

Abstract
The plasma protein β2GPI (β2-glycoprotein I) has been proposed to mediate phagocytosis of apoptotic cells and to play a role in the antiphospholipid syndrome. This suggestion is based mainly on the presumption that β2GPI has an appreciable interaction with PS (phosphatidylserine)-exposing cell membranes. However, quantitative data on the binding of β2GPI to PS-exposing cells under physiologically relevant conditions are scarce and conflicting. Therefore we evaluated the binding of β2GPI to PS-expressing blood platelets. Flow cytometry showed that binding of β2GPI is negligible at physiological ionic strength, in contrast with significant binding occurring at low ionic strength. Binding parameters of β2GPI and (for comparison) prothrombin were quantified by ellipsometric measurement of protein depletion from the supernatant following incubation with platelets. At low ionic strength (20 mM NaCl, no CaCl2), a dissociation constant (Kd) of 0.2 μM was found for β2GPI, with 7.4×105 binding sites per platelet. Under physiologically relevant conditions (120 mM NaCl and 3 mM CaCl2), binding of β2GPI was not detectable (extrapolated Kd>80 μM). Prothrombin binding (at 3 mM CaCl2) was much less affected by ionic strength: Kd values of 0.5 and 1.4 μM were observed at 20 and 120 mM NaCl respectively. The low affinity and the presence of many lipid-binding proteins in plasma that can compete with the binding of β2GPI suggest that only a small fraction (<5%) of the binding sites on PS-exposing blood cells are likely to be occupied by β2GPI. These findings are discussed in relation to the alleged (patho-)physiological functions of β2GPI.
Keywords: β2-glycoprotein I, binding site, dissociation constant, phagocytosis, platelet, prothrombin
Abbreviations: ACD, 0.052 M citric acid, 0.08 M trisodium citrate and 0.183 M glucose; APS, antiphospholipid syndrome; β2GPI, β2-glycoprotein I; PS, phosphatidylserine; PC, phosphatidylcholine
INTRODUCTION
β2GPI (β2-glycoprotein I), also known as apolipoprotein H, is a 50 kDa plasma protein of unknown physiological function. Besides a possible role in the metabolism of triacylglycerol-rich lipoproteins [1], β2GPI has been suggested to affect platelet function [2,3] and to modulate the activity of procoagulant cell surfaces by binding to anionic phospholipids [4,5]. Price et al. [6] have demonstrated selective binding of β2GPI to apoptotic cells that has been suggested to promote their scavenging by phagocytes [7–9].
β2GPI has gained much attention since the discovery of its role in APS (antiphospholipid syndrome) [10–12]. APS is defined by the presence of antiphospholipid antibodies in association with venous or arterial thrombosis. It is increasingly appreciated that so-called antiphospholipid antibodies in patients with APS are not directed to phospholipids, but bind to plasma proteins bound to anionic phospholipid surfaces. The most frequently observed antigens for antiphospholipid antibodies are β2GPI and prothrombin, although antibodies to other plasma proteins have been reported. Several mechanisms have been proposed to explain the relationship between antiphospholipid antibodies and increased risk of thromboembolic complications. Complexes of antiphospholipid antibodies with β2GPI or prothrombin on the lipid surface could impede the anticoagulant function of activated protein C and tissue factor pathway inhibitor [13–16]. It has also been suggested that binding of antiphospholipid-antibody–protein complexes to cell surfaces may promote or enhance cell activation [17].
Alternatively, it has been proposed that antibodies to β2GPI in patients with APS may be a consequence of persistent surface exposure of PS (phosphatidylserine) in the plasma membranes of blood cells or cells lining blood vessels [18–20]. Although PS is generally located in the cytoplasmic leaflet of the plasma membrane, conditions of cellular activation, apoptosis or certain pathological conditions may cause a reorganization of the transbilayer lipid distribution, resulting in its exposure at the cell surface. Normally, PS-exposing cells are removed rapidly by phagocytosis. A malfunction in one of these processes, however, could give rise to an increased number of PS-exposing cells, allowing the binding of plasma proteins with an affinity for anionic phospholipids. In this view, antiphospholipid antibodies could be considered to be the result of a natural immune response to (cryptic) epitopes exposed by lipid-bound plasma proteins.
In most studies on the pathophysiological role of β2GPI in APS as well as in studies on its function in phagocytosis, it is assumed that this protein has a substantial affinity for PS-exposing cell surfaces. This presumption, however, is not supported by data obtained in model membrane systems. Binding studies with β2GPI showed high-affinity binding at low ionic strength and on artificial lipid bilayers composed of 50–100% PS. However, since the mol fraction PS in the plasma membrane outer leaflet of activated platelets or apoptotic cells is more likely to be in the range 2–10 mol%, results from these model membrane studies may not reflect what occurs physiologically [21]. Indeed, when binding was assessed under physiological conditions, Kd values of 4 and 14 μM were found for lipid surfaces containing 20 and 10 mol% PS respectively [22]. Similar low affinities have been reported by Harper et al. [23]. It should be emphasized that antiphospholipid antibodies greatly enhance the binding of β2GPI due to the formation of multimeric antigen–antibody complexes on the lipid surface [22].
Nevertheless, early studies reported that β2GPI binds to non-stimulated washed human platelets with a dissociation constant of between 0.5 and 1 μM [24,25]. Indirect evidence for binding of β2GPI to activated platelets was obtained from studies on the inhibitory effect of the protein on platelet procoagulant activity [5], although a later study could not reproduce these results [26]. Other experiments also failed to demonstrate β2GPI binding to non-activated or activated platelets [27–29]. Nevertheless, β2GPI has been shown to bind to apoptotic, PS-exposing thymocytes [6,7], and immunization with thymocyte-bound β2GPI was shown to produce antiphospholipid antibodies [20].
In light of these contradictory observations, we have re-investigated the interaction of β2GPI with PS-exposing natural membranes. We have chosen as a model system human blood platelets treated with the calcium ionophore ionomycin, since this stimulus evokes a homogeneous population of PS-exposing cells, in contrast with more physiological agonists such as collagen or thrombin, which may cause only a minor fraction of the platelets to expose PS [21,30]. Because direct labelling of β2GPI appeared to abrogate lipid binding, binding was explored by flow cytometry using fluorescently labelled univalent Fab'- and bivalent anti-β2GPI IgG to evaluate the effect of antibody valency on affinity. Quantitative binding data were obtained by ellipsometric measurement of the depletion of β2GPI from the supernatant of incubations with ionomycin-activated blood platelets, using the same approach as we described previously to characterize the binding of annexin V to ionomycin-activated platelets [29]. For comparative purposes, and in view of it being the most frequent antigen for the production of antiphospholipid antibodies after β2GPI, binding of prothrombin was also measured. Our data indicate that, at physiologically relevant conditions, β2GPI has a low affinity for PS-exposing platelet membranes. In contrast, the affinity of prothrombin for the same cell surface and under the same conditions appears to be almost two orders of magnitude higher.
MATERIALS AND METHODS
Materials
BSA (essentially fatty acid free), ionomycin and FITC were from Sigma (St. Louis, MO, U.S.A.). 1,2-Dioleoyl-sn-glycero-3-phosphocholine [phosphatidylcholine (PC)] and 1,2-dioleoyl-sn-glycero-3-phosphoserine (PS) were obtained from Avanti Polar Lipids (Alabaster, AL, U.S.A.). FITC-conjugated annexin V was obtained from Molecular Probes (Leiden, The Netherlands). Unconjugated annexin V was kindly donated by Dr C. P. M. Reutelingsperger (Department of Biochemistry, Cardiovascular Research Institute Maastricht). Human prothrombin, purified according to Hendrix et al. [31], was a gift from Dr. T. M. Hackeng (Department of Biochemistry, Cardiovascular Research Institute Maastricht). Silicon slides for ellipsometry were purchased from Aurel GmbH (Landsberg, Germany).
Purification of β2GPI
Human β2GPI was purified according to previously described methods [32], with minor modifications. Briefly, perchloric acid (70%) was added to outdated pooled plasma to a final concentration of 1.2% (v/v) and stirred for 30 min at 0– 4 °C. The precipitate was discarded and the supernatant was adjusted to pH 8.0 with saturated Na2CO3, followed by extensive dialysis against 20 mM Tris, pH 8.0. This material was applied to a QAE column, and proteins were eluted using a gradient from 0 to 350 mM NaCl. β2GPI-containing fractions, detected by SDS/PAGE and silver staining, were pooled, dialysed against 20 mM Tris, pH 8.0, and applied to a heparin affinity column. Bound proteins were eluted using a gradient from 0 to 1.0 M NaCl. Finally, the β2GPI preparation was purified further by FPLC over Mono-Q, followed by Protein A–Sepharose to remove traces of IgG present in the preparation. The final preparation contained a homogeneous band at 50 kDa, as shown by non-reduced SDS/PAGE and silver staining.
Antibodies
Polyclonal antibodies against β2GPI were raised in rabbits using standard protocols and were purified from serum by Protein A affinity chromatography. Rabbit polyclonal antibodies against human prothrombin and FITC-conjugated swine anti-rabbit IgG were obtained from DAKO (Glostrup, Denmark). To prepare FITC-conjugated rabbit anti-β2GPI antibodies (anti-β2GPI–FITC), 50 μl of FITC (2 mg·ml−1 in DMSO) was added slowly under stirring to 3 ml of protein (1 mg·ml−1 in 0.1 M Na2CO3, pH 9) and the mixture was incubated at 25 °C in the dark. Additional 50 μl aliquots of FITC were added after 30 and 60 min. After 2 h, unbound FITC was removed by dialysis against PBS. This procedure resulted in labelling of 1.5–3 mol of FITC per mol of protein. FITC-labelled Fab' fragments were prepared by incubating anti-β2GP1 IgG (10 mg) with pepsin (0.1 mg) to produce F(ab')2 fragments. After a 20 h incubation at 37 °C, dithiothreitol was added (1 mM) and the reduced Fab' was isolated by gel filtration chromatography. The purified Fab' was then labelled immediately with a 10-fold excess of fluorescein-5-maleimide (Molecular Probes, Eugene, OR, U.S.A.). Unbound label was removed by dialysis. SDS/PAGE analysis confirmed the absence of IgG and F(ab')2 from the labelled preparation.
Platelet preparation and activation
Blood from healthy volunteers was collected into 1/6 volume of ACD (0.052 M citric acid, 0.08 M trisodium citrate and 0.183 M glucose). Platelet-rich plasma was obtained by centrifugation at 150 g for 15 min. Subsequently, platelets were sedimented by centrifugation at 750 g for 15 min. The platelet pellet was resuspended and washed twice in Hepes buffer at pH 6.6 (136 mM NaCl, 2.7 mM KCl, 2 mM MgCl2, 10 mM Hepes, 5 mM glucose and 0.5 mg·ml−1 BSA). Before each centrifugation, 5% (v/v) ACD was added to prevent platelet aggregation during sedimentation. Platelets were finally resuspended in Hepes buffer at pH 7.4 to 2×108 cells·ml−1 using a Coulter counter.
To induce exposure of PS, platelets were activated with 5 μM ionomycin in the presence of 3 mM CaCl2 for 10 min at 37 °C. After treatment with ionomycin, platelets were centrifuged at 1500 g and resuspended to the same cell concentration. This step was introduced to remove platelet microvesicles. Subsequently, the platelets were washed twice in 10 vol. of Hepes buffer and resuspended at a final concentration of 2×109 platelets·ml−1. This washing step appeared to be necessary to remove an unidentified component from the suspension that interfered with the binding of β2GPI (see the Results section). Ionomycin-treated platelet preparations were checked for surface exposure of PS by measuring the binding of annexin V using the ellipsometric approach [29]. To assess the effects of ionic strength on binding, platelets were resuspended in either physiological ‘high salt’ buffer (120 mM NaCl, 10 mM Hepes, 0.5 mg·ml−1 BSA and 50 mM glucose) or ‘low salt’ buffer (20 mM NaCl, 10 mM Hepes, 0.5 mg·ml−1 BSA and 250 mM glucose to maintain isotonicity). Identical results were obtained when glucose was replaced by sucrose in these buffers. All buffers were adjusted at pH 7.4.
Measurement of the binding of β2GPI to ionomycin-treated platelets by flow cytometry
Samples of 25 μl of ionomycin-activated platelets were incubated with β2GPI (final concentration 100 nM) and anti-β2GPI (final concentration 32 μg·ml−1), or prothrombin (final concentration 100 nM) and anti-prothrombin (final concentration 36 μg·ml−1) at ambient temperature. Binding was detected with FITC-conjugated swine anti-rabbit secondary antibody (final concentration 48 μg·ml−1). In some experiments, FITC-conjugated anti-β2GPI (final concentration 10 μg·ml−1) or FITC-labelled univalent anti-β2GPI Fab' fragments (final concentration 80 μg·ml−1) were used to detect binding. After 30 min, samples were diluted 10-fold with Hepes buffer and analysed in a Becton Dickinson FACSort flow cytometer, equipped with a 488 nm emitting laser. Light scatter and fluorescence channels were set at logarithmic gain. Fluorescence emission was monitored using a 530/30 bandpass filter. Listmode data were collected for 10.000 cells, measuring forward scatter, side scatter and green fluorescence for FITC. Listmode data were analysed with the WinMDI 2.8 software program (http://facs.scripps.edu). To detect binding under low ionic strength conditions, the standard sheath fluid for the flow cytometer was replaced with buffer composed of 20 mM NaCl, 10 mM Hepes and 250 mM glucose (to maintain isotonicity), pH 7.4.
Ellipsometric determination of β2GPI and prothrombin in the platelet supernatant
Planar phospholipid bilayers were deposited on silicon slides by adsorption of small unilamellar vesicles composed of 20 mol% PS and 80 mol% PC (PS/PC), as described in [22]. Ellipsometry was used to measure protein adsorption to these planar phospholipid bilayers as described previously [22,33]. Protein adsorption measurements were performed at ambient temperature (20–22 °C) under continuous stirring in a trapezoidal cuvette with Hepes buffer (10 mM Hepes, 75 mM NaCl, 0.5 mg·ml−1 BSA, pH 7.5) without CaCl2 for β2GPI and with 3 mM CaCl2 for prothrombin. This buffer was chosen to optimize the affinity of β2GPI and prothrombin for the PS/PC bilayers.
The protein concentration in samples of the supernatants of platelets incubated with β2GPI or prothrombin was determined by transferring samples to the ellipsometer cuvette to measure protein adsorption to a PS/PC bilayer deposited on the silicon slide. Sample size was adjusted to give a concentration in the range 0–5 nM for β2GPI and 0–10 nM for prothrombin. From Figure 1, which shows the time-dependent adsorption of various concentrations of β2GPI, it is apparent that the adsorption rate increases steeply with increasing concentration. A plot of initial adsorption rate against β2GPI concentration (inset of Figure 1) shows that adsorption is linearly related to concentration, indicating that this assay system can be used to determine protein concentration in supernatant samples. The same method was used to determine prothrombin in supernatant samples from ionomycin-activated platelets incubated with prothrombin. It should be emphasized that the adsorption as shown in Figure 1 is completed within 10 min.
Figure 1. Concentration-dependent binding of β2GPI to PS/PC bilayers containing 20 mol% PS.

The mass, Γ (μg·cm−2), of adsorbed protein measured by ellipsometry is plotted as function of time. At t=200 s, β2GPI (0.5, 1, 2, 3, 4 or 5 nM; top to bottom) was added to the cuvette and the adsorption was monitored by ellipsometry. The inset shows a plot of the initial adsorption rate, as estimated from the best fit of the regression line to the adsorption data measured between 20 and 120 s after addition of β2GPI to the cuvette. Experiments were performed at ambient temperature (20–22 °C) in Hepes buffer (75 mM NaCl, 10 mM Hepes, pH 7.5) containing 0.5 mg·ml−1 BSA but not containing CaCl2.
Measurement of protein binding to ionomycin-treated platelets
Washed, ionomycin-activated blood platelets were resuspended in Hepes buffers (10 mM Hepes, 0.5 mg·ml−1 BSA) with ionic strength varying from 20 to 120 mM NaCl, and glucose concentrations varying from 250 to 50 mM to maintain isotonicity. Platelets were incubated in these buffers with either prothrombin or β2GPI for 15 min in polycarbonate ultracentrifuge tubes, and subsequently centrifuged at 180000 g. The supernatant was collected quantitatively and transferred to another vial. After mixing, an aliquot was transferred to the ellipsometer cuvette and the resulting protein adsorption was used to determine the concentration of unbound β2GPI or prothrombin. Pilot experiments showed that maximal binding was obtained within 15 min, and hence this incubation period was used throughout the study. To determine binding as a function of the platelet concentration, 200 nM β2GPI (or 400 nM prothrombin) was incubated with platelet suspensions containing (0.3–6.0)×108 platelets·ml−1. Binding as a function of β2GPI concentration was measured by incubating increasing concentrations of β2GPI (0.1–2.4 μM) with a fixed platelet concentration of 6×108 platelets·ml−1. For prothrombin binding as a function of concentration, the prothrombin concentration was varied from 0.2 to 4.8 μM, while keeping the platelet concentration at 6×108 platelets·ml−1.
Analysis of binding data
The data were analysed using the Langmuir model for binding to independent binding sites:
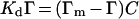 |
where Γm is the concentration of binding sites at the given platelet count (nM), Γ is the concentration of bound protein (=concentration of binding sites occupied) (nM), Kd is the dissociation constant (nM) and C is the concentration of unbound protein (nM). This model allows expression of Γ in terms of total added protein, Ct (Ct=C+Γ) and the binding parameters Kd and Γm:
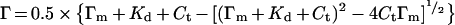 |
(1) |
RESULTS
A straightforward approach to measuring the binding of β2GPI to PS-exposing cell membranes would be to use fluorescently labelled or radiolabelled protein. Direct labelling of β2GPI with fluorescein, however, abrogated the binding of β2GPI to planar bilayers containing 20 mol% PS (results not shown). This is most probably due to reaction of FITC with lysine residues in domain V of β2GPI, known to be critical for binding to anionic lipid membranes. Alternative methods to provide β2GPI with a fluorescent tag via its carbohydrate residues, as well as radiolabelling using standard procedures (Iodogen), were also detrimental to the lipid-binding properties of the protein when analysed by ellipsometry. We therefore explored binding of β2GPI by flow cytometry using FITC-labelled antibodies.
Binding of β2GPI and prothrombin as detected by flow cytometry
Figure 2 shows that treatment of platelets with Ca2+ ionophore in the presence of 3 mM Ca2+ resulted in the surface exposure of PS, as seen by the binding of FITC-conjugated annexin V. Subsequently, binding of β2GPI was detected with polyclonal anti-β2GPI antibody from rabbit and a secondary FITC-conjugated swine anti-rabbit antibody. With unstimulated platelets, binding of β2GPI in the presence of anti-β2GPI could not be observed (results not shown). To investigate binding to PS-exposing platelets, initial experiments were performed with platelets that, after treatment with ionomycin, were centrifuged at 1500 g and resuspended to the same cell concentration. This step was introduced to remove platelet microvesicles. Figure 3 (left panels) shows that binding of β2GPI was dependent on platelet concentration; while the presence of β2GPI caused a 20-fold increase in fluorescence signal at 2×106 platelets·ml−1, the increase in fluorescence was only 2–3-fold when measured at a 100-fold higher platelet concentration. Since the lower binding at higher platelet concentrations was not due to depletion of either β2GPI or anti-β2GPI from the solution (results not shown), these results suggest that an inhibitor prevented the binding of β2GPI to platelets. This conclusion is supported by the finding that binding was also strongly diminished when platelets were resuspended to 2×106 platelets·ml−1 in an 18000 g supernatant obtained from a suspension of 109 platelets·ml−1. This supernatant also inhibited the binding of β2GPI and FITC-conjugated anti-β2GPI to liposomes composed of PS/phosphatidylethanolamine/PC (5:20:75) (results not shown). Together, these findings strongly suggest the presence of a substance derived from ionomycin-activated platelets that interferes with the binding or formation of the β2GPI immune complexes at the membrane surface. In contrast, binding of prothrombin–anti-prothrombin complexes was independent of platelet concentration (Figure 3, right panels). The slight decrease observed at the highest platelet concentration was attributed to protein depletion (results not shown). It therefore seems that the inhibitory effect on binding at high platelet concentration is specific for β2GPI. It should be emphasized that the inhibitor did not interfere with the ellipsometric measurements, since sample supernatants were diluted at least 40-fold and analysed on 20 mol% PS. Since the inhibitor was removed by washing, all subsequent experiments were performed on washed ionomycin-treated cells. The finding of an inhibitory component derived from ionomycin-treated platelets was somewhat surprising, and could explain the lack of β2GPI binding to ionomycin-treated platelet preparations in an earlier study where the inhibitor was not removed [29]. Since the effect was only observed at high platelet concentrations, the inhibitor is likely to be of low affinity. It is not known whether other platelet activation procedures produce a similar inhibitor.
Figure 2. Histograms of the binding of FITC-conjugated annexin V to ionomycin-activated platelets and control platelets.

Platelets were treated with ionomycin in the presence of Ca2+, and subsequently washed as described in the Materials and methods section. For detection of the binding of annexin V, 25 μl of platelets (2×107 platelets·ml−1) was incubated with 1 μl of FITC–annexin V (300 μg·ml−1) in the presence of 3 mM CaCl2. After 5 min, samples were diluted 10-fold with Hepes buffer (including 3 mM CaCl2) before being applied to the flow cytometer. Shaded area, ionomycin-activated platelets; non-shaded area, control platelets.
Figure 3. Histograms showing the binding of β2GPI and prothrombin in the presence of their respective antibodies to ionomycin-activated platelets: effect of platelet concentration.

Samples of 25 μl of ionomycin-treated platelets at platelet concentrations of 2×108, 2×107 and 2×106 platelets·ml−1 were incubated with 300 nM β2GPI or 100 nM prothrombin in the presence of 32 μg·ml−1 anti-β2GPI or 36 μg·ml−1 anti-prothrombin respectively. After a 30 min incubation at room temperature, FITC-conjugated swine anti-rabbit antibody was added to a final concentration of 48 μg·ml−1. After 15 min, the samples were diluted 10-fold and subsequently analysed by flow cytometry. All incubations and dilutions were performed in Hepes buffer containing 3 mM CaCl2.
Using washed ionomycin-treated platelets, the fluorescence signal obtained for FITC-conjugated anti-β2GPI showed an approx. 10-fold increase in the amount of bound protein (Figure 4, upper left panel). Since antibody valency is known to strongly affect the binding of β2GPI to planar PS/PC bilayers [22], it was important to determine the influence of antibody valency on binding. Binding could not be detected when univalent FITC-conjugated anti-β2GPI Fab' fragments were substituted for IgG (Figure 4, upper right panel). Since earlier studies indicated that binding of β2GPI to PS/PC planar bilayers is strongly dependent on the ionic strength of the incubation buffer [22,23], the binding of β2GPI to PS-exposing platelets was also assessed at low salt concentration (20 mM NaCl). Unlike the observations obtained in the presence of 120 mM NaCl, β2GPI bound to platelets in the presence of both intact antibody and univalent Fab' fragments in low-ionic-strength buffers (Figure 4, lower left and right panels respectively).
Figure 4. Histograms showing the binding of β2GPI to ionomycin-activated platelets in the presence of IgG or Fab': effect of ionic strength.

Platelets were resuspended at a concentration of 5×107 platelets·ml−1 in isotonic Hepes buffer at 20 mM NaCl (low ionic strength) or 120 mM NaCl (high ionic strength). From these suspensions, 25 μl aliquots were incubated with 100 nM β2GPI in the presence of 10 μg·ml−1 FITC-conjugated anti-β2GPI IgG or 10 μg·ml−1 FITC-conjugated Fab'. Before analysis, samples were diluted 10-fold in corresponding low- or high-ionic-strength buffers. For flow cytometry, the sheath fluid was also replaced with the same low- and high-salt buffers. Left panels, β2GPI in the presence of anti-β2GPI IgG; right panels, β2GPI in the presence of anti-β2GPI Fab'; upper panels, high-ionic-strength buffer; lower panels, low-ionic-strength buffer; shaded areas, presence of β2GPI; non-shaded areas, absence of β2GPI.
Surface exposure of PS can also be evoked by activation with thrombin in the presence of thapsigargin, an inhibitor of intracellular Ca2+-ATPase [34]. Similar binding data were obtained with these platelets, indicating that the interaction of β2GPI is not exclusive to ionomycin-treated platelets. Other platelet agonists, such as ADP and thrombin, did not result in appreciable binding, consistent with the notion that these agonists do not cause significant surface exposure of PS in platelets [21].
Quantitative determination of the binding parameters for binding of β2GPI to ionomycin-treated platelets
To quantify the binding of β2GPI to platelets, ionomycin-activated platelets were incubated with β2GPI for 15 min, the cells were removed by centrifugation, and residual β2GPI in the supernatant was measured by ellipsometry, as described in the Materials and methods section. Appreciable platelet-concentration-dependent binding was observed at low salt concentrations (20 mM NaCl); >70% of the added β2GPI bound at 6×108 platelets·ml−1 (Figure 5A). Binding as a function of β2GPI concentration was determined at a fixed platelet concentration of 6×108 platelets·ml−1. At this platelet concentration, the amount of β2GPI bound was a substantial fraction of the amount added. Figure 5(B) shows that binding was saturable. Eqn (1) (see the Materials and methods section) was used to analyse these data in order to account for the depletion of added protein. The solid line, representing the best fit of eqn (1) to these data, corresponds to a Γm of 7.8±0.5 nM (estimate±standard error of estimate) per 107 platelets·ml−1 and a Kd of 0.19±0.06 μM. There seems to be considerable variation in the number of binding sites, since a duplicate experiment resulted in estimations of Γm=16.8±1.9 nM per 107 platelets·ml−1 and Kd=0.23±0.08 μM. The difference in estimated number of binding sites (Γm) between these duplicate experiments is considerable and much larger than the confidence intervals of the estimated values. Therefore this difference is likely to reflect variation in the platelet preparations obtained from different donors.
Figure 5. Binding of β2GPI to ionomycin-activated blood platelets at low ionic strength.

(A) Binding of β2GPI as a function of platelet concentration. Platelets were incubated with 200 nM β2GPI for 15 min in isotonic Hepes buffer containing 20 mM NaCl. The incubation mixture was centrifuged and the concentration of free β2GPI in samples from the supernatant was determined by ellipsometry. Shown is the concentration of bound β2GPI as a function of the platelet concentration. (B) Binding of β2GPI as a function of the concentration of β2GPI. Platelets (6×108 platelets·ml−1) were incubated with various concentrations of β2GPI in isotonic Hepes buffer containing 20 mM NaCl. After 15 min, the incubation mixture was centrifuged and the concentration of free β2GPI in samples from the supernatant was determined by ellipsometry. Shown is the concentration of bound β2GPI as function of the added β2GPI concentration. The solid line represents the best fit of eqn (1) to these data.
At physiological ionic strength (10 mM Hepes, 120 nM NaCl and 50 mM glucose), incubation of β2GPI with ionomycin-activated platelets up to 109 platelets·ml−1 did not result in appreciable depletion of β2GPI from the supernatant, in agreement with the flow cytometry experiment shown in Figure 4 (upper right panel). Decreasing the ionic strength increased the binding of β2GPI to the ionomycin-activated platelets (Figure 6A). Assuming that the number of binding sites is unaffected by the NaCl concentration, the data presented in Figure 6(A) allow estimation of the Kd at 120 mM NaCl. Figure 6(B) shows that the Kd, calculated using a fixed mean value of Γm=12.3 nM per 107 platelets·ml−1, increases exponentially with increasing NaCl concentration. Best-fitting lines to these data resulted in estimates of the Kd at 120 mM NaCl of 13 μM and 30 μM (geometric mean 20 μM). Binding was not measurable in the presence of 3 mM CaCl2 with 120 mM NaCl. Since CaCl2 (2–3 mM) is known to increase the Kd by 4–10-fold [22], extrapolation of these data results in a Kd exceeding 80 μM for the binding of β2GPI to ionomycin-treated platelets.
Figure 6. Binding of β2GPI to ionomycin-activated blood platelets as a function of NaCl concentration.

Platelets, at a concentration of 6×108 cells·ml−1, were incubated with 200 nM β2GPI in isotonic Hepes buffers containing 20, 40, 80, 100 or 120 mM NaCl. After 15 min, the incubation mixture was centrifuged and the concentration of free β2GPI in samples from the supernatant was determined by ellipsometry. (A) The concentration of bound β2GPI is plotted as function of the NaCl concentration in the incubation buffer for two independent experiments. (B) From the data in (A), the dissociation constant was calculated using the formula Kd=(Γm−Γ)C/Γ (nM) and a value of Γm=738 nM at 6×108 platelets·ml−1. Shown is ln\(Kd) as function of the NaCl concentration. Solid lines represent the best fits to these data. The results of two independent experiments are shown.
Quantitative determination of binding parameters for the binding of prothrombin to ionomycin-treated platelets
For comparison, we also measured the binding of prothrombin to the same ionomycin-treated platelets at high and low ionic strength. Figure 7(A) shows considerable binding of prothrombin that was much less affected by ionic strength: increasing the NaCl concentration from 20 to 120 mM resulted in less than a 2-fold reduction in binding. Figure 7(B) shows the binding of prothrombin to 6×108 platelets·ml−1 as function of the prothrombin concentration at 20 mM NaCl. Binding was saturable and the data conformed to the Langmuir model (see eqn 1). Binding parameters estimated from the fit of eqn (1) to these data are Kd=0.54±0.15 μM and Γm=19.1±1.6 nM per 107 platelets·ml−1. From the data shown in Figure 7(A), a Kd of 1.4 μM was estimated for the affinity at 120 mM NaCl.
Figure 7. Binding of prothrombin to ionomycin-activated blood platelets.

(A) Binding of prothrombin (PT) as a function of platelet concentration. Platelets at various concentrations were incubated with 400 nM prothrombin for 15 min in isotonic Hepes buffer containing 3 mM CaCl2 and 20 mM NaCl (●) or 120 mM NaCl (■). The incubation mixture was centrifuged and the remaining (free) prothrombin concentration in the supernatant was determined by ellipsometry. Shown is the concentration of bound prothrombin as function of the platelet concentration. (B) Binding of prothrombin as a function of the prothrombin concentration. Platelets (6×108 platelets·ml−1) were incubated with various concentrations of prothrombin in isotonic Hepes buffer containing 3 mM CaCl2 and 20 mM NaCl. After 15 min, the incubation mixture was centrifuged and the free prothrombin concentration in samples from the supernatant was determined by ellipsometry. Shown is the concentration of bound prothrombin as function of the added prothrombin concentration. The solid line represents the best fit of eqn (1) to these data.
DISCUSSION
The present study shows that (i) at low ionic strength, β2GPI has a significant interaction with ionomycin-stimulated, but not with unstimulated, platelets; (ii) binding is greatly diminished upon increasing the ionic strength or adding Ca2+ ions; (iii) the Kd under physiological conditions (120 mM NaCl and 3 mM Ca2+) is at least 20-fold greater than the plasma concentration of β2GPI; (iv) the interaction of β2GPI with ionomycin-stimulated platelets is strongly enhanced by the presence of bivalent anti-β2GPI IgG, but not by its univalent Fab' fragments; and (v) the affinity of prothrombin for ionomycin-treated platelets under physiological conditions appears to be almost two orders of magnitude higher than that of β2GPI.
Flow cytometry data revealed that, under conditions of physiological ionic strength, the affinity of β2GPI binding was low, and the presence of bivalent anti-β2GPI antibodies was required to increase binding to measurable levels. These findings confirm results obtained with other lipid membranes, which indicated that the binding of β2GPI is strongly enhanced through formation of immune complexes at the membrane surface [22]. This phenomenon of an approx. 1000-fold increase in affinity of β2GPI for lipid membranes as a consequence of interactions with bivalent, but not univalent, antibodies has been studied extensively by our group [22] as well as by others (reviewed in [38]).
Consistent with the notion that the binding of β2GPI is determined mainly by electrostatic interactions [35–37], direct binding of β2GPI in the absence of antibodies was observed only at low salt concentrations. Analysis of the binding parameters indicated a concentration of ∼7.4×105 binding sites for β2GPI per platelet. In comparison, the total number of binding sites calculated for prothrombin was ∼11.5×105 per platelet. These numbers are comparable with the number of binding sites estimated for annexin V (∼30×105 per platelet) [29]. Taking into account the total amount of lipid per platelet (∼40 μM phospholipid for 108 platelets) and assuming that more than 80% is present in the plasma membrane of ionomycin-stimulated platelets (since these have undergone release of granule contents), one can estimate that there are ∼200 lipid molecules in the membrane bilayer per binding site. This number is consistent with the data obtained from previous studies on the interaction of β2GPI with PS/PC planar bilayers [22]. Taken together, these data support the notion that binding of β2GPI to platelets occurs predominantly through the lipid part of the plasma membrane. The lipidic nature of the β2GPI binding sites is also supported by observations that binding was completely blocked by annexin V [38]. Recently, β2GP1 was shown to interact with sulphatides [39]. However, platelet membranes contain <0.1% of this lipid [40], which would make it unlikely that sulphatides contribute to β2GP1 binding in this system.
The present data indicate that binding of β2GPI to platelets under physiological conditions is weak. Although not directly measurable, extrapolation of the data suggests a Kd exceeding 80 μM. This high Kd indicates that less than 5% of the binding sites on PS-expressing cells will be occupied by β2GPI at a plasma concentration of 4 μM. It should be emphasized, however, that this number is probably an overestimate, since (i) other plasma proteins, in particular prothrombin (Kd 1.4 μM), are likely to compete with β2GPI for binding to PS, and (ii) the extent of PS exposure in ionomycin-treated platelets exceeds the amount of PS exposed following other stimuli [21]. Nonetheless, binding of β2GPI to platelets was demonstrated two decades ago by Schousboe [24] and Nimpf et al. [25]. Both studies reported Kd values that are approx. 100-fold lower than the value estimated here. Schousboe [24], however, measured binding at low ionic strength, known to increase the affinity of β2GPI. It is also possible that enhanced binding was due to specific characteristics of the β2GPI preparation (possibly the presence of multimeric forms; see below) caused by subtle differences in its purification or handling. Indeed, the inhibitory effect of β2GPI on the procoagulant activity of activated platelets that we observed during the same period [5] was obtained using a preparation identical to the one used in [25]. An inhibitory effect on platelet prothrombinase activity cannot be detected with current β2GPI preparations.
As for β2GPI, reports on the binding of prothrombin to PS-exposing cells (i.e. activated blood platelets) are few. Scandura et al. [40a] have studied the binding of prothrombin to thrombin-activated platelets. Binding was saturable, reversible and Ca2+-dependent, with a total of 20500±1500 binding sites and a Kd of 0.47±0.11 μM. Although the affinity reported here is of the same order of magnitude (Kd ∼1.4 μM), the number of binding sites found by these authors was significantly lower than that presented here. This is due to the difference between ionomycin- and thrombin-activated platelets. It was shown recently that thrombin activation resulted in PS exposure in only a small (∼5%) subpopulation of the platelets [30]. As a consequence, the actual number of prothrombin binding sites per platelet as reported by Scandura et al. [40a] could have been 20-fold higher. Prothrombin has been shown to interact with the platelet integrin complex αIIbβ3 [41]. However, maximal binding was estimated at less than 10000 molecules per platelet. This would represent ∼1% of the number of sites reported here.
Surface-exposed PS plays an important role in the timely removal of apoptotic cells before they release pro-inflammatory cell contents. Several mechanisms have been proposed to be responsible for the clearance of PS-expressing cells. These include direct recognition through a PS-specific receptor [42–44] or indirectly through scavenger receptors [45–47]. Evidence is accumulating that plasma proteins may function as opsonins in this process [48]. In particular, Chonn et al. [49] injected mice with liposomes of different phospholipid composition and showed that β2GPI is a major protein associated with rapidly cleared PS/PC liposomes in vivo. This suggested that β2GPI could play a role in the clearance of liposomes and, by extension, senescent and apoptotic cells. Adsorption of β2GPI from plasma using PS/PC liposomes may seem inconsistent with its low-affinity binding to PS-exposing platelets shown here. It should be noted, however, that Chonn et al. [49] used liposomes containing 20 mol% PS, and the amount of lipid injected into the mice was more than 10 times the amount of lipid that can be presented by circulating platelets, assuming 100% activation. Under these conditions, considerable binding of β2GPI is to be expected. In vitro studies on the interaction between THP-1 cells and PS-containing phospholipid vesicles suggested that β2GPI plays a role in the removal of procoagulant PS-exposing cells from the circulation [9]. However, binding of phospholipid vesicles was only observed above 25 mol% PS; no interaction was found with vesicles containing 10 mol% PS. It should be emphasized that the total amount of PS in most, if not all, blood cell membranes does not exceed 15 mol%.
Considering its low affinity under physiological conditions, we conclude that binding of monomeric β2GPI to PS-exposing cell membranes in vivo is negligible. It is possible, though, that any in vivo function requiring high-affinity binding of β2GPI could be fulfilled by multivalent β2GPI–cell interactions. Indeed, multimeric structures of β2GPI, either artificial [50] or present in plasma [51,52], will bind even in the absence of antibodies to plasma membranes containing surface-exposed PS. The relatively high-affinity binding in the presence of antibody can be considered to mimic the multimeric form of the protein. Alternatively, multivalency may be accommodated by multiple β2GPI–cell-surface interactions [7,53] or by adaptor proteins that enhance the association of β2GPI with PS. Conceivably, binding of β2GPI to phagocytes via interaction with a non-lipid-binding domain of the protein could enhance its affinity for PS-expressing cells through clustering of accessible lipid-binding domains of β2GPI on the surface of the macrophage, enabling multiple interactions with PS-exposing membranes of apoptotic cells. In summary, we have quantitatively characterized the binding of β2GPI and prothrombin to PS-exposing platelets. The results demonstrate that, under physiological conditions, the binding of β2GPI to such platelets is of low affinity. This finding stresses the importance of multimeric complexes in the functioning of β2GPI in cellular interactions.
Acknowledgments
This work was supported, in part, by NIH grant GM-64610 (A.J.S.).
References
- 1.Wurm H., Beubler E., Polz E., Holasek A., Kostner G. Studies on the possible function of β2-glycoprotein-I: influence in the triglyceride metabolism in the rat. Metab. Clin. Exp. 1982;31:484–486. doi: 10.1016/0026-0495(82)90238-4. [DOI] [PubMed] [Google Scholar]
- 2.Schousboe I. Effect of β2-glycoprotein I on the activity of adenylate cyclase in platelet membranes. Thromb. Res. 1983;32:291–299. doi: 10.1016/0049-3848(83)90164-0. [DOI] [PubMed] [Google Scholar]
- 3.Nimpf J., Gries A., Wurm H., Kostner G. M. Influence of β2-glycoprotein I upon the content of cAMP and cGMP in human blood platelets. Thromb. Haemostasis. 1985;54:824–827. [PubMed] [Google Scholar]
- 4.Schousboe I. β2-glycoprotein I: a plasma inhibitor of the contact activation of the intrinsic blood coagulation pathway. Blood. 1985;66:1086–1091. [PubMed] [Google Scholar]
- 5.Nimpf J., Bevers E. M., Bomans P. H. H., Till U., Wurm H., Kostner G. M., Zwaal R. F. A. Prothrombinase activity of human platelets is inhibited by β2-glycoprotein I. Biochim. Biophys. Acta. 1986;884:142–149. doi: 10.1016/0304-4165(86)90237-0. [DOI] [PubMed] [Google Scholar]
- 6.Price B. E., Rauch J., Shia M. A., Walsh M. T., Lieberthal W., Gilligan H. M., O'Laughlin T., Koh J. S., Levine J. S. Anti-phospholipid autoantibodies bind to apoptotic, but not viable, thymocytes in a β2-glycoprotein I-dependent manner. J. Immunol. 1996;157:2201–2208. [PubMed] [Google Scholar]
- 7.Balasubramanian K., Chandra J., Schroit A. J. Immune clearance of phosphatidylserine-expressing cells by phagocytes – The role of β2-glycoprotein I in macrophage recognition. J. Biol. Chem. 1997;272:31113–31117. doi: 10.1074/jbc.272.49.31113. [DOI] [PubMed] [Google Scholar]
- 8.Balasubramanian K., Schroit A. J. Characterization of phosphatidylserine-dependent β2-glycoprotein I macrophage interactions. Implications for apoptotic cell clearance by phagocytes. J. Biol. Chem. 1998;273:29272–29277. doi: 10.1074/jbc.273.44.29272. [DOI] [PubMed] [Google Scholar]
- 9.Thiagarajan P., Le A., Benedict C. R. β2-glycoprotein I promotes the binding of anionic phospholipid vesicles by macrophages. Arterioscler. Thromb. Vasc. Biol. 1999;19:2807–2811. doi: 10.1161/01.atv.19.11.2807. [DOI] [PubMed] [Google Scholar]
- 10.McNeil H. P., Simpson R. J., Chesterman C. N., Krilis S. A. Anti-phospholipid antibodies are directed against a complex antigen that includes a lipid-binding inhibitor of coagulation: β2-glycoprotein I (apolipoprotein H) Proc. Natl. Acad. Sci. U.S.A. 1990;87:4120–4124. doi: 10.1073/pnas.87.11.4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galli M., Comfurius P., Maassen C., Hemker H. C., de Baets M. H., van Breda-Vriesman P. J., Barbui T., Zwaal R. F. A., Bevers E. M. Anticardiolipin antibodies (ACA) directed not to cardiolipin but to a plasma protein cofactor. Lancet. 1990;335:1544–1547. doi: 10.1016/0140-6736(90)91374-j. [DOI] [PubMed] [Google Scholar]
- 12.Matsuura E., Igarashi Y., Fujimoto M., Ichikawa K., Koike T. Anticardiolipin cofactor(s) and differential diagnosis of autoimmune disease. Lancet. 1990;336:177–178. doi: 10.1016/0140-6736(90)91697-9. [DOI] [PubMed] [Google Scholar]
- 13.Field S. L., Chesterman C. N., Hogg P. J. Dependence on prothrombin for inhibition of activated protein C activity by lupus antibodies. Thromb. Haemostasis. 2000;84:1132–1133. [PubMed] [Google Scholar]
- 14.Galli M., Ruggeri L., Barbui T. Differential effects of anti-β2-glycoprotein I and antiprothrombin antibodies on the anticoagulant activity of activated protein C. Blood. 1998;91:1999–2004. [PubMed] [Google Scholar]
- 15.Matsuda J., Gotoh M., Gohchi K., Kawasugi K., Tsukamoto M., Saitoh N. Resistance to activated protein C activity of an anti-β2-glycoprotein I antibody in the presence of β2-glycoprotein I. Br. J. Haematol. 1995;90:204–206. doi: 10.1111/j.1365-2141.1995.tb03401.x. [DOI] [PubMed] [Google Scholar]
- 16.Salemink I., Blezer R., Willems G. M., Galli M., Bevers E., Lindhout T. Antibodies to β2-glycoprotein I associated with antiphospholipid syndrome suppress the inhibitory activity of tissue factor pathway inhibitor. Thromb. Haemostasis. 2000;84:653–656. [PubMed] [Google Scholar]
- 17.Arnout J., Vermylen J. Current status and implications of autoimmune antiphospholipid antibodies in relation to thrombotic disease. J. Thromb. Haemostasis. 2003;1:931–942. doi: 10.1046/j.1538-7836.2003.00125.x. [DOI] [PubMed] [Google Scholar]
- 18.Bevers E. M., Galli M., Barbui T., Comfurius P., Zwaal R. F. A. Lupus anticoagulant IgG's (LA) are not directed to phospholipids only, but to a complex of lipid-bound human prothrombin. Thromb. Haemostasis. 1991;66:629–632. [PubMed] [Google Scholar]
- 19.Casciola-Rosen L., Rosen A., Petri M., Schlissel M. Surface blebs on apoptotic cells are sites of enhanced procoagulant activity: implications for coagulation events and antigenic spread in systemic lupus erythematosus. Proc. Natl. Acad. Sci. U.S.A. 1996;93:1624–1629. doi: 10.1073/pnas.93.4.1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levine J. S., Subang R., Koh J. S., Rauch J. Induction of anti-phospholipid autoantibodies by β2-glycoprotein I bound to apoptotic thymocytes. J. Autoimmun. 1998;11:413–424. doi: 10.1006/jaut.1998.0235. [DOI] [PubMed] [Google Scholar]
- 21.Bevers E. M., Comfurius P., Dekkers D. W., Zwaal R. F. A. Lipid translocation across the plasma membrane of mammalian cells. Biochim. Biophys. Acta. 1999;1439:317–330. doi: 10.1016/s1388-1981(99)00110-9. [DOI] [PubMed] [Google Scholar]
- 22.Willems G. M., Janssen M. P., Pelsers M. M., Comfurius P., Galli M., Zwaal R. F. A., Bevers E. M. Role of divalency in the high-affinity binding of anticardiolipin antibody-β2-glycoprotein I complexes to lipid membranes. Biochemistry. 1996;35:13833–13842. doi: 10.1021/bi960657q. [DOI] [PubMed] [Google Scholar]
- 23.Harper M. F., Hayes P. M., Lentz B. R., Roubey R. A. Characterization of β2-glycoprotein I binding to phospholipid membranes. Thromb. Haemostasis. 1998;80:610–614. [PubMed] [Google Scholar]
- 24.Schousboe I. Binding of β2-glycoprotein-I to platelets: effect of adenylate cyclase activity. Thromb. Res. 1980;19:225–237. doi: 10.1016/0049-3848(80)90421-1. [DOI] [PubMed] [Google Scholar]
- 25.Nimpf J., Wurm H., Kostner G. M. Interaction of β2-glycoprotein-I with human blood platelets: influence upon the ADP-induced aggregation. Thromb. Haemostasis. 1985;54:397–401. [PubMed] [Google Scholar]
- 26.Galli M., Bevers E. M., Comfurius P., Barbui T., Zwaal R. F. A. Effect of antiphospholipid antibodies on procoagulant activity of activated platelets and platelet-derived microvesicles. Br. J. Haematol. 1993;83:466–472. doi: 10.1111/j.1365-2141.1993.tb04672.x. [DOI] [PubMed] [Google Scholar]
- 27.Brighton T. A., Dai Y. P., Hogg P. J., Chesterman C. N. Microheterogeneity of β2-glycoprotein I: implications for binding to anionic phospholipids. Biochem. J. 1999;340:59–67. [PMC free article] [PubMed] [Google Scholar]
- 28.Long M., Thiagarajan P., Shapiro S. S. Quantitative measurement of binding of β2-glycoprotein I and IgG antiphospholipid antibodies to human platelets. Blood. 1995;86:550A. abstract. [Google Scholar]
- 29.Willems G. M., Janssen M. P., Comfurius P., Galli M., Zwaal R. F. A., Bevers E. M. Competition of annexin V and anticardiolipin antibodies for binding to phosphatidylserine containing membranes. Biochemistry. 2000;39:1982–1989. doi: 10.1021/bi9921081. [DOI] [PubMed] [Google Scholar]
- 30.London F. S., Marcinkiewicz M., Walsh P. N. A subpopulation of platelets responds to thrombin- or SFLLRN-stimulation with binding sites for factor IXa. J. Biol. Chem. 2004;279:19854–19859. doi: 10.1074/jbc.M310624200. [DOI] [PubMed] [Google Scholar]
- 31.Hendrix H., Lindhout T., Mertens K., Engels W., Hemker H. C. Activation of human prothrombin by stoichiometric levels of staphylocoagulase. J. Biol. Chem. 1983;258:3637–3644. [PubMed] [Google Scholar]
- 32.Wurm H. β2-Glycoprotein-I (apolipoprotein H) interactions with phospholipid vesicles. Int. J. Biochem. 1984;16:511–515. doi: 10.1016/0020-711x(84)90168-x. [DOI] [PubMed] [Google Scholar]
- 33.Cuypers P. A., Corsel J. W., Janssen M. P., Kop J. M., Hermens W. T., Hemker H. C. The adsorption of prothrombin to phosphatidylserine multilayers quantitated by ellipsometry. J. Biol. Chem. 1983;258:2426–2431. [PubMed] [Google Scholar]
- 34.Smeets E. F., Heemskerk J. W. M., Comfurius P., Bevers E. M., Zwaal R. F. Thapsigargin amplifies the platelet procoagulant response caused by thrombin. Thromb. Haemostasis. 1993;70:1024–1029. [PubMed] [Google Scholar]
- 35.Hunt J., Krilis S. The fifth domain of β2-glycoprotein I contains a phospholipid binding site (Cys281-Cys288) and a region recognized by anticardiolipin antibodies. J. Immunol. 1994;152:653–659. [PubMed] [Google Scholar]
- 36.Kertesz Z., Yu R. B., Steinkasserer A., Haupt H., Benham A., Sim R. B. Characterization of binding of human β2-glycoprotein 1 to cardiolipin. Biochem. J. 1995;310:315–321. doi: 10.1042/bj3100315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bouma B., de Groot P. G., van den Elsen J. M. H., Ravelli R. B. G., Schouten A., Simmelink M. J. A., Derksen R., Kroon J., Gros P. Adhesion mechanism of human β2-glycoprotein I to phospholipids based on its crystal structure. EMBO J. 1999;18:5166–5174. doi: 10.1093/emboj/18.19.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bevers E. M., Zwaal R. F. A., Willems G. M. The effect of phospholipids on the formation of immune complexes between autoantibodies and β2-glycoprotein I or prothrombin. Clin. Immunol. 2004;112:150–160. doi: 10.1016/j.clim.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 39.Merten M., Motamedy S., Ramamurthy S., Arnett F. C., Thiagarajan P. Sulfatides: targets for anti-phospholipid antibodies. Circulation. 2003;108:2082–2087. doi: 10.1161/01.CIR.0000095030.44185.6A. [DOI] [PubMed] [Google Scholar]
- 40.Kushi Y., Arita M., Ishizuka I., Kasama T., Fredman P., Handa S. Sulfatide is expressed in both erythrocytes and platelets of bovine origin. Biochim. Biophys. Acta. 1996;1304:254–262. doi: 10.1016/s0005-2760(96)00125-7. [DOI] [PubMed] [Google Scholar]
- 40a.Scandura J. M., Ahmad S. S., Walsh P. N. A binding site expressed on the surface of activated human platelets is shared by factor X and prothrombin. Biochemistry. 1996;35:8890–8902. doi: 10.1021/bi9525029. [DOI] [PubMed] [Google Scholar]
- 41.Byzova T. V., Plow E. F. Networking in the hemostatic system. Integrin αIIbβ3 binds prothrombin and influences its activation. J. Biol. Chem. 1997;272:27183–27188. doi: 10.1074/jbc.272.43.27183. [DOI] [PubMed] [Google Scholar]
- 42.Fadok V. A., Bratton D. L., Rose D. M., Pearson A., Ezekewitz R. A., Henson P. M. A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature (London) 2000;405:85–90. doi: 10.1038/35011084. [DOI] [PubMed] [Google Scholar]
- 43.Savill J., Fadok V. Corpse clearance defines the meaning of cell death. Nature (London) 2000;407:784–788. doi: 10.1038/35037722. [DOI] [PubMed] [Google Scholar]
- 44.Ramprasad M. P., Fischer W., Witztum J. L., Sambrano G. R., Quehenberger O., Steinberg D. The 94- to 97-kDa mouse macrophage membrane protein that recognizes oxidized low density lipoprotein and phosphatidylserine-rich liposomes is identical to macrosialin, the mouse homologue of human CD68. Proc. Natl. Acad. Sci. U.S.A. 1995;92:9580–9584. doi: 10.1073/pnas.92.21.9580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rigotti A., Acton S. L., Krieger M. The class B scavenger receptors SR-BI and CD36 are receptors for anionic phospholipids. J. Biol. Chem. 1995;270:16221–16224. doi: 10.1074/jbc.270.27.16221. [DOI] [PubMed] [Google Scholar]
- 46.Ren Y., Silverstein R. L., Allen J., Savill J. CD36 gene transfer confers capacity for phagocytosis of cells undergoing apoptosis. J. Exp. Med. 1995;181:1857–1862. doi: 10.1084/jem.181.5.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fukasawa M., Adachi H., Hirota K., Tsujimoto M., Arai H., Inoue K. SRB1, a class B scavenger receptor, recognizes both negatively charged liposomes and apoptotic cells. Exp. Cell Res. 1996;222:246–250. doi: 10.1006/excr.1996.0030. [DOI] [PubMed] [Google Scholar]
- 48.Balasubramanian K., Schroit A. J. Aminophospholipid asymmetry: A matter of life and death. Annu. Rev. Physiol. 2003;65:701–734. doi: 10.1146/annurev.physiol.65.092101.142459. [DOI] [PubMed] [Google Scholar]
- 49.Chonn A., Semple S. C., Cullis P. R. β2-glycoprotein I is a major protein associated with very rapidly cleared liposomes in vivo, suggesting a significant role in the immune clearance of “non-self” particles. J. Biol. Chem. 1995;270:25845–25849. doi: 10.1074/jbc.270.43.25845. [DOI] [PubMed] [Google Scholar]
- 50.Lutters B. C., Meijers J. C., Derksen R. H., Arnout J., de Groot P. G. Dimers of β2-glycoprotein I mimic the in vitro effects of β2-glycoprotein I-anti-beta 2-glycoprotein I antibody complexes. J. Biol. Chem. 2001;276:3060–3067. doi: 10.1074/jbc.M008224200. [DOI] [PubMed] [Google Scholar]
- 51.Galazka M., Keil L. B., Kohles J. D., Li J., Kelty S. P., Petersheim M., DeBari V. A. A stable, multi-subunit complex of β2-glycoprotein I. Thromb. Res. 1998;90:131–137. doi: 10.1016/s0049-3848(98)00047-4. [DOI] [PubMed] [Google Scholar]
- 52.Gushiken F. C., Le A., Arnett F. C., Thiagarajan P. Polymorphisms β2-glycoprotein I: phospholipid binding and multimeric structure. Thromb. Res. 2003;108:175–180. doi: 10.1016/s0049-3848(02)00392-4. [DOI] [PubMed] [Google Scholar]
- 53.Ma K., Simantov R., Zhang J. C., Silverstein R., Hajjar K. A., McCrae K. R. High affinity binding of β2-glycoprotein I to human endothelial cells is mediated by annexin II. J. Biol. Chem. 2000;275:15541–15548. doi: 10.1074/jbc.275.20.15541. [DOI] [PubMed] [Google Scholar]