

Highlights
This EHA–ESMO Clinical Practice Guideline provides key recommendations for managing HIV‐associated lymphomas.
The guideline covers clinical, imaging and pathological diagnosis; staging and risk assessment; treatment and follow‐up.
The author group encompasses a multidisciplinary group of experts from different institutions and countries in Europe.
Recommendations are based on available scientific data and the authors’ collective expert opinion.
INCIDENCE AND EPIDEMIOLOGY
Non‐Hodgkin lymphoma (NHL) remains the most common type of cancer and a leading cause of mortality in people who are living with human immunodeficiency virus (HIV). 1 This is despite a marked decrease in the incidence of HIV‐associated NHL (HIV–NHL) following the introduction of combination antiretroviral therapy (ART) in the mid‐1990s. 2 In contrast, the incidence of Hodgkin lymphoma (HL) increased slightly but has remained stable since 2000. 1 Compared with the age‐ and gender‐matched general population, the incidences of HIV–NHL and HIV‐associated HL (HIV–HL) are increased ~10‐ to 20‐fold. 3
The most common histological types of HIV‐associated lymphomas are diffuse large B‐cell lymphoma (DLBCL; 37%), HL (26%) and Burkitt lymphoma (BL; 20%). 4 Independent risk factors for DLBCL in people living with HIV (PLWH) include a low cluster of differentiation (CD)4 T‐cell count and an uncontrolled HIV‐1 viral load (VL). 5 The availability of ART and better management of opportunistic infections allow PLWH to receive the same treatments as people without HIV, including intensive therapies, such as autologous stem‐cell transplantation (ASCT), allogeneic stem‐cell transplantation (allo‐SCT) and chimeric antigen receptor T‐cell (CAR‐T) therapy. Patients with HIV‐associated lymphomas should be enrolled in clinical trials whenever possible.
The aim of this guideline is to provide practical clinical guidance and recommendations to clinicians who manage HIV‐associated lymphomas.
DIAGNOSIS, PATHOLOGY AND STAGING
Diagnostic procedures in patients with HIV‐associated lymphoma generally mirror those recommended for lymphoma in the general population and those necessary to assess the severity and complications of HIV and its treatment (see Supporting Information S1: Table S1).
Lymphoma should be diagnosed via tumour biopsy, preferably excisional, that is evaluated by an expert haematopathologist using immunohistochemistry (IHC) and molecular techniques. In exceptional cases when no tumour mass can be biopsied, diagnosis can be made by cytology and flow cytometry.
Lymphoma staging should involve a contrast‐enhanced computed tomography (CT) scan of the neck, chest, abdomen and pelvis and a bone marrow biopsy. A staging [18F]2‐fluoro‐2‐deoxy‐d‐glucose (FDG)–positron emission tomography (PET)–CT scan is more sensitive, especially for extranodal disease. FDG–PET–CT may, however, have a higher false‐positive rate in PLWH due to immune deficiency‐related lymphoid hyperplasia and non‐suppressed HIV infection. 6 Interim FDG–PET–CT (iFDG–PET–CT) results should, therefore, be interpreted cautiously if used to escalate treatment and when analysing end‐of‐treatment response; if there is doubt, FDG‐avid lesions should be re‐biopsied. Otherwise, response criteria do not differ from those used in immunocompetent individuals.
Magnetic resonance imaging (MRI) is the optimal method for staging and response assessment of central nervous system (CNS) lymphomas. Cerebral opportunistic infections may, however, mimic lymphoma in PLWH. Small case series suggest that FDG–PET–CT can differentiate between cerebral infections, such as toxoplasmosis, 7 and CNS lymphoma, but biopsy (preferably stereotactic) remains the gold standard for diagnosis.
All additional investigations for patients with newly diagnosed HIV should follow the annually updated, evidence‐graded European Acquired Immune Deficiency Syndrome Clinical Society guidelines, available at http://www.eacsociety.org/guidelines/eacs-guidelines/.
Recommendations
Diagnostic work‐up should follow recommendations for patients who are HIV‐negative and those necessary to assess the severity and complications of HIV [V, A].
Staging with FDG–PET–CT provides higher sensitivity than contrast‐enhanced CT [III, A], but MRI is the optimal method for staging CNS lymphomas [III, A]. These techniques may, however, give false‐positive results because of opportunistic infections or immune deficiency‐related lymphoid hyperplasia and should be interpreted with caution [IV, C].
Cytological and flow cytometry evaluation of cerebrospinal fluid (CSF) can be recommended in patients with aggressive NHL and when there is a high risk of CNS disease [IV, B].
CONCOMITANT THERAPIES
ART
All patients with HIV‐related lymphoma should be treated with effective ART, even those with relatively preserved immune function. ART should be initiated and continued during chemotherapy (ChT) to ensure sustained viral suppression. Several prospective studies have shown that ChT tolerability, immune recovery and patient outcomes are improved with ART; 8 however, pharmacokinetic and pharmacodynamic interactions between cytotoxic anticancer agents, ART and supportive medication [drug–drug interactions (DDIs)] must be carefully checked. A multidisciplinary approach is recommended and ART can be modified to avoid DDIs. Advice about specific ChT and ART DDIs is available at http://www.hiv-druginteractions.org and www.cancer-druginteractions.org.
Infection prophylaxis
When CD4 counts are <200 cells/µL, prophylaxis against Pneumocystis jirovecii pneumonia (PcP) and antiviral prophylaxis with acyclovir or valacyclovir in patients with a history of herpes simplex virus or varicella zoster virus infection is strongly recommended. 9 They should also be offered at higher CD4 counts, as ChT in combination with prednisolone usually causes a profound fall in these cells. PcP prophylaxis also provides protection against toxoplasmosis and some bacterial infections.
Routine primary prophylaxis against mucosal candidiasis is not recommended; however, in severely immunosuppressed patients (e.g., CD4 counts <100 cells/µL), in those with anticipated prolonged neutropenia or in patients receiving high‐dose (HD) methotrexate (MTX) or HD cytarabine (Ara‐C)‐containing regimens (most likely causing mucositis), antifungal prophylaxis with fluconazole may be considered, given its favourable DDI profile. Monitoring for cytomegalovirus is warranted if CD4 counts are <100 cells/µL.
Prophylactic fluoroquinolones are suggested for patients undergoing intensive ChT, who may experience prolonged neutropenia (>7 days) and mucositis.
Co‐infection with hepatitis B and C
Two European cohort studies conducted in the modern ART era have shown a consistent increase in the risk of NHL in PLWH who are also affected by chronic hepatitis C virus (HCV) infection. 10 , 11 HCV‐associated DLBCL is a therapeutic challenge in terms of liver injury during immunochemotherapy (ICT) and long‐term hepatic complications. Immediate delivery of direct‐acting antivirals (DAAs) is recommended during or after ICT. Concomitant treatment with ART, DAAs and ICT is feasible if particular attention is paid to DDIs.
The risk of reactivation of hepatitis B virus is markedly increased during and after treatment of HIV‐associated lymphomas, increasing the risk of lethal liver failure; thus, patients should receive tenofovir (often already included in the ART regimen) or entecavir.
Recommendations
Concomitant ART should be continued in patients receiving therapy for HIV‐associated lymphoma [I, A].
A multidisciplinary approach (including an HIV specialist) is strongly recommended to prevent DDIs [V, A].
Prophylaxis against PcP could be offered to all patients [III, B] and is strongly recommended when CD4 counts are <200 cells/µL [I, A].
Antiviral prophylaxis with acyclovir or valacyclovir should be offered to patients with a history of herpes simplex virus or varicella zoster virus infection and those with CD4 counts <200 cells/μL [I, A].
In the context of HIV and HCV co‐infection and DLBCL, DAAs can be recommended in patients established on ART [V, B].
DLBCL
Diagnosis, pathology and molecular biology
DLBCL is characterised by a diffuse infiltrate of large cells expressing the B‐cell markers CD19 and CD20. DLBCL can be classified as germinal centre B‐cell (GCB) or activated B‐cell subtypes based on the cell of origin (COO) determined by gene expression profiling (GEP). Although COO classification based on GEP is preferred, IHC‐based classification systems can be used if GEP is not available. The Hans algorithm is the most frequently used IHC‐based classification; this classifies DLBCL as GCB or non‐GCB subtypes, the latter being more frequent in HIV‐associated DLBCL (HIV–DLBCL). 12
Staging and risk assessment
The prognosis of HIV–DLBCL is determined by the International Prognostic Index (IPI). 13 The predictive value of HIV‐related factors such as CD4 count and VL on overall survival (OS) is not clear. Several tumour markers [i.e. proliferation index Ki‐67, CD44, CD20, Epstein–Barr virus (EBV) positivity, MYC translocation, BCL2 or BCL6 translocation, TP53 mutation and immunoglobulin (Ig) M expression] have shown prognostic value; 14 however, with the exception of CD20 positivity (which is crucial for application of the anti‐CD20 antibody rituximab), these markers currently have no impact on clinical management and need to be tested in prospective trials.
First‐line management of HIV–DLBCL
Studies carried out in the pre‐ART era suggest that patients with HIV–DLBCL tolerate standard ChT regimens poorly and have a poor prognosis. This changed with improvements in HIV therapy: phase II and real‐world studies have demonstrated that PLWH can receive similar regimens to those used in patients who are HIV‐negative. 14 , 15 Figure 1 shows a proposed treatment algorithm for patients with HIV–DLBCL.
Figure 1.
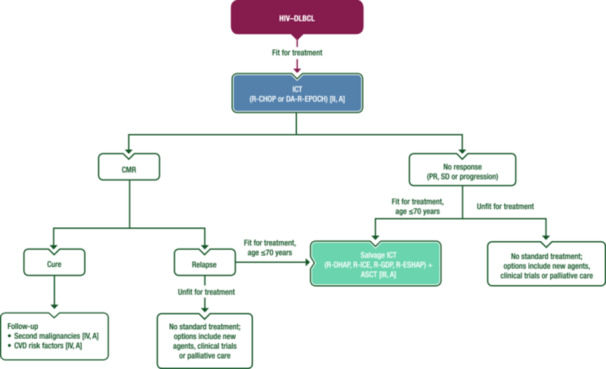
Management of HIV–DLBCL. Purple: algorithm title; blue: systemic anticancer therapy or their combination; turquoise: non‐systemic anticancer therapies or combination of treatment modalities; white: other aspects of management and non‐treatment aspects. ASCT, autologous stem‐cell transplantation; CMR, complete metabolic response; CVD, cardiovascular disease; DA‐R‐EPOCH, dose‐adjusted rituximab–etoposide–prednisone–vincristine–cyclophosphamide–doxorubicin; HIV–DLBCL, human immunodeficiency virus‐associated diffuse large B‐cell lymphoma; ICT, immunochemotherapy; PR, partial response; R‐CHOP, rituximab–cyclophosphamide–doxorubicin–vincristine–prednisone; R‐DHAP, rituximab–dexamethasone–cytarabine–cisplatin; R‐ESHAP, rituximab–etoposide–methylprednisolone–cytarabine–cisplatin; R‐GDP, rituximab–gemcitabine–dexamethasone–cisplatin; R‐ICE, rituximab–ifosfamide–carboplatin–etoposide; SD, stable disease.
Rituximab is beneficial if CD4 counts are ≥50 cells/µL and should be routinely combined with ChT. 14 , 15 In contrast, in patients with CD4 counts <50 cells/µL, there is an increased risk of lethal infections with rituximab, and a large retrospective analysis reported no benefit on overall outcome. 8 The most frequent regimens are rituximab–cyclophosphamide–doxorubicin–vincristine–prednisone (R‐CHOP) and infusional dose‐adjusted (DA) rituximab–etoposide–prednisone–vincristine–cyclophosphamide–doxorubicin (R‐EPOCH). A retrospective analysis of two consecutive phase II trials suggested that DA‐R‐EPOCH is more effective than R‐CHOP; however, the patient populations differed, 16 and a large retrospective pooled study did not report differences between the two regimens on multivariate analysis. 8 Another infusional regimen is rituximab–cyclophosphamide–doxorubicin–etoposide (R‐CDE); in a retrospective trial, both DA‐R‐EPOCH and R‐CDE provided outcomes comparable with R‐CHOP in patients with HIV–DLBCL, including those with MYC rearrangements. 17 In a phase III trial, the combination of polatuzumab vedotin with rituximab–cyclophosphamide–doxorubicin–prednisone (Pola‐R‐CHP) demonstrated improved progression‐free survival (PFS) versus R‐CHOP in patients without HIV. 18 Pola‐R‐CHP is used in patients who are HIV negative in some centres, but data in HIV–DLBCL are lacking. R‐CHOP should be administered using the standard schedule of six cycles given every 3 weeks. Whilst a reduction in the number of cycles and addition of radiotherapy (RT) in the rare subgroup of patients with low‐risk localised disease seems reasonable, there are no reliable data to support this approach. A National Cancer Institute study suggested that the number of DA‐R‐EPOCH cycles can be safely reduced to four in combination with two rituximab doses per cycle, 19 but most centres administer six cycles. There are no published data on the use of alternative regimens [e.g., rituximab–doxorubicin–cyclophosphamide–vindesine–bleomycin–prednisone (R‐ACVBP) and rituximab–cyclophosphamide–doxorubicin–vincristine–etoposide–prednisone (R‐CHOEP14)] in the ART era, but some European centres use these in high‐risk patients.
The incidence of subclinical leptomeningeal involvement in DLBCL seems higher than expected in risk‐matched patients with HIV. 20 CNS prophylaxis with intrathecal (IT) MTX with or without Ara‐C or intravenous (i.v.) MTX may be considered. The criteria used to determine whether patients require CNS prophylaxis should be the same as those used in people without HIV.
Management of relapsed or refractory HIV–DLBCL
There are no prospective randomised controlled trials for PLWH with relapsed or refractory (r/r) DLBCL. Indeed, HIV infection has been an exclusion criterion in most clinical trials of r/r DLBCL.
In general, if the HIV infection is well controlled with ART and there is no evidence of uncontrolled infections, transplant‐eligible patients (i.e., aged ≤70 years and fit) should be offered salvage ICT followed by ASCT if the disease is chemosensitive. Standard platinum‐based salvage regimens [e.g. rituximab–dexamethasone–Ara‐C–cisplatin (R‐DHAP), rituximab–ifosfamide–carboplatin–etoposide (R‐ICE), rituximab–gemcitabine–dexamethasone–cisplatin (R‐GDP) and rituximab–etoposide–methylprednisolone–Ara‐C–cisplatin (R‐ESHAP)] can be used, with response rates comparable to those observed in patients without HIV. Stem‐cell mobilisation has been proven feasible. Data from 118 PLWH who received ASCT for different types of lymphoma (including 47% with DLBCL) were reported in a retrospective study by the European Group for Blood and Marrow Transplantation. 21 The outcome after ASCT for HIV‐associated lymphoma in this series was determined by lymphoma‐dependent risk factors rather than HIV‐related characteristics. When allo‐SCT is indicated, a search for a matched donor who is homozygous for CCR5‐delta 32 deletion should be considered, following reports of long‐term HIV control in a small number of patients. 22 There is currently no standard of care (SoC) for non‐transplant‐eligible patients, but second‐line regimens such as rituximab–gemcitabine–oxaliplatin (R‐GemOx) can be used. There is limited evidence for novel agents such as polatuzumab vedotin, tafasitamab, brentuximab vedotin (BV), lenalidomide and proteasome inhibitors in this setting.
Based on randomised trials in patients without HIV, the use of CAR‐T therapy in early relapsed DLBCL is associated with a better outcome than ASCT. CAR‐T therapy is also approved following two lines of treatment. There is currently limited experience with this approach in HIV–DLBCL. Case reports have shown that anti‐CD19 CAR‐T therapy in PLWH is feasible, provided the VL is suppressed and CD4 counts are >200 cells/μL. 23 , 24 Given the expanded approved indications for patients with r/r DLBCL, this strategy could be considered for selected r/r HIV–DLBCL cases.
Follow‐up, long‐term implications and survivorship
The OS of patients with HIV–DLBCL who are on ART and receive first‐line therapy with standard ICT is approaching that of patients who are HIV‐negative and treated with similar regimens. 25 Around two‐thirds of patients are cured with first‐line therapy and are, therefore, susceptible to long‐term complications. PLWH and NHL have an increased risk of second primary malignancies 26 and thus prevention of other risk factors is warranted. Likewise, the incidence of cardiovascular disease (CVD) is increased in patients with HIV infection 27 so regular screening and control of additional CVD risk factors is mandatory.
Recommendations
Determination of COO by GEP or IHC may be recommended [IV, C].
IPI should be used to evaluate prognosis [IV, A].
Evaluation of MYC rearrangement may be carried out; if positive, investigation of BCL2 and BCL6 rearrangements may be carried out to exclude double‐ or triple‐hit lymphoma [IV, C].
Rituximab should be used routinely in combination with ChT if the CD4 count is ≥50 cells/µl [II, A].
When using R‐CHOP, six cycles every 3 weeks should be given [II, A].
In low‐risk patients, a reduction in the number of ChT cycles and addition of RT are optional [III, C].
When using DA‐R‐EPOCH, six cycles should be given [II, A].
Alternatively, four cycles of DA‐R‐EPOCH with two doses of rituximab per cycle can be given [III, B].
IT MTX with or without Ara‐C [III, C] or i.v. MTX [IV, A] are options for CNS prophylaxis.
In the relapse setting, salvage ICT and ASCT should be offered to fit patients with well‐controlled HIV infection [III, A].
Lifestyle measures to reduce the risk of a secondary malignancy should be discussed [IV, A].
CVD risk factors should be assessed and managed [IV, A].
BL
Diagnosis, pathology and molecular biology
Translocation of MYC with the Ig heavy‐chain loci (80%) or, less frequently, with kappa or lambda light chains, is the molecular hallmark of BL. In HIV‐associated BL (HIV–BL), TP53 mutations are present in a high percentage of patients. 28 Among cases with EBV infection, type I is the most common latency pattern. Some evidence suggests a direct role for HIV in the pathogenesis of HIV‐associated lymphomas, including BL. 28
Staging and risk assessment
The usual staging procedures for HIV–BL follow the general recommendations for BL. Assessment of the biochemical parameters that define clinical and biological tumour lysis syndrome [lactate dehydrogenase (LDH), potassium, calcium, phosphate, uric acid and creatinine clearance] must also be included. According to the recently proposed BL‐IPI, there are four variables [age ≥40 years, performance status (PS) ≥2, serum LDH >3 × upper limit of normal and CNS involvement] with independent prognostic value for PFS and OS. 29 Three risk groups were identified (see Supporting Information S1: Table S2). The index discriminated outcomes regardless of stage or first‐line ChT and, importantly, HIV status was not a prognostic factor.
First‐line management of HIV–BL
HIV–BL is now treated with the same multi‐agent regimens used in the HIV‐negative setting. These include rituximab–cyclophosphamide–vincristine–doxorubicin–MTX (R‐CODOX‐M) or rituximab–ifosfamide–etoposide–Ara‐C (R‐IVAC), rituximab–hyperfractionated cyclophosphamide–vincristine–doxorubicin–dexamethasone (R‐hyper‐CVAD), the German Multicentre Study Group for Adult Acute Lymphoblastic Leukaemia B‐cell acute lymphoblastic leukaemia (GMALL‐B‐ALL) protocol 30 , 31 , 32 or equivalent regimens and the less intensive DA‐R‐EPOCH regimen (Figure 2).
Figure 2.
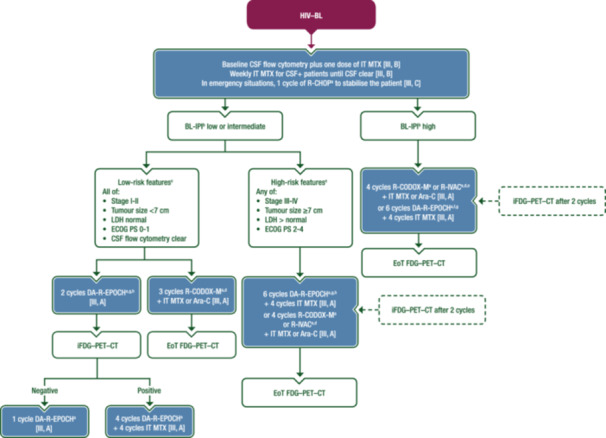
First‐line management of HIV–BL. Purple: algorithm title; blue: systemic anticancer therapy or their combination; white: other aspects of management and non‐treatment aspects; dashed lines: optional therapy. Ara‐C, cytarabine; BL, Burkitt lymphoma; CALGB, Cancer and Leukemia Group B; CNS, central nervous system; CSF, cerebrospinal fluid; CSF+, cerebrospinal fluid positive; CT, computed tomography; DA‐R‐EPOCH, dose‐adjusted rituximab–etoposide–prednisone–vincristine–cyclophosphamide–doxorubicin; ECOG, Eastern Cooperative Oncology Group; EMA, European Medicines Agency; EoT, end of treatment; FDA, Food and Drug Administration; FDG, [18F]2‐fluoro‐2‐deoxy‐D‐glucose; GMALL‐B‐ALL, German Multicentre Study Group for Adult Acute Lymphoblastic Leukaemia B‐cell acute lymphoblastic leukaemia; HIV–BL, human immunodeficiency virus‐associated Burkitt lymphoma; i, interim; IPI, International Prognostic Index; IT, intrathecal; LDH, lactate dehydrogenase; LMB, lymphomes malins B; MTX, methotrexate; PET, positron emission tomography; PS, performance status; R‐CHOP, rituximab–cyclophosphamide–doxorubicin–vincristine–prednisone; R‐CODOX‐M, rituximab–cyclophosphamide–vincristine–doxorubicin–methotrexate; R‐IVAC, rituximab–ifosfamide–etoposide–cytarabine; ULN, upper limit of normal. aRituximab is not EMA or FDA‐approved for the treatment of BL. bBL‐IPI factors are age ≥40 years, ECOG PS 2‐4, LDH >3 × ULN and CNS involvement, as per Olszewski et al. 29 cRisk definition as per Roschewski et al. 36 dOr equivalent regimens. Rituximab‐containing regimens such as LMB, GMALL‐B‐ALL and CALGB protocols are valid alternatives to R‐CODOX‐M or R‐IVAC. eR‐CODOX‐M or R‐IVAC (or equivalent regimens) are preferred for patients with high‐risk BL‐IPI and for those with CNS disease at diagnosis. fDA‐R‐EPOCH is an alternative for less fit patients or those aged >60 years. gConsider R‐CHOP plus immunotherapies for elderly and/or unfit patients with the aim of escalating if fitness improves. hDA‐R‐EPOCH may be preferred for patients with low‐ or intermediate‐risk BL‐IPI, especially those aged >60 years.
Few prospective trials have been conducted in the rituximab era to assess the efficacy and toxicity of R‐CODOX‐M, R‐IVAC or DA‐R‐EPOCH, with <35 PLWH recruited. 33 , 34 , 35 Two‐year OS rates were ~70% and treatment‐related mortality (TRM) rates were <5% for these regimens. 35 , 36 Short‐course EPOCH with a double dose of rituximab (SC‐RR‐EPOCH) is a less intensive regimen used successfully in a small group of patients with HIV. 35 R‐EPOCH has limited effect in the CNS and, therefore, is not recommended in HIV–BL with CNS involvement. In a retrospective series including 142 PLWH with BL in the United States, 19% had CNS involvement. 37 This was independently associated with HIV infection on multivariate analysis. DA‐R‐EPOCH had the highest CNS relapse rate when compared with other regimens, but only 45% of those receiving IT ChT adhered to the strict administration schedule in the original protocol. 37 In a large international retrospective analysis, which included 249 PLWH with BL, 11% had CNS relapse. The highest relapse rates were seen with DA‐R‐EPOCH compared with R‐hyper‐CVAD and R‐CODOX‐M or R‐IVAC (16%, 9% and 8%, respectively; p = 0.032). 33 Here, prognostic factors were associated with lymphoma characteristics rather than HIV control.
The phase II CARMEN study trialled a novel approach incorporating a dose‐dense regimen with ASCT as first‐line consolidation therapy for patients not achieving a complete response (CR) after induction. 38 Twenty PLWH were enrolled, with 5‐year PFS and OS rates of 70% and 75%, respectively, although the TRM rate was 10%. Further studies are warranted.
Management of r/r HIV–BL
There are no clinical trials specifically in r/r HIV–BL. Similar to patients not infected with HIV, the prognosis of r/r HIV–BL is extremely poor, especially for those with primary refractory disease; in a series of 249 patients with HIV–BL from the United Kingdom and United States, 84 of whom had relapsed, the mortality rate was 87%. 33 HD salvage ChT followed by ASCT and/or allo‐SCT is feasible in PLWH with DLBCL, but there is limited experience in BL. 21 , 39 Enrolment in a clinical trial is advised, but most trials (e.g., with immunotherapy such as CD19‐directed CAR‐T therapy or bispecific antibodies) have excluded PLWH. The kinetics of BL, with rapidly progressive disease, may not allow for the 4‐ to 6‐week waiting period during screening, apheresis and manufacturing necessary for CAR‐T therapy.
Follow‐up, long‐term implications and survivorship
BL survivors should be screened periodically for long‐term side effects related to the disease and its treatment, such as secondary malignancies, CVD, endocrine dysfunction, cognitive impairment and fatigue. A close collaboration with HIV teams is necessary. Cohort studies have not shown differences in survival rates between HIV–DLBCL and HIV–BL. 40 , 41
Recommendations
Diagnostic work‐up for HIV–BL should follow recommendations for patients who are HIV negative [IV, A].
Baseline CSF flow cytometry plus one dose of IT MTX can be recommended for all patients [III, B].
Weekly IT MTX should be given to CSF‐positive patients until CSF is clear [III, B].
In emergency situations, it is reasonable to give one cycle of R‐CHOP to stabilise the patient [III, C; rituximab is not European Medicines Agency (EMA) or Food and Drug Administration (FDA) approved for BL].
-
HIV–BL should be treated with the same multi‐agent regimens utilised in the HIV‐negative setting, noting that rituximab is not EMA or FDA approved for BL [III, A].
-
Treatment options for patients with BL‐IPI low or intermediate and low‐risk features:
-
▪
Two cycles of DA‐R‐EPOCH (preferred, especially in those aged >60 years) [III, A].
If iFDG–PET–CT negative: one cycle of DA‐R‐EPOCH [III, A].
If iFDG–PET–CT positive: four cycles of DA‐R‐EPOCH plus four cycles of IT MTX [III, A].
-
▪
Three cycles of R‐CODOX‐M plus IT MTX or Ara‐C [III, A].
-
▪
-
Treatment options for patients with BL‐IPI low or intermediate and high‐risk features:
-
▪
Six cycles of DA‐R‐EPOCH (preferred, especially for those aged >60 years) plus four cycles of IT MTX [III, A].
-
▪
Four cycles of R‐CODOX‐M or R‐IVAC plus IT MTX or Ara‐C [III, A].
-
▪
-
Treatment options for patients with BL‐IPI high:
-
▪
Four cycles of R‐CODOX‐M or R‐IVAC (preferred) plus IT MTX or Ara‐C [III, A].
-
▪
Six cycles of DA‐R‐EPOCH (alternative in less fit patients or those aged >60 years) plus four cycles of IT MTX [III, A].
-
▪
-
For patients not suitable for intensive therapy, DA‐R‐EPOCH should be considered, provided there is no CNS localisation [III, A].
Rituximab‐containing regimens, such as the lymphomes malins B (LMB), GMALL‐B‐ALL and Cancer and Leukemia Group B (CALGB) protocols are valid alternatives to R‐CODOX‐M or R‐IVAC [III, B; rituximab is not EMA or FDA approved for BL].
In the r/r setting, salvage ChT followed by ASCT or allo‐SCT may be considered for chemosensitive disease [IV, C].
PLASMABLASTIC LYMPHOMA (PBL)
Diagnosis, pathology and molecular biology
PBL is a rare and aggressive entity with diffuse proliferation of large neoplastic cells, mostly resembling B immunoblasts or plasmablasts. PBL typically has a plasma‐cell phenotype, which includes CD138, CD38, Vs38c and multiple myeloma 1/interferon regulatory factor 4 (MUM1/IRF4) expression and negativity or weak positivity for CD20 and paired box 5 (PAX5). The biological basis of PBL is not completely understood. In 75%‐80% of HIV‐positive cases, PBL is associated with EBV infection, which plays an anti‐apoptotic role in B cells. Overexpression of the MYC protein is frequently reported. The most frequent genetic alterations are translocations and/or rearrangement of (IG)/MYC in ~50% of cases, together with amplifications of MYC. Other recurrent mutations involve PRDM1 and the Janus kinase–signal transducer and activator of transcription (JAK‐STAT) and RAS‐mitogen‐activated protein kinase (MAPK) signalling pathways. 42
Staging and risk assessment
PBL is unique in its predilection for the oral cavity, but nodal or extranodal involvement may occur, frequently at gastrointestinal sites. Most patients present with either stage I or stage IV disease. Oral primary location is inversely associated with the presence of B symptoms and advanced Ann Arbor stage. 42 , 43 CSF analysis is advisable at initial staging.
First‐line management of HIV‐associated PBL
To date, there is no SoC for HIV‐associated PBL (HIV–PBL) and treatment recommendations are based on case reports, small series and expert opinions. A clinically aggressive course and unsatisfactory results are usually described. A variety of ChT combinations have been reported, including cyclophosphamide–doxorubicin–vincristine–prednisone (CHOP) or CHOP‐like regimens, hyperfractionated cyclophosphamide–vincristine–doxorubicin–dexamethasone–MTX–Ara‐C (hyper‐CVAD‐MA), cyclophosphamide–vincristine–doxorubicin–MTX (CODOX‐M), ifosfamide–etoposide–Ara‐C (IVAC) and etoposide–prednisone–vincristine–cyclophosphamide–doxorubicin (EPOCH). 42 , 44 CHOP is most commonly used and although published literature suggests that it achieves suboptimal results, there are no data demonstrating superior outcomes with more intensive regimens. 43 In patients with disseminated disease, CR rates with polyChT are >50%, but ~70% of patients die due to progressive disease [median event‐free survival (EFS) 22 months and OS 32 months]. 43 Among 16 patients treated with bortezomib−EPOCH, 94% achieved a CR and the 5‐year OS rate was 65%. 45 Patients with localised disease have a better prognosis and persistent disease control may be achieved with ChT plus local RT. Both CHOP and more aggressive regimens are acceptable approaches in HIV–PBL. Rituximab is not advised in the absence of CD20 expression. Consolidation ASCT may be considered for young patients with high‐risk disease. 46 Bortezomib in combination with ChT is an option. In early‐stage disease, doxorubicin‐based ChT may be combined with RT. Consensus opinion is that IT therapy should be given as CNS prophylaxis. New approaches with the addition of novel agents are required and participation in clinical trials is highly encouraged. Figure 3 shows a proposed treatment algorithm for the first‐line management of HIV–PBL.
Figure 3.
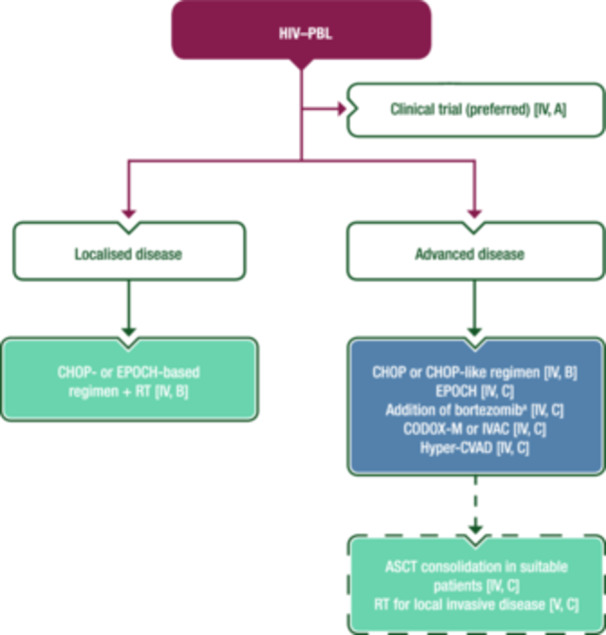
First‐line management of HIV–PBL. Purple: algorithm title; blue: systemic anticancer therapy or their combination; turquoise: non‐systemic anticancer therapies or combination of treatment modalities; white: other aspects of management and non‐treatment aspects; dashed lines: optional therapy. ASCT, autologous stem‐cell transplantation; CHOP, cyclophosphamide–doxorubicin–vincristine–prednisone; CODOX‐M, rituximab–cyclophosphamide–vincristine–doxorubicin–methotrexate; EMA, European Medicines Agency; EPOCH, etoposide–prednisone–vincristine–cyclophosphamide–doxorubicin; FDA, Food and Drug Administration; HIV–PBL, human immunodeficiency virus‐associated plasmablastic lymphoma; hyper‐CVAD, hyperfractionated cyclophosphamide–vincristine–doxorubicin–dexamethasone; IVAC, ifosfamide–etoposide–cytarabine; PBL, plasmablastic lymphoma; RT, radiotherapy. aNot EMA or FDA approved for the treatment of PBL.
Management of r/r HIV–PBL
There is even less evidence on which to base advice for the management of r/r HIV–PBL. Treatment of r/r HIV–PBL does not usually differ from treatment of other types of HIV–NHL. ASCT may be an option for those who did not receive it in first line, at least for young patients who achieve response with salvage ChT (usually platinum‐containing regimens). The use of antimyeloma agents, such as bortezomib and lenalidomide, alone or in combination with other treatments, has been attempted based on the plasmacytic differentiation of PBL cells, with some success in limited retrospective series. 47 Other agents with reported efficacy in case reports include BV and daratumumab. Figure 4 shows a proposed treatment algorithm for the management of r/r HIV–PBL.
Figure 4.
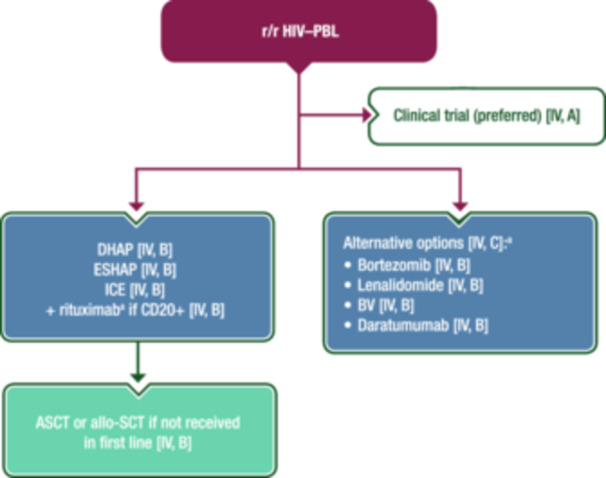
Management of r/r HIV–PBL. Purple: algorithm title; blue: systemic anticancer therapy or their combination; turquoise: non‐systemic anticancer therapies or combination of treatment modalities; white: other management and non‐treatment aspects. Allo‐SCT, allogeneic stem‐cell transplantation; ASCT, autologous stem‐cell transplantation; BV, brentuximab vedotin; CD, cluster of differentiation; DHAP, dexamethasone–cytarabine–cisplatin; EMA, European Medicines Agency; ESHAP, etoposide–methylprednisolone–cytarabine–cisplatin; FDA, Food and Drug Administration; HIV–PBL, human immunodeficiency virus‐associated plasmablastic lymphoma; ICE, ifosfamide–carboplatin–etoposide; PBL, plasmablastic lymphoma; r/r, relapsed or refractory. aNot EMA or FDA approved for the treatment of PBL.
Follow‐up, long‐term implications and survivorship
Although the prognosis of PBL is generally reported as poor, long‐term survival can be achieved. In an analysis of 135 patients, OS was better in PLWH than in HIV‐negative patients, which may be due to beneficial effects of ART on immune reconstitution. 43
Recommendations
Patients with HIV–PBL should be enrolled in clinical trials whenever possible [IV, A].
Patients with localised HIV–PBL should receive a CHOP‐ or EPOCH‐based regimen plus RT [IV, B].
-
Patients with disseminated HIV–PBL should receive polyChT (e.g., a CHOP‐based regimen) [IV, B].
-
∘
The addition of bortezomib is an option [IV, C; not EMA or FDA approved for PBL].
-
∘
Alternative regimens include EPOCH [IV, C], CODOX‐M or IVAC [IV, C] and hyper‐CVAD [IV, C].
-
∘
-
Consolidation with ASCT may be considered in suitable patients with advanced disease [IV, C].
-
∘
RT for local invasive disease may also be considered [V, C].
-
∘
In the relapse setting, platinum‐based salvage therapy [dexamethasone–Ara‐C–cisplatin (DHAP), etoposide–methylprednisolone–Ara‐C–cisplatin (ESHAP) or ifosfamide–carboplatin–etoposide (ICE)] plus rituximab if CD20 positive (not EMA or FDA approved for PBL), followed by ASCT or allo‐SCT (if not used in the first line) should be discussed [IV, B].
If the treatment above is not feasible, antimyeloma agents (bortezomib [IV, B], lenalidomide [IV, B], BV [IV, B] or daratumumab [IV, B], noting that none are EMA or FDA approved for PBL) are an option [IV, C].
PRIMARY EFFUSION LYMPHOMA
Diagnosis, pathology and molecular biology
The discovery of Kaposi sarcoma (KS) herpesvirus, or human herpesvirus 8 (HHV‐8), led to the recognition of primary effusion lymphoma (PEL) as a distinct lymphoproliferative disorder. PEL usually presents as serous effusions (ascites, pleuritis and/or pericarditis) without detectable tumour mass (60%–70% of cases) or as an extracavitary mass (extracavitary PEL, 30%–40% of cases). Diagnosis is based on cytological examination of serous fluid or pathological examination of a tumour biopsy specimen (see Supporting Information S1: Figure S1). Tumoural cells are polymorphic with immunoblasts, plasmablasts or anaplastic features. These cells are positive for the latency‐associated nuclear antigen (LANA) of HHV‐8 and are frequently (>70%) co‐infected by EBV (EBV‐encoded small RNAs), suggesting lymphomagenesis involves interplay between the two herpesviruses. The cells lack expression of most B‐cell markers except for a few terminal B‐cell differentiation markers (see Supporting Information S1: Table S3). The frequent aberrant expression of T‐cell markers may lead to misdiagnosis. In EBV‐negative cases, the differential diagnosis with HHV‐8‐positive large B‐cell lymphoma may be difficult.
Staging and risk assessment
As well as classical staging for lymphoma, staging of HIV‐associated PEL (HIV–PEL) should include evaluation of possible pericardial effusion (echocardiography and/or MRI). CSF analysis is also advisable.
Association with other HHV‐8‐related diseases, such as multicentric Castleman disease (MCD) and KS should be considered.
First‐line management of HIV–PEL
HIV–PEL is highly aggressive with a dismal prognosis and there is no established SoC. Based on retrospective studies and small case series, treatment options include ChT regimens used in other types of NHL in conjunction with ART. Figure 5 shows a proposed algorithm for the first‐line treatment of HIV–PEL.
Figure 5.
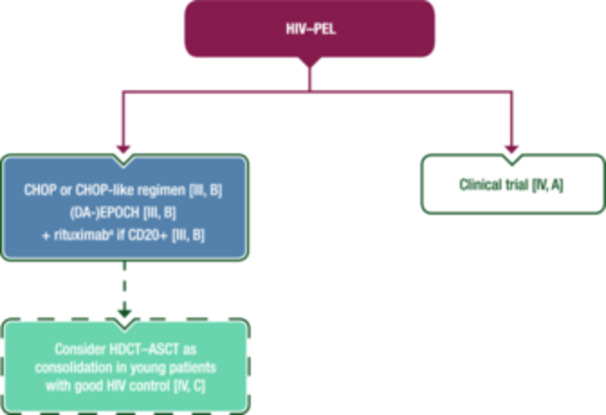
First‐line management of HIV–PEL. Purple: algorithm title; blue: systemic anticancer therapy or their combination; turquoise: non‐systemic anticancer therapies or combination of treatment modalities; white: other management and non‐treatment aspects; dashed lines: optional therapy. ASCT, autologous stem‐cell transplantation; CD, cluster of differentiation; CHOP, cyclophosphamide–doxorubicin–vincristine–prednisone; DA, dose‐adjusted; EMA, European Medicines Agency; EPOCH, etoposide–prednisone–vincristine–cyclophosphamide–doxorubicin; FDA, Food and Drug Administration; HDCT, high‐dose chemotherapy; HIV, human immunodeficiency virus; HIV–PEL, human immunodeficiency virus‐associated primary effusion lymphoma; PEL, primary effusion lymphoma. aNot EMA or FDA approved for the treatment of PEL.
CHOP‐based regimens and EPOCH provide similar CR rates (55%–57%), both with a median OS of <18 months. 48 , 49 Rituximab is not usually indicated, but some clinicians use it to treat concurrent MCD or to deplete the KS‐ or EBV‐associated herpesvirus‐infected B‐cell reservoir. 49
More aggressive ChT regimens or consolidation with ASCT might be considered in young patients with good HIV control, even in the absence of controlled studies. 50
Management of r/r HIV–PEL
r/r HIV–PEL is associated with a particularly poor prognosis. There is even less evidence in this setting than for first‐line treatment and no drug has been approved yet by the EMA or FDA. ASCT may be an option for patients who did not receive it in first line, as may allo‐SCT for those patients who respond to second‐line ChT. 51 , 52 Bortezomib and lenalidomide have been used alone or in combination with conventional ChT with some positive results. 53 Monoclonal antibodies directed at programmed death‐ligand 1, CD30 and CD38 are promising options and OS is expected to improve in the future. In a retrospective study, three of five patients with r/r HIV–PEL had a response with pembrolizumab with or without pomalidomide, including one CR and long‐term disease control. 54 Figure 6 shows a proposed algorithm for the treatment of r/r HIV–PEL.
Figure 6.
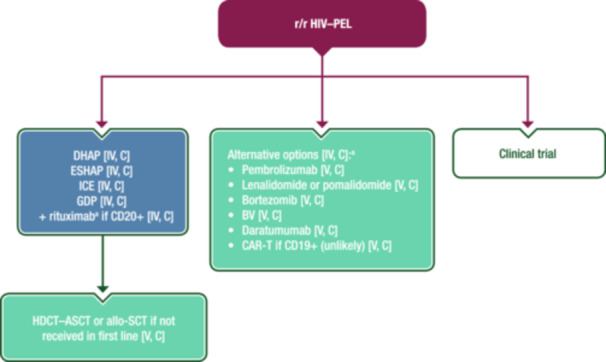
Management of r/r HIV–PEL. Purple: algorithm title; blue: systemic anticancer therapy or their combination; turquoise: non‐systemic anticancer therapies or combination of treatment modalities; white: other aspects of management and non‐treatment aspects. Allo‐SCT, allogeneic stem‐cell transplantation; ASCT, autologous stem‐cell transplantation; BV, brentuximab vedotin; CAR‐T, chimeric antigen receptor T‐cell therapy; CD, cluster of differentiation; DHAP, dexamethasone–cytarabine–cisplatin; EMA, European Medicines Agency; ESHAP, etoposide–methylprednisolone–cytarabine–cisplatin; FDA, Food and Drug Administration; GDP, gemcitabine–dexamethasone–cisplatin; HDCT, high‐dose chemotherapy; HIV–PEL, human immunodeficiency virus‐associated primary effusion lymphoma; ICE, ifosfamide–carboplatin–etoposide; PEL, primary effusion lymphoma; r/r, relapsed or refractory. aNot EMA or FDA approved for the treatment of PEL.
Recommendations
Detection of HHV‐8 and EBV using IHC, in situ hybridisation or quantitative PCR should be carried out on all specimens from patients with a suspected diagnosis of HIV–PEL [II, A].
CHOP‐based regimens and (DA‐)EPOCH can both be recommended with the addition of rituximab if CD20 positive (not EMA or FDA approved for PEL) [III, B].
HDCT–ASCT consolidation might be considered in young patients with good HIV control [IV, C].
In the relapse setting, salvage therapy [DHAP, ESHAP, ICE or gemcitabine–dexamethasone–cisplatin (GDP)] plus rituximab if CD20 positive (not EMA or FDA approved for PEL) [IV, C], followed by ASCT or allo‐SCT (if not used in first line) [V, C] could be discussed.
If the treatment above is not feasible, alternative options include pembrolizumab, lenalidomide or pomalidomide, bortezomib, BV, daratumumab or CAR‐T therapy if CD19 positive (unlikely) [V, C; noting that none are EMA or FDA approved for PEL].
CNS LYMPHOMA
Diagnosis, pathology and molecular biology
HIV‐associated primary CNS lymphoma (HIV–PCNSL) occurs in patients who are severely immunocompromised and differs in pathogenesis, natural history and treatment from primary CNS lymphoma (PCNSL) of the immunocompetent host. Nearly all PCNSLs are of B‐cell origin. In PLWH, the most frequent histology is DLBCL. Unlike PCNSL in immunocompetent hosts, EBV infection is essential to the pathobiology of HIV–PCNSL. Gene expression profiles in PCNSL are also different in PLWH compared with immunocompetent individuals. 55 PCNSL presents with acute or subacute neurological signs and symptoms depending on location, size of lesions and surrounding oedema; B symptoms are rare. Full medical and neurological evaluation and Mini‐Mental State Examination should be carried out alongside investigations including serum LDH, CSF analysis for EBV DNA using quantitative PCR, cytology and flow cytometry if lumbar puncture can be safely carried out, and contrast‐enhanced brain MRI. PCNSL can be multifocal or solitary and most commonly affects deep structures and white matter. 56 FDG–PET–CT scans support the diagnosis of PCNSL by excluding systemic involvement.
Staging and risk assessment
Ophthalmology assessment is essential as lymphomatous involvement may be identified in 5%–20% of cases. 57 In addition to FDG–PET–CT, testicular ultrasound is recommended in men to exclude occult systemic disease (present in ≤8% of cases), in which case the diagnosis is revised to secondary CNS lymphoma (SCNSL).
First‐line management of HIV–PCNSL
There is increasing evidence that HD‐MTX and ART can be used with curative intent, without whole brain RT (WBRT) or other consolidation. A retrospective study of 51 patients with HIV–PCNSL receiving a median of six infusions of HD‐MTX (3 g/m2) and ART demonstrated a median OS of 5.7 years and a 5‐year OS rate of 48%. 58 The only prospective trial conducted in this setting enrolled 12 patients treated with rituximab–HD‐MTX, with an estimated 5‐year OS rate of 67%. 59 Concurrent ART is essential for immune reconstitution and may contribute to long‐term disease control.
In the uncommon scenario of HIV–PCNSL in patients established on ART with well‐controlled HIV where the lymphoma pathogenesis and natural history resemble that of the immunocompetent host, clinicians should consider rituximab–MTX–Ara‐C–thiotepa (MATRix) 60 or a similar multi‐agent induction regimen and consolidation ASCT.
A proposed algorithm for first‐line treatment of HIV–PCNSL is shown in Figure 7.
Figure 7.
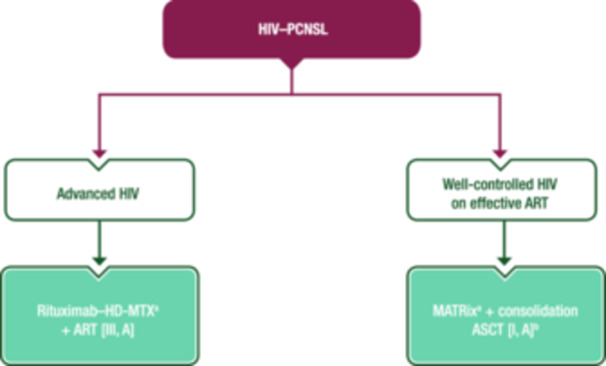
First‐line management of HIV–PCNSL. Purple: algorithm title; turquoise: non‐systemic anticancer therapies or combination of treatment modalities; white: other aspects of management and non‐treatment aspects. ART, combination antiretroviral therapy; ASCT, autologous stem‐cell transplantation; EMA, European Medicines Agency; FDA, Food and Drug Administration; HD‐MTX, high‐dose methotrexate; HIV, human immunodeficiency virus; HIV–PCNSL, human immunodeficiency virus‐associated primary central nervous system lymphoma; MATRix, rituximab–methotrexate–cytarabine–thiotepa; PCNSL, primary central nervous system lymphoma. aRituximab is not EMA or FDA approved for the treatment of PCNSL. bWhen the lymphoma resembles that of the immunocompetent host.
Management of r/r HIV–PCNSL
Both WBRT and HD‐MTX have been used in r/r HIV–PCNSL. WBRT remains a reasonable option in patients with chemorefractory disease, those who cannot tolerate HD‐MTX and for palliative intent in patients with poor functional status. Lenalidomide or Bruton tyrosine kinase inhibitors may also be considered if available. 61 Thiotepa–ICT, followed by ASCT if the disease is chemosensitive, is also an option.
Follow up, long‐term implications and survivorship
Brain MRI with contrast is the gold standard to assess treatment response 4–8 weeks after ChT. Thereafter, symptom‐driven investigations without routine rescanning are a reasonable approach as there is little evidence to support MRI brain surveillance in PLWH. Data on neurocognitive sequelae are scant. 59 , 62
Management of HIV‐associated SCNSL
Historically, SCNSL has been characterised by negative outcomes in both early (de novo presentation) and late (relapsed disease) settings. In the ART era, the frequency of CNS involvement in systemic HIV‐associated lymphomas is similar to that in patients who are HIV negative; 63 for this reason, treatment of secondary CNS DLBCL should not differ from that in the HIV‐negative population. Despite the exclusion of PLWH from the International Extranodal Lymphoma Study Group 42/MARIETTA study, the authors recommend consideration of the MARIETTA approach, which incorporates three courses of MATRix followed by three courses of R‐ICE and consolidation with carmustine–thiotepa and ASCT, for chemosensitive HIV‐associated SCNSL (HIV–SCNSL). 64 In patients with a first presentation of SCNSL, the 2‐year PFS rate (71%) was significantly improved compared with patients with CNS involvement at relapse (28%). CAR‐T therapy should also be considered in this setting, based on encouraging responses described in the HIV‐negative population. 65 , 66
Recommendations
HIV–PCNSL
Rituximab–HD‐MTX (≥3 g/m2) and fully active ART is recommended [III, A; rituximab is not EMA or FDA approved for PCNSL].
A multi‐agent induction regimen (e.g., MATRix) and consolidation ASCT should be considered for patients already established on effective ART, when the lymphoma resembles that of the immunocompetent host [I, A; rituximab is not EMA or FDA approved for PCNSL].
In the relapse setting, WBRT can be considered in patients who are chemorefractory or not suitable for ChT [III, B]. Thiotepa–ICT, followed by ASCT if the disease is chemosensitive, can also be considered [III, B].
HIV–SCNSL
Treatment for patients with HIV–SCNSL can be similar to that used for the HIV‐negative population and the MARIETTA approach or CAR‐T therapy should be considered [II, B].
HL
The main risk factor for HL in the HIV setting is a moderately lowered CD4 count. 5 In a recent study, the hazard ratio for HL was highest (6.36) among patients with CD4 counts of 100–200 cells/µL compared with the reference group (CD4 counts >500 cells/µL). 5 In contrast, higher HIV‐1 VL (>50 copies/mL) was not associated with an increased risk of HL compared with a VL of ≤50 copies/mL. 5
Diagnosis, pathology and molecular biology
HL is characterised by a small neoplastic infiltration of Hodgkin and Reed–Sternberg (HRS) cells against an inflammatory background of lymphocytes, histiocytes, plasma cells, eosinophils and neutrophils. The neoplastic HRS cells are typically CD30 and CD15 positive, with B‐cell markers such as CD20 and CD79a expressed in a minority of cells. EBV infection is almost universal in HIV–HL with a latency II type, expressing Epstein–Barr nuclear antigen 1, latent membrane protein 1 and 2 and EBV‐encoded RNA. Subtypes most frequently diagnosed in PLWH are mixed cellularity followed by nodular sclerosis. 67
Staging, risk assessment and response evaluation
In the ART era, initial staging and risk and response assessments are similar to those in the non‐HIV setting. Although PLWH display frequent adverse prognostic factors, HIV does not adversely affect OS per se. The Ann Arbor classification is used for initial staging and to subdivide patients according to the absence or presence of disease‐related symptoms. FDG–PET–CT is the gold standard for baseline and response assessments. A bone marrow aspirate or biopsy is no longer routinely required. 68 As in the non‐HIV setting, high metabolic tumour volume on initial FDG–PET–CT is associated with a worse prognosis. 69
FDG–PET–CT should be used for response assessment using the 5‐point scale. As in the non‐HIV setting, iFDG–PET–CT is prognostic in patients with HIV–HL treated with doxorubicin–bleomycin–vinblastine–dacarbazine (ABVD). 70 The Lymphoma Response to Immunomodulatory Therapy Criteria introduced the term ‘indeterminate response’ to describe lesions requiring biopsy or subsequent imaging to confirm pseudoprogression versus true progression. 71 These patterns deserve further evaluation in the HIV setting.
First‐line management of HIV–HL
Advanced‐stage disease is diagnosed in 66%–80% of HIV–HLs. As in the HIV‐negative setting, stage‐adapted treatment is recommended. A proposed treatment algorithm for HIV–HL is shown in Figure 8.
Figure 8.
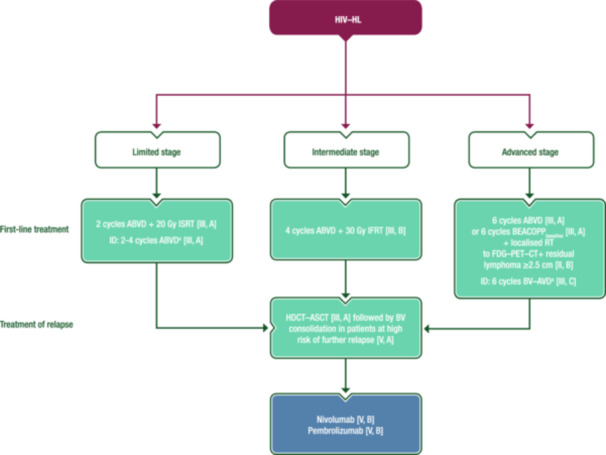
Management of HIV–HL. Purple: algorithm title; blue: systemic anticancer therapy or their combination; turquoise: non‐systemic anticancer therapies or combination of treatment modalities; white: other aspects of management and non‐treatment aspects. ABVD, doxorubicin–bleomycin–vinblastine–dacarbazine; ASCT, autologous stem‐cell transplantation; AVD, doxorubicin–vinblastine–dacarbazine; BEACOPP, bleomycin–etoposide–doxorubicin–cyclophosphamide–vincristine–procarbazine–prednisone; BV, brentuximab vedotin; CT, computed tomography; FDG, [18F]2‐fluoro‐2‐deoxy‐D‐glucose; FDG–PET–CT+, positive findings on FDG–PET–CT; HDCT, high‐dose chemotherapy; HIV–HL, human immunodeficiency virus‐associated Hodgkin lymphoma; ID, individual decision; IFRT, involved‐field radiotherapy; ISRT, involved‐site radiotherapy; PET, positron emission tomography; RT, radiotherapy. aNo data in HIV–HL.
Limited‐stage HL
Patients with limited‐stage HIV–HL, as defined by the German Hodgkin Study Group, should receive two cycles of ABVD followed by 20 Gy involved‐site RT (ISRT). 72 In a prospective study that included 23 patients with limited‐stage HIV–HL, two cycles of ABVD followed by 20 Gy involved‐field RT (IFRT) resulted in a CR rate of 96% and a 2‐year OS rate of 95.7% [95% confidence interval (CI) 87.7%–100%]. 73 As the use of 20 Gy and 30 Gy doses of RT proved equally effective in limited‐stage HL in the HIV‐negative setting, the lower dose should be preferred in limited‐stage HIV–HL. Moreover, there is accumulating evidence of excellent disease control with smaller ISRT fields. 74
If the long‐term risks of RT are thought to outweigh the benefit of improved PFS (e.g., in women ≤21 years of age with breast tissue in the radiation field), RT may be omitted or substituted with one or two additional cycles of ABVD in patients with a negative FDG–PET–CT scan after two cycles of ChT (i.e., PET2‐negative), although no data are available on the use of this approach in HIV–HL. 74
In patients who are HIV negative and have limited‐stage HL and a positive iFDG–PET–CT scan after two cycles of ABVD, the use of two cycles of escalated bleomycin–etoposide–doxorubicin–cyclophosphamide–vincristine–procarbazine–prednisone (BEACOPPescalated) before ISRT results in a reduced relapse rate. 75 This approach cannot, however, be generally recommended for limited‐stage HIV–HL as it has not been investigated in PLWH.
Intermediate‐stage HL
Limited data are available on the treatment of intermediate‐stage HIV–HL. Two‐year PFS and OS rates in 14 patients treated with four cycles of ABVD or bleomycin–etoposide–doxorubicin–cyclophosphamide–vincristine–procarbazine–prednisone (BEACOPPbaseline), followed by 30 Gy IFRT were 88% (95% CI: 67.3%–100%) and 100%, respectively. 73
In the HIV‐negative setting, the SoC for patients with intermediate‐stage HL is either four cycles of ABVD followed by 30 Gy ISRT or the 2 + 2 approach (two cycles of BEACOPPescalated followed by two cycles of ABVD), followed by 30 Gy ISRT if patients are PET4‐positive. 74 , 76 In patients initially treated with ABVD who have a positive FDG–PET–CT scan after two cycles, escalating treatment to two cycles of BEACOPPescalated (instead of ABVD) before ISRT results in significantly improved PFS. 75 As there is no data on these approaches in PLWH, they cannot be generally recommended for patients with intermediate‐stage HIV–HL.
Advanced‐stage HL
In a prospective trial including 71 PLWH with advanced‐stage HL, treatment with six to eight cycles of BEACOPPbaseline or ABVD was associated with a 2‐year PFS rate of 87.5% (95% CI: 79.1%–96.8%) and a 2‐year OS rate of 86.8% (95% CI: 79.0%–95.2%). 73 Four toxicity‐related deaths (5.6%) were reported among patients who received BEACOPPbaseline, three of which occurred after the seventh and eighth cycles. Thus, no more than six cycles of BEACOPPbaseline should be applied and caution is advised, especially in patients with low CD4 counts. This strategy is also supported by a retrospective study including 85 PLWH treated with six cycles of ABVD; there was no adverse effect of HIV infection on OS or EFS compared with patients who were HIV negative and treated with the same regimen. 77 A more recent report based on 12 patients with HIV–HL treated within the Southwest Oncology Group S0816 phase II trial showed that ABVD‐based FDG–PET–CT‐adapted therapy is feasible. 78 Patients with a negative PET2 subsequently received four additional cycles of ABVD, while those with a positive PET2 received six cycles of BEACOPPbaseline. Eleven of the 12 patients received six cycles of ABVD and three patients developed progressive disease (all PET2 negative), with an estimated 2‐year PFS rate of 83% (95% CI: 46.1%–95.3%). Data on the use of BEACOPPescalated in advanced‐stage HIV–HL are not available.
Based on prospective trials in the HIV‐negative setting, RT after ChT for advanced HL is restricted to patients with FDG–PET–CT‐positive residual disease; this approach is therefore also recommended in patients who are HIV positive. 74 BV is EMA and FDA approved in combination with doxorubicin–vinblastine–dacarbazine (AVD) in newly diagnosed stage IV HL, and six cycles of BV–AVD resulted in a 2‐year PFS rate of 87% in a phase I/II trial of patients with stage II–IV HIV–HL. 79 In this study, patients requiring ART that included strong cytochrome P450 3A4 inhibitors were excluded and there was a 10% incidence rate of grade 3‐4 peripheral sensory neuropathy.
There is increased use of BV–etoposide–doxorubicin–cyclophosphamide–dacarbazine–dexamethasone (BrECADD) instead of BEACOPP in patients without HIV aged ≤60 years, based on its more favourable toxicity profile; 80 however, data on BrECADD in HIV–HL are limited at this time.
Management of r/r HIV–HL
HD ChT (HDCT) followed by ASCT is SoC for r/r HL in the HIV‐negative setting and its feasibility in PLWH has been demonstrated in prospective trials and retrospective analyses. 21 , 81 PLWH with r/r HL who have a response to ART, adequate organ function and no active infections should, therefore, be treated with salvage ChT followed by HDCT–ASCT according to guidelines for the HIV‐negative setting. 74 Consolidation with BV following HDCT–ASCT was shown to improve tumour control in HIV‐negative patients with an increased risk of relapse. 82 Owing to the poor prognosis of patients with relapse after HDCT, BV consolidation should also be considered in PLWH with high‐risk r/r HL.
Patients relapsing after HDCT–ASCT should be treated with nivolumab or pembrolizumab, 83 and those not eligible for HDCT should be treated according to the recommendations for patients with HL who are HIV negative, favouring pembrolizumab over BV as data for pembrolizumab are available from a prospective phase III trial. 84
Recommendations
Patients with limited‐stage HIV–HL should receive two cycles of ABVD followed by 20 Gy ISRT [III, A].
Patients with intermediate‐stage HIV–HL can be treated with four cycles of ABVD followed by 30 Gy IFRT [III, B].
Patients with advanced‐stage HIV–HL should be treated with six cycles of ABVD or six cycles of BEACOPPbaseline [III, A] followed by RT for patients with FDG–PET–CT‐positive disease (residual lymphoma ≥2.5 cm) [II, B].
Six cycles of BV–AVD may be an individual option in newly diagnosed stage IV HIV–HL [III, C].
In the relapse setting, salvage ChT followed by ASCT is recommended in PLWH who are suitable for stem‐cell transplantation [III, A].
BV consolidation should be considered in patients with high‐risk r/r HIV–HL [V, A].
Patients relapsing after HDCT–ASCT can be treated with nivolumab [V, B] or pembrolizumab [V, B].
MCD
HHV‐8‐associated MCD (HHV‐8–MCD) is a rare lymphoproliferative disorder that is predominantly associated with HIV infection. Incidence does not correlate with CD4 count or HIV plasma viraemia.
Diagnosis, pathology and molecular biology
A combination of B symptoms, specific histopathological findings and detectable blood HHV‐8 is sufficient for diagnosis. 85 HHV‐8–MCD is characterised by episodic exacerbations and remissions. HHV‐8 encodes a viral homologue of interleukin (IL)‐6, the effects of which mirror those of its mammalian counterparts. Untreated HHV‐8–MCD can progress to a febrile, life‐threatening, acute illness with a rapid fatal course via multiple organ failure, haemophagocytic syndrome or evolution to NHL. The histological characteristics of affected lymph nodes include the presence of IgM lambda‐restricted plasmablasts with positive HHV‐8‐associated LANA staining.
Staging and risk assessment
Baseline CT or FDG–PET–CT is useful to identify affected nodes and to assess organ involvement. Eastern Cooperative Oncology Group PS, end‐organ involvement, haemophagocytic syndrome or haemolytic anaemia are helpful in defining severe cases for a risk‐stratified treatment approach. 86
First‐line management of HHV‐8–MCD
Although HHV‐8–MCD occurs mainly in patients with undetectable HIV RNA, ART should always be administered. Symptomatic HHV‐8–MCD usually requires prompt therapeutic intervention. Treatment evidence is based on two prospective studies 87 , 88 and a retrospective multicentre analysis, 89 which showed that rituximab is useful as monotherapy. In several cohort studies, OS and disease‐free survival improved with rituximab compared with historical controls. 87 , 88 , 89 High levels of HHV‐8 viraemia, serum C‐reactive protein, IL‐6 and IL‐10 rapidly decline after rituximab therapy. In severe cases, including those with haemophagocytic syndrome, etoposide can be added to first‐line rituximab to control the consequences of the cytokine storm. If KS is present at MCD diagnosis, the combination of rituximab with cytotoxic ChT, including etoposide or liposomal doxorubicin, is also considered as initial therapy for severe cases. 90 Other approaches such as antivirals, immunomodulators and IL‐6‐blocking agents are of limited value. In cases with severe refractory anaemia and thrombocytopenia, splenectomy could be considered despite providing only short‐term symptom resolution.
A proposed treatment algorithm for first‐line management of HHV‐8–MCD is shown in Figure 9.
Figure 9.
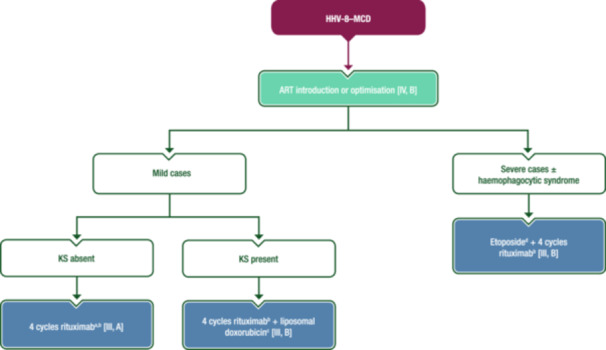
First‐line management of HHV‐8–MCD. Purple: algorithm title; blue: systemic anticancer therapy or their combination; turquoise: non‐systemic anticancer therapies or combination of treatment modalities; white: other management and non‐treatment aspects. ART, combination antiretroviral therapy; CD, cluster of differentiation; EMA, European Medicines Agency; FDA, Food and Drug Administration; HHV‐8–MCD, human herpesvirus 8‐associated multicentric Castleman disease; i.v., intravenous; KS, Kaposi sarcoma; MCD, multicentric Castleman disease. aEtoposide can be added to the first cycle to avoid a possible flare. bNot EMA or FDA approved for the treatment of MCD. Rituximab should be used with caution in patients with a CD4 count <50 cells/µL and/or delayed after a few courses of etoposide, but may be considered as it is associated with improved outcome in MCD and reduces the risk of lymphoma development. cThe number of liposomal doxorubicin cycles depends on the burden of KS disease, but the minimum number is four. dEtoposide can be administered orally or by i.v. injection; the i.v. route optimises bioavailability.
Management of r/r HHV‐8–MCD
With control of HHV‐8 VL, sustained CRs are often observed, but HHV‐8–MCD relapses are not infrequent and may occur after recovery of B‐cell counts. In case of relapse, multiple rechallenges with rituximab‐based ICT are safe and efficacious. 86 More aggressive ChT regimens (e.g., CHOP) can be considered in rituximab‐refractory disease.
Follow‐up, long‐term implications and survivorship
Clinical, virological and biochemical responses should be regularly monitored during acute episodes. Radiological response should be evaluated using CT or FDG–PET–CT after completion of ICT. Besides prolonged B‐cell depletion, the main serious adverse effect of rituximab is reactivation or progression of concomitant KS. In these settings, combination with liposomal doxorubicin should be considered.
NHL incidence is reduced with rituximab but remains high, especially for HHV‐8‐associated NHL (PEL, DLBCL). These entities should be considered during follow‐up. 86 , 91 Clinical follow‐up is recommended every 3–6 months. Patients should be made aware of the risk of relapse and should seek medical attention at the specialist centre if symptoms recur.
Recommendations
ART can be initiated or optimised [IV, B].
Rituximab is recommended as first‐line therapy [III, A; not EMA or FDA approved for MCD].
In severe cases, etoposide can be added to first‐line rituximab to control the consequences of the cytokine storm [III, B].
Combinations of rituximab with cytotoxic ChT, including etoposide or liposomal doxorubicin if KS is present at MCD diagnosis, are also considered as first‐line therapy for severe cases [III, B].
Antivirals, immunomodulators and IL‐6‐blocking agents are of limited value [IV, D].
In cases with severe refractory anaemia and thrombocytopenia, splenectomy could be considered [IV, C].
In case of relapse, multiple rechallenges with rituximab‐based ICT are safe and efficacious [III, B].
More aggressive ChT regimens (e.g., CHOP) can be considered in rituximab‐refractory disease [IV, B].
METHODOLOGY
This Clinical Practice Guideline (CPG) was developed in accordance with the ESMO standard operating procedures for CPG development (https://www.esmo.org/Guidelines/ESMO-Guidelines-Methodology). The relevant literature has been selected by the expert authors. The FDA/EMA or other regulatory body approval status of new therapies/indications is reported at the time of writing this CPG. Levels of evidence and grades of recommendation have been applied using the system shown in Supporting Information S1: Table S4. Statements without grading were considered justified standard clinical practice by the authors. The guideline uses people‐first terminology. For future updates to this CPG, including eUpdates and Living Guidelines, please see the ESMO Guidelines website: https://www.esmo.org/guidelines/guidelines-by-topic/haematological-malignancies.
AUTHOR CONTRIBUTIONS
All authors conceptualised, wrote and approved the final version.
CONFLICT OF INTEREST STATEMENT
KH reports personal fees for advisory board membership from Gilead, Hexal, Incyte, Miltenyi, Novartis, Recordati and Roche; personal fees as an invited speaker from BeiGene, Bristol Myers Squibb (BMS), Novartis, Recordati, Roche and Servier; personal fees for a writing engagement from Hexal; institutional fees as coordinating Principal Investigator (PI) from Incyte and Roche; a non‐renumerated role as working group chair of the German Society of Hematology and Medical Oncology (DGHO); non‐remunerated advisory role for the European Hematology Association (EHA) Education Committee; and a non‐remunerated leadership role for the German Lymphoma Alliance. MB reports personal fees as an invited speaker from BMS, EUSA Pharma, Gilead, Janssen, Merck and ViiV Healthcare. IA reports personal fees for advisory board membership from AbbVie, AstraZeneca, Eli Lilly, Genesis/Incyte, Janssen, Novartis, Roche, Sobi and Takeda; personal fees as an invited speaker from AbbVie, AstraZeneca, Eli Lilly, Genesis/Incyte, Janssen, Novartis, Roche, Sandoz, Sobi and Takeda; non‐remunerated leadership roles for the Croatian Cooperative Group for Hematologic Diseases and the EHA Lymphoma Specialized Working Group; and non‐renumerated membership of the Board of Directors of the Croatian Hematological Society and the European Lymphoma Institute. MBO reports personal fees for advisory board participation from Kite Pharma, Novartis and Roche; personal fees as an invited speaker from Janssen, Kite Pharma, Roche and Takeda; a personal and institutional research grant from Kite Pharma (Gilead/Kite Pharma award); and non‐renumerated leadership roles for the Spanish Lymphoma Group (GELTAMO; coordinator of the Aggressive Group and member of the Scientific Committee), the Madrid Hematology Society (member of the Board of Directors, treasurer) and the Spanish Society of Hematology and Hemotherapy (member of the Board of Directors, accountant). CBe reports personal fees for expert testimony from Janssen. UB reports personal fees for advisory board membership from Janssen‐Cilag; personal fees as an invited speaker from AstraZeneca and Janssen‐Cilag; personal fees for writing engagements from AbbVie and AstraZeneca; travel grants from AbbVie, BeiGene and Gilead; compensation for congress registration fees from Lilly; a non‐renumerated role as PI for Regeneron and Roche; and non‐renumerated membership of DGHO, the German Cancer Society (DKG) and EHA. CC reports no conflicts of interest. SC reports employment as a treatment advocate by the charity HIV i‐Base. KC reports personal fees for advisory board membership from AbbVie, Atara, Celgene, Incyte, Janssen, Kite, Roche and Takeda; personal fees as an invited speaker and personal travel grants/conference support from Celgene, Incyte, Kite, Roche and Takeda; personal fees for consulting from Atara, Celgene, Incyte and Kite. ADP reports institutional fees as an invited speaker from Gilead (non‐promotional teaching). MH reports personal fees for advisory board membership from Amgen, EUSA Pharma, Sanofi, Stemline and Takeda; and personal fees as an invited speaker from EUSA Pharma, Gilead, Janssen, Jazz Pharmaceuticals and Sanofi. CH reports personal fees for advisory board membership and as an invited speaker from EUSA Pharma, Gilead Sciences, Janssen‐Cilag, MSD and ViiV Healthcare; and institutional fees as local PI (for clinical studies conducted at institution) from Janssen‐Cilag, MSD and ViiV Healthcare. MJK reports institutional fees for advisory board membership from Adicet Bio, BMS/Celgene, Galapagos, Kite (a Gilead Company), Miltenyi Biotec and Novartis; institutional fees as an invited speaker from BeiGene, BMS/Celgene, Kite (a Gilead Company) and Novartis; institutional fees as local PI from BMS/Celgene; institutional fees as coordinating PI from Galapagos, Kite (a Gilead Company), Miltenyi Biotec and Novartis, and an institutional travel grant from AbbVie. SM reports personal fees for participation in a Data Monitoring Committee from Bayer. JTN reports personal fees for advisory board membership from Recordati Rare Diseases EUSA Pharma; personal fees as an invited speaker from Novartis and Recordati Rare Diseases EUSA Pharma; a personal research grant from Gilead Spain; and institutional funding from Recordati Rare Diseases EUSA Pharma. EO reports personal fees as an expert for grant attributions from CSL Behring; personal fees as a consultant from EUSA Pharma; an institutional research grant on unicentric Castleman disease pathogenesis from the Castleman Disease Collaborative Network (CDCN); and non‐renumerated membership of the advisory board of CDCN. AR reports personal fees for advisory board membership from Incyte, Italfarmaco and Takeda; personal fees as an expert discussant from Janssen and Servier; personal fees as an invited speaker from Sobi and Takeda; institutional fees as local PI from MSD, Pharmacyclis and Sanofi. JMR reports personal fees for advisory board membership from Amgen, Pfizer, Shire and Takeda; personal fees as an invited speaker from Amgen; and personal fees as local PI from Takeda. PS reports personal fees as an invited speaker from MSD and ViiV Healthcare; and a personal and institutional research grant from Gilead Sciences. BvT reports personal fees for advisory and consultancy roles for Allogene, Amgen, BMS/Celgene, Cerus, Gilead Kite, Incyte, IQVIA, Janssen‐Cilag GmbH, Lilly, Miltenyi, MSD, Noscendo, Novartis, Pentixapharm, Pfizer, Pierre Fabre, QualWorld, Roche, Sobi and Takeda; personal fees as an invited speaker from AbbVie, AstraZeneca, BMS/Celgene, Gilead Kite, Incyte, Lilly, MSD, Novartis, Roche Pharma AG and Takeda; institutional funding from Esteve, MSD, Novartis and Takeda; and travel support from AbbVie, AstraZeneca, Gilead Kite, Lilly, MSD, Pierre Fabre, Roche, Takeda and Novartis. CBu reports personal fees for advisory board membership from AbbVie, BeiGene, Celltrion, Gilead Sciences, Incyte, Janssen, Lilly Deutschland GmbH, MorphoSys, Novartis, Pfizer, Regeneron, Roche and Sobi; personal fees as an invited speaker from AbbVie, BeiGene, Celltrion, Gilead Sciences, Incyte, Janssen, Lilly Deutschland GmbH, MorphoSys, Novartis, Pfizer, Regeneron, Roche and Sobi; and institutional funding from AbbVie Amgen, Bayer, Celltrion, Janssen, MSD, Pfizer and Roche (all for investigator‐sponsored clinical trials and registries). MD reports personal fees as an advisory board member from AbbVie, AstraZeneca, BeiGene, BMS/Celgene, Gilead, Janssen, Lilly/Loxo, Novartis and Roche; personal fees as an invited speaker for AstraZeneca, BeiGene, Gilead/Kite, Janssen, Lilly, Novartis and Roche; institutional research grants from AbbVie, Bayer, Celgene, Janssen, Lilly and Roche; institutional funding from Gilead/Kite; and non‐renumerated membership of the American Society of Clinical Oncology, American Society of Hematology (subcommittee), DGHO (prior Board member), EHA (Executive Board), ESMO (Faculty) and the Lymphoma Research Foundation (Mantle Cell Lymphoma Consortium). AD reports personal fees as an advisory board member for AbbVie, Acerta Pharma, AstraZeneca, BMS/Celgene, Genmab, Gilead, Incyte, Karyopharm, Kite Pharma, Regeneron, Roche, Sobi and Takeda; personal fees as an invited speaker for AstraZeneca, Gilead and Roche; institutional research grants for conduct of commercial research and funding of IST from Acerta Pharma and Roche; institutional research grants for conduct of commercial research from ADC Therapeutics, AstraZeneca, BMS/Celgene, Gilead and Pfizer; institutional research grant from MSD (no financial interest); non‐renumerated leadership role, member and UK Board representative for the Precision Medicine in Aggressive Lymphoma Consortium of the International Extranodal Lymphoma Study Group; advisory role and international advisor for the Swiss SAKK Lymphoma Project Group and member of the UK National Cancer Research Institute's High Grade Lymphoma Study Group.
FUNDING
No external funding has been received for the preparation of this guideline. Production costs have been covered by ESMO (for Annals of Oncology) and EHA (for HemaSphere) central funds.
Supporting information
Supporting information.
ACKNOWLEDGMENTS
This article has been simultaneously copublished in HemaSphere and Annals of Oncology. The articles are identical except for minor stylistic and spelling differences in keeping with each journal's style. Either citation can be used when citing this article. Manuscript editing support was provided by Ioanna Ntai (ESMO Guidelines staff) and Nicky French, Sian‐Marie Lucas and Angela Corstorphine of Kstorfin Medical Communications Ltd (KMC); this support was funded by ESMO.
This guideline was developed by the EHA and ESMO. The two societies nominated authors to write the guidelines as well as reviewers to comment on them. This guideline was approved by the EHA Board and the ESMO Guidelines Committee in May 2024.
Contributor Information
Kai Hübel, Email: guidelines@ehaweb.org.
Mark Bower, Email: clinicalguidelines@esmo.org.
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
REFERENCES
- 1. Poizot‐Martin I, Lions C, Allavena C, et al. Spectrum and incidence trends of AIDS‐ and non‐AIDS‐defining cancers between 2010 and 2015 in the French Dat'AIDS cohort. Cancer Epidemiol Biomarkers Prev. 2021;30(3):554‐563. [DOI] [PubMed] [Google Scholar]
- 2. Hernandez‐Ramirez RU, Shiels MS, Dubrow R, et al. Cancer risk in HIV‐infected people in the USA from 1996 to 2012: a population‐based, registry‐linkage study. Lancet HIV. 2017;4(11):e495‐e504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kimani SM, Painschab MS, Horner MJ, et al. Epidemiology of haematological malignancies in people living with HIV. Lancet HIV. 2020;7(9):e641‐e651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carbone A, Vaccher E, Gloghini A. Hematologic cancers in individuals infected by HIV. Blood. 2022;139(7):995‐1012. [DOI] [PubMed] [Google Scholar]
- 5. Shepherd L, Ryom L, Law M, et al. Differences in virological and immunological risk factors for non‐Hodgkin and Hodgkin lymphoma. J Natl Cancer Inst. 2018;110(6):598‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mhlanga JC, Durand D, Tsai HL, et al. Differentiation of HIV‐associated lymphoma from HIV‐associated reactive adenopathy using quantitative FDG PET and symmetry. Eur J Nucl Med Mol Imaging. 2014;41(4):596‐604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Marcus C, Feizi P, Hogg J, et al. Imaging in differentiating cerebral toxoplasmosis and primary CNS lymphoma with special focus on FDG PET/CT. Am J Roentgenol. 2021;216(1):157‐164. [DOI] [PubMed] [Google Scholar]
- 8. Barta SK, Xue X, Wang D, et al. Treatment factors affecting outcomes in HIV‐associated non‐Hodgkin lymphomas: a pooled analysis of 1546 patients. Blood. 2013;122(19):3251‐3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Masur H, Brooks JT, Benson CA, et al. Prevention and treatment of opportunistic infections in HIV‐infected adults and adolescents: updated Guidelines from the Centers for Disease Control and Prevention, National Institutes of Health, and HIV Medicine Association of the Infectious Diseases Society of America. Clin Infect Dis. 2014;58(9):1308‐1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang Q, De Luca A, Smith C, et al. Chronic Hepatitis B and C virus infection and risk for non‐Hodgkin lymphoma in HIV‐infected patients: a cohort study. Ann Intern Med. 2017;166(1):9‐17. [DOI] [PubMed] [Google Scholar]
- 11. Besson C, Noel N, Lancar R, et al. Hepatitis C virus or hepatitis B virus coinfection and lymphoma risk in people living with HIV. AIDS. 2020;34(4):599‐608. [DOI] [PubMed] [Google Scholar]
- 12. Chao C, Silverberg MJ, Xu L, et al. A comparative study of molecular characteristics of diffuse large B‐cell lymphoma from patients with and without human immunodeficiency virus infection. Clin Cancer Res. 2015;21(6):1429‐1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Barta SK, Samuel MS, Xue X, et al. Changes in the influence of lymphoma‐ and HIV‐specific factors on outcomes in AIDS‐related non‐Hodgkin lymphoma. Ann Oncol. 2015;26(5):958‐966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Noy A. Optimizing treatment of HIV‐associated lymphoma. Blood. 2019;134(17):1385‐1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brunnberg U, Hentrich M, Hoffmann C, et al. HIV‐associated malignant lymphoma. Oncol Res Treat. 2017;40(3):82‐87. [DOI] [PubMed] [Google Scholar]
- 16. Barta SK, Lee JY, Kaplan LD, et al. Pooled analysis of AIDS malignancy consortium trials evaluating rituximab plus CHOP or infusional EPOCH chemotherapy in HIV‐associated non‐Hodgkin lymphoma. Cancer. 2012;118(16):3977‐3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pagani C, Rusconi C, Dalla Pria A, et al. MYC rearrangements in HIV‐associated large B‐cell lymphomas: EUROMYC, a European retrospective study. Blood Adv. 2024;8(4):968‐977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tilly H, Morschhauser F, Sehn LH, et al. Polatuzumab Vedotin in previously untreated diffuse large B‐cell lymphoma. N Engl J Med. 2022;386(4):351‐363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dunleavy K, Little RF, Pittaluga S, et al. The role of tumor histogenesis, FDG‐PET, and short‐course EPOCH with dose‐dense rituximab (SC‐EPOCH‐RR) in HIV‐associated diffuse large B‐cell lymphoma. Blood. 2010;115(15):3017‐3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hegde U, Filie A, Little RF, et al. High incidence of occult leptomeningeal disease detected by flow cytometry in newly diagnosed aggressive B‐cell lymphomas at risk for central nervous system involvement: the role of flow cytometry versus cytology. Blood. 2005;105(2):496‐502. [DOI] [PubMed] [Google Scholar]
- 21. Hubel K, Re A, Boumendil A, et al. Autologous stem cell transplantation for HIV‐associated lymphoma in the antiretroviral and rituximab era: a retrospective study by the EBMT Lymphoma Working Party. Bone Marrow Transplant. 2019;54(10):1625‐1631. [DOI] [PubMed] [Google Scholar]
- 22. Li J. Advances toward a cure for HIV: getting beyond n=2. Top Antivir Med. 2020;27(4):91‐95. [PMC free article] [PubMed] [Google Scholar]
- 23. Abramson JS, Irwin KE, Frigault MJ, et al. Successful anti‐CD19 CAR T‐cell therapy in HIV‐infected patients with refractory high‐grade B‐cell lymphoma. Cancer. 2019;125(21):3692‐3698. [DOI] [PubMed] [Google Scholar]
- 24. Hattenhauer ST, Mispelbaum R, Hentrich M, et al. Enabling CAR T‐cell therapies for HIV‐positive lymphoma patients—a call for action. HIV Med. 2023;24(9):957‐964. [DOI] [PubMed] [Google Scholar]
- 25. Coutinho R, Pria AD, Gandhi S, et al. HIV status does not impair the outcome of patients diagnosed with diffuse large B‐cell lymphoma treated with R‐CHOP in the cART era. AIDS. 2014;28(5):689‐697. [DOI] [PubMed] [Google Scholar]
- 26. Poizot‐Martin I, Lions C, Delpierre C, et al. Prevalence and spectrum of second primary malignancies among people living with HIV in the French Dat'AIDS cohort. Cancers (Basel). 2022;14(2):401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gooden TE, Gardner M, Wang J, et al. Incidence of cardiometabolic diseases in people with and without human immunodeficiency virus in the United Kingdom: a population‐based matched cohort study. J Infect Dis. 2022;225(8):1348‐1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dolcetti R, Gloghini A, Caruso A, et al. A lymphomagenic role for HIV beyond immune suppression? Blood. 2016;127(11):1403‐1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Olszewski AJ, Jakobsen LH, Collins GP, et al. Burkitt lymphoma international prognostic index. J Clin Oncol. 2021;39(10):1129‐1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hoelzer D, Walewski J, Dohner H, et al. Improved outcome of adult Burkitt lymphoma/leukemia with rituximab and chemotherapy: report of a large prospective multicenter trial. Blood. 2014;124(26):3870‐3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xicoy B, Ribera JM, Muller M, et al. Dose‐intensive chemotherapy including rituximab is highly effective but toxic in human immunodeficiency virus‐infected patients with Burkitt lymphoma/leukemia: parallel study of 81 patients. Leuk Lymphoma. 2014;55(10):2341‐2348. [DOI] [PubMed] [Google Scholar]
- 32. Ribera JM, Morgades M, Garcia‐Calduch O, et al. Feasibility and outcomes after dose reduction of immunochemotherapy in young adults with Burkitt lymphoma and leukemia: results of the BURKIMAB14 trial. Haematologica. 2024;109(2):543‐552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alderuccio JP, Olszewski AJ, Evens AM, et al. Prognostication, survival and treatment‐related outcomes in HIV‐associated Burkitt lymphoma (HIV‐BL): a US and UK Collaborative Analysis. Blood. 2020;136(Suppl 1):49‐50. [Google Scholar]
- 34. Noy A, Lee JY, Cesarman E, et al. AMC 048: modified CODOX‐M/IVAC‐rituximab is safe and effective for HIV‐associated Burkitt lymphoma. Blood. 2015;126(2):160‐166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dunleavy K, Pittaluga S, Shovlin M, et al. Low‐intensity therapy in adults with Burkitt's lymphoma. N Engl J Med. 2013;369(20):1915‐1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Roschewski M, Dunleavy K, Abramson JS, et al. Multicenter study of risk‐adapted therapy with dose‐adjusted EPOCH‐R in adults with untreated Burkitt lymphoma. J Clin Oncol. 2020;38(22):2519‐2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zayac AS, Evens AM, Danilov A, et al. Outcomes of Burkitt lymphoma with central nervous system involvement: evidence from a large multicenter cohort study. Haematologica. 2021;106(7):1932‐1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ferreri AJM, Cattaneo C, Lleshi A, et al. A dose‐dense short‐term therapy for human immunodeficiency virus/acquired immunodeficiency syndrome patients with high‐risk Burkitt lymphoma or high‐grade B‐cell lymphoma: safety and efficacy results of the “CARMEN” phase II trial. Br J Haematol. 2021;192(1):119‐128. [DOI] [PubMed] [Google Scholar]
- 39. Maramattom LV, Hari PN, Burns LJ, et al. Autologous and allogeneic transplantation for burkitt lymphoma outcomes and changes in utilization: a report from the center for international blood and marrow transplant research. Biol Blood Marrow Transplant. 2013;19(2):173‐179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gopal S, Patel MR, Yanik EL, et al. Temporal trends in presentation and survival for HIV‐associated lymphoma in the antiretroviral therapy era. J Natl Cancer Inst. 2013;105(16):1221‐1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schommers P, Hentrich M, Hoffmann C, et al. Survival of AIDS‐related diffuse large B‐cell lymphoma, Burkitt lymphoma, and plasmablastic lymphoma in the German HIV Lymphoma Cohort. Br J Haematol. 2015;168(6):806‐810. [DOI] [PubMed] [Google Scholar]
- 42. Lopez A, Abrisqueta P. Plasmablastic lymphoma: current perspectives. Blood Lymphat Cancer. 2018;8:63‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tchernonog E, Faurie P, Coppo P, et al. Clinical characteristics and prognostic factors of plasmablastic lymphoma patients: analysis of 135 patients from the LYSA group. Ann Oncol. 2017;28(4):843‐848. [DOI] [PubMed] [Google Scholar]
- 44. Hess BT, Giri A, Park Y, et al. Outcomes of patients with limited‐stage plasmablastic lymphoma: a multi‐institutional retrospective study. Am J Hematol. 2023;98(2):300‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Castillo JJ, Guerrero‐Garcia T, Baldini F, et al. Bortezomib plus EPOCH is effective as frontline treatment in patients with plasmablastic lymphoma. Br J Haematol. 2019;184(4):679‐682. [DOI] [PubMed] [Google Scholar]
- 46. Al‐Malki MM, Castillo JJ, Sloan JM, et al. Hematopoietic cell transplantation for plasmablastic lymphoma: a review. Biol Blood Marrow Transplant. 2014;20(12):1877‐1884. [DOI] [PubMed] [Google Scholar]
- 47. Ando K, Imaizumi Y, Kobayashi Y, et al. Bortezomib‐ and lenalidomide‐based treatment of refractory plasmablastic lymphoma. Oncol Res Treat. 2020;43(3):112‐116. [DOI] [PubMed] [Google Scholar]
- 48. Guillet S, Gerard L, Meignin V, et al. Classic and extracavitary primary effusion lymphoma in 51 HIV‐infected patients from a single institution. Am J Hematol. 2016;91(2):233‐237. [DOI] [PubMed] [Google Scholar]
- 49. Shanmugasundaram KRR, Widell A, Mangusan R, et al. Treatment outcome and prognostic factors on 40 patients with primary effusion lymphoma. Blood. 2021;138(Suppl 1):1437. [Google Scholar]
- 50. Mirza AS, Dholaria BR, Hussaini M, et al. High‐dose therapy and autologous hematopoietic cell transplantation as consolidation treatment for primary effusion lymphoma. Clin Lymphoma Myeloma Leuk. 2019;19(9):e513‐e520. [DOI] [PubMed] [Google Scholar]
- 51. Won JH, Han SH, Bae SB, et al. Successful eradication of relapsed primary effusion lymphoma with high‐dose chemotherapy and autologous stem cell transplantation in a patient seronegative for human immunodeficiency virus. Int J Hematol. 2006;83(4):328‐330. [DOI] [PubMed] [Google Scholar]
- 52. Bryant A, Milliken S. Successful reduced‐intensity conditioning allogeneic HSCT for HIV‐related primary effusion lymphoma. Biol Blood Marrow Transplant. 2008;14(5):601‐602. [DOI] [PubMed] [Google Scholar]
- 53. Shimada K, Hayakawa F, Kiyoi H. Biology and management of primary effusion lymphoma. Blood. 2018;132(18):1879‐1888. [DOI] [PubMed] [Google Scholar]
- 54. Lurain K, Ramaswami R, Mangusan R, et al. Use of pembrolizumab with or without pomalidomide in HIV‐associated non‐Hodgkin's lymphoma. J Immunother Cancer. 2021;9(2):e002097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gandhi MK, Hoang T, Law SC, et al. EBV‐associated primary CNS lymphoma occurring after immunosuppression is a distinct immunobiological entity. Blood. 2021;137(11):1468‐1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Thurnher MM, Thurnher SA, Schindler E. CNS involvement in AIDS: spectrum of CT and MR findings. Eur Radiol. 1997;7(7):1091‐1097. [DOI] [PubMed] [Google Scholar]
- 57. Maher EA, Fine HA. Primary CNS lymphoma. Semin Oncol. 1999;26(3):346‐356. [PubMed] [Google Scholar]
- 58. Moulignier A, Lamirel C, Picard H, et al. Long‐term AIDS‐related PCNSL outcomes with HD‐MTX and combined antiretroviral therapy. Neurology. 2017;89(8):796‐804. [DOI] [PubMed] [Google Scholar]
- 59. Lurain K, Uldrick TS, Ramaswami R, et al. Treatment of HIV‐associated primary CNS lymphoma with antiretroviral therapy, rituximab, and high‐dose methotrexate. Blood. 2020;136(19):2229‐2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ferreri AJM, Cwynarski K, Pulczynski E, et al. Long‐term efficacy, safety and neurotolerability of MATRix regimen followed by autologous transplant in primary CNS lymphoma: 7‐year results of the IELSG32 randomized trial. Leukemia. 2022;36(7):1870‐1878. [DOI] [PubMed] [Google Scholar]
- 61. Rubenstein JL, Geng H, Fraser EJ, et al. Phase 1 investigation of lenalidomide/rituximab plus outcomes of lenalidomide maintenance in relapsed CNS lymphoma. Blood Adv. 2018;2(13):1595‐1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nagai H, Odawara T, Ajisawa A, et al. Whole brain radiation alone produces favourable outcomes for AIDS‐related primary central nervous system lymphoma in the HAART era. Eur J Haematol. 2010;84(6):499‐505. [DOI] [PubMed] [Google Scholar]
- 63. Barta SK, Joshi J, Mounier N, et al. Central nervous system involvement in AIDS‐related lymphomas. Br J Haematol. 2016;173(6):857‐866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ferreri AJM, Doorduijn JK, Re A, et al. MATRix‐RICE therapy and autologous haematopoietic stem‐cell transplantation in diffuse large B‐cell lymphoma with secondary CNS involvement (MARIETTA): an international, single‐arm, phase 2 trial. Lancet Haematol. 2021;8(2):e110‐e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Karschnia P, Arrillaga‐Romany IC, Eichler A, et al. Neurotoxicity and management of primary and secondary CNS lymphoma after adoptive immunotherapy with CD19‐directed CAR T‐cells. Neuro Oncol. 2023;25(12):2239‐2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cook MR, Dorris CS, Makambi KH, et al. Toxicity and efficacy of CAR T‐cell therapy in primary and secondary CNS lymphoma: a meta‐analysis of 128 patients. Blood Adv. 2023;7(1):32‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Navarro JT, Molto J, Tapia G, et al. Hodgkin lymphoma in people living with HIV. Cancers (Basel). 2021;13(17):4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lawal IO, Nyakale NE, Harry LM, et al. The role of F‐18 FDG PET/CT in evaluating the impact of HIV infection on tumor burden and therapy outcome in patients with Hodgkin lymphoma. Eur J Nucl Med Mol Imaging. 2017;44(12):2025‐2033. [DOI] [PubMed] [Google Scholar]
- 69. Louarn N, Galicier L, Bertinchamp R, et al. First extensive analysis of (18)F‐labeled fluorodeoxyglucose positron emission tomography‐computed tomography in a large cohort of patients with HIV‐associated Hodgkin lymphoma: baseline total metabolic tumor volume affects prognosis. J Clin Oncol. 2022;40(12):1346‐1355. [DOI] [PubMed] [Google Scholar]
- 70. Okosun J, Warbey V, Shaw K, et al. Interim fluoro‐2‐deoxy‐D‐glucose‐PET predicts response and progression‐free survival in patients with Hodgkin lymphoma and HIV infection. AIDS. 2012;26(7):861‐865. [DOI] [PubMed] [Google Scholar]
- 71. Cheson BD, Ansell S, Schwartz L, et al. Refinement of the Lugano classification lymphoma response criteria in the era of immunomodulatory therapy. Blood. 2016;128(21):2489‐2496. [DOI] [PubMed] [Google Scholar]
- 72. Brockelmann PJ, Engert A. The GHSG approach to treating Hodgkin's lymphoma. Curr Hematol Malig Rep. 2015;10(3):256‐265. [DOI] [PubMed] [Google Scholar]
- 73. Hentrich M, Berger M, Wyen C, et al. Stage‐adapted treatment of HIV‐associated Hodgkin lymphoma: results of a prospective multicenter study. J Clin Oncol. 2012;30(33):4117‐4123. [DOI] [PubMed] [Google Scholar]
- 74. Eichenauer DA, Aleman BMP, Andre M, et al. Hodgkin lymphoma: ESMO clinical practice guidelines for diagnosis, treatment and follow‐up. Ann Oncol. 2018;29(Suppl 4):iv19‐iv29. [DOI] [PubMed] [Google Scholar]
- 75. Andre MPE, Girinsky T, Federico M, et al. Early positron emission tomography response‐adapted treatment in stage I and II Hodgkin lymphoma: final results of the randomized EORTC/LYSA/FIL H10 trial. J Clin Oncol. 2017;35(16):1786‐1794. [DOI] [PubMed] [Google Scholar]
- 76. Borchmann P, Plutschow A, Kobe C, et al. PET‐guided omission of radiotherapy in early‐stage unfavourable Hodgkin lymphoma (GHSG HD17): a multicentre, open‐label, randomised, phase 3 trial. Lancet Oncol. 2021;22(2):223‐234. [DOI] [PubMed] [Google Scholar]
- 77. Montoto S, Shaw K, Okosun J, et al. HIV status does not influence outcome in patients with classical Hodgkin lymphoma treated with chemotherapy using doxorubicin, bleomycin, vinblastine, and dacarbazine in the highly active antiretroviral therapy era. J Clin Oncol. 2012;30(33):4111‐4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Danilov AV, Li H, Press OW, et al. Feasibility of interim positron emission tomography (PET)‐adapted therapy in HIV‐positive patients with advanced Hodgkin lymphoma (HL): a sub‐analysis of SWOG S0816 Phase 2 trial. Leuk Lymphoma. 2017;58(2):461‐465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Rubinstein PG, Moore PC, Bimali M, et al. Brentuximab vedotin with AVD for stage II‐IV HIV‐related Hodgkin lymphoma (AMC 085): phase 2 results from an open‐label, single arm, multicentre phase 1/2 trial. Lancet Haematol. 2023;10(8):e624‐e632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Eichenauer DA, Plütschow A, Kreissl S, et al. Incorporation of brentuximab vedotin into first‐line treatment of advanced classical Hodgkin's lymphoma: final analysis of a phase 2 randomised trial by the German Hodgkin Study Group. Lancet Oncol. 2017;18(12):1680‐1687. [DOI] [PubMed] [Google Scholar]
- 81. Alvarnas JC, Le Rademacher J, Wang Y, et al. Autologous hematopoietic cell transplantation for HIV‐related lymphoma: results of the BMT CTN 0803/AMC 071 trial. Blood. 2016;128(8):1050‐1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Moskowitz CH, Nademanee A, Masszi T, et al. Brentuximab vedotin as consolidation therapy after autologous stem‐cell transplantation in patients with Hodgkin's lymphoma at risk of relapse or progression (AETHERA): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet. 2015;385(9980):1853‐1862. [DOI] [PubMed] [Google Scholar]
- 83. Serrao A, Canichella M, De Luca ML, et al. Nivolumab as a safe and effective treatment in an HIV patient with refractory Hodgkin lymphoma. Ann Hematol. 2019;98(6):1505‐1506. [DOI] [PubMed] [Google Scholar]
- 84. Kuruvilla J, Ramchandren R, Santoro A, et al. Pembrolizumab versus brentuximab vedotin in relapsed or refractory classical Hodgkin lymphoma (KEYNOTE‐204): an interim analysis of a multicentre, randomised, open‐label, phase 3 study. Lancet Oncol. 2021;22(4):512‐524. [DOI] [PubMed] [Google Scholar]
- 85. Bower M, Pria AD, Coyle C, et al. Diagnostic criteria schemes for multicentric Castleman disease in 75 cases. J Acquir Immune Defic Syndr. 2014;65(2):e80‐e82. [DOI] [PubMed] [Google Scholar]
- 86. Pria AD, Pinato D, Roe J, et al. Relapse of HHV8‐positive multicentric Castleman disease following rituximab‐based therapy in HIV‐positive patients. Blood. 2017;129(15):2143‐2147. [DOI] [PubMed] [Google Scholar]
- 87. Bower M, Powles T, Williams S, et al. Brief communication: rituximab in HIV‐associated multicentric Castleman disease. Ann Intern Med. 2007;147(12):836‐839. [DOI] [PubMed] [Google Scholar]
- 88. Gerard L, Berezne A, Galicier L, et al. Prospective study of rituximab in chemotherapy‐dependent human immunodeficiency virus associated multicentric Castleman's disease: ANRS 117 CastlemaB Trial. J Clin Oncol. 2007;25(22):3350‐3356. [DOI] [PubMed] [Google Scholar]
- 89. Hoffmann C, Schmid H, Muller M, et al. Improved outcome with rituximab in patients with HIV‐associated multicentric Castleman disease. Blood. 2011;118(13):3499‐3503. [DOI] [PubMed] [Google Scholar]
- 90. Uldrick TS, Polizzotto MN, Aleman K, et al. Rituximab plus liposomal doxorubicin in HIV‐infected patients with KSHV‐associated multicentric Castleman disease. Blood. 2014;124(24):3544‐3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Gerard L, Michot JM, Burcheri S, et al. Rituximab decreases the risk of lymphoma in patients with HIV‐associated multicentric Castleman disease. Blood. 2012;119(10):2228‐2233. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.