

Abstract
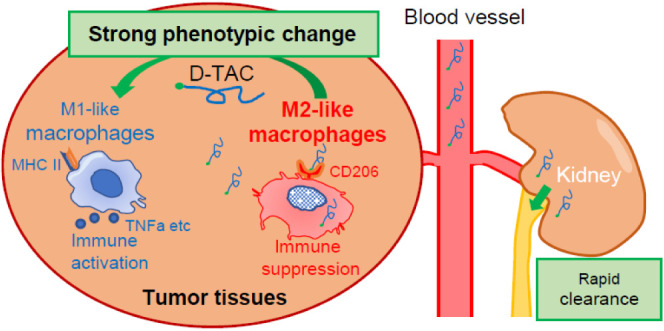
Toll-like receptors (TLRs) are a class of pattern recognition receptors (PRRs) crucial for the detection of infections and activation of downstream signaling pathways that lead to the production of pro-inflammatory cytokines and interferons. The TLR pathway is an attractive actively studied target pathway. Because of their strong immunostimulatory activity, TLRs are thought to be a “double-edged sword” for systemic treatment, even in the cancer field. To solve this, we have developed dextran-based TAM targeting activating conjugate (D-TAC) technology, which successfully uses tumor-associated macrophages (TAMs) to deliver the TLR7 agonist DSP-0509. We used low molecular weight dextran to target CD206 high M2-type macrophages, activate them, and induce a change in phenotype to antitumor M1-type macrophages with rapid clearance from the body and astonishing antitumor activity. We also demonstrated that the antitumor effect of our best drug candidate 5DEX-0509R is dependent on the abundance of TAMs, which is consistent with their mechanism of action. We believe that 5DEX-0509R generated by D-TAC technology can be a clinically applicable immunotherapy targeting the TLR signaling pathway.
Keywords: cancer immunology; tumor associated macrophages; nanomedicine, TLR7. tumor microenvironment
Introduction
Toll-like receptors (TLRs) are a class of pattern recognition receptors (PRRs) capable of recognizing pathogen-associated molecular patterns (PAMPs)1 and damage-associated molecular patterns (DAMPs)2 to initiate immune responses. Because TLRs are crucial for the detection of infections and activating downstream signaling pathways that lead to the production of pro-inflammatory cytokines and interferons, the TLR pathway is an attractive, actively studied target.3,4 Although several TLR agonists, such as bacillus Calmette–Guérin, monophosphoryl lipid A, and imiquimod, have been approved for cancer therapy, they are still being actively studied in the cancer field.5
TLR7 is predominantly expressed on the endosomes of plasmacytoid dendritic cells (pDCs), B cells, and monocytes.6 It recognizes viral single-stranded RNAs and activates signaling pathways that lead to innate immune responses.7 TLR7 signal activation triggers antitumor immunity by stimulating secretion of inflammatory cytokines such as IL-6, TNFα, and IL-12 in a NF-kB-dependent manner, and interferon regulatory factor 7 (IRF7) signaling leads to the secretion of type 1 interferons.8 So far, imiquimod is the only approved TLR7 agonist, and since then, several TLR7 agonists have been developed and studied in preclinical and clinical studies. Most of these compounds have been developed as intratumoral injectables, conjugated with tumor targeting antibodies or utilizing nanomolecules to reduce their systemic toxicity.9 Based on these facts, we have developed the small molecule TLR7 agonist, DSP-0509, which has physicochemical features that enable it to be administered systemically and endow it with a short half-life.10 On the other hand, shorter half-life may result in weaker antitumor immune responses.11
Tumor-associated macrophages (TAMs) are well-known to be major components and play a pivotal role in the tumor microenvironment. Classically, macrophages are divided into 2 types: M1 macrophages, which show a tumor-killing capacity and produce inflammatory cytokines to boost tumor immunity, and M2 macrophages, which show anti-inflammatory and immune suppressive phenotypes.12 Especially, the presence of M2-like macrophages, which suppress tumor immunity, is closely related to a poor prognosis. Indeed, several approaches targeting TAMs have been studied clinically and preclinically.13,14 Though TAMs are a very heterogeneous population, several major markers identify tumor-promoting macrophages such as SPP1, MARCO, CD163, CD204, and CD206.14,15 TLR7 is known to be expressed in TAMs,16 and a recent study demonstrated that TLR7/8 agonists strongly reverse the TAM M2 phenotype to M1 and show strong antitumor activity.17 Interestingly, some sugar particles such as cyclodextrin and dextran tend to accumulate and become incorporated by TAMs relatively specifically.17,18
Based on this evidence, we sought to deliver the TLR7 agonist DSP-0509 specifically to TAMs by using dextran, a biocompatible and safe molecule with stable formulation, to enhance antitumor activity without severe toxic side effects.
Results
Size-Dependent Distribution and Cell Uptake of Dextran Conjugate in Mice
In the previous study, DSP-0509 was designed as a systemically injectable TLR7 agonist which achieved safety by having a short half-life in the body.10 We first tested small molecular weight dextrans below 20 kDa, which are excreted from the kidney and have a short half-life comparable with DSP-050919−21. Both 6 kDa and 20 kDa AF750-labeled dextrans at the amino linker were successfully detected in tumor-bearing mouse organs (Figure 1A,B). There was not a very big difference in accumulation between the 6 and 20 kDa dextrans. In both cases, high kidney accumulation and reasonable tumor accumulation were observed, but with 20 kDa dextran, there was a tendency toward greater liver accumulation, too (Figure 1B). We next analyzed macrophage uptake by flow cytometry (Figure 1C). The uptake of both dextrans by macrophages was extensive and CD206-dependent, which is consistent with other reports that dextran is a CD206 ligand.22,23 Interestingly, 6 kDa dextran was not incorporated by other tumor microenvironment cells or splenic cells including macrophages, but 20 kDa dextran was slightly incorporated by splenic macrophages (Figure 1D,E).
Figure 1.

Tissue and cellular distributions of 6 kDa and 20 kDa aminodextran conjugates. (A, B) Organ distributions of 6 kDa (A) and 20 kDa (B) dextrans labeled with AF750 in EMT6 tumor-bearing mice (n = 3). Mice were inoculated with EMT6 tumors and then intravenously injected with AF750-labeled dextrans (1 mpk). After 24 h, the blood, heart, thymus, lymph node, spleen, lung, stomach, kidney, and colon were dissected, and their fluorescence intensities were measured. Tu: tumor, St: stomach, Col: colon, Li: liver, and Ki: kidney. (C–E) Cellular uptakes of 6 and 20 kDa dextran conjugates. Mice were inoculated with EMT6 tumors and then injected intravenously with AF750-labeled dextrans (1 mpk) (n = 2). After 24 h, the tumors (C, D) and spleens (E) were dissected, dissociated into single-cell suspensions, and then analyzed with flow cytometry. Macrophages were categorized as CD11b+ F4/80+ cells; MDSC as CD11b+ Gr1high cells; other myeloid cells as CD11b+ F4/80– cells, and T, B, NK, etc., cells as CD11b– F4/80– cells.
Size-Dependent Antitumor Effect of Dextran Conjugates in the Mouse Breast Tumor Model
We next investigated the molecular size dependence of the antitumor effect of aminodextran conjugates (Figure 2A) from 1 to 20 kDa (Figure 2B–D). Compared with the small molecule DSP-0509, all dextran conjugates showed better antitumor activity with 1/5 agonist equivalent dose. Concerning weight loss, there was not much difference between treatments, with a slightly higher loss at the first dose (Figure 2C, day 10) of dextran conjugates. Larger size conjugates tended to have stronger antitumor activity, but the activity with over 6 kDa conjugates was strong enough with a 2/5 complete response rate and no complete response with small molecule DSP-0509 and smaller DSP-0509-dextran conjugates (Figure 2D). During the experiment, we realized that several mice treated with 20 kDa aminodextran conjugates (20DEX-0509A), but none treated with 6DEX-0509A, exhibited anaphylactic shock at the second dose (Figure 2E,F). Several researchers have reported the occurrence of macrophage-dependent anaphylactic shock, which is caused by IgG and platelet-activating factor (PAF),24 and TLR signaling causes IgG-dependent anaphylactic shock.25 Compatible with these reports, the present study found that the shock caused by our dextran conjugates was inhibited by Fc receptor blocking and PAF inhibition (Figure S1). To avoid this, we optimized the method of DSP-0509 conjugation to dextran. We speculated that the free aminolinker might increase the immunogenicity because charges of the haptens are important for producing the antibody.26−28 We made two different 20 kDa conjugates, 20DEX-0509A and a reduced end type (20DEX-0509R), and found that only 20DEX-0509A caused the shock in mice (Figure 2F,G). We analyzed the antitumor activity and plasma cytokines of mice injected with 6DEX-0509RA, compared with DSP-0509 at a maximum effective dose at 5 mg per kilogram (mpk),10 and confirmed that 6DEX-0509A compared with DSP-0509 showed superior antitumor activity and induced the expression of similar levels of blood cytokines (Figure 2H,I). So, we named this conjugation technology D-TAC (dextran-based TAM-targeting activator conjugates) and applied it to the synthesis of 5 kDa dextran conjugated with DSP-0509 at the reduced end (5DEX-0509R), using the highest grade of dextran available to minimize the anaphylactic shock risk for further study.
Figure 2.

Antitumor effect and safety profiles of dextran conjugates. (A) Structure of DEX-0509A. (B–D) Antitumor effect of different sizes of dextran conjugates. EMT6-bearing mice (n = 5 per group) were intravenously injected once a week for 2 weeks with DSP-0509 (1 mpk) and different sizes of aminodextran-DSP0509 conjugates (0.2 mpk as the equivalent dose of DSP-0509). (B) Tumor sizes of each of the groups. Data are shown as mean ± SD. Statistical differences were evaluated by the parametric Dunnett test vs vehicle (PBS treatment) (*p < 0.01, **p < 0.001). (C) Weight changes of treated mice. (D) Individual tumor size in mice treated with vehicle, DSP-0509, 6 kDa aminodextran conjugates (6DEX-0509A), and 20 kDa aminodextran conjugates (20DEX-0509A). (E–G) Body temperature changes in tumor-bearing mice treated with 6DEX-0509A (E), 20DEX-0509A (F), and 20DEX-0509R (G). Mice were treated with each dextran (0.2 mpk as equivalent dose of DSP0509) at day 0 and day 7. Statistical differences were evaluated by the parametric Dunnett test. *p < 0.05 vs 0 min. (H, I) Comparison of the anti-EMT6 tumor effect of 6DEX-0509A and cytokine profiles in injected mice. EMT6-bearing mice (n = 6 per group) were intravenously injected once a week for 3 weeks with DSP-0509 (5 mpk) and 6DEX-0509A (0.2 mpk as equivalent dose of DSP-0509). (H) Tumor sizes of each of the groups. Data are shown as mean ± SD. Statistical differences were evaluated by the parametric Dunnett test vs vehicle (PBS treatment) (*p < 0.05). (I) Blood cytokine profiles. EMT6-bearing mice were intravenously injected once with DSP-0509 (5 mpk) and 6DEX-0509A (0.2 mpk as equivalent dose of DSP-0509), blood samples were collected 2 h after injection (n = 3), and plasma cytokines were measured. Data are shown as mean ± SD. Statistical differences were evaluated by the t-test.
In Vivo Distribution and Cellular Uptake of 5 kDa D-TAC and Biological Properties of 5DEX-0509R
We first analyzed the in vivo distribution in the same manner described in Figure 1A,B. Surprisingly, 5 kDa D-TAC showed superior in vivo distributions compared with the 6 kDa amino conjugate, and its accumulation was the highest in tumors compared with the organs, including the kidney (Figure 3A). Cellular uptakes were almost the same with 6 kDa aminodextran, with slight incorporation of 6 kDa aminodextran into the CD206 expressing myeloid-derived suppressor cell (MDSC) population (Figures 3B and S2A). We also confirmed CD206-dependent cellular uptakes of 5 kDa D-TAC using the blocking antibody with human monocyte-derived M2 macrophages (Figure S2B), the M0 and M2-differentiated macrophage cell lines (Figure S2C), and human CD206-transduced cells (Figure S2D). Our 5DEX-0509R (Figure 3C) showed dose-dependent antitumor activity, reaching saturation at 0.5 mpk, and was tolerated at doses of at least 3 mpk, without severe weight loss in the mouse EMT6 tumor model (Figure 3D). In the E0771 model, which is the same triple negative breast cancer mouse model with EMT6 but has low CD206 expressing TAMs, 5DEX-0509R did not show good antitumor efficacy (Figure S3A–C). This suggests that CD206 expression on TAMs is the essential for 5DEX-0509R activity. The combination of 5DEX0509 with anti-PD1 successfully overcame this. 5DEX-0509R strongly prevented tumor metastasis to the lung at 0.1 mpk in the EMT6 model (Figure S3D). In addition, 5DEX-0509R showed the strong antitumor activity with almost all tumors eliminated completely but no elimination with anti-PD1 antibody or DSP-0509 in the Colon26 model (Figure 3E). When the mice showed complete tumor elimination for more than 40 days, they were reinjected with the same tumor cells, but there was no tumor regrowth (Figure 3F), suggesting the establishment of the long-lasting antitumor activity. We next investigated whether the same antitumor activity is achieved by s.c. bolus, i.v. bolus, and i.v. infusion administration of 5DEX-0509R. Activity was the same regardless of the administration route (Figure 3G) but found that the infusion route lowered plasma inflammatory cytokines (Figure 3H), suggesting that administration by infusion has the benefit of lowering systemic cytokines. We further tested 0.5 mpk in the Colon26 model and obtained the same strong antitumor activity (Figure S3E). On the other hand, DSP0509 tended to show a weaker antitumor effect by the infusion route and not be completely eliminated. These results indicate that 5DEX-0509R is easy to handle in the clinic.
Figure 3.

In vivo profiles and antitumor effect of 5 kDa dextran conjugated with DSP-0509 at the reduced end. (A, B) Tissue and cellular distributions of 5 kDa dextran conjugates. Organ distribution of 5 kDa dextran labeled with AF750 in EMT6 tumor-bearing mice (n = 3). Mice were inoculated with EMT6 tumors and intravenously injected with the dextran-small molecule AF750 complex at 1 mg/kg of AF750 equiv. After 24 h, the blood, heart, thymus, lymph node (LN), spleen, lung, stomach, kidney, and colon were dissected, and their fluorescence intensities were measured. Data are shown as mean ± SD (B) Cellular uptake of 5 kDa dextran conjugates. Mice were inoculated with EMT6 tumors and then intravenously injected with the dextran-AF750 complex 1 mpk of AF750 (n = 3). After 24 h, the tumors and spleens were dissected, dissociated into single -cell suspensions, and then analyzed with flow cytometry. Macrophages were categorized as CD11b+ F4/80+ cells. (C) Chemical structure of DEX-0509R. (D) Dose-dependent antitumor effect of 5DEX-0509R and weight changes in mice injected with 5DEX-0509R. EMT6-bearing mice (n = 6 per group) were intravenously injected once a week for 2 weeks with different doses of 5DEX-0509R (indicated as equivalent dose of DSP-0509). Data are shown as mean ± SD. Statistical differences were evaluated by the parametric Dunnett test vs vehicle (PBS treatment) (*p < 0.05, **p < 0.01). (E) Comparison of the antitumor effect of 5DEX-0509R with that of anti-PD1 and DSP-0509. Colon26-bearing mice (n = 6 per group) were injected with vehicle (PBS, i.v., once a week), anti-PD1 (10 mg/kg, i.p., twice a week), DSP-0509 (5 mg/kg, i.v., once a week), and 5DEX-0509R (0.5 mpk as equivalent dose of DSP-0509, once a week) for 3 weeks. Data are shown as mean ± SD. Statistical differences were evaluated by the parametric Dunnett test vs vehicle (PBS treatment) (*p < 0.01). (F) Reinjection of EMT6 tumor cells into cured mice. Mice showing complete remission of EMT6 tumors by 5DEX-0509R treatment, and naïve mice were injected with EMT6 tumor cells (0.5 × 106 cells/mouse) and monitored. (G, H) Antitumor effect and cytokine profiles of mice injected with 5DEX-0509R via different routes. EMT6-bearing mice (n = 6 per group) were injected once a week for 2 weeks with 5DEX-0509R (0.2 mg/kg as equivalent dose of DSP-0509) by the i.v. bolus, s.c. bolus, and i.v. infusion routes. Data are shown as mean ± SD. Statistical differences were evaluated by the parametric Dunnett test vs vehicle (PBS treatment) (*p < 0.05, **p < 0.01). (H) Plasma cytokine levels of mice injected with 5DEX-0509R via the i.v. bolus and i.v. infusion routes at 2 h after injection. Data are shown as mean ± SD. Statistical differences were evaluated by t-tests. (I, J) Plasma pharmacokinetics and cytokine profiles of 5DEX-0509R. Mouse plasma samples (n = 3) were collected at 10 min, 30 min, 1 h (60 min), 2 h (120 min), 4 h (240 min), 24 h (1440 min), 48 h (2880 min), and 12 h (7200 min) and subjected to (I) pharmacokinetic analysis and (J) cytokine analysis. (K) Antitumor Effect of 5DEX-0509R and DSP-0509. Colon26-bearing nu/nu mice (n = 6 per group) were treated at day 7 with vehicle (PBS, i.v.), DSP-0509 (5 mg/kg, i.v.), or 5DEX-0509R (0.5 mpk as equivalent dose of DSP-0509, i.v.). Data are shown as mean ± SD. Statistical differences were evaluated by the parametric Dunnett test vs vehicle (PBS treatment) (**p < 0.01). (L) Antitumor effect of 5DEX-0509R in the B2M KO CT26 tumor model. B2M knockout of the CT26 cell line was confirmed by Western blotting (left panel). The cells were inoculated, and the tumor-bearing mice (n = 6 per group) were treated with vehicle (PBS, i.v., once a week), DSP-0509 (5 mpk, i.v., once a week), or 5DEX-0509R (1 mpk as equivalent dose of DSP-0509, i.v., once a week) for 3 weeks. Data are shown as mean ± SD.
PK/PD Profiles of 5DEX-0509R
We next investigated the plasma PK profiles in mice. We developed a bioanalytical method to measure the total amount of DSP-0509 in mouse plasma by the hydrolytic degradation of the amide bond of 5DEX-0509R. (Figure 3I). 5DEX-0509R showed rapid clearance from the body with a t1/2 of 0.461 h. Without the alkaline treatment of plasma, DSP-0509 was not detected, suggesting that conjugation is highly stable within the mouse body. We also analyzed time course plasma cytokine profiles to see whether 5DEX-0509R caused a cytokine storm. 5DEX-0509R at 1 mpk caused rapid elevation of cytokines to a maximum at 1–2 h, but levels disappeared rapidly by 24 h (1440 min), suggesting that the elevation of inflammatory plasma cytokines by 5DEX-0509R was not long-lasting (Figure 3J). Interestingly, IFNγ was re-elevated at day 5, suggesting 5DEX-0509R also induces a later-phase antitumor immunity, probably T cell immunity. We also tested different tumor-type syngeneic tumor models and found that 5DEX-0509R exerts significant tumor growth inhibition in most of them (Table 1 and Figure S4).
Table 1. Summary of In Vivo Antitumor Efficacy by 5DEX-0509R Treatment.
| cancer types | mouse strain | cell line | CR (%) | TGI (%) |
|---|---|---|---|---|
| colon | Balb | Colon26 | 100 | 100 |
| colon | Balb | CT26 | 83.3 | 96.3 |
| osteosarcoma | Balb | K7M2 | 60 | 94.5 |
| bladder | B6C3HF1 | MBT2 | 37.5 | 93.9 |
| breast | Balb | EMT6 | 60 | 93 |
| colon | B6 | MC38 | 50 | 89 |
| B cell | Balb | A20 | 60 | 85.3 |
| pancreatic | B6 | Pan02 | 0 | 81.1 |
| liver | B6 | Hepa1–6 | 40 | 70.4 |
| kidney | Balb | Renca | 0 | 67.7 |
| HNSCC | C3H | SCCVII | 0 | 67.2 |
| prostate | B6 | RM-1 | 0 | 63.9 |
| lung | B6 | LLC | 0 | 63.3 |
| ovary | B6C3HF1 | HM-1 | 0 | 55.8 |
| AML | SCID/Beige | MV4;11 | 0 | 51.7 |
| T cell lymphoma | Balb | EG7 | 0 | 46.8 |
| melanoma | B6 | B16_BL6 | 0 | 40.9 |
T Cell-Independent Immunity of 5DEX-0509R
Several reports show that macrophages directly kill tumor cells without T cells.14,15,29 To investigate T cell-independent antitumor effect of 5DEX-0509R, we used the in vivo Colon26 tumor-bearing mouse model established by injecting athymic nude mice with Colon26 tumor cells (Figure 3K). Although we could not see complete elimination, 5DEX-0509R clearly showed an antitumor effect, suggesting that 5DEX-0509R modulates tumor suppressive activity in a T cell-independent manner. We next used B2M knockout CT26 tumor cells to see whether 5DEX-0509R exerts an antitumor effect under conditions of immune checkpoint resistance and CD8 T cell dysfunction.30 As shown in Figure 3L, 5DEX-0509R, but not anti-PD-1 antibody, still elicited strong antitumor immunity.
Comparison of 5DEX-0509R with the TLR7 Agonist-Loaded Nanomicelle Formulation, MBS-8
There are several TLR7 agonist-based nanomedicines in the clinical stage of testing. One of them is MBS-8, which is a formulation of the TLR7 agonist loaded into relatively small nanomicelles (∼10 nm diameter). The formulation has a longer blood PK half-life and strong antitumor activity similar to that of 5DEX-0509R (WO 2021/053163 Al). We made this MBS-8 nanomicellar formulation according to the patent information and compared it with our 5DEX-0509R. 5DEX-0509R showed similar antitumor activity at 0.1 mpk with MBS-8 at 100 nmol/mouse (Figure 4A). We next investigated the blood cytokine profiles to assess systemic inflammation. At this dose, 5DEX-0509R showed a lower peak amount and shorter period of elevation of inflammatory cytokines compared with MBS-8 (Figure 4B). That may reflect a shorter blood pharmacokinetic half-life of 5DEX-0509R than MBS-8 (WO 2021/053163AI).
Figure 4.

Comparison of 5DEX-0509R with the clinical stage agent MBS-8 and its effect on human PBMCs and monocyte-derived M2 macrophages. (A) Antitumor effect and blood cytokine profiles of MBS-8 and 5DEX-0509R. LLC-bearing mice (n = 6 per group) were intravenously injected once a week with 5DEX-0509R (0.1 mpk) and MBS-8 (100 nmol/mouse). Data show the average tumor size in each of the groups. Data are shown as mean ± SD. Statistical differences were evaluated by the parametric Dunnett test vs vehicle (PBS treatment, *p < 0.05). (B) Cytokine profiles of 5DEX-0509R (0.1 mpk as equivalent dose of DSP-0509) and MBS-8 (100 nmol/mouse). Data are shown as mean ± SD. Statistical differences were evaluated by t-tests. (C, D) CD206 expression and TNFα production from PBMCs (C) and monocyte-derived M2 macrophages (D). Each of the cells (PBMCs and M2 macrophages) was stimulated with 1 μM DSP0509 and 5DEX-0509R, and their supernatants were subjected to TNFα measurement.
5DEX-0509R Induced TNFα Release from CD206-Expressing Human Monocyte-Derived M2 Macrophages
Our prior study basically focused on the mouse immune system. Thus, we sought to examine whether 5DEX-0509R, compared with DSP-0509, preferentially activates and induces phenotypic changes in human M2-type macrophages. We first used peripheral blood mononuclear cells (PBMCs) to compare agonistic ability between 5DEX-0509R and DSP0509 and found that DSP-0509 induced more TNFα production from PBMCs than 5DEX-0509R (Figure 4C). That is consistent with the CD206 expression profiles in PBMCs where few or no cells express CD206 molecules on their surface. On the other hand, in vitro differentiated M2 macrophages from monocytes, which express that strong CD206 only produced TNFα when stimulated by 5DEX-0509R (Figure 4D), suggesting that because of its targeting ability, 5DEX-0509R is also able to change the M2-like macrophage phenotype to the M1-like one.
Gene Expression Profiling of Immune Cells and Their Changes Induced by 5DEX-0509R Treatment
Next, in order to examine the overall effects of the compound on immune status in the tumor microenvironment, we first measured the bulk gene expression profiles of CD45+ cells from tumors by RNA-seq. 5DEX-0509R was found to greatly affect CD45+ cells since expression levels of over 1000 genes were significantly changed at early (4 h) and later time points (days 5 and 7) (Figure S5A). The early changes included upregulation of interferon-responsive genes, which is a well-established effect of TLR7 agonism. The latter changes included elevation of marker genes for various immune cell types (Figure S5B,C). Intratumor abundance of immune cell types estimated from gene expression profiles suggested that 5DEX-0509R increased frequencies of macrophages and granulocytes in the tumor from 4 h after administration and also clearly increased CD8+ T and B cells at a later time point (day 7) (Figure 5A,B). Time course changes in expression of individual genes also suggested an increase in the number of macrophages, CD8+ T cells, and B cells (Figure 5C–E). Consistent with this, immunohistochemistry and flow cytometry analysis suggest the increase of B cells and CD8 T cells and the decrease of Foxp3+ Tregs (Figure S6). Although we detected markedly increased expression of marker genes for M1-type macrophages (Cd86, Stat1, and class II MHC [H2-Aa]) (Figure 5C), expected decreases of genes for M2-type macrophages (Mrc1/CD206 and Cd163) were not statistically significant, which were then re-examined at single-cell resolution.
Figure 5.

Bulk RNA-seq analysis of tumor tissues. (A) Potentially increased and decreased abundance of cell types at indicated time points after 5DEX-0509R administration (n = 3). Estimated intratumoral abundance (scores) for each cell type in compound-treated tumors was compared to scores in vehicle-treated tumors. Significantly increased or decreased abundance scores for cell types (q < 0.25, two-sided Welch t-test) are shown as red or blue points. (B) Abundance scores for representative, significantly changed cell types. (C–E) Expression levels of representative marker genes for macrophages (C), T cells (D), and B cells (E). Asterisk denotes significant (q < 0.25) changes compared to the vehicle by the two-sided Welch t-test.
Single-Cell Gene Expression Profiling
To examine detailed compound-related changes in the composition and status of tumor CD45+ cells, we carried out single-cell RNA-seq. Single cells were roughly classified into major cell types (Figures 6A and S7A–D). Monocytes, macrophages, and cDCs were classified into 22 clusters (Figure 6B). Among the clusters, the C17 cluster showed prominently high expression of Mrc1/CD206 (Figure 6B,C). When compared among treatment arms, the CD206-high C17 cluster was found to have almost disappeared at 2 and 7 days after compound treatment, and new groups of cells (C1 and C8) had emerged on day 7 (Figures 6D,E and S7E). The newly emerged cells showed high expression of genes known to be upregulated or downregulated in M1-type macrophages (Figures 6F,H, S7B and D), and the disappeared C17 cluster had high expression of genes for markers of M2-type macrophages (Figures 6F, S7B and D).
Figure 6.

Single-cell RNA-seq analysis of tumor infiltrating lymphocytes. (A, B) A uniform manifold approximation and projection (UMAP) representation of CD45+ cells (A) and monocyte, macrophage, and classical dendritic cell (cDC) subsets (B). All treatment groups were merged. Distinct cell clusters are shown in colors (B). (C) Gene expression levels of Mrc1 (CD206) in monocyte, macrophage, and cDC populations. (D) Separate UMAP representation for monocytes and macrophages at day 7 after vehicle administration or 0.2 or 0.5 mg/kg 5DEX-0509R administration. Clusters are colored as in (B). Prominently increased or decreased clusters are highlighted with red letters. Intermediate cells between these clusters are shown by arrowheads. (E) Cellular frequency changes of clusters in the 5DEX-0509R-administered group compared with the vehicle-administered group. Asterisks denote significant difference (q < 0.05) in the cluster frequency of 5DEX-0509R versus vehicle, compared by the Fischer exact test. (F) Expression levels of representative marker genes for each cluster. Sizes of dots indicate percent cells expressing the gene. (G) Correlation of Mrc1 (CD206) expression among clusters to cluster frequency changes associated with administration (at day 7). Pearson correlation coefficients (r) are shown. (H, I) Over- and under-expressed genes in cluster 1 and 8 (H) or cluster 9 (I).
Expression levels of CD206 and compound-induced frequency changes were inversely correlated among clusters (Figure 6G). Furthermore, cells having intermediate phenotypes between CD206-high cells (C17) and M1-like cells (C1 and C8) were observed (Figure 6D, red arrowheads). These data may indicate that CD206-targeted 5DEX-0509R causes CD206-high macrophages to gradually shift phenotypes from the M2-like phenotype into the M1-like phenotype. One of the increased clusters, C9, showed a different profile from those of M1-like C1 and C8 and had higher expression of B cell-related genes Cd72 and Cd79b (Figure 6F,I). Since this cluster did not show upregulation of other genes characteristic of B cells (such as CD19 or CD20) or genes suggestive of macrophages functional subtypes, the significance of this treatment-specific cluster in mediation of the antitumor effect remains unknown.
Subclustering of CD8+ T cells yielded several cell clusters (Figure S8A,B) showing high expression of näve and memory genes (Tcf7 and Ccr7) or effector genes (Gzmb and Prf1) (Figure S8C). Cells having upregulated expression of effector genes were markedly increased 7 days after 5DEX-0509R administration (Figure S8D). Comparison of gene expression in the 5DEX-0509R arm to that in the vehicle arm also detected significant upregulation of effector genes such as interferon gamma (Ifng), Gzmb, and Prf1 (Figure S8E).
Efficacy-Related Gene Signature
Large variation in efficacy between different syngeneic models provides an opportunity to identify molecular biomarkers predictive of efficacy. To explore these, we utilized RNA-seq profiles of tumors obtained from each model. As in earlier RNA-seq analysis, we estimated the abundance of immune cell types from the profiles and examined the relation between the abundance of each of the cell types and the degree of efficacy calculated as tumor growth inhibition (TGI). This approach found significant a correlation between the estimated abundance of macrophages and TGI (r = 0.8, q = 0.0423) and between the abundance of CD8+ T cells and TGI (r = 0.59, q = 0.249) (Figure 7A,B). This good correlation with macrophages supports our macrophage-targeting strategy and motivated the further selection of biomarker genes from macrophage-related genes. Gene set enrichment analysis using cell type-specific gene sets from various cell types throughout the body (MSigDB gene set collection C8) confirmed that macrophage-related gene sets from various tissues were most significantly associated with TGI (Figure 7C). The 27 genes included in 8 of the top 10 associated gene sets (8/10) were selected for predictive signature construction to reduce tissue bias. The 27 genes were all well-correlated with TGI (Figure 7D). By summarizing the expression levels of the 27 genes and comparing them among the Cancer Genome Atlas (TCGA) cancer types, we estimated potentially efficacious cancers with a high expression of macrophage-related and TGI-correlated genes (Figure 7E).
Figure 7.

Predictive biomarkers. (A) Correlation of estimated abundance of each cell type to efficacy of 5DEX-0509R. Cell types positively or negatively correlated to tumor growth inhibition (TGI) (q < 0.25) are shown as red or blue points. Pearson correlation coefficients (r) are shown on the horizontal axis. (B) Relation between the abundance of macrophages or CD8+ T cells and TGI. (C) Cell type-specific gene sets associated with efficacy. Gene sets showing significant association between their expression and efficacy (q < 0.25, Kolmogorov–Smirnov test) are shown as colored points. The top 10 strongly associated gene sets in terms of the nominal enrichment score (NES) are shown in red. (D) Correlation between the expression of individual genes and efficacy (TGI). Signature genes are shown in red. (E) Summarized expression levels (geneset variation analysis scores) of signature genes for each patient’s tumor from the Cancer Genome Atlas database. Cancer types are shown on the horizontal axis. The boxplot extends from the 25th to 75th percentile, with whiskers extending to the minimum and maximum values within a 1.5 × interquartile range.
Discussion
TLR agonists are known to be very strong innate immune activators. That makes them attractive targets for cancer immunotherapy.4 However, only 2 drugs have been approved so far because of their strong systemic inflammatory effects. We sought TLR7 agonists as good targets for cancer immunotherapy because of their druggability, induction of high levels of TLR7 expression on dendritic cells and macrophages in tumor microenvironments, and induction of type I IFN.31 Dextran was obtained by Louis Pasteur in 1861 and has been used for several medicinal purposes such as plasma expanders, blood viscosity reducers, and erythrocyte aggregators.32 Our fluorescent dextran data showed that the uptake of small dextran conjugates is more specific to TAMs. Although we and others showed that dextran is a CD206 ligand, its binding might not be very specific since CD206 is known to bind collagen and mannose, and specificity of binding to larger polymers is higher.33 Interestingly, other reports demonstrated that mannose- or galactose-conjugated dextran further accumulates in liver tissues.34 This may suggest that low molecular weight dextran is good enough to bind high CD206-expressing TAMs. In addition, small-size dextrans also have the advantage of penetrating tumor tissues.35,36 Considering these reports, our dextran used for D-TAC is a suitable size for tumor tissue penetration and targeting TAMs resident deep within tumor tissues.
Dextran sometimes causes anaphylactic shock in animals and humans, and larger molecular size dextrans tend to be more immunogenic.37 Since anaphylactic shock and related infusion reactions are huge matters for clinical application of nanomedicines,38,39 it is important to lower risks of this toxicity, especially of targeted immune activators like 5DEX-0509R.
5DEX-0509 is rapidly cleared from the body, and plasma cytokines disappear rapidly from the plasma, indicating 5DEX-0509R has similar or better safety than DSP-0509 and MBS-8, both of which are now cleared investigational new drugs that have been tested in humans. These facts suggest that 5DEX-0509R has a good safety profile, at least one worth testing clinically.
Bulk- and single-cell analyses clearly demonstrated that 5DEX-0509R activates macrophages efficiently. Interestingly, bulk tissue sequencing showed clear induction of CD8 T cells and B cells and a decrease of the Foxp3 gene at a later phase. This may reflect that TAM-dependent immune activation sequentially elicits antitumor immunity including acquired immunity. Single-cell RNA-seq showed the disappearance of the CD206-high cluster in macrophages. It is possible that 5DEX-0509R is mainly incorporated by CD206-high M2-like TAMs and changes its phenotypes. In the previous study, we did not observe obvious B cell induction and Treg decrease by the DSP-0509 treatment in the tumor microenvironment.10 This may be the reason 5DEX-0509R has a stronger antitumor effect than DSP-0509. Since TAMs and Tregs are closely interacting in tumor tissues,40 phenotypic change of TAMs may further inhibit Treg recruitment, induction, and/or proliferation. In the single-cell analysis, we surprisingly found that 5DEX-0509R treatment induced a unique macrophage population, which expresses the B cell markers CD72 and CD79b. Its function is still unclear because there are no reports concerning such macrophages so far, but it may be an intermediate transient phenotype that develops as M2 transitions to M1 phenotype, because this population was decreased in size at day 7, when the M1-type clusters appeared (C1 and C8).
In recent cancer therapies, it is important to identify biomarkers that can be used to select patients suitable for that therapy. Since 5DEX-0509R specifically targets TAMs, the population of TAMs in tumors can be a good biomarker. We successfully screened human tumor types that have high gene signature expression in our preclinical model. Further screening to select the right patient within the tumor type may be needed, but CD206-high M2-type macrophages in tumor tissues would be a good marker to select those patients.
Study Limitations
In this study, we have unveiled the potential of 5DEX-0509R as the TAM-targeting agent, demonstrating rapid clearance from the body alongside strong antitumor efficacy. We optimized the molecular size, linker, and conjugation method and successfully improved its distribution and immunotoxicity without losing the antitumor activity. However, precise chemical properties such as molecular diameter, molecular weight distribution, and charges, which are important for a “nanomedicine” approach, were not thoroughly examined and optimized in this study. In future studies, it is important to analyze and define the precise molecular properties through biological analysis, focusing on parameters crucial for effective TAM targeting, improved body distribution, and enhanced antitumor activity and enhancing antitumor activity. Additionally, we optimized our D-TAC through a mouse in vivo study. To the best of our knowledge, excretion of dextrans from the body is mostly the same across animal species. Therefore, we are confident that our technology will be effective in humans, but further translational studies, including clinical trials, are desired. We think that additional safety studies are necessary to assess the feasibility of safely administering D-TAC to humans.
Conclusion
In summary, we have developed the D-TAC technology TLR7 agonist, 5DEX-0509R, which has a short half-life, a good safety profile, and strong antitumor activity. By changing the tumor microenvironment, monotherapy with 5DEX-0509R shows excellent TAM-targeting ability and strong antitumor activity with complete tumor elimination. We also demonstrated the dependence of the antitumor effect of 5DEX-0509R on the abundance of TAMs, which is consistent with its mechanism of action. We believe 5DEX-0509R can be the clinically applicable immunotherapy targeting TLR signaling.
Methods
Reagents
DSP-0509 was chemically synthesized by Sumitomo Pharma Co., Ltd. For in vitro studies, 10 mM solutions were prepared by dissolution in DMSO followed by dilution of the DMSO up to a final concentration of 0.1%. For in vivo studies, the compounds were dissolved in a 2.5 mM glycine buffer solution of pH 10.2. Dextran-DSP-0509 conjugates were synthesized as described in Supporting Information. Aminodextrans and fluorescent dextrans were purchased at Fina Biosolutions (Rockville, MD, USA). All dextran conjugates were dissolved in PBS at 10 mg/mL and maintained at 4 °C. MBS-8 was generated in the house as described in the patent (WO 2021/053163 Al).
Cells and Cultures
Sources of cells, culture medium, and the number of tumor cells inoculated are listed below (Table 2). The Renca cell line was kindly provided by Dr. Fujioka, Iwate Medical University School of Medicine. The cells were maintained in the culture by passage 1–2 times a week.
Table 2. Tumor Cell Lines Used in This Study.
| cell line | mouse strain | medium | cell number | source |
|---|---|---|---|---|
| Colon26 | Balb | RPMI1640 + 10% FBS | 1 × 106 | Riken |
| CT26 | Balb | RPMI1640 + 10% FBS | 1 × 106 | ATCC |
| K7M2 | Balb | DMEM + 10% FBS + 0.8 mg/mL G-418 | 1 × 106 | ATCC |
| MBT2 | B6C3HF1 | MEM + 10% FBS | 1 × 106 | JCRB |
| EMT6 | Balb | Waymouth’sMB752/1 + 2 mM l-glutamine +15% FBS | 1 × 106 | ATCC |
| Renca | Balb | RPMI1640 + 10% FBS | 1 × 106 | Dr. Fujioka |
| SCCVII | C3H | MEM + 10% FBS | 1 × 106 | Kyoto U |
| LLC | B6 | DMEM + 10% FBS | 1 × 106 | Riken |
| HM-1 | B6C3HF1 | MEM- α + 10% FBS | 1 × 106 | Riken |
| MV4;11 | SCID/Beige | IMDM + 10% FBS | 10 × 106 | ATCC |
| EG7 | Balb | ATCC RPMI1640 + 10% FBS + G418 | 1 × 106 | ATCC |
| E0771 | B6 | DMEM + 10% FBS | 0.5 × 106 | ATCC |
Mice
Balb/c and C57BL/6 mice were purchased from Jackson Laboratory Japan or SLC Japan. and 6- to 10-week-old C3H/HeN and B6C3HF1 mice were purchased from Charles River Japan. All animal studies were conducted in compliance with the Sumitomo Pharma Animal Ethics Code.
Organ Distribution of Dextran Conjugates
EMT6 cells were suspended in HBSS and implanted subcutaneously into Balb/c mice (1 × 106 cells/mouse). When the tumors reached approximately 100 mm3, the mice were randomly divided into groups, and AF750-labeled dextrans were injected intravenously. The organs were then extracted at each time point described, and fluorescence intensity was measured using an IVIS Lumina II imaging system (PerkinElmer).
In Vivo Antitumor Study
All tumor cells were suspended in HBSS and implanted subcutaneously into the mice. The number of implanted cells per mouse varied from 0.1 × 106 to 10 × 106, summarized below unless otherwise performed at Crownbio Sciences. When the tumors reached approximately 50–100 mm3, the mice were randomly divided into groups and treated as indicated. For the rechallenge study, 0.5 × 106 EMT6 cells were inoculated into the dorsal flanks of completely cured or naïve mice. For the s.c. injection, neck flank skin was injected with 100 μL volume. For the i.v. infusion, an i.v. catheter (TERUMO) connected with an extension tube (JMS) was inserted into the tail vein, and 5DEX-0509R and DSP-0509 were injected at a 500 μL/10 mL flow rate using a Microsyringe pump (AS ONE, Osaka, Japan). The tumor volume was calculated using the formula (L × W2)/2, where L and W refer to the length and width dimensions, respectively.
Pharmacokinetics
Blood samples were collected from EMT6-bearing mice at 5 min, 20 min, 1, 2, 4, 24, 48, and 120 h and centrifuged at 5000 rpm for 15 min for plasma collection and storage at −80 °C. After thawing, the plasma samples were treated with sodium hydroxide (50 μL plasma +450 μL 5 M NaOH), boiled for 8 h, and treated with 66% acetic acid (220 μL). 50 μL of each sample was transferred to another tube, treated with acetonitrile (50 μL), and centrifuged. The supernatant was subjected to LC-MS/MS (AB Qtrap5500, Sciex and Nexera X2, Shimadzu).
Cytokine Measurement
Whole blood samples were obtained at indicated time points after intravenous administration of the compounds described. The plasma was separated from the collected blood after centrifugation. The cytokine levels were measured using a mouse Milliplex mouse cytokine/chemokine panel (Merck Millipore) for the various other cytokines according to the manufacturer’s instructions. For human cytokine measurement, Quantkine human TNFα ELISA kits (R and D Systems) were used according to the manufacturer’s instructions. Luminex 200 (Luminex) and Elx808 (BioTek) analyzers were used for detection in each assay.
Flow Cytometry Analysis
For TILs, tumors were dissected and minced in digestion buffer (Tumor Dissociation Kit II, Myltenyi Biotech) as described in the manufacturer’s instructions using a Gentle MACS dissociator (Myltenyi Biotech). Then, cells were filtered through a cell strainer (Falcon), stained with live/dead fixable V450 (Invitrogen) for 10 min at 4 °C, incubated with anti-CD16/32 (BD Bioscience), and stained with anti-CD11b BV510 (M1/70, BD), anti-CD45 FITC (30-F11), anti-CD19 PE (1D3), anti-Gr1 PerCP (RB6-8C5), anti-CD206 PE-Cy7 (C068C2), and anti-F4/80 APC (BM8) antibodies for myeloid and B cell markers and anti-CD8 BV510 (53-6-7), anti-CD4 FITC (GK1.5), anti-KI67 PE (16A8), anti-CD45 perCP (30-F11), and anti-CD3 PE-Cy7 (17A2) antibodies for T cell markers. Antibodies were obtained from Biolegend or BD Bioscience unless otherwise indicated, for example, anti-Foxp3 (eBioscience). After 20 min of incubation on ice, the cells were fixed with a Foxp3 staining buffer set (eBioscience) for T cell staining with Foxp3-APC (FJK-16s) and anti-Ki67 PE, as detailed in the manufacturer’s instructions, and analyzed with a MACS Quant analyzer 10 (Myltenyi). Fluorescence-activated cell sorting (FACS) data were analyzed with FlowJo software (Tree Star). For the dextran distribution, EMT6 tumor-bearing mice were injected with dextran-AF750 (1 mg/kg of AF750). After 1 day, tumors were dissected and analyzed, as described above.
Bulk Tissue RNA-Seq
EMT6 tumors were dissociated into single cells using a gentleMACS Octo dissociator and a mouse tumor dissociation kit (Miltenyi Biotec). CD45+ cells were isolated using a FACS Aria Fusion cell sorter (BD Biosciences) after presorting with antimouse CD45 MicroBeads (Miltenyi Biotec). Total RNA was extracted from cells using an RNeasy Mini kit (Qiagen). An RNA-seq library was prepared by using a TruSeq Stranded mRNA kit (Illumina) and sequenced using a NovaSeq 6000 sequencer (Illumina). Mapping of the sequence data to the genome and the counting of reads per feature were performed using STAR (ver. 2.7.10a).41 Count data were analyzed in the R environment (ver 4.1.0). Differentially expressed genes in the treatment group versus the vehicle group were selected using the exactTest function of the edgeR package (ver 3.36.0){Robinson, 2010 #2}. Abundance of immune and other cell types was estimated using the mMCP-counter package (ver 1.1.0).42 Estimated abundance scores of cell types or expression levels of individual genes (transcripts per million [TPM]) were compared between compound and vehicle-treated groups using the Welch t-test. p values from multiple testing were adjusted by the Benjamini and Hochberg method, from which false discovery rates (shown as q values) were calculated.
ScRNA-Seq
CD45+ single cells were prepared from tumors as in RNA-seq. The cDNA library was prepared from cells using Chromium Controller and Chromium Next GEM Single Cell 3′ GEM, Library and Gel Bead Kit v3.1 (10× Genomics). The cDNA library was sequenced using a NovaSeq 6000 sequencer (Illumina). Mapping of the sequence data to the genome and the counting of reads per feature were performed using CellRanger software (ver 6.1.2, 10× Genomics). Count data were analyzed in the R environment (ver 4.1.0) mainly using the Seurat package (ver. 4.9.9).43 Initial normalization and scaling were performed with the default settings. Clustering of cells was performed using the FindNeighbors function with the top 10 principal components and using the FindClusters function with a resolution of 0.5 for whole cells or 1 for the monocyte-macrophage-cDC or CD8+T cells fraction. In order to annotate cell types, whole cells were first clustered, and gene expression levels of cell type markers were manually examined to identify T, NK, B, the macrophage-cDC fraction, pDCs, granulocytes, and mast cells. Similarly, the macrophage-cDC population was separated into macrophages, cDCs, and osteoclasts, respectively, based on clustering and manual examination of markers. T cells were further separated into CD8+ or CD4+ T cells based on nonzero expression of Cd8a or Cd8b1 genes and zero expression of the Cd4 gene for CD8+T cells and vice versa for CD4+T cells. Differentially expressed genes between clusters or between treatment groups were selected using the FindMarkers function with parameters: test.use = “wilcox”, logfc.threshold = 0, and min.pct = 0.2. The expression level of a gene is shown as the unique molecular identifier (UMI) count per 10000 cellular UMI count. Expression scores for gene sets of macrophage markers were calculated by using the AddModuleScore function.
Efficacy-Related Gene Signature
TGI data of 5DEX-0509R were obtained by Crown Biosciences with eight different in vivo tumor assays (MuScreen). RNA sequencing data of each of the tumor models were obtained from Crown Biosciences. From RNA-seq profiles, frequencies of immune and other cell types were estimated using the mMCP-counter package (ver 1.1.0). Pearson correlation between efficacy and cell frequencies or expression was evaluated using the cor.test function in R. Gene sets showing strong association between their expression and efficacy were examined with the gene set enrichment analysis (GSEA) method44 using the clusterProfiler package (ver 4.2.2)45 and MSigDB46 8 gene sets (https://www.gsea-msigdb.org/gsea/msigdb/). For the top 10 efficacy-associated gene sets in terms of the GSEA nominal enrichment score, 27 common genes constituting leading edges of at least 8 of the 10 gene sets were selected as signature genes: C1QA, C1QB, C1QC, C3AR1, CCR1, CD14, CD163, CD83, CD86, CLEC7A, CSF1R, CYBB, FGD2, FOLR2, FPR1, GPR34, HCK, IGSF6, LGMN, MPEG1, MS4A7, NLRP3, P2RY13, PLD4, PTAFR, RNASE6, and SPI1. Expression levels of signature genes in clinical tumors were summarized into gene set variation analysis (GSVA) scores using the GSVA package (ver 1.42.0)47 and RNA-seq TPM data from the TCGA database (obtained using the TCGAbiolinks package, ver 2.22.4).48
Anaphylactic Shock
Dextran conjugates were injected into Balb/c mice at days 0 and 7. After the second dosing, the rectal temperature was measured using a digital thermometer (TD-300, Shibaura Electronics, Saitama, Japan) just before the challenge and then every 10 min for 1 h. For the blocking experiment, the PAF inhibitor WEB-2086 (3 mg/kg) was injected 10 min before, and the anti-CD16/32 antibody (500 μg/mouse, 2.4G2, BioXcell) was injected 1 day before the second injection.
Establishment of the B2M KO CT26 Cell Line
The B2M KO CT26 cell line was established by the CRISPR-Cas9 system. Alt-R S.p. HiFi Cas9 Nuclease V3 (Integrated DNA Technologies) was mixed with Alt-R CRISPR-Cas9 sgRNA (Integrated DNA Technologies). Then, the mixture above was transfected to CT26 cells by a NEPA21 Type II electroporator (Neppa Gene, Tokyo, Japan). The sequence of sgRNA was FW 5-CTGGTGCTTGTCTCACTGAC-3 and RV 5-GTCAGTGAGACAAGCACCAGC-3 described in ref.49 Single clones of transfected CT26 cells were isolated by limiting the dilution. The knockout of B2M was confirmed by Western blot using the anti-B2M antibody (Cell Signaling Technology, #59035).
Human PBMC Stimulation and Monocyte-Derived M2 Macrophage Differentiation and Activation
Human PBMCs were obtained from the CTLs. The cells were thawed and preincubated with the 5% AIM-V medium (Invitrogen) supplemented with 5% human serum (BioWest). After recovery, the cells were seeded in 96-well flat bottom plates (Falcon) at 1 × 105 cells/well and stimulated with DSP-0509 and 5DEX-0509R (1 μM for 2 h). For the monocyte-derived M2 macrophage differentiation, the PBMCs were collected, and monocytes were separated by using EasySep Human CD14 Positive Selection Kit II (StemCell). Cells were then suspended with the AIM-V medium supplemented with 10% fetal bovine serum (FBS) and recombinant human macrophage colony-stimulating factor (M-CSF; 40 ng/mL, R and D Systems) and cultured for 6 days with a half-medium change at day 3. The cells were then induced to differentiate by the AIM-V medium supplemented with 10% FBS, recombinant human M-CSF (40 ng/mL), recombinant human IL-4, and IL-10 (20 ng/mL each, Peprotech). After the differentiation, the medium was changed to RPMI supplemented with 10% FBS and then stimulated with DSP-0509 and 5DEX-0509R (1 μM for 6 h).
In Vitro Dextran Uptake Assay
Monocyte-derived M2 macrophages were differentiated as described above. After differentiation, the cells were incubated with the antihuman CD206 antibody (10 μg/mL, Biolegend, 15-2) for 10 min. Subsequently, AF750-5 kDa dextran (5 μg/mL) was added to the wells and further incubated for 60 min. After incubation, the cells were washed with PBS and subjected to dextran uptake analysis by Odyssey CLx (LI-COR). For cell line-derived M2 macrophages, M0-like macrophage cell line DH82 cells (ATCC) were differentiated into M2-type macrophages as described prebiously.50 Briefly, the cells were incubated with recombinant canine IL-4 and IL-10 (R and D Systems, 20 ng/mL) for 2 days in the growth medium. Then, the cells were incubated with AF750-5 kDa dextran (1 μg/mL) for 1 h, and uptake was analyzed as described above. CD206 expression was confirmed by flow cytometry using antihuman CD206-APC (Biolegend, 15-2).
For CD206 transfection, 293T cells (ATCC) were transfected with MRC1_OHu11098C_pcDNA3.1 (Genscript) or control GFP vector (pmaxGFP, LONZA) using Lipofectamine 3000 (Thermo) according to the manufacturer’s instruction. After 72 h of transfection, the cells were incubated with AF750-5 kDa dextran (5 μg/mL) for 1 h and transferred into 384-well plates (Black clear bottom, Falcon), and uptake was detected as described above.
Data and Statistical Analysis
Stat Preclinica Client (SAS 9.4, Takumi Information Technology Inc.) was used for statistical analysis in the in vivo antitumor studies. To compare differences between the two groups, unpaired two-tailed t-tests were conducted. In the in vivo tumor study, one-way repeated ANOVA was used, followed by post hoc Dunnett tests, unless otherwise indicated. All data are presented as mean ± SD. Sequence data was deposited to NCBI SRA with BioProject accession ID PRJNA1095666.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.4c08811.
Figure 1: body temperature changes of mice treated with 20DEX-0509A and PAF inhibitor WEB-2086 or anti-CD16/32; Figure 2: cellular uptakes of 5 kDa dextran conjugated with AF750 at the reduced end; Figure 3: in vivo antitumor profiles of 5DEX-0509R; Figure 4: in vivo efficacy data of 5DEX-0509R summarized in Table 1; Figure 5: bulk RNA-seq of tumor tissues A; Figure 6: changes in the tumor-infiltrating lymphocyte (TIL) population by 5DEX-0509R treatment; Figure 7: single-cell RNA-seq clustering; Figure 8: single-cell RNA-seq of tumor infiltrating CD8+ T cells; methods: synthesis of dextran conjugates (PDF)
Author Contributions
# Y.O., R.I., and Y. T. contributed equally to this work. Y.O. performed in vivo and in vitro experiments, interpreted the results, and wrote the manuscript. R.I. performed bulk- and single-cell RNA-seq analysis, interpreted the results, and wrote the manuscript. D.H. generated dextran conjugates, interpreted the results, and wrote the manuscript. K.M. performed in vivo experiments, interpreted the results, and wrote the manuscript. M.M. generated MBS-8 and analyzed dextran conjugate profiles. M.H. established the method of PK analysis and measured blood. PK., R.T., and Y.I. performed the in vivo antitumor experiment. M.N. established the method for in vitro macrophage differentiation. M.G. and H.B. helped in the conceptual organization of the project. Y.T. generated dextran conjugates and helped in the conceptual organization of the project. Y.N. performed in vivo and in vitro experiments, wrote the manuscript, interpreted the results, designed experiments, and organized this project.
The authors declare the following competing financial interest(s): Authors are employed by Sumitomo Pharma Co., Ltd. Osaka, Japan.
Supplementary Material
References
- Janeway C. A. Jr; Medzhitov R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- Matzinger P. The danger model: A renewed sense of self. Science 2002, 296 (5566), 301–305. 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- Beutler B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature 2004, 430 (6996), 257–263. 10.1038/nature02761. [DOI] [PubMed] [Google Scholar]
- Hoden B.; Nagai Y.; Schuettpelz L.; Zhang D. Editorial: Updates on toll-like receptors in cancer immunity and immunotherapy. Front. Immunol. 2023, 14, 1331317. 10.3389/fimmu.2023.1331317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Li H.; Fotopoulou C.; Cunnea P.; Zhao X. Toll-like receptor-targeted anti-tumor therapies: Advances and challenges. Front. Immunol. 2022, 13, 1049340. 10.3389/fimmu.2022.1049340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barchet W.; Wimmenauer V.; Schlee M.; Hartmann G. Accessing the therapeutic potential of immunostimulatory nucleic acids. Curr. Opin. Immunol. 2008, 20 (4), 389–395. 10.1016/j.coi.2008.07.007. [DOI] [PubMed] [Google Scholar]
- Lee J.; Chuang T. H.; Redecke V.; She L.; Pitha P. M.; Carson D. A.; Raz E.; Cottam H. B. Molecular basis for the immunostimulatory activity of guanine nucleoside analogs: Activation of Toll-like receptor 7. Proc. Natl. Acad. Sci. U. S. A. 2003, 100 (11), 6646–6651. 10.1073/pnas.0631696100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggadike K.; Ahmed M.; Ball D. I.; Coe D. M.; Dalmas Wilk D. A.; Edwards C. D.; Gibbon B. H.; Hardy C. J.; Hermitage S. A.; Hessey J. O.; et al. Discovery of 6-Amino-2-[(1S)-1-methylbutyl]oxy-9-[5-(1-piperidinyl)pentyl]-7,9-dihydro-8H-purin-8-one (GSK2245035), a Highly Potent and Selective Intranasal Toll-Like Receptor 7 Agonist for the Treatment of Asthma. J. Med. Chem. 2016, 59 (5), 1711–1726. 10.1021/acs.jmedchem.5b01647. [DOI] [PubMed] [Google Scholar]
- Bhagchandani S.; Johnson J. A.; Irvine D. J. Evolution of Toll-like receptor 7/8 agonist therapeutics and their delivery approaches: From antiviral formulations to vaccine adjuvants. Adv. Drug Delivery Rev. 2021, 175, 113803. 10.1016/j.addr.2021.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota Y.; Nagai Y.; Hirose Y.; Hori S.; Koga-Yamakawa E.; Eguchi K.; Sumida K.; Murata M.; Umehara H.; Yamamoto S. DSP-0509, a systemically available TLR7 agonist, exhibits combination effect with immune checkpoint blockade by activating anti-tumor immune effects. Front. Immunol. 2023, 14, 1055671. 10.3389/fimmu.2023.1055671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins S. R.; Vasilakos J. P.; Deschler K.; Grigsby I.; Gillis P.; John J.; Elder M. J.; Swales J.; Timosenko E.; Cooper Z.; et al. Intratumoral immunotherapy with TLR7/8 agonist MEDI9197 modulates the tumor microenvironment leading to enhanced activity when combined with other immunotherapies. J. Immunother. Cancer 2019, 7 (1), 244. 10.1186/s40425-019-0724-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruni D.; Angell H. K.; Galon J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 2020, 20 (11), 662–680. 10.1038/s41568-020-0285-7. [DOI] [PubMed] [Google Scholar]
- Pathria P.; Louis T. L.; Varner J. A. Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol. 2019, 40 (4), 310–327. 10.1016/j.it.2019.02.003. [DOI] [PubMed] [Google Scholar]
- Khan S. U.; Khan M. U.; Azhar Ud Din M.; Khan I. M.; Khan M. I.; Bungau S.; Hassan S. S. U. Reprogramming tumor-associated macrophages as a unique approach to target tumor immunotherapy. Front. Immunol. 2023, 14, 1166487. 10.3389/fimmu.2023.1166487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Wang X.; Zhang X.; Xu W. The promising role of tumor-associated macrophages in the treatment of cancer. Drug Resist. Updates 2024, 73, 101041. 10.1016/j.drup.2023.101041. [DOI] [PubMed] [Google Scholar]
- Cassetta L.; Fragkogianni S.; Sims A. H.; Swierczak A.; Forrester L. M.; Zhang H.; Soong D. Y. H.; Cotechini T.; Anur P.; Lin E. Y.; et al. Human Tumor-Associated Macrophage and Monocyte Transcriptional Landscapes Reveal Cancer-Specific Reprogramming, Biomarkers, and Therapeutic Targets. Cancer Cell 2019, 35 (4), 588–602.E10. 10.1016/j.ccell.2019.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodell C. B.; Arlauckas S. P.; Cuccarese M. F.; Garris C. S.; Li R.; Ahmed M. S.; Kohler R. H.; Pittet M. J.; Weissleder R. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng. 2018, 2 (8), 578–588. 10.1038/s41551-018-0236-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aghighi M.; Theruvath A. J.; Pareek A.; Pisani L. L.; Alford R.; Muehe A. M.; Sethi T. K.; Holdsworth S. J.; Hazard F. K.; Gratzinger D.; et al. Magnetic Resonance Imaging of Tumor-Associated Macrophages: Clinical Translation. Clin. Cancer Res. 2018, 24 (17), 4110–4118. 10.1158/1078-0432.CCR-18-0673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneo Y.; Uemura T.; Tanaka T.; Kanoh S. Polysaccharides as drug carriers: Biodisposition of fluorescein-labeled dextrans in mice. Biol. Pharm. Bull. 1997, 20 (2), 181–187. 10.1248/bpb.20.181. [DOI] [PubMed] [Google Scholar]
- Arturson G.; Groth T.; Grotte G. Human glomerular membrane porosity and filtration pressure: Dextran clearance data analysed by theoretical models. Clin. Sci. 1971, 40 (2), 137–158. 10.1042/cs0400137. [DOI] [PubMed] [Google Scholar]
- Arturson G.; Wallenius G. The Renal Clearance of Dextran of Different Molecular Sizes in Normal Humans. Scand. J. Clin. Lab. Invest. 1964, 16, 81–86. 10.3109/00365516409060486. [DOI] [PubMed] [Google Scholar]
- Allavena P.; Chieppa M.; Bianchi G.; Solinas G.; Fabbri M.; Laskarin G.; Mantovani A. Engagement of the mannose receptor by tumoral mucins activates an immune suppressive phenotype in human tumor-associated macrophages. Clin. Dev. Immunol. 2010, 2010, 547179. 10.1155/2010/547179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlt A.; von Bonin F.; Rehberg T.; Perez-Rubio P.; Engelmann J. C.; Limm K.; Reinke S.; Dullin C.; Sun X.; Specht R.; et al. High CD206 levels in Hodgkin lymphoma-educated macrophages are linked to matrix-remodeling and lymphoma dissemination. Mol. Oncol. 2020, 14 (3), 571–589. 10.1002/1878-0261.12616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escribese M. M.; Rosace D.; Chivato T.; Fernandez T. D.; Corbi A. L.; Barber D. Alternative Anaphylactic Routes: The Potential Role of Macrophages. Front. Immunol. 2017, 8, 515. 10.3389/fimmu.2017.00515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y.; Nagai Y.; Kuroishi T.; Endo Y.; Sugawara S. Stimulation of Ly-6G on neutrophils in LPS-primed mice induces platelet-activating factor (PAF)-mediated anaphylaxis-like shock. J. Leukocyte Biol. 2011, 91 (3), 485–494. 10.1189/jlb.1210697. [DOI] [PubMed] [Google Scholar]
- Fromen C. A.; Robbins G. R.; Shen T. W.; Kai M. P.; Ting J. P.; DeSimone J. M. Controlled analysis of nanoparticle charge on mucosal and systemic antibody responses following pulmonary immunization. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (2), 488–493. 10.1073/pnas.1422923112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromen C. A.; Rahhal T. B.; Robbins G. R.; Kai M. P.; Shen T. W.; Luft J. C.; DeSimone J. M. Nanoparticle surface charge impacts distribution, uptake and lymph node trafficking by pulmonary antigen-presenting cells. Nanomedicine 2016, 12 (3), 677–687. 10.1016/j.nano.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertekin O.; Akcael E.; Kocaaga H.; Ozturk S. Biological Activity of the Carrier as a Factor in Immunogen Design for Haptens. Molecules 2018, 23 (11), 2977. 10.3390/molecules23112977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackerman S. E.; Pearson C. I.; Gregorio J. D.; Gonzalez J. C.; Kenkel J. A.; Hartmann F. J.; Luo A.; Ho P. Y.; LeBlanc H.; Blum L. K.; et al. Immune-stimulating antibody conjugates elicit robust myeloid activation and durable antitumor immunity. Nat. Cancer 2021, 2 (1), 18–33. 10.1038/s43018-020-00136-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfeld A. J.; Hellmann M. D. Acquired Resistance to Immune Checkpoint Inhibitors. Cancer Cell 2020, 37 (4), 443–455. 10.1016/j.ccell.2020.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H.; Li Y.; Zhang P.; Xing H.; Zhao S.; Song Y.; Wan D.; Yu J. Targeting toll-like receptor 7/8 for immunotherapy: Recent advances and prospectives. Biomarker Res. 2022, 10 (1), 89. 10.1186/s40364-022-00436-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q.; Lu Y.; Luo Y. Recent advances in dextran-based drug delivery systems: From fabrication strategies to applications. Carbohydr. Polym. 2021, 264, 117999. 10.1016/j.carbpol.2021.117999. [DOI] [PubMed] [Google Scholar]
- Feinberg H.; Jegouzo S. A. F.; Lasanajak Y.; Smith D. F.; Drickamer K.; Weis W. I.; Taylor M. E. Structural analysis of carbohydrate binding by the macrophage mannose receptor CD206. J. Biol. Chem. 2021, 296, 100368. 10.1016/j.jbc.2021.100368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa M.; Kamijo A.; Fujita T.; Takakura Y.; Sezaki H.; Hashida M. Synthesis and pharmacokinetics of a new liver-specific carrier, glycosylated carboxymethyl-dextran, and its application to drug targeting. Pharm. Res. 1993, 10 (9), 1253–1261. 10.1023/A:1018949109004. [DOI] [PubMed] [Google Scholar]
- Dreher M. R.; Liu W.; Michelich C. R.; Dewhirst M. W.; Yuan F.; Chilkoti A. Tumor vascular permeability, accumulation, and penetration of macromolecular drug carriers. J. Natl. Cancer Inst. 2006, 98 (5), 335–344. 10.1093/jnci/djj070. [DOI] [PubMed] [Google Scholar]
- Li Y.; Qiao Y.; Chen H.; Bai R.; Staedtke V.; Han Z.; Xu J.; Chan K. W. Y.; Yadav N.; Bulte J. W. M.; et al. Characterization of tumor vascular permeability using natural dextrans and CEST MRI. Magn. Reson. Med. 2018, 79 (2), 1001–1009. 10.1002/mrm.27014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter W. Hapten inhibition of passive antidextran dextran anaphylaxis in guinea pigs. Role of molecular size in anaphylactogenicity and precipitability of dextran fractions. Int. Arch. Allergy Appl. Immunol. 2004, 41 (6), 826–844. 10.1159/000230575. [DOI] [PubMed] [Google Scholar]
- Hedin H.; Ljungstrom K. G. Prevention of dextran anaphylaxis. Ten years experience with hapten dextran. Int. Arch. Allergy Immunol. 2004, 113 (1–3), 358–359. 10.1159/000237601. [DOI] [PubMed] [Google Scholar]
- Moghimi S. M. Nanomedicine safety in preclinical and clinical development: Focus on idiosyncratic injection/infusion reactions. Drug Discovery Today 2018, 23 (5), 1034–1042. 10.1016/j.drudis.2017.11.006. [DOI] [PubMed] [Google Scholar]
- Pan Y.; Yu Y.; Wang X.; Zhang T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. 10.3389/fimmu.2020.583084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A.; Davis C. A.; Schlesinger F.; Drenkow J.; Zaleski C.; Jha S.; Batut P.; Chaisson M.; Gingeras T. R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29 (1), 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitprez F.; Levy S.; Sun C. M.; Meylan M.; Linhard C.; Becht E.; Elarouci N.; Tavel D.; Roumenina L. T.; Ayadi M.; et al. The murine Microenvironment Cell Population counter method to estimate abundance of tissue-infiltrating immune and stromal cell populations in murine samples using gene expression. Genome Med. 2020, 12 (1), 86. 10.1186/s13073-020-00783-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y.; Hao S.; Andersen-Nissen E.; Mauck W. M. 3rd; Zheng S.; Butler A.; Lee M. J.; Wilk A. J.; Darby C.; Zager M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184 (13), 3573–3587.e29. 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A.; Tamayo P.; Mootha V. K.; Mukherjee S.; Ebert B. L.; Gillette M. A.; Paulovich A.; Pomeroy S. L.; Golub T. R.; Lander E. S.; Mesirov J. P. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (43), 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G.; Wang L. G.; Han Y.; He Q. Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS: J. Integr. Biol. 2012, 16 (5), 284–287. 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A.; Birger C.; Thorvaldsdottir H.; Ghandi M.; Mesirov J. P.; Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1 (6), 417–425. 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzelmann S.; Castelo R.; Guinney J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 2013, 14, 7. 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colaprico A.; Silva T. C.; Olsen C.; Garofano L.; Cava C.; Garolini D.; Sabedot T. S.; Malta T. M.; Pagnotta S. M.; Castiglioni I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44 (8), e71 10.1093/nar/gkv1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrejon D. Y.; Abril-Rodriguez G.; Champhekar A. S.; Tsoi J.; Campbell K. M.; Kalbasi A.; Parisi G.; Zaretsky J. M.; Garcia-Diaz A.; Puig-Saus C.; et al. Overcoming Genetically Based Resistance Mechanisms to PD-1 Blockade. Cancer Discovery 2020, 10 (8), 1140–1157. 10.1158/2159-8290.CD-19-1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann I.; Gotovina J.; Fazekas-Singer J.; Fischer M. B.; Hufnagl K.; Bianchini R.; Jensen-Jarolim E. Canine macrophages can like human macrophages be in vitro activated toward the M2a subtype relevant in allergy. Dev. Comp. Immunol. 2018, 82, 118–127. 10.1016/j.dci.2018.01.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.