

Key Points
-
•
Venetoclax resistance involves reduced mitochondrial priming and changes in BCL-2 family protein expression.
-
•
Venetoclax-resistant cells retain sensitivity to immunotherapeutic treatments.
Visual Abstract
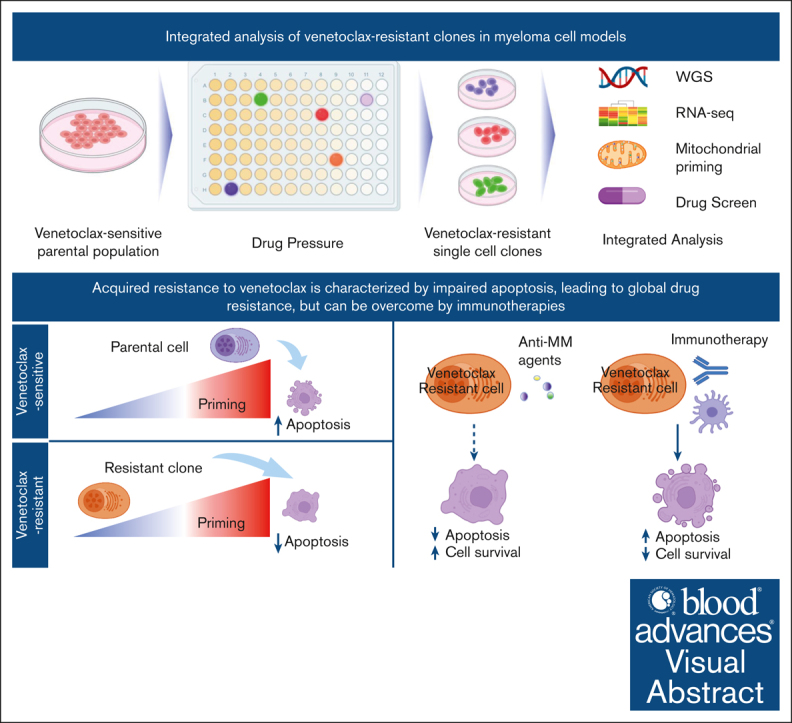
Abstact
To our knowledge, venetoclax is the first example of personalized medicine for multiple myeloma (MM), with meaningful clinical activity as a monotherapy and in combination in patients with myeloma harboring the t(11:14) translocation. However, despite the high response rates and prolonged progression-free survival, a significant proportion of patients eventually relapse. Here, we aim to study adaptive molecular responses after the acquisition of venetoclax resistance in sensitive t(11:14) MM cell models. We therefore generated single-cell venetoclax-resistant t(11:14) MM cell lines and investigated the mechanisms contributing to resistance as well as the cells’ sensitivity to other treatments. Our data suggest that acquired resistance to venetoclax is characterized by reduced mitochondrial priming and changes in B-cell lymphoma-2 (BCL-2) family proteins’ expression in MM cells, conferring broad resistance to standard-of-care antimyeloma drugs. However, our results show that the resistant cells are still sensitive to immunotherapeutic treatments, highlighting the need to consider appropriate sequencing of these treatments after venetoclax-based regimens.
Introduction
The B-cell lymphoma-2 (BCL-2) family of proteins forms the crux of the mitochondrial apoptotic pathway. The dynamic interplay between antiapoptotic members and proapoptotic ligands is integral to controlling the apoptotic threshold of cells, and changes in this balance are frequently responsible for the evasion of apoptosis observed in cancer.1
Venetoclax binds competitively to the BCL2 homology 3 (BH3) domain of BCL2, restricting BCL2 from sequestering and inactivating proapoptotic proteins, leading to apoptotic cell death.2, 3, 4 Venetoclax paved the way for a biomarker-driven intervention in multiple myeloma (MM) based on its meaningful clinical activity as a monotherapy or in combination with other agents in patients with t(11;14)-positive relapsed/refractory MM.5, 6, 7, 8, 9, 10, 11, 12 Several studies have attempted to understand the biology underlying this drug sensitivity, showing that numerous intrinsic and extrinsic factors contribute to the therapeutic response in t(11:14) MM cells. This subgroup of patients generally displays upregulated BCL-2 expression and higher BCL2/BCL2L1 and BCL2/myeloid cell leukemia-1 (MCL1) ratios.13,14 Moreover, other predictive factors, such as a B-cell–like epigenetic signature and energy metabolism, were identified as potential influencers of the response to venetoclax, even in the absence of t(11;14).14, 15, 16
Nonetheless, venetoclax is not universally effective across all patients with t(11;14), and resistance may manifest either de novo or during the progression of the disease, ultimately resulting in relapse. For instance, the occurrence of resistance to venetoclax in individuals with t(11;14) MM has been associated with the acquisition of canonical plasma cell transcription factors, along with copy number gains in MCL1 and BCL2L1, as well as structural rearrangements.12
In the BELLINI study, despite the significant improvement in progression-free survival in patients with t(11;14) compared with patients without the translocation and/or low BCL2 expression, a lack of improvement in overall survival with venetoclax was observed.7
Ongoing research is therefore crucial to address resistance issues and refine the optimal treatment timing and strategies. In a retrospective analysis by the Emory group, 32 patients who progressed on venetoclax were treated with the next line of therapy.17 The analysis suggests the benefits of using venetoclax earlier; however, the small sample size and the treatment heterogeneity indicate the need for additional studies to refine optimal treatment strategies.
Here, we aim to study adaptive molecular responses after acquisition of venetoclax resistance in t(11;14) MM cell models and investigate whether acquired venetoclax resistance could lead to global resistance to multiple subsequent anti-MM therapies, which in turn can explain the lack of overall survival benefit observed in the venetoclax-treated arm. We report that although standard-of-care agents, regardless of the mechanism of action, demonstrated a reduced ability to induce cell death in venetoclax-resistant cells, cytotoxicity was still observed with immunotherapeutic approaches.
Methods
Development of DTEP cells
Resistant clones were developed from the KMS12PE and KMS27 cell lines as previously described,18 with some changes. Briefly, a total of 1000 cells were plated in 96-well plates (1 cell per well) and continuously treated with 10× half maximal inhibitory concentration (IC50) of venetoclax. After 2 weeks of exposure, drug-tolerant expanded persister (DTEP) single-cell clones were selected and expanded. The DTEP clones were maintained in 100 nM venetoclax for 2 months. Parental and resistant clones were authenticated by Short Tandem Repeat (STR) analysis.
BH3 profiling
Cells were permeabilized with digitonin and exposed to BH3 peptides. Mitochondrial transmembrane potential loss was monitored using the JC-1 dye.
Cell viability assay
Cell viability was assessed using CellTiter-Glo Luminescent Cell Viability Assay (no. G7572; Promega).
B-cell maturation antigen (BCMA) chimeric antigen receptor (CAR) T-cell killing
A second-generation anti-BCMA CAR construct containing an anti-BCMA single-chain variable fragment was custom made at GenScript. CAR-T cell cytotoxic activity was assessed by lactate dehydrogenase (LDH) release using an LDH-Glo Cytotoxicity Assay kit (Promega).
Antibody-dependent cellular cytotoxicity (ADCC)
MM cells were stained with calcein AM (Invitrogen) and incubated with or without daratumumab (1 μg/mL). NK effector cells were added at different effector-to-target cell ratios. After a 3-hour incubation, the calcein released in the supernatant by dying tumor cells was measured using a fluorescence plate reader.
Written informed consent was obtained from the participant included in the study, in accordance with the Declaration of Helsinki.
Results
Acquired resistance to venetoclax in t(11;14) MM models is characterized by reduced mitochondrial priming and changes in BCL2-family protein expression
We modeled in vitro the development of an acquired venetoclax-tolerant/resistant phenotype in MM cells. We exposed t(11;14) venetoclax-sensitive myeloma cell lines (KMS12PE and KMS27) to high-dose venetoclax treatment and generated monoclonal DTEP clones from single cells (4-5 single cell clones for each cell line) (Figure 1A-B), characterized by threefold to 10-fold increase in IC50 compared with that in the parental cells (Figure 1C).
Figure 1.

Development of t(11:14) venetoclax-resistant clones from single cells. (A) A panel of t(11:14) MM cell lines were treated with several doses of venetoclax for 72 hours. Cell viability was assessed by CellTiter-Glo (CTG) and IC50 was calculated using GraphPad prism. (B) Schema of development of DTEP/resistant clones. (C) Cell viability was evaluated using CTG after 72 hours of treatment with venetoclax in parental cells and clones and represented as percentage of viable cells compared with each untreated control. IC50 is also shown.
To determine whether venetoclax resistance in DTEP clones is mediated by genomic or transcriptional adaptation, we conducted whole-genome sequencing and RNA-seq analysis of the clones and compared them with the parental cell line. Parental and resistant cells did not show any shared difference in mutational frequency or copy number variation, including alterations in the BCL-2 gene, such as the Gly101Val mutation,19 1q21 gain, or 8p loss (supplemental Figure 1A-B).20 Moreover, each resistant clone had a unique transcriptomic signature with minimal overlap with the others’ (supplemental Figure 1C-D). Without any large differences to explain resistance, we hypothesized that it develops from alterations in the apoptosis pathway.
Antiapoptotic members of the BCL-2 family all perform a similar function, sequestering proapoptotic ligands. Targeting one can result in increased expression of other antiapoptotic members as a compensatory measure.21,22 We therefore evaluated the expression of proapoptotic (BIM, BID, BAD, PUMA, and NOXA), antiapoptotic (BCL2, MCL1, BCL-W, and BCL-XL), or pore-forming effectors (BAX and BAK) at the RNA and protein level. Although we did not detect consistent and statistically significant alterations in gene expression (supplemental Figure 1E-F), at the protein level, we observed a significant upregulation of antiapoptotic members across all clones in both cellular models, leading to an overall reduction in the BCL-2 ratio, particularly significant for MCL1 (Figure 2A-B). Furthermore, we observed increased binding and sequestration of BIM by MCL-1 in resistant clones than in parental cells (Figure 2C-D), including in the KMS27 cells, where the expression of BIM isoforms was detected. Because the downregulation of proapoptotic members is associated with the development of resistance to venetoclax,12,23,24 we evaluated their protein expression in our 2 cellular models. Among all proapoptotic members, PUMA was significantly downregulated in all the clones tested, suggesting a potential involvement of PUMA loss in the acquisition of venetoclax resistance (supplemental Figure 2).
Figure 2.

Increased expression of antiapoptotic proteins characterizes the acquisition of venetoclax resistance. (A-B) Western Blot (WB) analysis of BCL-2, MCL-1, BCL-XL, and BCL-W in parental cells and clones of KMS12PE model (A) and KMS27 model (B). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. Protein expression densitometry values were calculated using ImageJ. Ratios of BCL-2 vs other antiapoptotic proteins in parental and resistant clones are also illustrated. Error bars represent the standard deviation (SD) of at least duplicate results. (C) Immunoprecipitation (IP) for BIM in parental cells and clones 12A and 12D of KMS12PE model, followed by western blot for BIM, BCL2, MCL-1, BCL-W, and BCL-XL. GAPDH was used as a loading control. (D) IP for BIM in parental cells and clone 27B of KMS27 model. (E-H) BH3 profiling was performed on parental cells and resistant cells using a plate-based BH3 profiling assay and several doses of indicated peptides. Each experiment was performed in triplicate. The heat map for panels E,G represents mean of % depolarization from 1 experiment performed in triplicate in all indicated cells. The bar graph for panels F,H represents percentage of depolarization in parental and representative clones (clone 12A in panel F and clone 27B in panel H). Two-way analysis of variance (ANOVA) test was used to calculate statistical significance. ∗P ≤ .05; ∗∗P ≤ .01; ∗∗∗P ≤ .001; ∗∗∗∗P ≤ .0001. BAD, BCL2 and BCL-XL dependency; HRK, BCL-XL dependency; MS1, MCL1 dependency; PUMA/BIM, promiscuous peptides.
To investigate how these differences affect apoptotic priming, we performed baseline BH3 profiling. In both cellular models, resistant clones exhibited a diminished response to the promiscuous peptides compared with parental cells, suggesting reduced mitochondrial priming and impaired apoptotic sensitivity in the resistant cells (Figure 2E-H).
In the KMS12PE model, comparable apoptotic responses were observed in the presence of the BCL-XL-peptide HRK; moreover, some resistant clones displayed heightened mitochondrial sensitivity to the MCL1 peptide MS-1, whereas others predominantly maintained rather than increased their mitochondrial sensitivity (Figure 2E-F). In the KMS27 model, although no heightened sensitivity to the MCL1 peptide was observed, there was a discernible trend toward increased sensitivity to the BCL-XL peptide, albeit not reaching statistical significance (Figure 2G-H).
Simultaneous inhibition of BCL2 antiapoptotic members overcomes acquired venetoclax resistance
We explored the impact of heightened MCL1 expression in fostering venetoclax resistance by using 2 different MCL1 antagonists. We noted a divergence in the response to MCL1 inhibition among different clones, with some maintaining sensitivity and others displaying increased resistance (Figure 3A). Next, resistant clones were exposed to increasing concentrations of venetoclax in the presence or absence of the MCL1 antagonist S63845. Although single inhibition did not rescue the resistant phenotype, combination with venetoclax synergistically inhibited MM cell growth (Figure 3B), inducing apoptosis (Figure 3E-F; supplemental Figure 3B). The synergistic interaction between the drugs was determined by calculating combination index values, demonstrating strong synergism (combination index <0.3) in the resistant cells (Figure 3D). Interestingly, dual BCL2 and BCL-xL inhibition (venetoclax plus A1155463) also resulted in synergistic activity (Figure 3C-E,G) compared with single inhibition (supplemental Figure 3A). Altogether, these data suggest that combined inhibition of BCL2 family members represents an effective strategy to overcome venetoclax resistance. However, the detection of significant toxicity with combination therapies observed in normal peripheral blood mononuclear cells (data not shown) suggests an unfavorable therapeutic index and the need for alternative therapeutic options.
Figure 3.

Dual targeting of BCL2 antiapoptotic members synergistically inhibits cell proliferation of venetoclax-resistant cells. (A) Parental cells and venetoclax-resistant clones were treated with increasing doses of MCL-1 inhibitors (S63845 and AZD5991) for 48 hours. Cell viability was evaluated using CTG and it is expressed as a percentage of cell viability from untreated cells. IC50 values are also shown. (B-C) Venetoclax-resistant clones (KMS12PE clone 12A and KMS27 clone 27B) were treated for 24 hours with a combination of venetoclax and MCL1 inhibitor, S63845 (B), or BCL-XL inhibitor, A-1155463 (C). Cell viability was assessed by CTG assay and represented as percentage of cell viability compared with each untreated control. Two-way ANOVA test was used to calculate statistical significance compared to venetoclax single agent. Data represent mean ± SD; n = 3. (D) Synergism analysis was performed with the CalcuSyn software. Combination index (CI) is represented in the heat map. CI = 1 additive, CI < 1 synergistic, and CI > 1 antagonistic. (E-G) Venetoclax-resistant clone (KMS27 model) were treated with a combination of different BH3 mimetic drugs for 24 hours and apoptosis was measured by flow cytometry after annexin V and propidium iodide staining. One representative experiment in resistant clones is shown in panel E. The bar graph in panels F-G represents the mean percentage of apoptosis (annexin V-positive cells) from 2 independent experiments. SV100: S63845 100 nM + venetoclax 100 nM, SV500: S63845 100 nM + venetoclax 500 nM, AV100: A-1155463 100 nM + venetoclax 100 nM, AV500: A-1155463 100 nM + venetoclax 500 nM. Two-way ANOVA test was used to calculate statistical significance between combination treatment with venetoclax single agent.
Venetoclax-resistant clones displayed cross-resistance toward standard antimyeloma agents
Triggering apoptosis is a crucial mechanism for numerous anticancer agents. Consequently, we postulate that the diminished sensitivity to apoptosis in venetoclax-resistant clones could similarly affect their responsiveness to other chemotherapeutic agents, as suggested in other studies.23, 24, 25 In fact, we observed that, compared with parental cells, venetoclax-resistant clones displayed cross-resistance toward most standard-of-care anti-MM agents, including alkylating agents (melphalan and bendamustine), proteasome inhibitors (bortezomib and carfilzomib), and dexamethasone (Figure 4; supplemental Figure 4A-B). We additionally examined the impact of immunomodulatory drugs (lenalidomide and pomalidomide) on both parental and resistant clones. Although the parental cells exhibited intrinsic resistance to lenalidomide, we demonstrated that, in the resistant clones, the IC50 was achieved at an even higher dose (Figure 4B-D; supplemental Figure 4A-B). Moreover, combinations of these agents (eg, bortezomib plus dexamethasone or bortezomib plus lenalidomide) were also not effective in the resistant clones (supplemental Figure 4C-D), implying these therapies will not be as efficacious after the development of venetoclax resistance.
Figure 4.

Venetoclax-resistant clones display a broader resistance to standard-of-care antimyeloma agents. (A,C) Drug sensitivity screening in the KMS12PE (A) and KMS27 (C) cell models. Cell viability was assessed by CTG after 48 hours of treatment and represented as percentage of cell killing compared with untreated cells. The parental bar represents the median cell killing of triplicated data. The resistant clones represent the median cell killing of all the clones tested with triplicated data (12A, 12B, 12C, and 12D in KMS12PE model; and 27B, 27D in KMS27 model). Two-way ANOVA was used to calculate P values. (B,D) IC50 ratios of venetoclax-resistant clones to parental cells for the indicated drugs are depicted (<1: sensitive than parental and >1: resistant than parental).
Immunotherapeutic strategies successfully overcome venetoclax resistance
Because immunotherapies do not rely exclusively on traditional apoptotic signaling to mediate cell death, we further investigated the efficacy of immunotherapeutic approaches to overcome venetoclax resistance. Anti-CD38 antibody induced a similar extent of antibody-mediated cellular cytotoxicity in both parental and resistant clones (Figure 5A). Similarly, we observed comparable cytotoxicity of BCMA CAR-T cells between parental and venetoclax-resistant clones (Figure 5B-C). We further investigated the ability of CAR-T cells to overcome venetoclax resistance ex vivo in primary MM cells obtained from a patient with t(11;14) MM progressing on venetoclax, carfilzomib, and dexamethasone combination. We confirmed the ability of BCMA CAR-T cells to induce significant cytotoxicity in vitro against these primary resistant MM cells (Figure 5D). Importantly, the patient was subsequently treated with the commercial cilta-cel product and achieved a complete response in 2 months (Figure 5E), confirming the efficacy of BCMA CAR-T cells in overcoming venetoclax resistance.
Figure 5.

Antibody-based and cellular immunotherapies are effective against venetoclax-resistant cells. (A) NK cell–mediated ADCC with or without daratumumab (1 μg/mL) in venetoclax-resistant clones compared with parental cells (KMS12PE model). (B-C) KMS12PE (B) and KMS27 (C) parental and resistant clones were incubated with either untransduced T cells or BCMA CAR-T cells for 4 hours at the effector-to-target ratio of 2:1 and 5:1. Killing was assessed by LDH release using LDH-Glo Cytotoxicity Assay kit. The percentage of target cell cytotoxicity was calculated using 10% Triton X-100 as a control. Data represent mean of 3 experiments performed in triplicates. (D) BCMA CAR-T cells were incubated with CD138+ primary cells isolated from a bone marrow aspirate of a patient with t(11;14) MM progressing on venetoclax. (E) Changes in the difference between involved and uninvolved serum-free light chains (dFLC) and κ/λ FLC ratio (rFLC) of the patient with venetoclax resistance before and after CAR-T cell therapy.
Discussion
Understanding and addressing the heterogeneity in mechanisms of resistance is crucial for developing strategies to overcome or prevent resistance to venetoclax, thereby enhancing the long-term effectiveness of this targeted therapy in cancer treatment. Our research aligns with a growing body of evidence indicating heterogeneity in the mechanisms involved in intrinsic and acquired resistance to venetoclax. These include mutations in BCL2 or BAX, upregulation of MCL1 due to chromosome 1 amplification or NF-κB activation, and downregulation of PUMA or NOXA.19,23,26, 27, 28, 29, 30, 31
We found that MCL1 and PUMA protein levels were recurrently affected in all the clones with acquired resistance to venetoclax, providing a potential molecular explanation for the reduction in priming after the acquisition of resistance. However, despite its substantial elevation in all resistant cell lines, MCL1 protein levels did not serve as a distinguishing factor for responders to the MCL1 inhibitor. Indeed, we noted a divergence in the response to MCL1 inhibition among different clones, with some maintaining sensitivity and others displaying increased resistance, consistent with recent findings by Thomalla et al.23
Although the molecular mechanisms driving intrinsic and extrinsic resistance appear to be distinct, an altered expression of the antiapoptotic regulators MCL1 and BCL-XL in MM cells was a common factor contributing to primary resistance to venetoclax in MM cells, irrespective of the presence of t(11:14).14,32, 33, 34, 35, 36 Attacking this mutual interplay through coinhibition of BCL-2 with MCL-1 or BCL-XL demonstrated robust synergistic activity in preclinical studies.22,26,37 In our investigation, we observed a synergistic effect of venetoclax when combined with MCL1 or BCL-XL antagonists, effectively impeding growth and inducing apoptosis in resistant cells.
Importantly, we show that venetoclax resistance displayed cross-resistance toward DNA-damaging drugs and other agents used in MM therapy. The diminished effectiveness of most standard antimyeloma agents after venetoclax resistance acquisition indicates that although venetoclax-specific mechanisms may be involved, broad resistance to anticancer agents results from a general selection for reduced apoptotic signaling. These data suggest using venetoclax at a later stage of the disease, after conventional agents are used. Therefore, further investigation in patients experiencing progression while on venetoclax is warranted.
Notably, assessing drug sensitivity in resistant clones after venetoclax discontinuation revealed the reacquisition of drug sensitivity (data not shown). This suggests the possibility of implementing a limited-duration or intermittent schedule for venetoclax therapy to prevent the emergence of resistance and achieve remission in patients.
Because resistance mechanisms to venetoclax involve regulators of the intrinsic apoptotic pathway, therapeutic approaches not relying exclusively on traditional apoptotic signaling to mediate cell death, such as immunotherapies, have the potential to be effective in the context of acquired resistance to venetoclax. Here, we demonstrated that resistant cells remain sensitive to both antibody-based and cellular immunotherapies. This was also confirmed ex vivo in primary MM cells obtained from a patient with t(11;14) MM progressing on venetoclax, carfilzomib, and dexamethasone combination. Our data, implying the use of immunotherapies to overcome venetoclax resistance, complement the promising early results observed in patients with relapsed/refractory MM treated with a combination of daratumumab and venetoclax.9,38 Although we acknowledge the importance of expanding the patient cohort for a more exhaustive interpretation, our data still offer crucial translational insights and may also be applicable to a non-t(11;14) setting.
In conclusion, our results demonstrate continued sensitivity to immunotherapeutic treatments despite the acquisition of resistance to venetoclax in myeloma cells and highlight the need to consider appropriate sequencing of these treatments after venetoclax-based regimens.
Conflict-of-interest disclosure: K.C.A. has received consulting fees from Bristol Myers Squibb (BMS), Celgene, Gilead, Johnson & Johnson Innovative Medicine, Precision Biosciences, Sanofi-Aventis, Takeda, and Tolero; and serves on the board of directors of, and has stock options in, OncoPep. N.C.M. is a consultant for BMS, Johnson & Johnson Innovative Medicine, OncoPep, Amgen, Karyopharm, Legend, AbbVie, Takeda, and GlaxoSmithKline (GSK); and serves on the board of directors of, and stock options in, OncoPep. M.K.S is a consultant for AbbVie and serves as an advisor for NCGM. The remaining authors declare no competing financial interests.
Acknowledgments
The authors thank Christina Usher for comments on the manuscript and Jeremy A. Ryan for sharing reagents and expertise for the BH3 profiling.
This study was supported by the National Institutes of Health (NIH)/National Cancer Institute (NCI) P01 grant (CA155258-10 [M.F., N.M., and K.A.]); a Department of Veterans Affairs I01 grant (BX001584-09 [N.M.]); a NIH/NCI R01 grant (CA207237-05) and a Paula and Rodger Riney Foundation grant (K.A.); and an NIH/NCI SPORE grant (P50-CA100707-18 [N.M. and K.A]).
Authorship
Contribution: M.F. and N.C.M. designed and conducted the study and wrote the manuscript; S. Deng performed the experiments and analyzed the data; S. Derebail generated the drug-tolerant expanded persister clones; V.J.W. and J.F.N helped with the experiments and performed gene expression analysis; E.M.-M. applied natural killer cells and guided the antibody-dependent cellular cytotoxicity experiment; M.C., M.S., and R.P. performed the CAR T-cell experiment; G.G., C.C., P.K., T.D., and Y.Y. helped with the cell culture and immunoprecipitation; A.A.S. and M.K.S. analyzed the gene expression data; A.G., L.Q., and K.C.A. provided critical evaluation of experimental data and the manuscript; and all authors critically reviewed and approved the manuscript before submission.
Footnotes
M.F. and N.C.M. contributed equally to this study.
Sequencing data have been deposited in the Gene Expression Omnibus database (accession number GSE269245).
Data are available on request from the corresponding author, Mariateresa Fulciniti (mariateresa_fulciniti@dfci.harvard.edu).
The full-text version of this article contains a data supplement.
Supplementary Material
References
- 1.Delbridge AR, Grabow S, Strasser A, Vaux DL. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer. 2016;16(2):99–109. doi: 10.1038/nrc.2015.17. [DOI] [PubMed] [Google Scholar]
- 2.Del Gaizo Moore V, Letai A. Rational design of therapeutics targeting the BCL-2 family: are some cancer cells primed for death but waiting for a final push? Adv Exp Med Biol. 2008;615:159–175. doi: 10.1007/978-1-4020-6554-5_8. [DOI] [PubMed] [Google Scholar]
- 3.Roberts AW, Davids MS, Pagel JM, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):311–322. doi: 10.1056/NEJMoa1513257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DiNardo CD, Pratz KW, Letai A, et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-randomised, open-label, phase 1b study. Lancet Oncol. 2018;19(2):216–228. doi: 10.1016/S1470-2045(18)30010-X. [DOI] [PubMed] [Google Scholar]
- 5.Boccon-Gibod C, Talbot A, Le Bras F, et al. Carfilzomib, venetoclax and dexamethasone for relapsed/refractory multiple myeloma. Br J Haematol. 2020;189(3):e73–e76. doi: 10.1111/bjh.16483. [DOI] [PubMed] [Google Scholar]
- 6.Costa LJ, Davies FE, Monohan GP, et al. Phase 2 study of venetoclax plus carfilzomib and dexamethasone in patients with relapsed/refractory multiple myeloma. Blood Adv. 2021;5(19):3748–3759. doi: 10.1182/bloodadvances.2020004146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar SK, Harrison SJ, Cavo M, et al. Venetoclax or placebo in combination with bortezomib and dexamethasone in patients with relapsed or refractory multiple myeloma (BELLINI): a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2020;21(12):1630–1642. doi: 10.1016/S1470-2045(20)30525-8. [DOI] [PubMed] [Google Scholar]
- 8.Mateos MV. Paper presented at: Proceedings of the 20th International Myeloma Workshop 2023; 27 September-31 December 2023. Results from the randomized, open-label phase 3 CANOVA study of venetoclax-dexamethasone versus pomalidomide-dexamethasone in patients with t(11;14)-positive relapsed/refractory multiple myeloma. Athens, Greece. Abstract OA-52. [Google Scholar]
- 9.Bahlis NJ, Baz R, Harrison SJ, et al. Phase I study of venetoclax plus daratumumab and dexamethasone, with or without bortezomib, in patients with relapsed or refractory multiple myeloma with and without t(11;14) J Clin Oncol. 2021;39(32):3602–3612. doi: 10.1200/JCO.21.00443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumar S, Kaufman JL, Gasparetto C, et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood. 2017;130(22):2401–2409. doi: 10.1182/blood-2017-06-788786. [DOI] [PubMed] [Google Scholar]
- 11.Touzeau C, Dousset C, Le Gouill S, et al. The Bcl-2 specific BH3 mimetic ABT-199: a promising targeted therapy for t(11;14) multiple myeloma. Leukemia. 2014;28(1):210–212. doi: 10.1038/leu.2013.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leblay N, Ahn S, Tilmont R, et al. Integrated epigenetic and transcriptional single-cell analysis of t(11;14) multiple myeloma and its BCL2 dependency. Blood. 2024;143(1):42–56. doi: 10.1182/blood.2023020276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cleynen A, Samur M, Perrot A, et al. Variable BCL2/BCL2L1 ratio in multiple myeloma with t(11;14) Blood. 2018;132(26):2778–2780. doi: 10.1182/blood-2018-09-876433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gupta VA, Barwick BG, Matulis SM, et al. Venetoclax sensitivity in multiple myeloma is associated with B-cell gene expression. Blood. 2021;137(26):3604–3615. doi: 10.1182/blood.2020007899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bajpai R, Matulis SM, Wei C, et al. Targeting glutamine metabolism in multiple myeloma enhances BIM binding to BCL-2 eliciting synthetic lethality to venetoclax. Oncogene. 2016;35(30):3955–3964. doi: 10.1038/onc.2015.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bajpai R, Sharma A, Achreja A, et al. Electron transport chain activity is a predictor and target for venetoclax sensitivity in multiple myeloma. Nat Commun. 2020;11(1):1228. doi: 10.1038/s41467-020-15051-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maples KT, Nooka AK, Gupta V, et al. Natural history of multiple myeloma patients refractory to venetoclax: a single center experience. Am J Hematol. 2021;96(3):E68–E71. doi: 10.1002/ajh.26064. [DOI] [PubMed] [Google Scholar]
- 18.Zhao X, Ren Y, Lawlor M, et al. BCL2 amplicon loss and transcriptional remodeling drives ABT-199 resistance in B cell lymphoma models. Cancer Cell. 2019;35(5):752–766.e9. doi: 10.1016/j.ccell.2019.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blombery P, Anderson MA, Gong JN, et al. Acquisition of the recurrent Gly101Val mutation in BCL2 confers resistance to venetoclax in patients with progressive chronic lymphocytic leukemia. Cancer Discov. 2019;9(3):342–353. doi: 10.1158/2159-8290.CD-18-1119. [DOI] [PubMed] [Google Scholar]
- 20.Khalsa JK, Cha J, Utro F, et al. Genetic events associated with venetoclax resistance in CLL identified by whole-exome sequencing of patient samples. Blood. 2023;142(5):421–433. doi: 10.1182/blood.2022016600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Letai A, Bhola P, Welm AL. Functional precision oncology: testing tumors with drugs to identify vulnerabilities and novel combinations. Cancer Cell. 2022;40(1):26–35. doi: 10.1016/j.ccell.2021.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramsey HE, Fischer MA, Lee T, et al. A novel MCL1 inhibitor combined with venetoclax rescues venetoclax-resistant acute myelogenous leukemia. Cancer Discov. 2018;8(12):1566–1581. doi: 10.1158/2159-8290.CD-18-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomalla D, Beckmann L, Grimm C, et al. Deregulation and epigenetic modification of BCL2-family genes cause resistance to venetoclax in hematologic malignancies. Blood. 2022;140(20):2113–2126. doi: 10.1182/blood.2021014304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dousset C, Maïga S, Gomez-Bougie P, et al. BH3 profiling as a tool to identify acquired resistance to venetoclax in multiple myeloma. Br J Haematol. 2017;179(4):684–688. doi: 10.1111/bjh.14251. [DOI] [PubMed] [Google Scholar]
- 25.Thijssen R, Tian L, Anderson MA, et al. Single-cell multiomics reveal the scale of multilayered adaptations enabling CLL relapse during venetoclax therapy. Blood. 2022;140(20):2127–2141. doi: 10.1182/blood.2022016040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhatt S, Pioso MS, Olesinski EA, et al. Reduced mitochondrial apoptotic priming drives resistance to BH3 mimetics in acute myeloid leukemia. Cancer Cell. 2020;38(6):872–890.e6. doi: 10.1016/j.ccell.2020.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blombery P, Lew TE, Dengler MA, et al. Clonal hematopoiesis, myeloid disorders and BAX-mutated myelopoiesis in patients receiving venetoclax for CLL. Blood. 2022;139(8):1198–1207. doi: 10.1182/blood.2021012775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choudhary GS, Al-Harbi S, Mazumder S, et al. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis. 2015;6(1):e1593. doi: 10.1038/cddis.2014.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alford SE, Kothari A, Loeff FC, et al. BH3 inhibitor sensitivity and Bcl-2 dependence in primary acute lymphoblastic leukemia cells. Cancer Res. 2015;75(7):1366–1375. doi: 10.1158/0008-5472.CAN-14-1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020;383(7):617–629. doi: 10.1056/NEJMoa2012971. [DOI] [PubMed] [Google Scholar]
- 31.Chen X, Glytsou C, Zhou H, et al. Targeting mitochondrial structure sensitizes acute myeloid leukemia to venetoclax treatment. Cancer Discov. 2019;9(7):890–909. doi: 10.1158/2159-8290.CD-19-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morales AA, Kurtoglu M, Matulis SM, et al. Distribution of Bim determines Mcl-1 dependence or codependence with Bcl-xL/Bcl-2 in Mcl-1-expressing myeloma cells. Blood. 2011;118(5):1329–1339. doi: 10.1182/blood-2011-01-327197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gong JN, Khong T, Segal D, et al. Hierarchy for targeting prosurvival BCL2 family proteins in multiple myeloma: pivotal role of MCL1. Blood. 2016;128(14):1834–1844. doi: 10.1182/blood-2016-03-704908. [DOI] [PubMed] [Google Scholar]
- 34.Punnoose EA, Leverson JD, Peale F, et al. Expression profile of BCL-2, BCL-XL, and MCL-1 predicts pharmacological response to the BCL-2 selective antagonist venetoclax in multiple myeloma models. Mol Cancer Ther. 2016;15(5):1132–1144. doi: 10.1158/1535-7163.MCT-15-0730. [DOI] [PubMed] [Google Scholar]
- 35.Touzeau C, Ryan J, Guerriero J, et al. BH3 profiling identifies heterogeneous dependency on Bcl-2 family members in multiple myeloma and predicts sensitivity to BH3 mimetics. Leukemia. 2016;30(3):761–764. doi: 10.1038/leu.2015.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gupta VA, Matulis SM, Conage-Pough JE, et al. Bone marrow microenvironment-derived signals induce Mcl-1 dependence in multiple myeloma. Blood. 2017;129(14):1969–1979. doi: 10.1182/blood-2016-10-745059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leverson JD, Phillips DC, Mitten MJ, et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci Transl Med. 2015;7(279) doi: 10.1126/scitranslmed.aaa4642. [DOI] [PubMed] [Google Scholar]
- 38.Regidor BS, Siddiqi D, Beatty BM, et al. Efficacy of venetoclax plus anti-CD38 monoclonal antibody-containing therapies among t(11;14) positive multiple myeloma patients regardless of timing of prior treatment with this antibody. Eur J Haematol. 2023;110(2):222–223. doi: 10.1111/ejh.13896. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.