

Abstract
The Philippines is a high-incidence country for tuberculosis, with the increasing prevalence of multi- (MDR-TB) and extensively-drug (XDR-TB) resistant Mycobacterium tuberculosis strains posing difficulties to disease control. Understanding the genetic diversity of circulating strains can provide insights into underlying drug resistance mutations and transmission dynamics, thereby assisting the design of diagnostic tools, including those using next generation sequencing (NGS) platforms. By analysing genome sequencing data of 732 isolates from Philippines drug-resistance survey collections spanning from 2011 to 2019, we found that the majority belonged to lineages L1 (531/732; 72.5%) and L4 (European-American; n = 174; 23.8%), with the Manila strain (L1.2.1.2.1) being the most prominent (475/531). Approximately two-thirds of isolates were found to be at least MDR-TB (483/732; 66.0%), and potential XDR-TB genotypic resistance was observed (3/732; 0.4%), highlighting an emerging problem in the country. Genotypic resistance was highly concordant with laboratory drug susceptibility testing. By finding isolates with (near-)identical genomic variation, five major clusters containing a total of 114 isolates were identified: all containing either L1 or L4 isolates with at least MDR-TB resistance and spanning multiple years of collection. Closer inspection of clusters revealed transmission in prisons, some involving isolates with XDR-TB, and mutations linked to third-line drug bedaquiline. We have also identified previously unreported mutations linked to resistance for isoniazid, rifampicin, ethambutol, and fluoroquinolones. Overall, this study provides important insights into the genetic diversity, transmission and circulating drug resistance mutations of M. tuberculosis in the Philippines, thereby informing clinical and surveillance decision-making, which is increasingly using NGS platforms.
Subject terms: Epidemiology, Population screening, Tuberculosis, Genomics
Introduction
Tuberculosis (TB), caused by Mycobacterium tuberculosis, is a prevalent infectious disease in the Philippines. With over 741 K cases and 61 K deaths in 2021 alone1, the Philippines is the country with the second highest active disease burden, after China1. Approximately 70 Filipinos die daily from TB. Worryingly, increasing HIV prevalence and a high burden of multi-drug resistance (MDR-TB) to isoniazid (INH) and rifampicin (RIF) treatments pose serious challenges for effective control3. These challenges are exacerbated by a serious gap between those expected to have MDR-TB (2% new, 21% re-treatment cases) and those detected and subsequently put on treatment, with detrimental consequences including poorer treatment outcomes and strains progressing to extensively drug-resistance (XDR-TB)2. Novel applications of whole genome (WGS) and targeted (candidate) amplicon sequencing (Amp-seq) using next generation (NGS) technologies can provide insights into the underlying drug resistance mutations and profiles for clinical and surveillance decision-making. However, there is a lack of WGS studies of M. tuberculosis from low- and middle-income countries, such as the Philippines, where the prevalence and burden of TB tend to be greatest.
In the Philippines, genomic data for local M. tuberculosis isolates is scarce, but previous work has shown that ‘ancient’ (lineage L1) and ‘modern’ lineages (L2 and L4) are present in the country3. Members of the EA12-Manila clade (L1.2.1.2.1) are known to be highly associated with the Filipino population3, with a molecular barcode established to rapidly identify this strain type4. Here, we present the results of sequencing 732 isolates recently collected between 2011 and 2019, including from prison populations. We analysed the temporal evolution of drug resistance and clustering by geography and found evidence of the transmission of L4 and MDR-TB strains across the islands. More generally, TB transmission in prison poses a challenge to infection control, with the potential spread of infections within and outside the institution, and several studies have used sequencing to understand links between isolates5–8, and inform a public health response. Within confined correctional spaces, TB thrives, driven by overcrowding and potentially limited ventilation and healthcare access, thereby amplifying the risk of transmission. The flow of individuals into and out of prisons poses a broader public health concern, and the response necessitates targeted screening, rapid diagnosis, and tailored treatments.
Mixed infections in TB introduce complex genetic interactions that impact disease severity, treatment response, and transmission dynamics. The co-existence of diverse clones within a host complicates treatment regimes, and genetic investigation of the bacterial population can inform patient management9. Platforms such as Illumina and the development of portable devices, such as Oxford Nanopore Technology, have ushered in a transformative era, making WGS and Amp-seq a cornerstone in deciphering the genome variation and diversity of M. tuberculosis. WGS is now widely used to identify drug resistance markers to guide treatment, determine phylogenetic relatedness and potential transmission events, inform surveillance and infection control decision-making, and discover new targets for drugs and vaccines. Applying such technologies and assays will have the greatest impact in settings with high TB burden, such as The Philippines. In this study, we utilised WGS data from a convenience dataset to explore the dynamics of drug resistance development, transmission, and the complexities of mixed infections in the Philippines. By conducting an integrated analysis of WGS data, we aimed to establish a baseline characterisation of genomic diversity within the country. This foundational insight will inform future routine applications of NGS, guiding public health decision-making and helping to reduce the high burden of TB.
Results
Study population
The study analysed a convenience sample of 732 M. tuberculosis isolates, all with sequencing coverage more than 25-fold (range: 27- to 2191-fold) and collected from 2011 to 2019 across three studies (see “Materials and methods”). They were collected from 671 TB patients, but eight of the isolates were found to be sourced from mixed infections and subsequently excluded from further population genomic analysis (Table S1). The study patients (n = 671) were predominantly male (70.5%), with a median age of 42 (range 14–83) years, mostly from Luzon Island (74.4%), and 18 (2.7%) contributed more than one isolate at different time points (range: 2 to 14 isolates) (Table 1). The 724 isolates belong predominantly to lineage 1 (L1; 531, 73.3%), with the Manila strain (lineage 1.2.1.2.1) being the most common (89.5%; 475/531) and were found in all three major islands (Luzon, Visayas, and Mindanao) (Table 2). Lineage 4 (L4) was the second most common lineage (24.0%, n = 174), followed by lineage 2 (L2; 2.5%; n = 18) and lineage 3 (L3; 0.1%, n = 1). To avoid bias in some population-based analyses, we used a single representative isolate for each patient (n = 671), whereas for the 18 patients with multiple isolates, we used the most recent sample.
Table 1.
Mycobacterium tuberculosis individual (N = 671).
| Characteristic | N (median) | % (range) |
|---|---|---|
| Age (years) | 42 | (14-83) |
| Gender | ||
| Male | 473 | 70.5 |
| Female | 149 | 22.2 |
| Unknown | 49 | 7.3 |
| Location* | ||
| National Capital Region, Luzon | 200 | 29.8 |
| Calabarzon, Luzon | 125 | 18.6 |
| Other Luzon | 174 | 25.9 |
| Mindanao | 113 | 16.8 |
| Visayas | 53 | 7.9 |
| Unknown | 6 | 0.9 |
| No. of isolates per individual** | ||
| 1 | 653 | 97.3 |
| 2 | 8 | 1.2 |
| 3 | 2 | 0.3 |
| 4 | 3 | 0.4 |
| 5 | 1 | 0.1 |
| 6 | 3 | 0.4 |
| 14 | 1 | 0.1 |
*Based on the Island of collection for sequenced isolates.
**At different time points.
Table 2.
Mycobacterium tuberculosis samples (N = 724).
| Characteristic | N | % |
|---|---|---|
| Year of collection | ||
| 2011 | 61 | 8.4 |
| 2012 | 113 | 15.6 |
| 2013 | 60 | 8.3 |
| 2014 | 37 | 5.1 |
| 2015 | 116 | 16.0 |
| 2016 | 267 | 36.9 |
| 2017–2019 | 70 | 9.7 |
| Lineage | ||
| L1 | 531 | 73.3 |
| L4 | 174 | 24.0 |
| L2 | 18 | 2.5 |
| L3 | 1 | 0.1 |
| Drug resistance** | ||
| Sensitive | 119 | 16.4 |
| HR-TB | 79 | 10.9 |
| RR-TB | 36 | 5.0 |
| MDR-TB | 374 | 51.7 |
| Pre-XDR | 98 | 13.5 |
| XDR-TB | 3 | 0.4 |
| Other | 15 | 2.1 |
| Individual resistance | ||
| Rifampicin | 511 | 70.6 |
| Isoniazid | 554 | 76.5 |
| Ethambutol | 317 | 43.8 |
| Ethionamide | 282 | 39.0 |
| Pyrazinamide | 223 | 30.8 |
| Streptomycin | 288 | 39.8 |
| Fluoroquinolones | 102 | 14.1 |
| Aminoglycosides | 72 | 9.9 |
| Amikacin | 72 | 9.9 |
| Capreomycin | 92 | 12.7 |
| Kanamycin | 76 | 10.5 |
| Cycloserine | 3 | 0.4 |
| Para-aminosalicylic acid | 19 | 2.6 |
| Bedaquiline | 3 | 0.4 |
HR-TB isoniazid resistant, MDR-TB multidrug-resistant, XDR extensive-drug-resistant.
*This does not include 8 mixed infection samples (see Table S1).
**Genotypic; isolates included only in one category.
Drug resistance
Genotypic drug resistance characterisation was performed using TB-Profiler software (v2) on WGS data from 724 M. tuberculosis isolates. The drug-resistance profiles generated span 16 drugs, with resistance prevalent for first-line drugs such as rifampicin (70.6%), isoniazid (76.5%), ethambutol (43.8), and pyrazinamide (30.8%) (Table 2). More than half of all strains (n = 374; 51.7%) were multidrug-resistant tuberculosis (MDR-TB), with high concordance between phenotypic (drug susceptibility testing) and genotypic resistance for rifampicin (93.5%) and isoniazid (91.7%). Furthermore, genotypic resistance to streptomycin (39.8%), fluoroquinolones (14.1%), aminoglycosides (9.9%), and ethionamide (39.0%) was detected. Pre-extensively drug-resistant tuberculosis (pre-XDR-TB), defined as MDR-TB with additional resistance to any fluoroquinolone, was also found in 98 (13.5%) isolates. Similar proportions of isolates with MDR-TB were observed across all lineages except L3 (Fig. 1).
Figure 1.
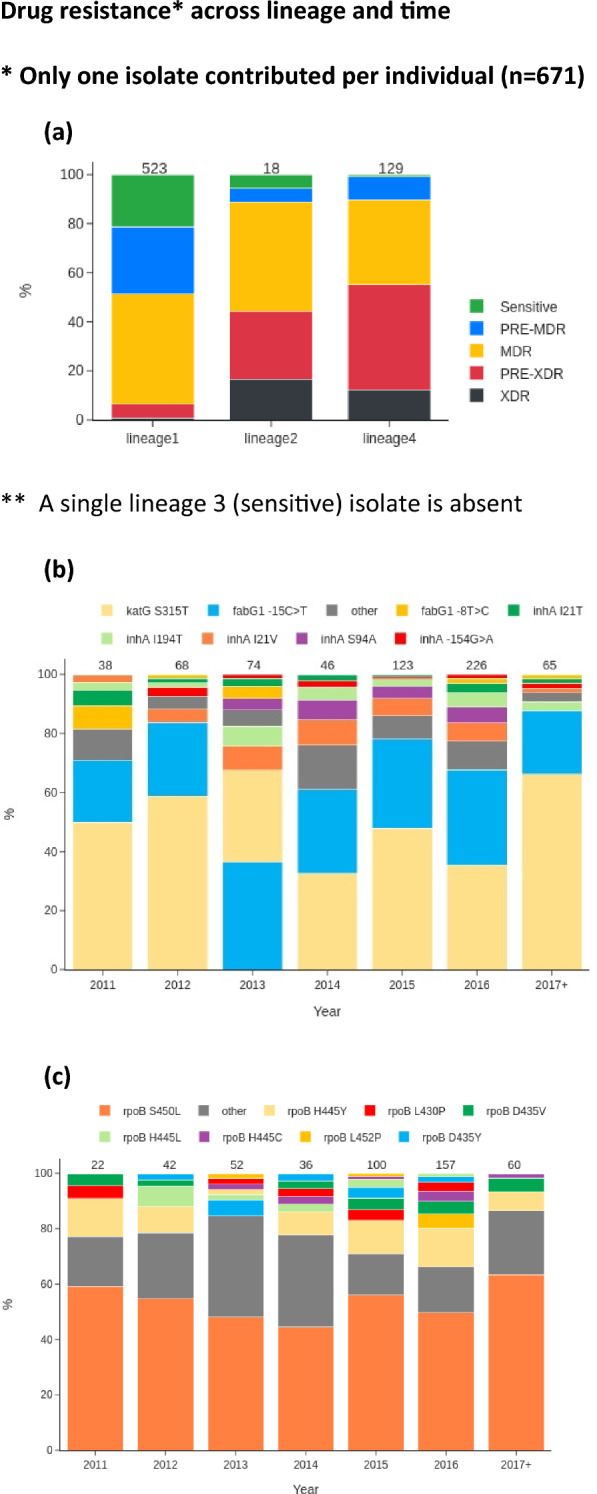
Drug resistance* across lineage and time. *Only one isolate contributed per individual (n = 671). (a) Types by lineage. A single lineage 3 (sensitive) isolate is absent. (b) Mutations linked to resistance to isoniazid. (c) Mutations linked with resistance to rifampicin.
The most common mutations underlying MDR-TB were in katG (S315T; 321/554) for isoniazid, fabG1 promoter region (− 15C>T; 222/554) for isoniazid, and rpoB (D435G/F/V/E, 51/511; S450L/F/W, 303/511) for rifampicin (Table 3). Moreover, compensatory mutations were also identified linked to isoniazid (ahpC − 48G>A, n = 4; − 52C>T, 3; − 54C>T, 2; − 74G>A, 1; − 81C>T, 4; − 51G>A, 4; − 52C>A, 3) and rifampicin (rpoC D485N, n = 2; I491T, 1; N698K, 8; N698S, 5; L516P, 6; F452L, 1; V483A, 13; V483G, 11). Other common mutations included embB (G406A/S/D, 19/343; M306I/L/V, 88/343; Q497R/P, 6/343) linked to ethambutol and gyrA (A90V, 32/103; S91P, 3/103; D94G/A/H/Y/N, 65/103) linked to fluoroquinolones. Mutations associated with para-aminosalicylic acid (PAS) resistance were identified in 21 isolates folC E40G (n = 18), folC R49P (n = 2), and thyA R126Q (n = 1). Several resistance mutations, rare in a global dataset (“Global50k”; n = 50,722) (< 1%) were found, including those for isoniazid (inhA I21V, I21T), pyrazinamide (pncA 316_317dupTT, H57P), and streptomycin (rrs 514A>T). Most established resistance mutations were present from 2011 (e.g., katG S315T, rpoB S450L), while others emerged in later years (e.g., inhA − 154G>A, rpoB H445T in 2012) (Fig. 1; Fig. S1). Interestingly, some isolates with known resistance mutations had a sensitive phenotype, often due to reported “disputed” mutations other than the canonical rpoB S450L10. For example, only five (38.5%) of 13 isolates with the rpoB L430P mutation had a rifampicin-resistant phenotype. In total, seven known rifampicin resistance mutations (across 38 isolates) had resistant proportions above 50%, but mutation frequencies were uncommon (n < 14) compared to rpoB S450L (n = 266/671; 39.6%), H445Y (n = 57/671; 8.5%), and D435V (19/671; 2.8%).
Table 3.
Common mutations linked to drug resistance.
| Drug resistance | Gene name | Change | Our study (n = 724) N (%) |
Our study one isolate per individual (n = 671) N (%) |
Global 50 k %* |
|---|---|---|---|---|---|
| Isoniazid | katG | S315T | 333 (46.0) | 291 (43.4) | 61.2 |
| Rifampicin | rpoB | S450L | 297 (41.0) | 266 (39.6) | 57.4 |
| Isoniazid | fabG1 | − 15C>T | 236 (32.6) | 203 (30.3) | 17.9 |
| Ethambutol | embB | M306V | 121 (16.7) | 94 (14.0) | 29.2 |
| Streptomycin | rrs | 514A>C | 102 (14.1) | 83 (12.4) | 8.4 |
| Ethambutol | embB | M306I | 82 (11.3) | 68 (10.1) | 22.4 |
| Kanamycin | rrs | 1401A>G | 72 (9.9) | 53 (7.9) | 55.8 |
| Pyrazinamide | pncA | L172P | 62 (8.6) | 46 (6.9) | 0.6 |
| Rifampicin | rpoB | H445Y | 57 (7.9) | 57 (8.5) | 3.7 |
| Streptomycin | rpsL | K43R | 41 (5.7) | 39 (5.8) | 45.8 |
| Isoniazid | inhA | I21V | 41 (5.7) | 40 (6.0) | 0.5 |
| Ofloxacin | gyrA | A90V | 32 (4.4) | 22 (3.3) | 25.6 |
| Isoniazid | inhA | I194T | 29 (4.0) | 25 (3.7) | 1.3 |
| Ofloxacin | gyrA | D94G | 28 (3.9) | 22 (3.3) | 35.0 |
| Ethambutol | embB | G406S | 28 (3.9) | 12 (1.8) | 1.9 |
| Rifampicin | rpoB | D435V | 25 (3.5) | 19 (2.8) | 7.0 |
| Isoniazid | inhA | S94A | 24 (3.3) | 24 (3.6) | 1.4 |
| Ethambutol | embB | G406D | 24 (3.3) | 20 (3.0) | 3.0 |
| Streptomycin | gid | 102delG | 22 (3.0) | 22 (3.3) | 4.2 |
| Rifampicin | rpoB | H445D | 22 (3.0) | 9 (1.3) | 3.2 |
| Streptomycin | rrs | 514A>T | 20 (2.8) | 8 (1.2) | 0.3 |
| Streptomycin | rpsL | K88R | 20 (2.8) | 20 (3.0) | 8.7 |
| Rifampicin | rpoB | Q432K | 16 (2.2) | 3 (0.4) | 0.2 |
| Pyrazinamide | pncA | 316_317dupTT | 16 (2.2) | 3 (0.4) | 0 |
| PAS | folC | E40G | 16 (2.2) | 3 (0.4) | 0 |
| Streptomycin | gid | 351delG | 15 (2.1) | 15 (2.2) | 2.0 |
| Streptomycin | gid | 48delT | 15 (2.1) | 13 (1.9) | 0.2 |
| Isoniazid | inhA | I21T | 15 (2.1) | 15 (2.2) | 0.8 |
| Ofloxacin | gyrA | D94Y | 15 (2.1) | 2 (0.3) | 3.9 |
| Pyrazinamide | pncA | H57P | 15 (2.1) | 4 (0.6) | 0.1 |
| Rifampicin | rpoB | L430P | 13 (1.8) | 13 (1.9) | 2.0 |
| Isoniazid | fabG1 | − 8T>C | 13 (1.8) | 13 (1.9) | 1.6 |
| Rifampicin | rpoB | D435Y | 13 (1.8) | 13(1.9) | 2.7 |
| Ethambutol | embB | D1024N | 12 (1.7) | 12 (1.8) | 2.1 |
| Streptomycin | rrs | 517C>T | 12 (1.7) | 12 (1.8) | 4.8 |
| Capreomycin | tlyA | 269dupG | 12 (1.7) | 1 (0.1) | 0 |
| Ethambutol | embB | M306L | 11 (1.5) | 11 (1.6) | 1.4 |
| Rifampicin | rpoB | H445L | 11 (1.5) | 11 (1.6) | 1.1 |
*Frequency in isolates with associated drug resistance in the Global50k database (n = 50,722); PAS Para-aminosalicylic acid.
Putative novel drug resistance genes
Although there was a high concordance between phenotypic resistance and genotypic predictions (accuracy: rifampicin: 93.5%, isoniazid: 91.7%), some isolates presented a resistant phenotype with no known resistance mutation. These isolates were further analysed to identify potentially novel mutations that could explain resistance. For isoniazid, three mutations were identified across five samples, one in inhA (I21M) and two in katG (K143E, D419Y) (Table S3). Moreover, the characterization of rare and unknown-association mutations in candidate genes and phenotypic testing of selected strains led to the detection of potential novel resistance mutations for ethambutol (11 mutations across embA/B/C), pyrazinamide (two mutations in pncA), streptomycin (27 mutations in gid and one in rrs), fluoroquinolones (one in gyrA) and capreomycin (two in tlyA). While some of the isolates with these mutations were classified as sensitive according to phenotypic methods, the majority exhibited a resistant phenotype. Additionally, four frameshift mutations across three samples were identified in mmpR5 (Rv0678), which is strongly associated with bedaquiline resistance. While phenotype testing was not available for bedaquiline, frameshift mutations are widely accepted to cause resistance. Additionally, these mutations were acquired on an MDR-TB and fluoroquinolone background, making them XDR-TB.
Phylogenetic and clustering analysis
The study detected 34,260 SNPs across all 724 isolates examined, of which 73.8% were unique to a single sample. Phylogenetic tree construction using all SNPs confirmed the expected grouping of the isolates based on their evolutionary lineage. Clades with similar drug-resistance profiles were found, including large clusters of MDR and pre-XDR isolates in L4 (Fig. 2) spanning up to eight years. Prison-sourced isolates (n = 71) from 18 inmates across different time points (range: 2 to 14 isolates per person; from 2013 to 2019 with a median time span of 2 years) explained some of this clustering (Table S4). The prison-sourced isolates were mostly L4 (55/71), predominantly L4.3.4.1 and L4.3.4.2 strains, with some additional representation from L1 (16/71), largely consisting of the Manila strain (L1.2.1.2.1). Using all serially sampled isolates without mixed infections (n = 69), the estimated crude mutation rate was 0.66 SNPs per isolate per year (Fig. S2). To control for potential cryptic reinfections, we excluded isolates with more than one SNP difference and a time interval of less than one year (n = 60), resulting in an estimate of 0.34. An analysis of L4 isolates (48/60) led to a crude rate of 0.35 SNPs per isolate per year. The largest sublineage in our dataset was L1.2.1.2.1 (Manila family), which has been predominantly observed in isolates collected from the Philippines3. To characterise the molecular clock rate in this sublineage, a time-based phylogenetic tree was reconstructed using BEAST2 software with parameters similar to those described elsewhere11. It revealed a clock rate of 0.63 (95% highest posterior density (HPD): 0.17–1.11) mutations per genome per year.
Figure 2.

Phylogenetic tree of the 724 M. tuberculosis study isolates constructed using 34,260 SNPs. The colour scheme label on the phylogenetic tree from the innermost to outermost ring indicates: Drug Resistance (DR) type, Lineage, Islands, and Year of Collection. The 5 red-coloured clades of highly similar isolates are explored in Fig. 3.
The distribution of pairwise SNP differences across the 671 isolates (one per individual) (median 368; range: 1–2217 SNPs) was multimodal, with modes representing differences within and between lineages (Fig. S3). A genetic distance threshold of 12 SNPs for defining potential transmission was established by evaluating a range of SNP cut-offs (0 to 30) (Fig. S3, Table S5). This cut-off resulted in 32 clusters containing a total of 120 isolates (Table S5), with a maximum of 45 samples in a single cluster (Fig. S4).
An analysis of the pairwise most recent common ancestor across the clusters led to median and mean values of 9 and 8 years, respectively, ranging from 0.6 to 16 years. The 120 isolates in clusters were found in all three islands (Luzon, 89; Visayas, 6; and Mindanao, 25) and were predominantly MDR-TB and pre-XDR (89/120). Logistic regression analysis revealed a greater risk of transmissibility (compared to L1) for L4 (odds ratio (OR) 12.87) and L2 strains (OR 4.02), as well as more advanced drug-resistance (OR 2.16) (all P < 0.001) (Table S6). There was some suggestive evidence of increased transmissibility for isolates from Mindanao compared to those from Luzon (OR 2.18, P = 0.015) (Table S6). A positive association between overall SNP distance and geographical distance was observed (ANOVA F = 346.2, P < 0.001) (Fig. S5). The largest cluster (n = 45) spans 19 cities, seven regions, and two islands, with a high proportion in Luzon, and contains MDR-TB (n = 36) and pre-XDR (n = 9) strains (Fig. S4; Fig. 3). Time-based Bayesian phylogenetic trees of the five largest clusters (from the analysis of 724 isolates) were generated using BEAST2 software12, revealing clusters of isolates collected from the same island and geographic units (Fig. 3). Interestingly, the clusters included serial samples from the same patients, which were found to have different levels of drug resistance at different periods while interspaced with the samples from unique sample hosts, suggesting potential direct transmission events between individuals. Additionally, the microevolution of drug resistance was observed within and between hosts, with the progression of MDR-TB to pre-XDR and then to XDR-TB.
Figure 3.

Clustered isolates. Major clusters from Fig. 2, and includes individuals with > 1 sample. Strip colour scheme from left to right: Drug resistance (DR) type, Lineage, Islands, and year of collection. Sample IDs that are highlighted with colours indicate isolates from the same patients. The numbers on the nodes of the tree show the maximum SNP distance between samples in the bifurcating branches. The timeframes of clusters are indicated (earliest and latest years).
A genome-wide association study (GWAS) approach (n = 671) was applied to identify genetic loci associated with transmissibility. This identified signals in the following genes: Rv0425c, rrs, Rv2828A, Rv3198c, Rv0766c, and Rv0825c genes (Table S7) (all ORs > 3; P < 0.0001). The detected rrs mutation 514A>C is associated with streptomycin resistance, while 1401A>G is linked to resistance against kanamycin, capreomycin, and amikacin, according to GWAS analysis. The rrs gene encodes 16S ribosomal RNA, which is involved in metabolism and xenobiotic detoxification13. The rrs 1401A>G is present in three clusters, while rrs 514A>C is found in two clusters in L4. The Rv0425c (mutation M689V) gene is a metal cation transporting P-type ATPase (CtpH), suggesting a role in membrane maintenance and ion transport14. The Rv2828A R89W mutation has been previously linked to TB survival and virulence15, and it was found in all isolates in the largest cluster, which consists exclusively of L4 isolates. The Rv0825c gene is responsible for fatty acid metabolism16, and the Gln178* mutation was found in two L4 clusters. The Rv3198c gene (D420V mutation) is linked to a probable ATP-dependent DNA helicase II known as UvrD2, which maintains bacterial genome integrity17. The Rv0766c (G337C mutation) gene is linked to cytochrome P450, Cyp123, which is involved in cellular metabolism and xenobiotic detoxification18. Rv3198c D420V and Rv0766c G337C mutations are only found in the large L4 cluster. Lastly, the Rv3092c gene (P250L mutation) may influence the function of a conserved integral membrane protein19.
Discussion
WGS is increasingly being used to diagnose and track TB infections, and the Philippines, a high-burden TB country, has growing investments in such genomic technologies. WGS of isolates from previous Philippine TB prevalence surveys revealed circulating L1 and L4 strains, including MDR-TB and XDR-TB forms in clustered sequences. While “ancient” L1 Manila strains are considered the most prevalent circulating strains, evidence of “modern” L4 drug-resistant strains within prisons was found, consistent with a previous study (n = 25) that characterised strain types using spoligotyping and MIRU-VNTR typing20. Although our M. tuberculosis isolates were pseudo-randomly selected and considered a convenience sample of mostly drug-resistant isolates, which may not reflect circulating allele frequencies, much-needed insights into resistance mutations and related transmission events were gained. Five large clusters of highly similar isolates were identified on the islands of Luzon and Mindanao, and the underlying isolates clustered by geography through the Bayesian dated phylogenetic reconstruction. Using a GWAS approach, several loci (e.g., Rv0425c, rrs, Rv2828A, Rv3198c, Rv0766c, and Rv0825c) associated with the “transmissibility” phenotype were identified, primarily in L4 clusters, which could be linked to increased M. tuberculosis fitness and transmission. Whilst the Rv2828A locus has been previously linked to TB survival and virulence15, the relevance of the identified genes on transmission should be investigated through prospective collections, analysis of other populations, and experiments on gene function.
Using serially sampled isolates, we inferred a crude mutation rate of 0.41 SNPs per year. It has been shown that different lineages of M. tuberculosis evolve at different rates11. Serial isolates can be from re-infections of independent but closely related strains in the patients rather than evolution of the bacteria within a single patient, both leading to inaccurate calculations11. By excluding isolates with > 1 SNP and less than one year difference, the estimate was 0.66 SNPs per isolate per year, whilst an analysis of L4 isolates revealed a rate of 0.35. Using BEAST2 software, the molecular clock rate of the predominant L1.2.1.2.1 Manila family was estimated at 0.63 (95% HPD: 0.17–1.11) mutations per genome per year. Although, the clock rates are consistent with estimates of other lineages, the Manila family could be faster than L4 and may contribute to the observed relatively higher transmission rates of the L1 strain type4,11. A more comprehensive analysis involving other L1 strain types is required to confirm any molecular clock differences in the Manila family. We used the most recent isolate to avoid bias from using serial isolates in some population-based analyses, including when estimating mutation frequencies.
WGS data also revealed the presence of XDR-TB strains with genotypic resistance to isoniazid, rifampicin, fluoroquinolones and bedaquiline for the first time in the Philippines. Three isolates from two hosts had frameshift mutations in mmpR5 (144dupC, 198dupG, and 135delG) strongly associated with bedaquiline resistance. Interestingly, all three mutations were not found at fixed frequencies in the population, suggesting that they may have been collected while the genetic heterogeneity was still present in the host bacterial population. Remarkably, all three mutations were found across two samples from the same host in the same year, with one sample containing 144dupC at 17% within-sample abundance, and the other isolate containing 198dupG at 60% and 135delG at 32% abundance. This observation indicates that three independent acquisitions of bedaquiline resistance mutations occurred in the same host. The other host had a sample containing 144dupC at 55%. Both hosts were in prison at the time of collection. This finding suggests that XDR-TB is developing in Philippine prisons, with the potential to spread to the community. Indeed, Bayesian phylogenetic reconstruction indicated samples from prisons clustering closely and sometimes interspaced with those collected from community settings. Additionally, two of the eight mixed-strain infections identified were sourced from prisoners. These observations suggest that prisons are a potential reservoir of highly resistant and transmissible TB. A previous study of TB in Filipino prisons (n = 25) used genotyping methods to identify two potential clusters and 23 genotypes20, but WGS provides a much higher resolution of transmission4. Public health measures for adequately managing such cases are imperative to prevent onward transmission.
Although phenotypic drug susceptibility testing (DST) and genotypic predictions for MDR-TB were highly concordant (> 90%), our analysis of discordant cases revealed three putative novel markers for isoniazid (inhA I21M, katG K143E and D419Y). The three markers are currently classified as having uncertain significance. However, the findings from this study bolster the confidence in a potential association between these three SNPs and their corresponding drugs21. We also identified a number of potentially novel resistance mutations in candidate genes for other drugs, including streptomycin (n = 27 novel mutations), ethambutol (n = 11), pyrazinamide (n = 2), capreomycin (n = 2), and fluoroquinolones (n = 1). These rare mutations were supported by phenotypic DST data and the large Global50k database (n = 50,722) of strains, ruling out phylogenetic-specific mutations. Interestingly, rifampicin DST data showed that samples with established resistance mutations in rpoB other than S450L had much higher odds of presenting a sensitive phenotype. This result is consistent with previous reports22, and has been linked to slower growth on Mycobacteria growth indicator tube (MGIT) assays. This observation implies that individuals could be prescribed suboptimal regimens with rifampicin that are not effective, highlighting the strengths of using NGS to identify these cases.
Overall, this study confirms the advantages of using whole genome approaches to characterise drug resistance profiles and transmission patterns. With the advent of affordable and accessible rapid WGS or targeted amplicon sequencing diagnostics, the generated sequences and identified mutations provide a baseline set of comparative data for future applications. These include integrating machine learning algorithms and databases with informative drug resistance markers. A deeper understanding of transmission dynamics across time and geography through routine surveillance will help prioritise infection control resources and activities. Surveillance programs should also record and share novel drug resistance SNPs to coordinate a global response. Ultimately, these insights will inform clinical and public health decision-making, contributing to significant reductions in the burden of TB.
Materials and methods
DNA extraction and sequencing
A total of 475 M. tuberculosis short-term cultured isolates from sputum samples collected by the Research Institute for Tropical Medicine (RITM) in the Philippines between 2012 and 2019 were pseudo-randomly selected for the study. Informed consent was obtained from all subjects and/or their legal guardian(s). This study was given authorisation by the Institutional Review Board of the RTIM (ID No. RITM-IRB 2017–05). Drug susceptibility testing was performed as part of routine TB culture and phenotypic assessments in BSL3 laboratories at the RITM for rifampicin and isoniazid, and additionally for some isolates for ethambutol, streptomycin, amikacin, kanamycin, capreomycin and levofloxacin drugs (see protocols elsewhere3). Total genomic DNA was extracted using a phenol–chloroform extraction procedure. DNA extract concentrations and quality were measured using a Qubit fluorometer (Life Technologies Holdings Pte Ltd, Singapore) and were visualised with a 1% agarose gel. Library preparation of the DNA samples was performed using a QIAseq FX DNA library kit, following the manufacturer’s protocol. We quantified the libraries using dsDNA Qubit Assay, while the libraries’ sizes were measured using Agilent Tapestation 2200 DNA 1000 assay kit. At the Philippine Genome Center (Manila), we normalized to 4 nM, pooled and sequenced all H37Rv libraries using Illumina Novaseq 6000 (2 × 151 base pair reads). WGS data was also available from previous studies (n = 257), including DRS2 (Second National TB Drug Resistance Survey in 2012) (n = 166; years 2011–2012)3 and NTPS (National Tuberculosis Control Program Survey) (n = 91, year 2016)23. The combined dataset consisted of 732 isolates with WGS data. All raw sequencing data is available (see Supplementary Data 1 for a list of accession numbers). All methods were performed in accordance with the relevant guidelines and regulations.
Bioinformatic and statistical analyses
Sequence reads were inspected using fastQC (www.bioinformatics.babraham.ac.uk/projects/fastqc/) as a primary data quality assessment. The reads were trimmed using trimmomatic24 (v0.38; LEADING:3 TRAILING:3 SLIDINGWINDOW:4:20 MINLEN:36) to remove low-quality sequences, and then mapped against the H37Rv reference genome (AL123456) using BWA-mem25 (v0.7.17). SNPs were called using the BCF/VCF tool suite (v1.8)26 in regions with at least 10 reads. SNPs were removed from non-unique regions of the genome (e.g., ppe genes). Full-length consensus genomes were created by inserting SNPs for each sequence into the H37Rv reference using the bcftools consensus tool. Consensus genomes were masked at low-coverage positions (< tenfold depth) and within genomic regions that are difficult to characterise with short-read sequencing (e.g. pe/ppe genes). Consensus genomes were concatenated and used as input to iq-tree (v2.2.2.7)27 to reconstruct the phylogeny. BEAST2 software12 was used to construct an MCMC phylogenetic trees, with parameter settings that calibrated the time scale using alignments without invariant positions28. Lastly, ITOL software was used to visualise the trees29. Drug resistance profiles and lineages were predicted in-silico using TB-Profiler software (v2.0). Variant annotations were labelled using SnpEff software30.
Mixed infections were found using Gaussian mixture modelling30,31 of SNP allele coverage data, leading to eight samples being removed and full analysis being performed on 724 isolates. The distribution of SNP genotype differences between isolates (pairwise) was used to determine a transmission cut-off (of 12), which was sufficiently stringent to avoid expected (sub-)lineage differences (Fig. S3). The association of the presence in clustered isolates (yes/no) with lineages and drug resistance was explored using logistic regression models, leading to odds ratios. To determine SNPs linked to potential transmissibility, GWAS was performed using a logistic regression model in Plink2 software32, which adjusted for lineage and drug resistance. This approach has been applied previously33. Mutation frequencies for SNPs of interest were compared to those from an M. tuberculosis database (“Global50k”; n = 50,72234), which covers all lineages across > 100 countries. We used the current definitions of drug-resistant TB: we defined MDR-TB as TB resistant to isoniazid and rifampicin, pre-XDR TB as MDR-TB with resistance to any fluoroquinolone, and XDR-TB as MDR-TB with additional resistance to any fluoroquinolone and another WHO group A drug (bedaquiline or linezolid)35.
Supplementary Information
Acknowledgements
We thank Dr. Edelwisa S. Mercado for supporting the study. LW is funded by a BBSRC LIDO studentship (Reference no. BB/T008709/1). The project was funded by an MRC UK – PCHRD—Newton Agham award (Grant no. MR/R025576/1) and a British Council – CHED – Newton Institutional links grant (Ref. 261868591). TGC and SC are funded by the UKRI MRC (Ref: MR/M01360X/1, MR/N010469/1, MR/R025576/1, MR/R020973/1, and MR/X005895/1) and EPSRC (EP/Y018842/1) grants.
Author contributions
JEP, JCH, SC, MLH, CGA, EMC-DLP, and TGC conceived the project and were involved in securing its funding. DRL, JEP, CGA, RPB, EMC-DLP, and TGC directed the project. DRL, LTR, AGP, MGCS, LAAA, MAAT, IAPM, CMAD, CGA, and RPB coordinated sample and data collection, data and sample processing, DNA extraction, library building and sensitivity testing. J-HSL coordinated sequencing. SM, YMu, YMo, CFA, JCMM and JCM contributed sequence data. LW performed bioinformatic and statistical analyses under the supervision of JEP and TGC. All authors interpreted the results. LW wrote the first draft of the manuscript. All authors commented and edited various drafts of the manuscript and approved the final version. LW, JEP and TGC compiled the final manuscript.
Data availability
Previously published and newly generated data can be found on the ENA using the Run accession codes in Supplementary Data 1. The newly generated data can be found under the ENA study accession number ERP114520.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Linfeng Wang, Dodge R. Lim, Jody E. Phelan, Ramon P. Basilio, Eva Maria Cutiongoco-De La Paz and Taane G. Clark.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-70471-x.
References
- 1.WHO. Global Tuberculosis Report 2021 (WHO, 2021). [Google Scholar]
- 2.Ragonnet, R., Trauer, J. M., Denholm, J. T., Marais, B. J. & McBryde, E. S. High rates of multidrug-resistant and rifampicin-resistant tuberculosis among re-treatment cases: Where do they come from?. BMC Infect. Dis.17, 1–10 (2017). 10.1186/s12879-016-2171-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Phelan, J. E. et al. Mycobacterium tuberculosis whole genome sequencing provides insights into the Manila strain and drug-resistance mutations in the Philippines. Sci. Rep.9, 1–6 (2019). 10.1038/s41598-019-45566-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Napier, G. et al. Robust barcoding and identification of Mycobacterium tuberculosis lineages for epidemiological and clinical studies. Genome Med12, 1–10 (2020). 10.1186/s13073-020-00817-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roycroft, E. et al. The largest prison outbreak of TB in Western Europe investigated using whole-genome sequencing. Int. J. Tuberc. Lung Dis.25, 491–497 (2021). 10.5588/ijtld.21.0033 [DOI] [PubMed] [Google Scholar]
- 6.Sanabria, G. E. et al. Phylogeography and transmission of Mycobacterium tuberculosis spanning prisons and surrounding communities in Paraguay. Nat. Commun.14, 303 (2023). 10.1038/s41467-023-35813-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anselmo, L. M. P. et al. New insights on tuberculosis transmission dynamics and drug susceptibility profiles among the prison population in Southern Brazil based on whole-genome sequencing. Rev. Soc. Bras. Med. Trop.56, e0181 (2023). 10.1590/0037-8682-0181-2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Utpatel, C. et al. Prison as a driver of recent transmissions of multidrug-resistant tuberculosis in Callao, Peru: A cross-sectional study. The Lancet31, 100674 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arnold, A. et al. XDR-TB transmission in London: Case management and contact tracing investigation assisted by early whole genome sequencing. J. Infect.73, 210–218 (2016). 10.1016/j.jinf.2016.04.037 [DOI] [PubMed] [Google Scholar]
- 10.Lin, W.-H., Lee, W.-T., Tsai, H.-Y. & Jou, R. Disputed rpoB mutations in Mycobacterium tuberculosis and tuberculosis treatment outcomes. Antimicrob. Agents Chemother.65, 0157320 (2021). 10.1128/AAC.01573-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Menardo, F., Duchêne, S., Brites, D. & Gagneux, S. The molecular clock of Mycobacterium tuberculosis. PLoS Pathog.15, e1008067 (2019). 10.1371/journal.ppat.1008067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bouckaert, R. et al. BEAST 25: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol.15, e1006650 (2019). 10.1371/journal.pcbi.1006650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cuevas-Córdoba, B. et al. rrs and rpsL mutations in streptomycin-resistant isolates of Mycobacterium tuberculosis from Mexico. J. Microbiol. Immunol. Infect.46, 30–34 (2013). 10.1016/j.jmii.2012.08.020 [DOI] [PubMed] [Google Scholar]
- 14.Ioerger, T. R. et al. Variation among genome sequences of H37Rv strains of Mycobacterium tuberculosis from multiple laboratories. J. Bacteriol.192, 3645 (2010). 10.1128/JB.00166-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Souza, G. A., Leversen, N. A., Målen, H. & Wiker, H. G. Bacterial proteins with cleaved or uncleaved signal peptides of the general secretory pathway. J. Proteomics75, 502–510 (2011). 10.1016/j.jprot.2011.08.016 [DOI] [PubMed] [Google Scholar]
- 16.Gu, S. et al. Comprehensive proteomic profiling of the membrane constituents of a Mycobacterium tuberculosis strain. Mol. Cell. Proteomics2, 1284–1296 (2003). 10.1074/mcp.M300060-MCP200 [DOI] [PubMed] [Google Scholar]
- 17.Williams, A. et al. UvrD2 is essential in Mycobacterium tuberculosis, but its helicase activity is not required. J. Bacteriol.193, 4487 (2011). 10.1128/JB.00302-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ouellet, H., Johnston, J. B. & Ortiz de Montellano, P. R. The Mycobacterium tuberculosis Cytochrome P450 System. Arch. Biochem. Biophys.493, 82 (2010). 10.1016/j.abb.2009.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Agarwal, N., Woolwine, S. C., Tyagi, S. & Bishai, W. R. Characterization of the Mycobacterium tuberculosis sigma factor SigM by assessment of virulence and identification of SigM-dependent genes. Infect. Immun.75, 452 (2007). 10.1128/IAI.01395-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Montoya, J. C., Borja, M. P., Ang, C. F. & Murase, Y. Molecular epidemiologic analysis of Mycobacterium tuberculosis among prison inmates in selected prisons in the Philippines. Philipp. J. Sci.150, 417–427 (2021). 10.56899/150.02.07 [DOI] [Google Scholar]
- 21.WHO. Catalogue of Mutations in Mycobacterium Tuberculosis Complex and Their Association with Drug Resistance 2nd edn. (WHO, 2023). [Google Scholar]
- 22.Miotto, P., Cabibbe, A. M., Borroni, E., Degano, M. & Cirilloa, D. M. Role of disputed mutations in the rpoB gene in interpretation of automated liquid MGIT culture results for rifampin susceptibility testing of Mycobacterium tuberculosis. J. Clin. Microbiol.56, 01599 (2018). 10.1128/JCM.01599-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Montoya, J. C. et al. Molecular characterization of drug-resistant Mycobacterium tuberculosis among Filipino patients derived from the national tuberculosis prevalence survey Philippines 2016. Tuberculosis135, 102211 (2022). 10.1016/j.tube.2022.102211 [DOI] [PubMed] [Google Scholar]
- 24.Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics30, 2114–2120 (2014). 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Figshare 10.6084/M9.FIGSHARE.963153.V1 (2013). 10.6084/M9.FIGSHARE.963153.V1 [DOI]
- 26.Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics27, 2987–2993 (2011). 10.1093/bioinformatics/btr509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Minh, B. Q. et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol.10.1093/molbev/msaa015 (2020). 10.1093/molbev/msaa015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu, Y. et al. Transmission analysis of a large tuberculosis outbreak in London: A mathematical modelling study using genomic data. Microb. Genom.6, mgen000450 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res.49, W293–W296 (2021). 10.1093/nar/gkab301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly6, 80 (2012). 10.4161/fly.19695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang, L., Campino, S., Phelan, J. & Clark, T. G. Mixed infections in genotypic drug-resistant Mycobacterium tuberculosis. Sci. Rep.13, 1–8 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang, C. C. et al. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience4, 1–16 (2015). 10.1186/s13742-015-0047-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Napier, G. et al. Characterisation of drug-resistant Mycobacterium tuberculosis mutations and transmission in Pakistan. Sci. Rep.12, 7703 (2022). 10.1038/s41598-022-11795-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Napier, G. et al. Comparison of in silico predicted Mycobacterium tuberculosis spoligotypes and lineages from whole genome sequencing data. Sci. Rep.13, 11368 (2023). 10.1038/s41598-023-38384-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.WHO TEAM Global Tuberculosis Programme (GTB). Meeting Report of the WHO Expert Consultation on the Definition of Extensively Drug-Resistant Tuberculosis. https://www.who.int/publications/i/item/9789240018662 (2021).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Figshare 10.6084/M9.FIGSHARE.963153.V1 (2013). 10.6084/M9.FIGSHARE.963153.V1 [DOI]
Supplementary Materials
Data Availability Statement
Previously published and newly generated data can be found on the ENA using the Run accession codes in Supplementary Data 1. The newly generated data can be found under the ENA study accession number ERP114520.