

Abstract
Gastric H+/K+-ATPase is a P-type ATPase responsible for acid secretion in the stomach. This protein adopts mainly two conformations called E1 and E2. Even though two high-resolution structures for a P-ATPase in these conformations are available, little structural information is available about the transition between these two conformations. In the present study, we used two experimental approaches to investigate the structural differences that occur when gastric ATPase is placed in the presence of various ligands and ligand combinations. We used attenuated total reflection–Fourier-transform IR experiments under a flowing buffer to modify the environment of the protein inside the measurement cell. The high accuracy of the results allowed us to demonstrate that the E1–E2 transition induces a net change in the secondary structure that concerns 10–15 amino acid residues of a total of 1324 in the proteins. The E2.K+ structure is characterized by a decreased β-sheet content and an increase in the disordered structure content with respect to the E1 form of the enzyme. Modifications in the absorption of the side chain of amino acids are also suggested. By using hydrogen/deuterium-exchange kinetics, we show that tertiary-structure modifications occurred in the presence of the same ligands, but these changes involved several hundreds of residues. The present study suggests that conformational changes in the catalytic cycle imply secondary-structure rearrangements of small hinge regions that have an impact on large domain re-organizations.
Keywords: conformation, coupling ATR–FTIR (attenuated total reflection–Fourier-transform infrared), gastric ATPase, hydrogen/deuterium exchange
Abbreviations: ATR–FTIR, attenuated total reflection–Fourier-transform infrared; COBSI index, change of backbone structure and interaction index; IRE, internal reflection element
INTRODUCTION
Gastric H+/K+-ATPase is the protein responsible for acid secretion in the stomach. It is an electro-neutral pump that transports protons from the cytoplasm of stomach parietal cells and creates a large pH gradient in exchange for the internalization of potassium [1,2]. Energy for these transportations comes from ATP hydrolysis. H+/K+-ATPase belongs to the family of ubiquitous P-type ATPases [3]. These proteins are particularly characterized by the formation of a covalent aspartylphosphate bond during their catalytic cycle. They are also called E1,E2-ATPases since they adopt, mainly, two conformations during their catalytic cycle. Several subconformations have also been isolated [4,5].
Most P-ATPases are composed of one polypeptide chain, but H+/K+-ATPase is composed of two subunits, α (114 kDa) and β (33 kDa) [6]. The large α-subunit contains the phosphorylation site (Asp385) and the nucleotide- and cation-binding sites. This multi-spanning membrane polypeptide is the catalytic subunit responsible for the coupling between ATP hydrolysis and transport of cations. The β-subunit is a single-membrane-spanning polypeptide that is heavily glycosylated [7]. This subunit plays a role in the proper assembly and targeting of the protein in the parietal cell. A possible role for the β-subunit in the cation specificity of the protein was also suggested [8].
Recently, the structure of SERCA1A (sarcoplasmic/endoplasmic-reticulum Ca2+-ATPase 1A) in two conformations has been published [9,10]; an NMR structure of the nucleotide-binding domain of Na+,K+-ATPase in the presence or absence of ATP has also been published [11]. Even though these two high-resolution structures of a P-type ATPase are now available, the mechanism that is responsible for ATP hydrolysis and ion pumping remains elusive. A structural description of the enzyme in its major conformations E1 and E2 and in intermediate conformations should shed some light on the enzyme mechanism at a submolecular level.
IR spectroscopy can provide structural information for large membrane proteins [12]. The easiest way to obtain information about a protein under different conditions is to prepare a new sample under each of those conditions and record the corresponding spectra. Yet, in the early work by Mitchell et al. [13] and later by other authors [14], it was found that, to obtain a high sensitivity, the environment of the protein must be modified inside the measurement cell. Difference IR spectroscopy consists in recording a first spectrum of the sample, modifying the environment of the protein in situ and then recording a new spectrum. The difference between these two spectra gives an IR difference spectrum that is characteristic of the changes that appear in the sample [15]. The spectral contribution of the unmodified part of the sample is cancelled out in the subtraction. Positive bands in the new spectrum are characteristic of the final state of the sample and negative ones of the initial state. Different methods exist to modify the environment of a protein inside the cell. Some proteins such as bacteriorhodopsin are light-sensitive and a simple illumination can modify the conformation of the protein [16]. Other proteins are sensitive to redox reactions and a particular spectroelectrochemical cell was designed to control the redox state of a protein such as cytochrome c [17]. Caged compounds are photosensitive precursors that are capable of releasing a specific ligand after a UV illumination. The use of caged compounds has increased since more caged molecules are available [18]. Finally, ATR–FTIR (attenuated total reflection–Fourier-transform infrared) experiments under a flowing buffer can be used to change the environment of a membrane protein attached to an IRE (internal reflection element). This method has been shown previously to be useful for the nicotinic acetylcholine receptor [19,20].
In the present study, two experimental approaches have been used to investigate the conformational changes that occur when gastric H+/K+-ATPase is placed in the presence of various ligands and ligand combinations. We first used a flow system in an ATR–FTIR mode to modify the environment of the protein. In the second approach, we recorded hydrogen/deuterium (H/2H)-exchange kinetics to investigate tertiary-structure changes. We conclude that only a few amino acid residues are involved in the conformational change of the H+/K+-ATPase during its catalytic cycle and very limited secondary-structure changes are necessary to induce a large movement of domains in the protein.
EXPERIMENTAL
Isolation and purification of gastric vesicles
Gastric vesicles were isolated from hog gastric fundus by differential centrifugation and discontinuous sucrose-density-gradient ultracentrifugation as described previously [21]. The material collected at the 8–30% sucrose interface will be referred to as tubulovesicles hereafter. H+/K+-ATPase is the principal protein component of tubulovesicles. Coomassie Blue-stained SDS/polyacrylmide gels essentially revealed a major band at 95 kDa corresponding to the α-subunit and a smear at 60–90 kDa corresponding to the β-subunit of the H+/K+-ATPase. Protein concentration was estimated with the BCA kit (Pierce) using BSA as a standard. The hydrolysis activity of H+/K+-ATPase was measured in 50 mM Hepes/Tris (pH 7.2), 2 mM ATP-Na2 and 2 mM MgCl2 in the presence or absence of 20 mM KCl. The reaction was performed at 37 °C for 15 min and stopped by the addition of 1.75% (w/v) SDS. Pi generated by the hydrolysis of ATP was quantified essentially by the method of Stanton [22], except that coloration was developed by ascorbate. Typical values of ATPase activity in tubulovesicles are 21±1 μmol of Pi·h−1·(mg of protein)−1 without KCl and 119±3 μmol of Pi·h−1·(mg of protein)−1 in the presence of KCl.
FTIR measurements
For ATR–FTIR experiments, tubulovesicles were suspended in 2 mM Hepes/Tris (pH 7.2) and centrifuged at 125000 g for 35 min at 4 °C. The pellet was washed by another step of suspension followed by centrifugation under the same conditions. The final pellet was suspended in 2 mM Hepes/Tris (pH 7.2) to obtain a final concentration of 5 mg/ml in proteins. Depending on the experiments, NaCl or KCl was added at a final concentration of 20 mM to the same buffer. Film preparation is described in Figure 1.
Figure 1. Experimental procedure for recording spectra during a flowing buffer ATR–FTIR measurement.

(A) Schematic representation of the pumping system and buffer selection under computer control. The measurement cell is shown in (B). A 50×20×2 mm Ge monocrystal with bevelled edge (45°) allowed 25 internal reflections of the IR beam. The sample was deposited on the Ge plate by evaporating the solvent under a stream of nitrogen. The experimental procedure is schematically shown in (C). A first buffer was used to hydrate completely the sample. Typically, 4 h were needed to obtain a fully hydrated stable film. The first spectrum was recorded in buffer A. After 1 min, a second spectrum was recorded in the same buffer. The valve was then switched to pump in buffer B for 1 min. A third spectrum was then recorded. The valve was then switched again and buffer A flowed at the surface of the sample for 1 min and a fourth spectrum was recorded. After a 20 min wash, a new cycle of recording the spectra was started.
Flowing buffer in the liquid cell
For flowing buffer experiments, an assembly was created that allows a buffer to flow inside an IR liquid cell at the surface of a Ge plate. The geometry of the IRE allowed 25 internal reflections. A peristaltic pump adjusted the flow rate at 1.5 ml/min and a computer-controlled valve selected the buffer. All experiments were performed at 20 °C on a Bruker Equinox 55 spectrophotometer equipped with a nitrogen-cooled MCT (mercury cadmium telluride) detector. Before starting the experiments, a reference spectrum was recorded with the assembly alone. For experiments in the presence of H+/K+-ATPase, a film of 100 μg of tubulovesicle proteins suspended in 2 mM Hepes/Tris (pH 7.2) was deposited on the Ge plate. The procedure for a typical experiment is shown in Figure 1. The first buffer flowed for 4 h in the cell to obtain a stable IR signal. A first spectrum was recorded in buffer A at a resolution of 4 cm−1 and resulted from 1024 co-added scans. A new spectrum was recorded 1 min later in the same buffer. The IR difference spectrum obtained by subtracting these two spectra was used as a control for the stability of the film. We assumed that the sample was fully hydrated and stable when the difference between these two spectra did not present any absorption band that arose above the noise. A threshold for the amplitude was thus set above which the deviation in difference IR spectra can be considered as significant.
When the film was stable under the flowing buffer, the valve was activated to pump in the second solution. A spectrum was recorded after 1 min and the IR difference spectrum was calculated as the difference between the spectrum under condition B and the spectrum under condition A. This difference reflected the changes that appeared after changing the buffer. The valve was then switched again to allow the first solution to flow into the cell. In some cases, a spectrum was recorded after washing for 1 min to verify the reversibility of spectral changes. After 20 min of washing, a new cycle was started. This procedure was repeated at least ten times for each experiment and the difference spectra were averaged. Collecting a large number of difference spectra and computing the average spectrum further increased the signal-to-noise ratio. To obtain an estimate for the noise of a spectrum, we considered the 2800–2200 cm−1 region, because this region is free of absorption. In this region, a linear baseline was fitted and subtracted. The S.D. was then computed over a segment of 12 cm−1. The segment was then shifted by 1 cm−1 and the operation was repeated. For a single IR difference spectrum, the mean±S.D. over the 2800–2200 cm−1 region reached 7.4×10−6 a.u. (absorbance units). A mean spectrum of ten difference spectra had a typical S.D. of 2.0×10−6 a.u. over the same region. This improvement was as expected, since noise decreases in proportion to the square root of the number of experiments. Multiple experiments have been performed with four different extractions of the tubulovesicles. Compositions of the buffers used in this study are presented in Table 1.
Table 1. Composition of the solutions used in the present study.
The presence of ligands induced a specific conformation of the H+,K+-ATPase. We used the sample names reported previously [37]. All the solutions were prepared with the same buffer (50 mM Hepes/Tris, pH 7.2), but different ligands were added at the indicated concentrations (in mM).
| Sample name | NaCl | KCl | ATP-Na2 | CaCl2 | H+,K+-ATPase conformation |
|---|---|---|---|---|---|
| Na condition | 20 | − | − | − | E1 |
| K condition | − | 20 | − | − | E2 |
| CaNa condition | 20 | − | − | 2 | E1 |
| CaK condition | − | 20 | − | 2 | E1 |
| Ca condition | 4 | − | − | 2.1 | E1 |
| Ca-ATP condition | − | − | 2 | 4 | E1-P |
Calculation of the COBSI (change of backbone structure and interaction) index
The COBSI index has been introduced for quantitative estimation of the conformational change of the Ca-ATPase during its catalytic cycle [23].
 |
where Abs is the total protein absorbance and |ΔAbs| the sum of positive and negative bands in the difference IR spectra. Spectra of the hydrated film in the presence of buffer were obtained by subtracting the contribution of the spectrum of the buffer alone from the spectrum of the protein overlaid by the buffer, after normalization of both spectra on the 2500–1900 cm−1 water band.
Spectral simulation
The difference spectrum expected for the E1–E2 transition of Ca2+-ATPase was computed on the basis of the secondary-structure difference between both states derived from the three-dimensional structures [9,10] using the DSSP program of Kabsch and Sander [24]. The number of residues in the α-helix structure was found to be 406 and 401, in β-sheet structures 145 and 121, in β-turn structures 117 and 124 and in undefined structures 178 and 203 for 1EUL [9] and 1IWO [10] respectively. Band shapes typical for the different secondary-structure contributions were computed by the method described recently in [25].
Polarized measurements
Experiments were performed as described previously, but spectra were recorded with the incident light polarized parallel or perpendicular to the incidence plane. Spectra were corrected for the absorption of the buffer as described above. The dichroic spectrum is the difference between spectra recorded with parallel and perpendicular polarizations. Before the subtraction, the perpendicular spectrum was normalized with the parallel spectrum by scaling both spectra on the 1750–1710 cm−1 region associated with ν(C=O) of lipids to take into account the differences in the relative power of the evanescent field as described previously [26].
Hydrogen/deuterium-exchange kinetics
For kinetics of hydrogen/deuterium exchange, spectra were recorded on a PerkinElmer 1720X FTIR spectrophotometer equipped with a liquid nitrogen-cooled MCT detector at a resolution of 4 cm−1. The spectrophotometer was continuously purged with dry air. Measurements were performed at 20 °C. Thin films were obtained by evaporating slowly a sample containing 50 μg of tubulovesicle proteins [suspended in 2 mM Hepes/Tris (pH 7.2) and 20 mM of either KCl or NaCl] on one side of the ATR plate under a stream of nitrogen. The ATR plate was then sealed in a universal sample holder (PerkinElmer). Before starting the deuteration, ten spectra of each sample were recorded (co-addition of 12 scans each) to test the stability of the measurements.
To perform the hydrogen/deuterium exchange, nitrogen gas was saturated with 2H2O by bubbling through a series of four vials containing liquid 2H2O. The flow rate of 75 ml/min was controlled by a Brooks flow meter. Bubbling was started 1 h before starting the experiments. At zero time, the tubing was connected to the cavity of the sealed chamber surrounding the film. For each kinetic time point, 12 scans were recorded and averaged at a resolution of 4 cm−1.
A computer program was written to drive the spectrophotometer during the kinetic measurements. At the beginning, spectra were recorded every 15 s. After the first 2 min, the time interval was exponentially increased. After 16 min, the interval between the scans was large enough to allow the inter-digitation of a second kinetic measurement. A second sample placed on another ATR set up on the PerkinElmer shuttle was then analysed with the same time sampling, but with 16 min offset by connecting the 2H2O-saturated N2 flow in series with the first sample. From this time onwards, our program changed the shuttle position to follow both the kinetic measurements. Typically, one of the samples placed on the shuttle was prepared in the presence of NaCl and the other was prepared in the presence of KCl to compare both conditions under identical experimental conditions.
All the spectra of the kinetics were corrected for atmospheric water absorption. This was done automatically by an in-house-developed software that computed the subtraction coefficient as the ratio of the atmospheric water band area between 1565 and 1551 cm−1 on the sample spectrum and on a reference atmospheric water spectrum. As described previously, deuteration of protein side chains induced modifications in the amide I (1700–1600 cm−1) and amide II (1600–1500 cm−1) regions. Several parameters modulate their contribution including the ionization state of the carboxylic amino acid and the fraction of deuterated and undeuterated amino-acid side chains for every spectrum of the kinetics. We used an in-house-developed software that can compute the contribution of the amino acid side chains as a function of the extent of deuteration. This was done using the published contributions of the side chains in their protonated and deuterated forms [27]. The fraction of deuterated side chains and of the subtraction coefficient at any time of the kinetics was determined as described in [27].
The areas of amides I and II were obtained by integration between 1700–1600 and 1570–1510 cm−1 respectively. For each spectrum, the area of amide II was divided by the area of amide I. This allowed us to take into account small but significant variations in the overall spectral intensity, which were partly due to the presence of 2H2O, which induced a minor swelling of the sample layer. This swelling increased the average distance between the protein sample and the Ge interface. The ATR spectrum intensity exponentially decays with the distance; this resulted in a loss of a small percentage of the band intensity for all the measured bands. Undeuterated spectra were recorded before the kinetic measurement experiments as explained above, and we assumed that fully deuterated samples have an amide II area of 0. The area of amide II relative to the area of amide I was finally expressed between 100% (undeuterated form) and 0% (deuterated form) for each kinetic time point.
RESULTS
The particular set-up and experimental procedure used here are schematically represented in Figure 1 and in the Experimental section. Typical spectra obtained during the present study are presented in Figure 2. Figure 2(A) presents a spectrum of the buffer alone (50 mM Hepes/Tris, pH 7.2/20 mM KCl) flowing at the surface of the Ge IRE. Broad bands near 3375 and 1640 cm−1 can be assigned respectively to water ν(OH) and δ(OH). Figure 2(B) shows the spectrum of a multilayer film of tubulovesicle membranes deposited on the IRE before passaging the aqueous solution through the cell. In this spectrum, bands at 1652 cm−1 (amide I) and 1544 cm−1 (amide II) arise mainly from the absorption of proteins; some bands, between 3000 and 2800 cm−1, at 1739 cm−1 and 1466 cm−1 are due to different levels of absorption of the lipids. Comparing the spectra of Figures 2(A) and 2(B) reveals the large degree of overlap between the absorption of the film and that of the buffer. Figure 2(C) presents a spectrum of a tubulovesicle-membrane multilayer film in the presence of a flowing buffer. Absorption bands due to water are mainly observed, but also weak bands associated with tubulovesicle molecules are seen. Figure 2(D) presents the spectrum of a film of tubulovesicles in the presence of the previous buffer, but prepared in 2H2O. In this spectrum, the bands of the buffer do not overlap the absorption bands of proteins and lipids. This approach reveals, therefore, the lipid and protein spectra in the presence of the solution. Alternatively, it is possible to subtract mathematically the contribution of the spectrum of the buffer alone (Figure 2A) from the spectrum of the protein in the presence of a flowing buffer (Figure 2C). This was achieved after normalization of both spectra on the 2500–1900 cm−1 band associated with water. The resulting spectrum is presented in Figure 2(E). By comparing the spectra of Figures 2(B) and 2(E) with the initial spectrum of the film in Figure 2(B), a significant decrease in the intensity is observed. This can be rationalized as the consequence of the swelling of the film in the presence of an excess of buffer, which results in an increase in the film thickness. The amount of sample that can interact with the exponentially decaying evanescent field of the IR beam is, therefore, significantly decreased. The inset in Figure 2 presents the time dependence of the decrease in the amide I area when a buffer flows on the surface of a film of 100 μg of proteins from tubulovesicles deposited on the Ge IRE. In the presence of the buffer, a rapid decrease in the intensity of the amide I band appears. After 4 h, the amide I area is stable. Surprisingly, the membrane stack stayed attached to the Ge plate without a significant loss of material during 19 h. This was demonstrated by a recovery of the spectral intensity after re-drying the film after the experiment (results not shown).
Figure 2. ATR–FTIR spectra obtained during a typical experiment under flowing buffer.

(A) ATR–FTIR spectrum of 50 mM Hepes/Tris (pH 7.2) and 20 mM KCl flowing at the surface of the Ge plate in the IR measurement cell. (B) ATR–FTIR spectrum of a dry film of 100 μg of proteins from tubulovesicles deposited on the surface of the crystal and dried. (C) ATR–FTIR spectrum obtained with the previous buffer (A) flowing in the IR cell when tubulovesicles were spread on the crystal (B). (D) Spectrum of the same film of tubulovesicles (A), but the buffer was prepared in 2H2O. (E) Spectrum of the same film of tubulovesicles under a flowing buffer without the spectral contribution of the solution. This spectrum was calculated as [(spectrum C)−c×(spectrum A)] where c is the subtraction coefficient obtained on zeroing the area under the 2500–1900 cm−1 band in the difference. For the clarity of the Figure, the spectra were offset along the absorbance axis. The inset presents the decrease in amplitude of the amide I region (% of the initial area) under a flowing buffer during 20 h.
In a first experiment, we compared the influence of NaCl and KCl on the IR spectrum of the tubulovesicles. We used 50 mM Hepes/Tris (pH 7.2) and 20 mM NaCl (Na condition) as buffer A (see the Experimental section). After flowing this buffer for 4 h to obtain a stable film, we started the cycles of recording as presented in Figure 1. We considered that the film was stable when the difference between two spectra recorded in buffer A with an interval of 1 min gave an IR difference spectrum with no absorption band above the noise level. The scheme presented in Figure 1 was then followed, alternating the Na condition with the K condition (50 mM Hepes/Tris, pH 7.2/20 mM KCl). The IR difference spectrum was obtained by subtraction of the Na condition spectra from the K condition spectra.
Figure 3(A) presents the average of 72 experiments performed with different extractions of tubulovesicles. Figure 3(B) shows the IR difference spectrum obtained with the same buffers but without any protein on the Ge IRE. The latter difference is probably due to the different effects of Na+ and K+ on the structure of water [28]. To compensate for the ion effect on the water spectrum, we subtracted the spectrum of Figure 3(B) from that of Figure 3(A). The resulting spectrum is presented in Figure 3(C). The stars on this latter spectrum indicate the wave numbers where both mean spectra (Figures 3A and 3B) are significantly different (Student's t test; α<0.01). This result shows that changing the buffer induced statistically significant spectral modification in the 1800–1400 cm−1 region. The whole experiment was then repeated, but in the additional presence of calcium in both solutions. The difference IR spectrum was therefore obtained by subtracting the spectra recorded in the presence of 50 mM Hepes/Tris (pH 7.2), 2 mM CaCl2 and 20 mM NaCl (CaNa condition) from the spectra recorded in the presence of 50 mM Hepes/Tris (pH 7.2), 2 mM CaCl2 and 20 mM KCl (CaK condition). The average difference spectrum obtained after 33 experiments and after correction for the buffer-induced change is presented in Figure 4(B). The presence of calcium abolishes the spectral changes observed previously in Figure 3(C) or 4(A).
Figure 3. Difference IR spectra between Na and K conditions.

Difference IR spectra were calculated as (K condition)−(Na condition) (see text) obtained in the presence (A) or absence (B) of 100 μg of tubulovesicles. Spectrum A is the average of 72 experiments and spectrum B that of 24 experiments. All 96 spectra were used for statistical comparison (Student's t test, α<0.01). Spectrum C is calculated as (spectrum A)−(spectrum B). The stars point to wave numbers where both spectra A and B are significantly different. (D) Computed difference spectrum for the Ca2+-ATPase according to the secondary-structure difference found in both the available high-resolution three-dimensional structures (see the Experimental section for more details). (E) Same as (D), but the simulation was performed with bandwidths reduced to 1/3 of their published values for all the structures. For clarity, the spectra were offset along the absorbance axis.
Figure 4. Difference IR spectra obtained in the presence of different specific ligands.

Difference IR spectra obtained under different conditions (see Table 1 for the complete composition of buffers). The contribution of the change due to the buffer alone has been subtracted. (A) The difference of K condition and Na condition, identical with spectrum C in Figure 3. (B) The difference between CaK condition and CaNa condition. (C) The difference between Ca-ATP condition and Ca condition. For clarity, the spectra were offset along the absorbance axis.
In a second series of experiments, we tested the effect of ATP on the IR spectrum of tubulovesicles. For this purpose, we used 50 mM Hepes/Tris (pH 7.2), 2.1 mM CaCl2 and 4 mM NaCl as buffer A (Ca condition) and 50 mM Hepes/Tris (pH 7.2), 4 mM CaCl2 and 2 mM ATP-Na2 as buffer B (Ca-ATP condition). We performed 33 experiments as described previously. The mean difference spectrum is presented in Figure 4(C).
Although this is the first time secondary-structure modifications are observed in gastric H+/K+-ATPase, differences in the tertiary structure of H+/K+-ATPase have been reported previously in the presence of Ca, Ca-ADP and Ca-ATP [29]. It was shown that the presence of the nucleotide induced a closed conformation of the protein with fewer amino acid residues accessible from the solvent. No information has appeared so far about the Na+/K+-induced transition. We decided to determine whether tertiary-structure modifications occur in H+/K+-ATPase by monitoring the hydrogen/deuterium-exchange kinetics of the amide protons in the presence of KCl and NaCl, which are shown in the present study to affect the secondary structure of only a very small number of residues.
Hydrogen/deuterium exchange was recorded in the presence of KCl or NaCl. The decreasing area of amide II, computed between 0 and 100% (see the Experimental section), is presented in Figure 5 for these experimental conditions. It clearly appears that the exchange is significantly faster in the presence of Na+ than in the presence of K+. Since the hydrogen/deuterium exchange is a first-order reaction, the fraction of non-exchanged amide protons, H(t), can be described as the sum of individual exponential functions. Considering only a few classes of exchange rates (three classes in the present case) allows the description of the data within the experimental error limits. Each exponential corresponds to a group i of amide bonds characterized by a common time constant Ti as follows:
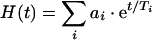 |
where ai is the proportion of residues belonging to group i. For three populations of amide protons i=1–3, least-squares curve fitting yields three time constants T1, T2 and T3 and three proportions a1, a2 and a3 for each condition (Na+ or K+). For a better comparison of the two conditions, the time constants have been set to their average value:
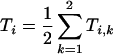 |
where k is the experimental condition (Na+ or K+). We found that T1=0.94 min, T2=9.69 min and T3=2374 min. A new fit with these fixed values of Ti was made and the results are reported in Table 2. Selecting either the individually determined time constants or the average time constants yielded nearly identical proportions of each class under the different experimental conditions, demonstrating the validity of the approach (results not shown).
Figure 5. Evolution of the proportion of unexchanged amide bonds computed between 100% (no exchange) and 0% (total exchange) as a function of the deuteration time (between 0 and 100 min).

Measurement of the hydrogen/deuterium exchange for a film of 50 μg of proteins from a suspension of tubulovesicles in 2 mM Hepes/Tris (pH 7.2)/20 mM KCl (○) or 20 mM NaCl (×). Curves represent the best approximation of the experimental points (means±S.D. for four experiments) by a sum of three exponential decays.
Table 2. The proportions of the three populations of amide protons that describe the deuteration of H+,K+-ATPase in the presence of NaCl or KCl.
Analysis of hydrogen/deuterium-exchange curves for a film of 50 μg of tubulovesicle proteins prepared on the surface of a Ge plate in the presence of 20 mM of either KCl or NaCl. The experimental curves were fitted with a sum of three exponentials with fixed constants, T1=0.94 min, T2=9.69 min and T3=2374 min. This yielded the proportions a1(rapid component), a2(intermediate component) and a3(slow component) of each contribution for each condition (results are expressed as means±S.D.).
| a1 | a2 | a3 | |
|---|---|---|---|
| 20 mM NaCl | 39±4 | 13±2 | 52±1 |
| 20 mM KCl | 27±3 | 15±1 | 61±3 |
It is well established that drying the tubulovesicle membranes on a Ge crystal results in the formation of an orientated multilayer stack with the membrane plane preferentially orientated parallel to the Ge surface [12,30]. In turn, orientation of the membrane molecules can be analysed by IR linear dichroism [31]. Spectra were recorded with a light polarized parallel or perpendicular to the incidence plane. The difference between the spectra yielded a dichroic spectrum that reflected the preferred orientation of the sample molecules. Figure 6(A) presents the dichroic spectrum obtained with the film of tubulovesicles dried on the surface of the Ge plate. Positive deviations in the dichroic spectrum pointed to transitions polarized parallel to the normal of the membrane. In Figure 6(A), the major band at 1655 cm−1 in the amide I region can be assigned to orientated α helices that are inserted into the membrane [30,32,33]. The negative bands at 2919 and 2850 cm−1, assigned to νas(CH2) and νs(CH2) respectively, confirmed the orientation of the membrane plane parallel to the Ge surface, since the transition moments associated with these bands are normal to the lipid acyl chain [34]. Figures 6(B) and 6(C) present the dichroic spectra of the same film obtained in the presence of a flowing buffer of 50 mM Hepes/Tris (pH 7.2)+20 mM KCl and 20 mM NaCl respectively. Spectra are presented with the same scale. Vertical dotted lines point to the major orientated bands in the dichroic spectra of the tubulovesicle membranes. In the dichroic spectra of the film in the presence of a flowing buffer shown in Figures 6(B) and 6(C), only bands below 1700 cm−1 appeared. The position of the band at 1655 cm−1 remained unmodified but no bands associated with lipid vibration near 2900 cm−1 were visible. Moreover, the difference between the dichroic spectra obtained in the presence of NaCl or KCl did not reveal any statistically significant difference. This means that, at this point, no difference in the orientation of secondary structures could be detected between the E1 and E2 conformations of the H+/K+-ATPase.
Figure 6. Dichroic spectra of a film of tubulovesicle proteins.

ATR–FTIR spectra of a film of 160 μg of proteins from tubulovesicles were recorded with a light polarized parallel or perpendicular to the incidence plane of the IR beam. The difference between those two spectra yields the dichroic spectrum of the film. (A) Dichroic spectrum of the film deposited on the Ge plate before hydration. Vertical dotted lines point to major bands in this spectrum. (B) Dichroic spectrum of the same film in the presence of a flowing buffer containing Na+ (50 mM Hepes/Tris, pH 7.2/20 mM NaCl). (C) Dichroic spectrum of the same film but in the presence of a flowing buffer containing K+ (50 mM Hepes/Tris, pH 7.2/20 mM KCl).
DISCUSSION
The high sensitivity required to put forward secondary-structure changes in proteins remains a bottleneck for the understanding of membrane enzyme molecular mechanisms. The possibility of attaching membrane multilayers to a Ge IRE and modulate the buffer composition solves these problems and opens a new avenue of research.
Potential pitfalls of the method
Although the approach described in the present study appears to be quite straightforward, a number of potential problems must be considered. A major potential problem is the stability of the film. Although the material loss in the flowing buffer was found to be very small, film swelling and shrinking due to changes in the ionic strength may be of significant concern for the sensitivity required (see below). In particular, when bivalent cations are compared with univalent ones (e.g. Ca2+ and Na+), care must be taken to compute the ionic strength thoroughly to adjust the concentrations accordingly. A second problem that must be considered is the effect of the solute change on the spectrum of water. In this respect, using NaCl instead of KCl has a significant impact that must be taken into account (see e.g. Figure 3).
Protein state in the multilamellar films
First, it is important to assess the quality of the multilayer stack model used in the present study, particularly the capability of the H+/K+-ATPase to modify its conformation in such a system. In a first experiment (results not shown), tubulovesicles were left at room temperature (25 °C) for 5 days and aliquots were periodically withdrawn. The loss of ATPase activity was extremely limited, with more than 90% of the activity retained after 5 days. It was also shown previously that at least 70% of its specific activity was recovered after resuspending a multilayer film of tubulovesicles prepared on a Ge plate as described here [14]. The 30% loss was probably due to the formation of multilamellar vesicles with a part of the protein not accessible to the ATP added from the outside. Other previous work used a system of tubulovesicle membrane multilayer stacks immobilized on a thin glass plate and placed in a quartz cell to monitor the fluorescence quenching of FITC-labelled H+/K+-ATPase [14]. The fluorescence change associated with the conformational change was recorded when an NaCl-containing buffer was replaced by a KCl-containing buffer. It was found that the amplitude of the fluorescence quenching observed in the multilayer stack was the same as that measured on vesicle suspensions. This result demonstrated that H+/K+-ATPase undergoes a similar conformational change both in the multilayer stacks and in the vesicle suspensions, and all the ATPase molecules were accessible to the ligands. It also demonstrates that the multilayer stack is fully permeable to the ligands even though the mean number of membranes can be computed to be nearly 200 under our experimental conditions. It is surprising that such a thick deposit can be simultaneously permeable to the ligands and stable under the buffer flow. Yet, such amazing properties have been reported earlier [14,35]. The present study also definitively demonstrates the stability of a tubulovesicle multilayer stack built on a Ge IRE when submitted to a constant buffer flow. This stability allows the conformation of a protein to be monitored on-line as the buffer composition is modulated. It also allows the repeatability of the measurements, a necessary condition to monitor reversibility, build statistics and improve the signal-to-noise ratio.
Sensitivity
Estimating the noise level in the 2800–2200 cm−1 frequency range (see the Experimental section) is usually not sufficient to evaluate correctly the signal detection limit over the amide I range. Taking into account the film stability, ionic strength effects and the effect of the ligands on the spectrum of water, we found that an S.D. of approx. 2×10−6 a.u. can be reached (10 spectra of 1024 scans each). It is evident from Figure 3 that the statistically significant bands in the difference spectra over the amide I–amide II frequency range have an absorbance of at least 2×10−5 a.u. Since the amide I intensity is approx. 40×10−3 a.u. in the presence of the film overlaid by the buffer (Figure 2E), we suggest that this approach can allow the observation of a conformational change of approx. 6 residues or more. This is a large improvement compared with previous reports [14,36], which failed to identify secondary-structure changes in gastric ATPase.
E1 (Na+)–E2 (K+) transition
A Student's t test highlighted some particular bands that were significantly different (α<0.01) after subtracting the Na condition spectra from the K condition spectra as shown in Figure 3(C). Moreover, these bands were reproducible for four different extractions of tubulovesicles. All spectral changes were reversible as the NaCl- replaced the KCl-containing buffer. The presence of K+ induced the E2 conformation, whereas Na+ favoured the E1 conformation of H+/K+-ATPase [37–40]; hence, we can assume that the bands in the IR difference spectrum are due to a conformational change in the protein. This conclusion is strengthened by the experiment in which calcium was added to both the buffers. Calcium is known to inhibit the E1–E2 conformational change in gastric H+/K+-ATPase even in the presence of 20 mM KCl [39]. When Ca2+ was added in the presence of NaCl or KCl, the spectral differences observed previously disappeared, as shown in Figure 4. This important result indicates that the bands that appear after subtracting a spectrum recorded in the presence of Na+ from a spectrum recorded in the presence of K+ can be associated with a conformational change of the H+/K+-ATPase specific to the E1–E2 transition. These bands were located in the amide I and II regions, indicating changes in the secondary structure of the protein and in the side chains of the amino acids. The present study is the first to demonstrate that H+/K+-ATPase is capable of modifying its secondary structure in the course of the E1–E2 transition. Major bands appeared at 1677, 1671, 1654 and 1640 cm−1 (Figures 3 and 4). According to the usual assignments [41,42], the negative contributions at 1640 and 1677 cm−1 can be safely assigned to a decrease in the β-sheet content after E2.K+ formation. The positive deviation at 1654 cm−1 can be assigned to either helical or disordered structures, and the positive 1671 cm−1 band could be related to an increase in the β-turn content. It is tempting to relate the features described here with the spectral difference expected for the E1–E2 transition for the Ca2+-ATPase for which both the high-resolution structures are available [9,10]. The difference spectrum was computed as described in the Experimental section and is reported in Figure 3(D). Although the bands presented in Figure 3(D) are significantly broader than the experimental ones (Figure 3C), they are located at the same position and have the same polarity (positive or negative). Their relative broadness can be understood since the ‘typical’ parameters describing the IR contribution of the individual secondary structures have been determined empirically on large proteins and include all kinds of structures (length, curvature, defects) and intra-structure regions (centre, edges). To simulate more localized changes, the bandwidth of the different structural components was arbitrarily divided by three for a new simulation (Figure 3E). The similarity to the experimental curve (Figure 3C) is now striking. Only the 1671 cm−1 band assigned to turns is not resolved, but is in agreement with the DSSP analysis of the Ca2+-ATPase structures (see the Experimental section). This comparison allows the tentative assignment of the 1654 cm−1 band to disordered structures rather than to helical structures (see the Experimental section). For the sake of comparison, it could be pointed out that similar bands were observed in the IR difference spectra for the E1–E2 transition of Ca2+-ATPase [23], where the authors used caged compounds, and were assigned to the E1-to-E2 transition. In their work, the E2-P formation spectrum in the amide I range is very similar to Figure 3(C). The maxima found here at 1671 and 1654 cm−1 are located at 1670 and 1652 cm−1 for the Ca2+-ATPase, and the minima found here at 1677 and 1640 cm−1 are located at 1680 and 1641 cm−1 for the Ca2+-ATPase. It turns out that, qualitatively, both spectral simulations and direct comparison with the Ca2+-ATPase strongly suggest a similar structural mechanism involving a secondary structure change highly specific to the E1–E2 transition. These modifications could originate from the movement of the N and A domains towards the P domain.
A quantitative estimate of the extent of secondary-structure change can be obtained using the COBSI index introduced to quantify the conformational changes of the Ca2+-ATPase [23]. This index is determined by measuring the area of the amide I region that is redistributed after the conformational change. By comparing the area of the difference IR spectrum for the conversion of H,K-ATPase from the E1 to the E2 conformation (Figure 3C) with the intensity of the film in the flowing buffer (Figure 2E), a COBSI index of 1.1±0.7×10−3 was obtained. This value is similar to the COBSI index of 0.8×10−3 obtained for Ca-ATPase in the presence of water or 2H2O [23]. Taking into account the total number of residues in H+/K+-ATPase (1324 for both α- and β-subunits), it turns out that the difference in the secondary structure of the protein involves 10–15 amino acids. It can be concluded at this stage that the secondary-structure changes observed in Ca2+-ATPase and gastric H+/K+-ATPase are very similar in nature and extent.
The results described so far indicate that only 10–15 residues change their secondary structure after the transition of the protein from the E1 to the E2 conformation. On the other hand, we have reported previously that this transition could involve movement of large domains of the protein. Differential scanning calorimetry suggested cytoplasmic domain rearrangements [37]. When monitored by hydrogen/deuterium-exchange kinetics (Figure 5), it appears that these two conformations are characterized by different rates of deuteration, showing that the E1 conformation, favoured in the presence of NaCl, is more accessible from the solvent than the E2 state obtained in the presence of KCl. Figure 5 and Table 2 indicate that 10% of the amide protons are less exchangeable in the presence of K+ than in the presence of Na+. Qualitative information could be obtained by multiplying the proportions ai by the total number of residues in H+/K+-ATPase (1324 amino acid residues for both α- and β-subunits) to obtain the number of residues in each population. The population associated with the rapid exchange in the presence of Na+ involved 516 (0.39×1324) residues, but only 357 (0.27×1324) residues in the presence of K+. It turns out that approx. 160 amino acids modified their accessibility to the solvent. These changes obviously reflect tertiary-structure changes, whereas a surprisingly small number of residues experienced a secondary-structure change in the course of the same transition. No overall reorientation of secondary structures with respect to the membrane could be detected by linear dichroism.
E1-ATP–E1-P transition
In a previous report [29], we have used the kinetics of hydrogen/deuterium exchange to show that a large part of the H+/K+-ATPase accessibility is sensitive to the presence of nucleotide. In the presence of Ca-ADP and Ca-ATP, the protein adopted a conformation with fewer residues accessible from the solvent compared with a condition in the presence of Ca alone. In the present study, we used the ATR flow technique to investigate this point. As shown in Figure 4(C), positive bands were observed at 1675, 1655 and 1630 cm−1 and negative ones appeared near 1666 and 1643 cm−1. As the presence of Ca2+ and ATP induced the phosphorylation of the H+/K+-ATPase [39], the Ca-ATP condition favoured the E1-P conformation of the protein. In the presence of K+, the wild-type ATPase is quickly dephosphorylated (see e.g. [43,44]), but in the additional presence of Ca2+, the kinetics of dephosphorylation is slow. We can therefore tentatively assume that the protein remained phosphorylated in the course of the whole experiment. This is not a crucial hypothesis as phosphorylation by itself does not bring significant changes in the amide I range of the spectrum [23]. In turn, the bands present in the difference IR spectra must be assigned to the binding of the nucleotide on H+/K+-ATPase. The simulation (Figure 3D) emphasizes the broadness of the difference lines for consensus bandwidths associated with different secondary structures. It is clear that the width of the water contribution is still larger. In turn, it is very unlikely that the contributions that appear near 1643 cm−1 could be related to water. As before, we assign it to a decrease in the β-sheet content, whereas the strong positive band at 1630 cm−1 indicates the rise of a new β-sheet type with stronger hydrogen bonds [45]. The latter band must probably be associated with the positive 1675 cm−1 deviation. Finally, we suggest that some turns (1666 cm−1) disappear, whereas some disordered or helical structures appear at 1655 cm−1. We can compare the results of the present study with the bands observed on binding of AMP-PNP [46] and other ATP derivatives [46,47] to Ca2+-ATPase. Clearly, all the features, notably the strong negative band at 1643 cm−1 surrounded by a positive feature at 1655 (weak) and 1630 (strong) cm−1 as observed here (Figure 4C), are similar to those observed on the Ca2+-ATPase after nucleotide binding. The phosphorylation bands near 1721 and 1549 cm−1 could not be identified [48,49].
Our results demonstrate that changes appear in the secondary structure after the binding of the nucleotide to the protein. Importantly, these changes are limited to a few residues and are very similar in nature to those observed on the Ca2+-ATPase [46,47], supporting again the idea of a very similar molecular mechanism for these two pumps.
The description of both conformational transitions investigated in the present study supports models of the catalytic cycle of H+/K+-ATPase, where subtle secondary-structure changes in some local site in the protein, such as a hinge region, induce large conformational changes and movements of domains relative to each other.
Acknowledgments
E.G. and V.R. are supported by Research Associateships from the National Fund for Scientific Research (Brussels, Belgium). We thank the Communauté Française de BelgiqueActions de Recherches Concertées for financial support. We also thank Dr J. Baenziger (University of Ottawa, Ottawa, ON, Canada) for his advice at the early stages of this work.
References
- 1.Sachs G., Chang H. H., Rabon E., Schackman R., Lewin M., Saccomani G. A nonelectrogenic H+ pump in plasma membranes of hog stomach. J. Biol. Chem. 1976;251:7690–7698. [PubMed] [Google Scholar]
- 2.Yao X. B., Forte J. G. Cell biology of acid secretion by the parietal cell. Annu. Rev. Physiol. 2003;65:103–131. doi: 10.1146/annurev.physiol.65.072302.114200. [DOI] [PubMed] [Google Scholar]
- 3.Axelsen K. B., Palmgren M. G. Evolution of substrate specificities in the P-type ATPase superfamily. J. Mol. Evol. 1998;46:84–101. doi: 10.1007/pl00006286. [DOI] [PubMed] [Google Scholar]
- 4.Moller J. V., Juul B., le Maire M. Structural organization, ion transport, and energy transduction of P-type ATPases. Biochim. Biophys. Acta. 1996;1286:1–51. doi: 10.1016/0304-4157(95)00017-8. [DOI] [PubMed] [Google Scholar]
- 5.Jorgensen P. L., Hakansson K. O., Karlish S. J. D. Structure and mechanism of Na,K-ATPase: functional sites and their interactions. Annu. Rev. Physiol. 2003;65:817–849. doi: 10.1146/annurev.physiol.65.092101.142558. [DOI] [PubMed] [Google Scholar]
- 6.Toh B. H., Gleeson P. A., Simpson R. J., Moritz R. L., Callaghan J. M., Goldkorn I., Jones C. M., Martinelli T. M., Mu F. T., Humphris D. C., et al. The 60- to 90-kDa parietal cell autoantigen associated with autoimmune gastritis is a β subunit of the gastric H+/K+-ATPase (proton pump) Proc. Natl. Acad. Sci. U.S.A. 1990;87:6418–6422. doi: 10.1073/pnas.87.16.6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reuben M. A., Lasater L. S., Sachs G. Characterization of a β subunit of the gastric H+/K+-transporting ATPase. Proc. Natl. Acad. Sci. U.S.A. 1990;87:6767–6771. doi: 10.1073/pnas.87.17.6767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geering K. The functional role of β subunits in oligomeric P-type ATPases. J. Bioenerg. Biomembr. 2001;33:425–438. doi: 10.1023/a:1010623724749. [DOI] [PubMed] [Google Scholar]
- 9.Toyoshima C., Nakasako M., Nomura H., Ogawa H. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 Å resolution. Nature (London) 2000;405:647–655. doi: 10.1038/35015017. [DOI] [PubMed] [Google Scholar]
- 10.Toyoshima C., Nomura H. Structural changes in the calcium pump accompanying the dissociation of calcium. Nature (London) 2002;418:605–611. doi: 10.1038/nature00944. [DOI] [PubMed] [Google Scholar]
- 11.Hilge M., Siegal G., Vuister G. W., Guntert P., Gloor S. M., Abrahams J. P. ATP-induced conformational changes of the nucleotide-binding domain of Na,K-ATPase. Nat. Struct. Biol. 2003;10:468–474. doi: 10.1038/nsb924. [DOI] [PubMed] [Google Scholar]
- 12.Goormaghtigh E., Raussens V., Ruysschaert J. M. Attenuated total reflection infrared spectroscopy of proteins and lipids in biological membranes. Biochim. Biophys. Acta. 1999;1422:105–185. doi: 10.1016/s0304-4157(99)00004-0. [DOI] [PubMed] [Google Scholar]
- 13.Mitchell R. C., Haris P. I., Fallowfield C., Keeling D. J., Chapman D. Fourier transform infrared spectroscopic studies on gastric H+/K+-ATPase. Biochim. Biophys. Acta. 1988;941:31–38. doi: 10.1016/0005-2736(88)90210-6. [DOI] [PubMed] [Google Scholar]
- 14.Vander Stricht D. V., Raussens V., Oberg K. A., Ruysschaert J. M., Goormaghtigh E. Difference between the E1 and E2 conformations of gastric H+/K+-ATPase in a multilamellar lipid film system. Characterization by fluorescence and ATR-FTIR spectroscopy under a continuous buffer flow. Eur. J. Biochem. 2001;268:2873–2880. doi: 10.1046/j.1432-1327.2001.02173.x. [DOI] [PubMed] [Google Scholar]
- 15.Zscherp C., Barth A. Reaction-induced infrared difference spectroscopy for the study of protein reaction mechanisms. Biochemistry. 2001;40:1875–1883. doi: 10.1021/bi002567y. [DOI] [PubMed] [Google Scholar]
- 16.Gerwert K., Souvigner G., Hess B. Simultaneous monitoring of light-induced changes in protein side-group protonation, chromophore isomerization, and backbone motion of bacteriorhodopsin by time-resolved Fourier-transform infrared spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 1990;87:9774–9778. doi: 10.1073/pnas.87.24.9774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moss D., Nabedryk E., Breton J., Mantele W. Redox-linked conformational changes in proteins detected by a combination of infrared spectroscopy and protein electrochemistry. Evaluation of the technique with cytochrome c. Eur. J. Biochem. 1990;187:565–572. doi: 10.1111/j.1432-1033.1990.tb15338.x. [DOI] [PubMed] [Google Scholar]
- 18.Gurney A. M. Photolabile caged compounds. In: Mason W. T., editor. Fluorescent and Luminescent Probes for Biological Activity. London: Academic Press; 1999. pp. 335–348. [Google Scholar]
- 19.Ryan S. E., Demers C. N., Chew J. P., Baenziger J. E. Structural effects of neutral and anionic lipids on the nicotinic acetylcholine receptor. An infrared difference spectroscopy study. J. Biol. Chem. 1996;271:24590–24597. doi: 10.1074/jbc.271.40.24590. [DOI] [PubMed] [Google Scholar]
- 20.Ryan S. E., Baenziger J. E. A structure-based approach to nicotinic receptor pharmacology. Mol. Pharmacol. 1999;55:348–355. doi: 10.1124/mol.55.2.348. [DOI] [PubMed] [Google Scholar]
- 21.Soumarmon A., Abastado M., Bonfils S., Lewin M. J. C1-transport in gastric micorsomes. An ATP-dependent influx sensitive to membrane potential and to protein kinase inhibitor. J. Biol. Chem. 1980;255:11682–11687. [PubMed] [Google Scholar]
- 22.Stanton M. G. Colorimetric determination of inorganic phosphate in the presence of biological material and adenosine triphosphate. Anal. Biochem. 1968;22:27–34. doi: 10.1016/0003-2697(68)90255-8. [DOI] [PubMed] [Google Scholar]
- 23.Barth A., von-Germar F., Kreutz W., Mantele W. Time-resolved infrared spectroscopy of the Ca2+-ATPase. The enzyme at work. J. Biol. Chem. 1996;271:30637–30646. doi: 10.1074/jbc.271.48.30637. [DOI] [PubMed] [Google Scholar]
- 24.Kabsch W., Sander S. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 25.Raussens V., Ruysschaert J. M., Goormaghtigh E. Analysis of 1H/2H exchange kinetics using model infrared spectra. Appl. Spectrosc. 2004;58:68–82. doi: 10.1366/000370204322729496. [DOI] [PubMed] [Google Scholar]
- 26.Bechinger B., Ruysschaert J. M., Goormaghtigh E. Membrane helix orientation from linear dichroism of infrared attenuated total reflection spectra. Biophys. J. 1999;76:552–563. doi: 10.1016/S0006-3495(99)77223-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goormaghtigh E., de Jongh H., Ruysschaert J. M. Relevance of protein thin films prepared for ATR-FTIR spectroscopy: significance of the pH. Appl. Spectrosc. 1996;50:1519–1527. [Google Scholar]
- 28.Goormaghtigh E., Cabiaux V., Ruysschaert J. M. Determination of soluble and membrane protein structure by Fourier transform infrared spectroscopy. II. Experimental aspects, side chain structure, and H/D exchange. Subcell. Biochem. 1994;23:363–403. doi: 10.1007/978-1-4615-1863-1_9. [DOI] [PubMed] [Google Scholar]
- 29.Scheirlinckx F., Buchet R., Ruysschaert J. M., Goormaghtigh E. Monitoring of secondary and tertiary structure changes in the gastric H+/K+-ATPase by infrared spectroscopy. Eur. J. Biochem. 2001;268:3644–3653. doi: 10.1046/j.1432-1327.2001.02266.x. [DOI] [PubMed] [Google Scholar]
- 30.Raussens V., Ruysschaert J. M., Goormaghtigh E. Fourier transform infrared spectroscopy study of the secondary structure of the gastric H+,K+-ATPase and of its membrane-associated proteolytic peptides. J. Biol. Chem. 1997;272:262–270. doi: 10.1074/jbc.272.1.262. [DOI] [PubMed] [Google Scholar]
- 31.Goormaghtigh E., Ruysschaert J. M. Polarized attenuated total reflection infrared spectroscopy as a tool to investigate the conformation and orientation of membrane components. In: Brasseur R., editor. Molecular Description of Biological Membranes by Computer Aided Conformational Analysis. Brussels: 1990. pp. 285–329. [Google Scholar]
- 32.Raussens V., de Jongh H., Pézolet M., Ruysschaert J. M., Goormaghtigh E. Secondary structure of the intact H+,K+-ATPase and of its membrane-embedded region. An attenuated total reflection infrared spectroscopy, circular dichroism and Raman spectroscopy study. Eur. J. Biochem. 1998;252:261–267. doi: 10.1046/j.1432-1327.1998.2520261.x. [DOI] [PubMed] [Google Scholar]
- 33.Raussens V., le Maire M., Ruysschaert J. M., Goormaghtigh E. Secondary structure of the membrane-bound domains of H+,K+-ATPase and Ca2+-ATPase, a comparison by FTIR after proteolysis treatment of the native membranes. FEBS Lett. 1998;437:187–192. doi: 10.1016/s0014-5793(98)01225-3. [DOI] [PubMed] [Google Scholar]
- 34.Fringeli U. P., Günthard H. H. Infrared membrane spectroscopy. Mol. Biol. Biochem. Biophys. 1981;31:270–332. doi: 10.1007/978-3-642-81537-9_6. [DOI] [PubMed] [Google Scholar]
- 35.Acha V., Ruysschaert J. M., Goormaghtigh E. Stacks of close to 100 phospholipid bilayers fully accessible to proteins –an ATR-FTIR-based chemometric analysis on hydrated phospholipid films. Anal. Chim. Acta. 2001;435:215–226. [Google Scholar]
- 36.Raussens V., Pézolet M., Ruysschaert J. M., Goormaghtigh E. Structural difference in the H+,K+-ATPase between the E1 and E2 conformations. An attenuated total reflection infrared spectroscopy, UV circular dichroism and Raman spectroscopy study. Eur. J. Biochem. 1999;262:176–183. doi: 10.1046/j.1432-1327.1999.00365.x. [DOI] [PubMed] [Google Scholar]
- 37.Gasset M., Laynez J., Menendez M., Raussens V., Goormaghtigh E. Structural domain organization of gastric H+,K+-ATPase and its rearrangement during the catalytic cycle. J. Biol. Chem. 1997;272:1608–1614. doi: 10.1074/jbc.272.3.1608. [DOI] [PubMed] [Google Scholar]
- 38.Helmich de Jong M. L., van Emst de Vries S. E., de Pont J. J. Conformational states of (K++H+)-ATPase studied using tryptic digestion as a tool. Biochim. Biophys. Acta. 1987;905:358–370. doi: 10.1016/0005-2736(87)90464-0. [DOI] [PubMed] [Google Scholar]
- 39.Jackson R. J., Mendlein J., Sachs G. Interaction of fluorescein isothiocyanate with the (H++K+)-ATPase. Biochim. Biophys. Acta. 1983;731:9–15. doi: 10.1016/0005-2736(83)90391-7. [DOI] [PubMed] [Google Scholar]
- 40.Baeyens N., Wattiez R., Raussens V., Ruysschaert J. M., Goormaghtigh E. Structural modifications in the membrane-bound regions of the gastric H+/K+-ATPase upon ligand binding. Eur. J. Biochem. 2001;268:5135–5141. doi: 10.1046/j.0014-2956.2001.02443.x. [DOI] [PubMed] [Google Scholar]
- 41.Susi H., Byler D. M. Protein structure by Fourier transform infrared spectroscopy: second derivative spectra. Biochem. Biophys. Res. Commun. 1983;115:391–397. doi: 10.1016/0006-291x(83)91016-1. [DOI] [PubMed] [Google Scholar]
- 42.Goormaghtigh E., Cabiaux V., Ruysschaert J. M. Determination of soluble and membrane protein structure by Fourier transform infrared spectroscopy. III. Secondary structures. Subcell. Biochem. 1994;23:405–450. doi: 10.1007/978-1-4615-1863-1_10. [DOI] [PubMed] [Google Scholar]
- 43.Hermsen H. P. H., Swarts H. G. P., Koenderink J. B., de Pont J. J. H. H. The negative charge of glutamic acid-820 in the gastric H+,K+-ATPase α-subunit is essential for K+ activation of the enzyme activity. Biochem. J. 1998;331:465–472. doi: 10.1042/bj3310465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Swarts H. G. P., Koenderink J. B., Hermsen H. P. H., Willems P. H. G. M., de Pont J. J. H. H. K+-independent gastric H+,K+-ATPase activity –dissociation of K+-independent dephosphorylation and preference for the E-1 conformation by combined mutagenesis of transmembrane glutamate residues. J. Biol. Chem. 2001;276:36909–36916. doi: 10.1074/jbc.M103945200. [DOI] [PubMed] [Google Scholar]
- 45.Cabiaux V., Brasseur R., Wattiez R., Falmagne P., Ruysschaert J. M., Goormaghtigh E. Secondary structure of diphtheria toxin and its fragments interacting with acidic liposomes studied by polarized infrared spectroscopy. J. Biol. Chem. 1989;264:4928–4938. [PubMed] [Google Scholar]
- 46.Liu M., Barth A. TNP-AMP binding to the sarcoplasmic reticulum Ca2+-ATPase studied by infrared spectroscopy. Biophys. J. 2003;85:3262–3270. doi: 10.1016/S0006-3495(03)74744-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu M., Barth A. Mapping interactions between the Ca2+-ATPase and its substrate ATP with infrared spectroscopy. J. Biol. Chem. 2003;278:10112–10118. doi: 10.1074/jbc.M212403200. [DOI] [PubMed] [Google Scholar]
- 48.Barth A., Mantele W. ATP-induced phosphorylation of the sarcoplasmic reticulum Ca2+ ATPase: molecular interpretation of infrared difference spectra. Biophys. J. 1998;75:538–544. doi: 10.1016/S0006-3495(98)77543-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barth A. Phosphoenzyme conversion of the sarcoplasmic reticulum Ca2+- ATPase. Molecular interpretation of infrared difference spectra. J. Biol. Chem. 1999;274:22170–22175. doi: 10.1074/jbc.274.32.22170. [DOI] [PubMed] [Google Scholar]