, microbes, and geographic regions, the importance of collecting paired AST data cannot be understated. The merging and binary coding of this data is challenging, particularly for microbe-antimicrobial combinations that do not have breakpoints available, or when the dilutions on commercial panels do not have a sufficient range for an accurate MIC determination. Different laboratories also use different platforms, methods, and criteria for interpretation. Additionally, some breakpoints in veterinary medicine are intentionally set low to discourage use. Here, we conservatively chose to include only the definitive interpretations provided to clinicians, omitting the intermediate phenotypes and the MIC or disk diffusion diameters where no interpretation was provided to the clinician. Due to the limitations imposed by the aforementioned factors, we chose to focus on genes associated with eight or more antimicrobials after correction for repeated measures. Interestingly, with this strict criterion, we found different versions of the same gene (wza) to be associated with either susceptibility or resistance (Table S1). The strong association with cefovecin observed may have been facilitated by heavy demand for susceptibility testing of this third-generation cephalosporin in dogs (and cats). As we also saw resistance to antimicrobials of human medical importance in these same dog strains, stewardship of these treatments in our companion animals is of utmost importance.
ACKNOWLEDGMENTS
The authors are grateful to Chris Whitfield and Michael Feldgarden for helpful discussions. Trevor L. Alexander, Melanie Prarat, Ashley Johnson, Katherine Shiplett, Dominika Jurkovic,and Jing Cui provided excellent technical support.
This work was supported (FOA PAR-18–604) and performed in collaboration with the US Food and Drug Administration’s Veterinary Laboratory Investigation and Response Network under grants 1U18FD006993 to Cornell; 1U18FD006442 to Louisiana State; 1U18FD006712 to Ohio ADDL; 1U18FD006862 and 1U18FD007244-to University of Guelph; 1U18FD006558 to South Dakota, 1U18FD006453- to Washington State.
The views expressed in this article are those of the authors and do not necessarily reflect the official policy of the Department of Health and Human Services, the U.S. Food and Drug Administration, or the U.S. Government. Reference to any commercial materials, equipment, or process does not in any way constitute approval, endorsement, or recommendation by the Food and Drug Administration.
Contributor Information
Laura B. Goodman, Email: laura.goodman@cornell.edu.
Edward G. Dudley, The Pennsylvania State University, University Park, Pennsylvania, USA
DATA AVAILABILITY
All genomes are available in the following NCBI BioProjects: PRJNA318591, PRJNA324565, PRJNA324573, PRJNA481346, PRJNA503851, and PRJNA318589, all under the umbrella BioProject PRJNA314609. All RNA-seq data have been deposited to NCBI Sequence Read Archive (SRA) under the BioProject accession PRJNA1053349. All alignments, inferred phylogenies, and raw output files are available at the Cornell University eCommons Repository under https://doi.org/10.7298/pdzk-rq02.2.
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/aem.00354-24.
Figures S1 to S9.
Summary of gene groups associated with resistance or susceptibility to 8 or more antibiotics.
Summary of strains carrying wza2 including metadata and antibiotic susceptibility phenotypes based on MIC testing with VET01S interpretations. Where dog breakpoints were not available, the human breakpoint was applied.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Ventola CL. 2015. The antibiotic resistance crisis: part 1: causes and threats. P T 40:277–283. [PMC free article] [PubMed] [Google Scholar]
- 2. Ceric O, Tyson GH, Goodman LB, Mitchell PK, Zhang Y, Prarat M, Cui J, Peak L, Scaria J, Antony L, et al. 2019. Enhancing the one health initiative by using whole genome sequencing to monitor antimicrobial resistance of animal pathogens: Vet-LIRN collaborative project with veterinary diagnostic laboratories in United States and Canada. BMC Vet Res 15:130. doi: 10.1186/s12917-019-1864-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harrison L, Tyson GH, Strain E, Lindsey RL, Strockbine N, Ceric O, Fortenberry GZ, Harris B, Shaw S, Tillman G, Zhao S, Dessai U. 2022. Use of large-scale genomics to identify the role of animals and foods as potential sources of extraintestinal pathogenic Escherichia coli that cause human illness. Foods 11:1975. doi: 10.3390/foods11131975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Elankumaran P, Cummins ML, Browning GF, Marenda MS, Reid CJ, Djordjevic SP. 2022. Genomic and temporal trends in canine ExPEC reflect those of human ExPEC. Microbiol Spectr 10:e0129122. doi: 10.1128/spectrum.01291-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brynildsrud O, Bohlin J, Scheffer L, Eldholm V. 2016. Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary. Genome Biol 17:238. doi: 10.1186/s13059-016-1108-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beavan AJS, Domingo-Sananes MR, McInerney JO. 2024. Contingency, repeatability, and predictability in the evolution of a prokaryotic pangenome. Proc Natl Acad Sci U S A 121:e2304934120. doi: 10.1073/pnas.2304934120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tantoso E, Eisenhaber B, Kirsch M, Shitov V, Zhao Z, Eisenhaber F. 2022. To kill or to be killed: pangenome analysis of Escherichia coli strains reveals a tailocin specific for pandemic ST131. BMC Biol 20:146. doi: 10.1186/s12915-022-01347-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Denamur E, Condamine B, Esposito-Farèse M, Royer G, Clermont O, Laouenan C, Lefort A, de Lastours V, Galardini M, COLIBAFI, SEPTICOLI groups . 2022. Genome wide association study of Escherichia coli bloodstream infection isolates identifies genetic determinants for the portal of entry but not fatal outcome. PLoS Genet 18:e1010112. doi: 10.1371/journal.pgen.1010112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li X, Lin J, Hu Y, Zhou J. 2020. PARMAP: a pan-genome-based computational framework for predicting antimicrobial resistance. Front Microbiol 11:578795. doi: 10.3389/fmicb.2020.578795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Elankumuran P, Browning GF, Marenda MS, Kidsley A, Osman M, Haenni M, Johnson JR, Trott DJ, Reid CJ, Djordjevic SP. 2023. Identification of genes influencing the evolution of Escherichia coli ST372 in dogs and humans. Microb Genom 9:mgen000930. doi: 10.1099/mgen.0.000930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bourély C, Cazeau G, Jarrige N, Leblond A, Madec JY, Haenni M, Gay E. 2019. Antimicrobial resistance patterns of bacteria isolated from dogs with otitis. Epidemiol Infect 147:e121. doi: 10.1017/S0950268818003278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hata A, Fujitani N, Ono F, Yoshikawa Y. 2022. Surveillance of antimicrobial-resistant Escherichia coli in sheltered dogs in the Kanto Region of Japan. Sci Rep 12:773. doi: 10.1038/s41598-021-04435-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li Y, Fernández R, Durán I, Molina-López RA, Darwich L. 2020. Antimicrobial resistance in bacteria isolated from cats and dogs from the Iberian Peninsula. Front Microbiol 11:621597. doi: 10.3389/fmicb.2020.621597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Osman M, Albarracin B, Altier C, Gröhn YT, Cazer C. 2022. Antimicrobial resistance trends among canine Escherichia coli isolated at a New York veterinary diagnostic laboratory between 2007 and 2020. Prev Vet Med 208:105767. doi: 10.1016/j.prevetmed.2022.105767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. CDC . 2022. Antibiotic resistance is a national priority. Centers for Disease Control and Prevention. Available from: https://www.cdc.gov/drugresistance/us-activities.html. Retrieved 23 Sep 2023. [Google Scholar]
- 16. Timme RE, Wolfgang WJ, Balkey M, Venkata SLG, Randolph R, Allard M, Strain E. 2020. Optimizing open data to support one health: best practices to ensure interoperability of genomic data from bacterial pathogens. One Health Outlook 2:20. doi: 10.1186/s42522-020-00026-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Souvorov A, Agarwala R, Lipman DJ. 2018. SKESA: strategic k-mer extension for scrupulous assemblies. Genome Biol 19:153. doi: 10.1186/s13059-018-1540-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. doi: 10.1093/bioinformatics/btu153 [DOI] [PubMed] [Google Scholar]
- 19. Tonkin-Hill G, MacAlasdair N, Ruis C, Weimann A, Horesh G, Lees JA, Gladstone RA, Lo S, Beaudoin C, Floto RA, Frost SDW, Corander J, Bentley SD, Parkhill J. 2020. Producing polished prokaryotic pangenomes with the Panaroo pipeline. Genome Biol 21:180. doi: 10.1186/s13059-020-02090-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. doi: 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334 [DOI] [PubMed] [Google Scholar]
- 22. Talavera G, Castresana J. 2007. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol 56:564–577. doi: 10.1080/10635150701472164 [DOI] [PubMed] [Google Scholar]
- 23. Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32:268–274. doi: 10.1093/molbev/msu300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Letunic I, Bork P. 2021. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res 49:W293–W296. doi: 10.1093/nar/gkab301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Seemann T. 2023. Mlst tool. https://github.com/Tseemann/Mlst.
- 26. Jolley KA, Maiden MCJ. 2010. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11:595. doi: 10.1186/1471-2105-11-595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bessonov K, Laing C, Robertson J, Yong I, Ziebell K, Gannon VPJ, Nichani A, Arya G, Nash JHE, Christianson S. 2021. ECTyper: in silico Escherichia coli serotype and species prediction from raw and assembled whole-genome sequence data. Microb Genom 7:000728. doi: 10.1099/mgen.0.000728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lam MMC, Wick RR, Watts SC, Cerdeira LT, Wyres KL, Holt KE. 2021. A genomic surveillance framework and genotyping tool for Klebsiella pneumoniae and its related species complex. 1. Nat Commun 12:4188. doi: 10.1038/s41467-021-24448-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tonkin-Hill G, Lees JA, Bentley SD, Frost SDW, Corander J. 2018. RhierBAPS: an R implementation of the population clustering algorithm hierBAPS. Wellcome Open Res 3:93. doi: 10.12688/wellcomeopenres.14694.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Feldgarden M, Brover V, Gonzalez-Escalona N, Frye JG, Haendiges J, Haft DH, Hoffmann M, Pettengill JB, Prasad AB, Tillman GE, Tyson GH, Klimke W. 2021. AMRFinderPlus and the Reference Gene Catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence. 1. Sci Rep 11:12728. doi: 10.1038/s41598-021-91456-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Andreopoulos WB, Geller AM, Lucke M, Balewski J, Clum A, Ivanova NN, Levy A. 2022. Deeplasmid: deep learning accurately separates plasmids from bacterial chromosomes. Nucleic Acids Res 50:e17. doi: 10.1093/nar/gkab1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wertheim JO, Murrell B, Smith MD, Kosakovsky Pond SL, Scheffler K. 2015. RELAX: detecting relaxed selection in a phylogenetic framework. Mol Biol Evol 32:820–832. doi: 10.1093/molbev/msu400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kosakovsky Pond SL, Posada D, Gravenor MB, Woelk CH, Frost SDW. 2006. GARD: a genetic algorithm for recombination detection. Bioinformatics 22:3096–3098. doi: 10.1093/bioinformatics/btl474 [DOI] [PubMed] [Google Scholar]
- 34. Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A. 2019. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35:4453–4455. doi: 10.1093/bioinformatics/btz305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shank SD, Weaver S, Kosakovsky Pond SL. 2018. phylotree.js - a JavaScript library for application development and interactive data visualization in phylogenetics. BMC Bioinformatics 19:276. doi: 10.1186/s12859-018-2283-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kosakovsky Pond SL, Poon AFY, Velazquez R, Weaver S, Hepler NL, Murrell B, Shank SD, Magalis BR, Bouvier D, Nekrutenko A, Wisotsky S, Spielman SJ, Frost SDW, Muse SV. 2020. HyPhy 2.5—a customizable platform for evolutionary hypothesis testing using phylogenies. Mol Biol Evol 37:295–299. doi: 10.1093/molbev/msz197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, et al. 2021. Highly accurate protein structure prediction with AlphaFold. Nature 596:583–589. doi: 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M. 2022. ColabFold: making protein folding accessible to all. Nat Methods 19:679–682. doi: 10.1038/s41592-022-01488-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, Ferrin TE. 2021. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci 30:70–82. doi: 10.1002/pro.3943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dong C, Beis K, Nesper J, Brunkan-Lamontagne AL, Clarke BR, Whitfield C, Naismith JH. 2006. Wza the translocon for E. coli capsular polysaccharides defines a new class of membrane protein. Nature 444:226–229. doi: 10.1038/nature05267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xu J, Zhang Y. 2010. How significant is a protein structure similarity with TM-score = 0.5?. Bioinformatics 26:889–895. doi: 10.1093/bioinformatics/btq066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang Y, Skolnick J. 2004. Scoring function for automated assessment of protein structure template quality. Proteins: Structure, Function, and Bioinformatics 57:702–710. doi: 10.1002/prot.20264 [DOI] [PubMed] [Google Scholar]
- 43. CLSI . 2020. Performance standards for antimicrobial disk and dilution susceptibility tests for bacteria isolated from animals. 5th ed. Clinical and Laboratory Standards Institute, USA. [Google Scholar]
- 44. Krueger F. 2024. Trimgalore tool. https://github.com/FelixKrueger/TrimGalore.
- 45. Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet j 17:10. doi: 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- 46. Andrews S. 2010. FastQC: a quality control tool for high throughput sequence data. Available from: https://www.bioinformatics.babraham.ac.uk/projects/fastqc
- 47. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. doi: 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Varet H, Brillet-Guéguen L, Coppée J-Y, Dillies M-A. 2016. SARTools: a DESeq2- and EdgeR-based R pipeline for comprehensive differential analysis of RNA-Seq data. PLoS ONE 11:e0157022. doi: 10.1371/journal.pone.0157022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MTG, Fookes M, Falush D, Keane JA, Parkhill J. 2015. Roary: Rapid large-scale Prokaryote pan genome analysis. Bioinformatics 31:3691–3693. doi: 10.1093/bioinformatics/btv421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cusumano CK, Hung CS, Chen SL, Hultgren SJ. 2010. Virulence plasmid harbored by uropathogenic Escherichia coli functions in acute stages of pathogenesis. Infect Immun 78:1457–1467. doi: 10.1128/IAI.01260-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Riley LW. 2014. Pandemic lineages of extraintestinal pathogenic Escherichia coli. Clin Microbiol Infect 20:380–390. doi: 10.1111/1469-0691.12646 [DOI] [PubMed] [Google Scholar]
- 53. Forde BM, Roberts LW, Phan M-D, Peters KM, Fleming BA, Russell CW, Lenherr SM, Myers JB, Barker AP, Fisher MA, Chong T-M, Yin W-F, Chan K-G, Schembri MA, Mulvey MA, Beatson SA. 2019. Population dynamics of an Escherichia coli ST131 lineage during recurrent urinary tract infection. Nat Commun 10:3643. doi: 10.1038/s41467-019-11571-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Elankumaran P, Browning GF, Marenda MS, Reid CJ, Djordjevic SP. 2022. Close genetic linkage between human and companion animal extraintestinal pathogenic Escherichia coli ST127. Curr Res Microb Sci 3:100106. doi: 10.1016/j.crmicr.2022.100106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wijetunge DSS, Gongati S, DebRoy C, Kim KS, Couraud PO, Romero IA, Weksler B, Kariyawasam S. 2015. Characterizing the pathotype of neonatal meningitis causing Escherichia coli (NMEC). BMC Microbiol 15:211. doi: 10.1186/s12866-015-0547-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Whitfield C, Wear SS, Sande C. 2020. Assembly of bacterial capsular polysaccharides and exopolysaccharides. Annu Rev Microbiol 74:521–543. doi: 10.1146/annurev-micro-011420-075607 [DOI] [PubMed] [Google Scholar]
- 57. Whitfield C. 2006. Biosynthesis and assembly of capsular polysaccharides in Escherichia coli. Annu Rev Biochem 75:39–68. doi: 10.1146/annurev.biochem.75.103004.142545 [DOI] [PubMed] [Google Scholar]
- 58. Stegemann MR, Passmore CA, Sherington J, Lindeman CJ, Papp G, Weigel DJ, Skogerboe TL. 2006. Antimicrobial activity and spectrum of cefovecin, a new extended- spectrum cephalosporin, against pathogens collected from dogs and cats in Europe and North America. Antimicrob Agents Chemother 50:2286–2292. doi: 10.1128/AAC.00077-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sun E, Graves ML, Oliver DC. 2020. Propelling a course-based undergraduate research experience using an open-access online undergraduate. Front Microbiol 11:589025. doi: 10.3389/fmicb.2020.589025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Murail S, de Vries SJ, Rey J, Moroy G, Tufféry P. 2021. SeamDock: an interactive and collaborative online docking resource to assist small compound molecular docking. Front Mol Biosci 8:716466. doi: 10.3389/fmolb.2021.716466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Robert X, Gouet P. 2014. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42:W320–W324. doi: 10.1093/nar/gku316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nemser SM, Doran T, Grabenstein M, McConnell T, McGrath T, Pamboukian R, Smith AC, Achen M, Danzeisen G, Kim S, Liu Y, Robeson S, Rosario G, McWilliams Wilson K, Reimschuessel R. 2014. Investigation of Listeria, Salmonella, and toxigenic Escherichia coli in various pet foods. Foodborne Pathog Dis 11:706–709. doi: 10.1089/fpd.2014.1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Martins LRL, Pina SMR, Simões RLR, de Matos AJF, Rodrigues P, da Costa PMR. 2013. Common phenotypic and genotypic antimicrobial resistance patterns found in a case study of multiresistant E. coli from cohabitant pets, humans, and household surfaces. J Environ Health 75:74–81. [PubMed] [Google Scholar]
- 64. Cole SD, Perez-Bonilla D, Hallowell A, Redding LE. 2022. Carbapenem prescribing at a veterinary teaching hospital before an outbreak of carbapenem-resistant Escherichia coli. J Small Anim Pract 63:442–446. doi: 10.1111/jsap.13481 [DOI] [PubMed] [Google Scholar]
- 65. Menard J, Goggs R, Mitchell P, Yang Y, Robbins S, Franklin-Guild RJ, Thachil AJ, Altier C, Anderson R, Putzel GG, McQueary H, Goodman LB. 2022. Effect of antimicrobial administration on fecal microbiota of critically ill dogs: dynamics of antimicrobial resistance over time. Anim Microbiome 4:36. doi: 10.1186/s42523-022-00178-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Flament-Simon S-C, Toro M de, García V, Blanco JE, Blanco M, Alonso MP, Goicoa A, Díaz-González J, Nicolas-Chanoine M-H, Blanco J. 2020. Molecular characteristics of extraintestinal pathogenic E. coli (ExPEC), uropathogenic E. coli (UPEC), and multidrug resistant E. coli isolated from healthy dogs in Spain. whole genome sequencing of canine ST372 isolates and comparison with human isolates causing extraintestinal infections. Microorganisms 8:1712. doi: 10.3390/microorganisms8111712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wassenaar TM. 2016. Insights from 100 years of research with probiotic E. coli. EuJMI 6:147–161. doi: 10.1556/1886.2016.00029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rivera-Betancourt M, Keen JE. 2001. Murine monoclonal antibodies against Escherichia coli O4 lipopolysaccharide and H5 flagellin. J Clin Microbiol 39:3409–3413. doi: 10.1128/JCM.39.9.3409-3413.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Liu CM, Aziz M, Park DE, Wu Z, Stegger M, Li M, Wang Y, Schmidlin K, Johnson TJ, Koch BJ, Hungate BA, Nordstrom L, Gauld L, Weaver B, Rolland D, Statham S, Hall B, Sariya S, Davis GS, Keim PS, Johnson JR, Price LB. 2023. Using source-associated mobile genetic elements to identify zoonotic extraintestinal E. coli infections. One Health 16:100518. doi: 10.1016/j.onehlt.2023.100518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Slack MP, Nichols WW. 1982. Antibiotic penetration through bacterial capsules and exopolysaccharides. J Antimicrob Chemother 10:368–372. doi: 10.1093/jac/10.5.368 [DOI] [PubMed] [Google Scholar]
- 71. Nickerson NN, Mainprize IL, Hampton L, Jones ML, Naismith JH, Whitfield C. 2014. Trapped translocation intermediates establish the route for export of capsular polysaccharides across Escherichia coli outer membranes. Proc Natl Acad Sci U S A 111:8203–8208. doi: 10.1073/pnas.1400341111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Su A, Wang A, Yeo L. 2017. Deletion of the group 1 capsular gene Wza in Escherichia coli E69 confers resistance to the antibiotic erythromycin on solid media but not in liquid media | jemi.microbiology.ubc.ca. JEMI 3:1–8. [Google Scholar]
- 73. Yuen B, Ting J, Kang K, Wong T. 2017. Investigation of Wza in erythromycin sensitivity of Escherichia coli K30 E69 by genetic complementation. JEMI 21:52–57. [Google Scholar]
- 74. Reid AN, Whitfield C. 2005. Functional analysis of conserved gene products involved in assembly of Escherichia coli capsules and exopolysaccharides: evidence for molecular recognition between Wza and Wzc for colanic acid biosynthesis. J Bacteriol 187:5470–5481. doi: 10.1128/JB.187.15.5470-5481.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Collins RF, Beis K, Dong C, Botting CH, McDonnell C, Ford RC, Clarke BR, Whitfield C, Naismith JH. 2007. The 3D structure of a periplasm-spanning platform required for assembly of group 1 capsular polysaccharides in Escherichia coli. Proc Natl Acad Sci U S A 104:2390–2395. doi: 10.1073/pnas.0607763104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Liu B, Furevi A, Perepelov AV, Guo X, Cao H, Wang Q, Reeves PR, Knirel YA, Wang L, Widmalm G. 2020. Structure and genetics of Escherichia coli O antigens. FEMS Microbiol Rev 44:655–683. doi: 10.1093/femsre/fuz028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Liu B, Knirel YA, Feng L, Perepelov AV, Senchenkova SN, Reeves PR, Wang L. 2014. Structural diversity in Salmonella O antigens and its genetic basis. FEMS Microbiol Rev 38:56–89. doi: 10.1111/1574-6976.12034 [DOI] [PubMed] [Google Scholar]
- 78. Larson MR, Biddle K, Gorman A, Boutom S, Rosenshine I, Saper MA. 2021. Escherichia coli O127 group 4 capsule proteins assemble at the outer membrane. PLoS One 16:e0259900. doi: 10.1371/journal.pone.0259900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Peleg A, Shifrin Y, Ilan O, Nadler-Yona C, Nov S, Koby S, Baruch K, Altuvia S, Elgrably-Weiss M, Abe CM, Knutton S, Saper MA, Rosenshine I. 2005. Identification of an Escherichia coli operon required for formation of the O-antigen capsule. J Bacteriol 187:5259–5266. doi: 10.1128/JB.187.15.5259-5266.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Rahn A, Drummelsmith J, Whitfield C. 1999. Conserved organization in the cps gene clusters for expression of Escherichia coli group 1 K antigens: relationship to the colanic acid biosynthesis locus and the cps genes from Klebsiella pneumoniae. J Bacteriol 181:2307–2313. doi: 10.1128/JB.181.7.2307-2313.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Fuentes-Castillo D, Navas-Suárez PE, Gondim MF, Esposito F, Sacristán C, Fontana H, Fuga B, Piovani C, Kooij R, Lincopan N, Catão-Dias JL. 2021. Genomic characterization of multidrug-resistant ESBL-producing Escherichia coli ST58 causing fatal colibacillosis in critically endangered Brazilian merganser (Mergus octosetaceus). Transbound Emerg Dis 68:258–266. doi: 10.1111/tbed.13686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fuentes-Castillo D, Esposito F, Cardoso B, Dalazen G, Moura Q, Fuga B, Fontana H, Cerdeira L, Dropa M, Rottmann J, González-Acuña D, Catão-Dias JL, Lincopan N. 2020. Genomic data reveal international lineages of critical priority Escherichia coli harbouring wide resistome in Andean condors (Vultur gryphus Linnaeus, 1758). Mol Ecol 29:1919–1935. doi: 10.1111/mec.15455 [DOI] [PubMed] [Google Scholar]
- 83. Sellera FP, Cardoso B, Fuentes-Castillo D, Esposito F, Sano E, Fontana H, Fuga B, Goldberg DW, Seabra LAV, Antonelli M, Sandri S, Kolesnikovas CKM, Lincopan N. 2022. Genomic analysis of a highly virulent NDM-1-producing Escherichia coli ST162 infecting a pygmy sperm whale (Kogia breviceps) in South America. Front Microbiol 13:915375. doi: 10.3389/fmicb.2022.915375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Moffatt JH, Harper M, Harrison P, Hale JDF, Vinogradov E, Seemann T, Henry R, Crane B, St Michael F, Cox AD, Adler B, Nation RL, Li J, Boyce JD. 2010. Colistin resistance in Acinetobacter baumannii is mediated by complete loss of lipopolysaccharide production. Antimicrob Agents Chemother 54:4971–4977. doi: 10.1128/AAC.00834-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1 to S9.
Summary of gene groups associated with resistance or susceptibility to 8 or more antibiotics.
Summary of strains carrying wza2 including metadata and antibiotic susceptibility phenotypes based on MIC testing with VET01S interpretations. Where dog breakpoints were not available, the human breakpoint was applied.
Data Availability Statement
All genomes are available in the following NCBI BioProjects: PRJNA318591, PRJNA324565, PRJNA324573, PRJNA481346, PRJNA503851, and PRJNA318589, all under the umbrella BioProject PRJNA314609. All RNA-seq data have been deposited to NCBI Sequence Read Archive (SRA) under the BioProject accession PRJNA1053349. All alignments, inferred phylogenies, and raw output files are available at the Cornell University eCommons Repository under https://doi.org/10.7298/pdzk-rq02.2.