

Abstract
Purpose:
To identify the accurate clinical diagnosis of rare syndromic inherited retinal diseases (IRDs) based on the combination of clinical and genetic analyses.
Methods:
Four unrelated families with various autosomal recessive syndromic inherited retinal diseases were genetically investigated using whole-exome sequencing (WES).
Results:
Two affected subjects in family MOL0760 presented with a distinctive combination of short stature, developmental delay, congenital mental retardation, microcephaly, facial dysmorphism and retinitis pigmentosa (RP). Subjects were clinically diagnosed with suspected Kabuki syndrome. WES revealed a homozygous nonsense mutation (c.5492dup, p.Asn1831Lysfs*8) in VPS13B that is known to cause Cohen syndrome. The index case of family MOL1514 presented with both RP and liver dysfunction, suspected initially to be related. WES identified a homozygous frameshift mutation (c.1787_1788del, p.His596Argfs*47) in AGBL5, associated with nonsyndromic RP. The MOL1592 family included three affected subjects with crystalline retinopathy, skin ichthyosis, short stature and congenital adrenal hypoplasia, and were found to harbour a homozygous nonsense mutation (c.682C>T, p.Arg228Cys) in ALDH3A2, reported to cause Sjögren-Larsson syndrome (SLS). In the fourth family, SJ002, two siblings presented with hypotony, psychomotor delay, dysmorphic facial features, pathologic myopia, progressive external ophthalmoplegia and diffuse retinal atrophy. Probands were suspected to have atypical Kearns-Sayre syndrome, but were diagnosed with combined oxidative phosphorylation deficiency-20 due to a novel suspected missense variant (c.1691C>T, p.Ala564Val) in VARS2.
Conclusion:
Our findings emphasize the important complement of WES and thorough clinical investigation in establishing precise clinical diagnosis. This approach constitutes the basis for personalized medicine in rare IRDs.
Keywords: inherited rare disease, precise clinical diagnosis, syndromic retinal disease, whole-exome sequencing
Introduction
Inherited retinal diseases (IRDs) are a group of heterogeneous genetic ocular phenotypes. Retinitis pigmentosa (RP) is the most common IRD in humans (MIM #268000) with an estimated worldwide prevalence of 1:4500 (Bundey & Crews 1984; Haim 2002). The disease is both clinically and genetically heterogeneous, including multiple patterns of inheritance: autosomal recessive (AR) (50–60% of cases), autosomal dominant (AD) (30–40%) and X-linked (XL) (5–15%) (Hartong et al. 2006). Mostly, RP presents as non-syndromic, while only 20–30% of cases are considered syndromic (Hartong et al. 2006) and are associated with additional clinical features such as: sensorineural hearing loss (Usher syndrome); ataxia, developmental delay, hyperpnea, hypotonia and polydactyly (Joubert syndrome); obesity, polydactyly, renal failure and hypogonadism (Bardet-Beidl syndrome); and renal failure (Senior-Loken syndrome) (Hartong et al. 2006). Usher syndrome is the most common syndromic phenotype for RP.
To date, 49 genes and three additional loci were identified to cause AR syndromic RP, 8 causing AD (1 locus) and 2 causing XL (1 locus) (The Retinal Information Network Retnet: https://sph.uth.edu/retnet/sum-dis.htm, in the public domain, accessed August 2018). Due to the high prevalence of consanguinity among the Israeli and Palestinian populations present in our study cohort, extremely rare diseases can be found at a relatively higher prevalence compared to other populations (Machado et al. 2013). Of note, significant progress has been made in imaging techniques and laboratory tests leading to increased numbers of clinical diagnoses, making the genetic workup difficult to perform using classical methods. On the other hand, the development of high capacity sequencing techniques including whole-exome (WES) and whole-genome (WGS) sequencing has significantly improved our ability to identify the causative mutations for rare inherited diseases in general (Gilissen et al. 2011; Dolled-Filhart et al. 2012) and for retinal diseases in particular (Özgül et al. 2011; Züchner et al. 2011; Shen et al. 2015; Bacchelli & Williams 2016; Fernandez-Marmiesse et al. 2018) in already known genes. In addition, an increasing number of novel disease-causing genes have been identified using these tools, significantly extending our genetic knowledge (Zeitz et al. 2013; El Shamieh et al. 2014; Khateb et al. 2014, 2018; Corton et al. 2016; Namburi et al. 2016).
Here, we report the role of genetic analysis using WES in the establishment of accurate clinical diagnosis of four index cases suffering from rare syndromic IRDs, demonstrating a personalized attitude for genetic analysis.
Patient and Methods
Patient recruitment
Four index cases suffering from rare inherited syndromic retinal diseases and their relatives were recruited for the study. The tenets of the Declaration of Helsinki were followed; the study was approved by the Institutional Review Board (IRB) of our respective institutions, and prior to donation of a blood sample, a written informed consent was obtained from all individuals who participated in this study.
Clinical evaluation
Ocular evaluation included a comprehensive ophthalmologic examination, Goldmann perimetry, full-field electroretinography (ffERG), electrooculography (EOG), colour vision testing using the Ishihara 38-panel and Farnsworth-Munsell D-15 tests, colour and pseudocolour fundus photographs, spectral domain-optical coherence tomography (SD-OCT) and short-wave fundus autofluorescence (SWAF) imaging. Systemic workup was performed in the Hadassah-Hebrew University Medical Center, and clinical information was summarized based on clinical records.
Genetic analyses
Genomic DNA was extracted from peripheral blood using the FlexiGene DNA kit (Qiagen, Venlo, The Netherlands) or the Maxwell® 16 Blood DNA purification kit and Maxwell® 16 instrument (Promega; Madison, WI, USA, AS1010 and AS2000, respectively). Homozygosity mapping was performed using whole-genome single nucleotide polymorphism (SNP) microarrays (10K, Affymetrix 6.0 and Illumina; Santa Clara, CA, USA) and analysed by the Homozygositymapper online program (http://www.homozygositymapper.org/, in the public domain). Whole-exome sequencing (WES) analysis was performed in several platforms (Otogenetics Corporation; Pronto diagnostics; Illumina and Neurobiology-Neurodegeneration & Repair Laboratory, NEI, NIH, MD, USA). Sequence readings were aligned to the human genome reference sequence (build hg19), and variants were named and annotated using the DNAnexus software package. Dataset files including the annotated information were analysed using ANNOVAR (http://wannovar.wglab.org/, in the public domain) according to the dbSNP database (build 135) and filtering of the non-pathogenic variants was performed as detailed previously (Khateb et al. 2014).
Results
Our major long-term aim was to identify the genetic cause of disease in our cohort of patients. Efficient genetic analysis depends on accurate clinical diagnosis, allowing one to focus on a specific set of candidate genes. For the current study, we analysed patients from four families, where each had a specific diagnosis based on clinical examination. Below, we provide the clinical and genetic information on each of the families studied.
Family MOL0760
MOL0760 is an Arab Muslim family from the Jerusalem area with multiple consanguineous marriages, and the parents are first-degree cousins (Fig. 1). The family consisted of five siblings with two affected subjects. The index case MOL0760 IV:1 was a 30-year-old (YO) female with uneventful pregnancy and delivery. She was monitored for many years in our institute due to low vision caused by RP. Systemically, she was known to suffer from short stature, developmental delay, microcephaly, mental retardation and facial dysmorphism. Best-corrected visual acuity (BCVA) was 0.13 and 0.10 Snellen with refraction of −5.00(−4.50)180° and −6.50(−4.50)180° in the right (RE) and left (LE) eye, respectively. The anterior segments were unremarkable with clear lenses in both eyes (BE). Fundoscopy showed a slightly waxy optic disc, narrow retinal vessels, diffuse retinal atrophy extending from the far periphery centrally to involve the fovea combined with few bone-spicules like pigmentations (BSPs) in both eyes (Fig. 2). Short-wave autofluorescence (SWAF) showed a parafoveal hyper-autofluorescent ring, while SD-OCT showed retinal thinning due to ellipsoid zone atrophy (Fig. 2A). full-field electroretinography (ffERG), at the age of 21, was recorded under sedation due to limited co-operation. The test revealed severely reduced cone response and delayed implicit time of the 30 Hz flicker stimulation in BE, with the mixed response being barely detectable. Pattern visual evoked potentials (PVEP) using 50′ grid showed a slightly delayed implicit time in the LE and normal responses in the RE compatible with low BCVA. Amplitudes were within normal limits. This combination of systemic and ocular pathologies was diagnosed clinically as Kabuki syndrome.
Fig. 1.
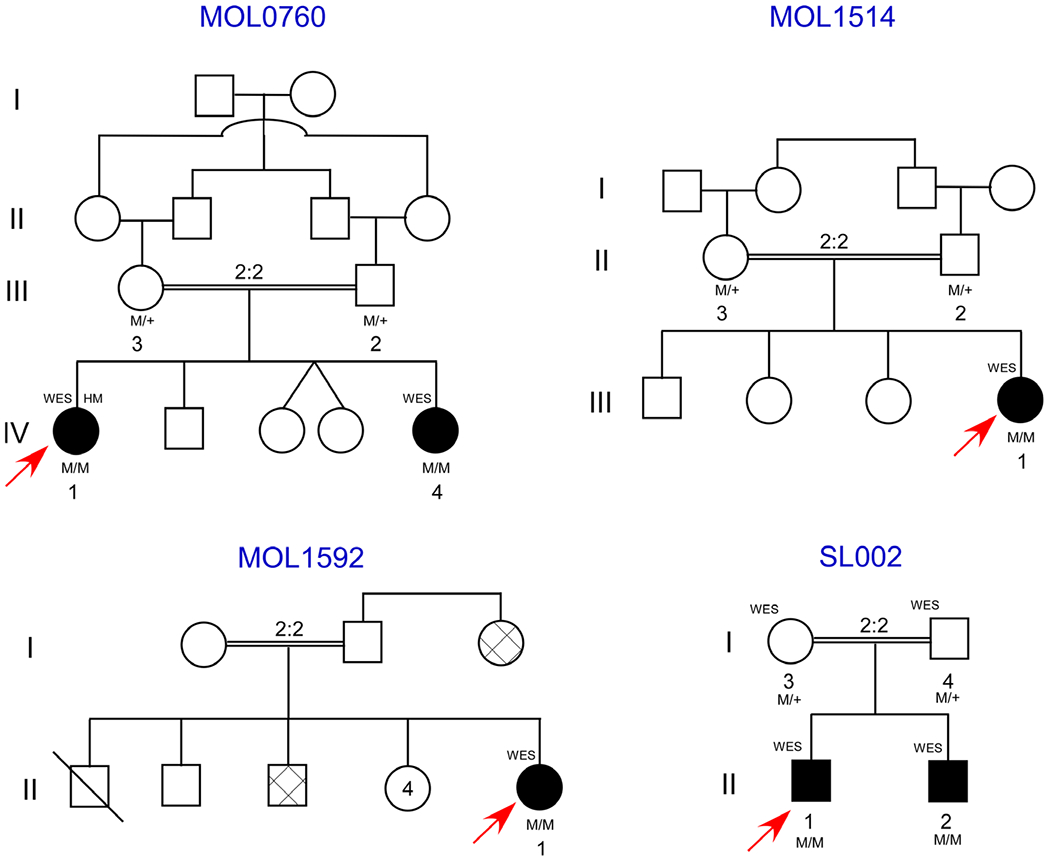
Pedigrees of the included families. Affected subjects are indicated as black-filled objects, and genotype is written under the recruited subject symbol. Gray-filled shapes indicate patients with similar phenotype as detailed in the results section per anamnesis without examination. Arrows indicate index cases and deceased subject is indicated by oblique line.
Fig. 2.

Colour, autoflourescence (FAF) and optical coherence tomography (OCT) images of affected subjects. (A) MOL0760 IV:1 and MOL0760 IV:4 RE colour fundus photograph show normal looking optic disc, narrow retinal vessels and peripheral retinal atrophy. Corresponding FAF images show a hyper autoflourescent ring surrounding the fovea. SD-OCT horizontal cross sections demonstrate atrophy of the outer retinal layers while MOL0760 IV:4 present with cystoid macular oedema (CME). (B) BE ultra-wide field pseudocolour and FAF Optos photographs of MOL1514 III:1 show peripheral retinal atrophy encroaching the fovea combined with patches of pigmentation. SD-OCT horizontal sections show a preserved island of outer nuclear layers and ellipsoid zone within the fovea, complicated by CME. Retinal atrophy is surrounding the macula peripherally. (C) BE colour and near infrared reflectance photographs of MOL1592 II:1 show dot crystalline deposits within the macula with pigmentary changes, presented as hyperreflective dots along the inner nuclear layer and outer plexiform layers with preservation of the outer retinal layers.
Her sister, MOL0760 IV:4, was a 20-year-old female, with uneventful pregnancy and delivery history. She had been followed at our clinic since 12 years old when her BCVA was measured as 0.20 Snellen in BE, with refraction of −6.00(−2.50)5° and −5.50(−2.75)160° in RE and LE, respectively. Full eye movements with the combination of the head to the right but the gaze to the left were observed. Anterior segments were unremarkable including a clear lens, while fundoscopy revealed attenuated retinal vessels and peripheral retinal atrophy encroaching the fovea in addition to few BSPs outside the arcades, although the optic discs looked normal. Short-wave fundus autofluorescence (SWAF) showed a parafoveal hyper-autofluorescent ring. Horizontal SD-OCT cross sections showed disruption of the ellipsoid zone with hyporeflective cysts in the inner retina compatible with cystoid macular oedema (CME) (Fig. 2A). The BCVA was stable until age 14, but then deteriorated to 0.10 in BE. Full-field electroretinography (ffERG) testing at age 1 year old under sedation demonstrated severely reduced mixed responses and moderately diminished cone response. Repeated tests at age 12 years old showed extinct rod response with severely diminished cone response compatible with rod-cone dystrophy. Pattern visual evoked potentials (PVEP) was within normal limits. Due to intellectual and developmental delay, microcephaly and facial dysmorphism; she underwent a brain computed tomography (CT) at age 1 year old which showed agenesis of corpus callosum. She also had a short stature that was treated with growth hormone replacement therapy (Norditropin 0.03–0.04 mg/kg/day) since early childhood, in addition to constant treatment with Methylphenidate (Retalin 2.5–5 mg/day). This distinctive combination of structural and intellectual phenotypes was also suspected as being Kabuki syndrome, similar to her sister’s clinical diagnosis.
Genetic analysis of the two affected sisters and their parents (Fig. 1) included Sanger sequencing screen of founder mutations, which were negative, followed by homozygosity mapping of the index case, which revealed multiple homozygous regions ranging from 3.1 to 36.8 Mb, which is as expected in families with multiple consanguineous loops (Fig. 3A). Subsequently, WES analysis was performed on DNA samples of the two affected subjects (MOL0760 IV:1 and −4) and their unaffected father (MOL0760 III:2; Fig. 1 and Table 1). Whole-exome sequencing (WES) revealed a homozygous frameshift variant (c.5492dup) in exon 34 of VPS13B (NM_017890.4), causing a premature termination of the protein [p.(Asn1831Lysfs*8)] (Tables 2 and 3, Fig. 3B). This homozygous variant is located on chromosome 8 at position g.100654235, surrounded by a large homozygous area (chr8: 64445521 to 117192505) as evident by homozygosity mapping of the SNP array, as well as the WES data. Complete co-segregation of the frameshift variant was verified by Sanger sequencing in the affected subjects and their parents. VPS13B recessive mutations are known to cause Cohen syndrome (Kolehmainen et al. 2003; Mochida et al. 2004), and therefore, the clinical diagnosis of the two affected individuals was revised accordingly.
Fig. 3.
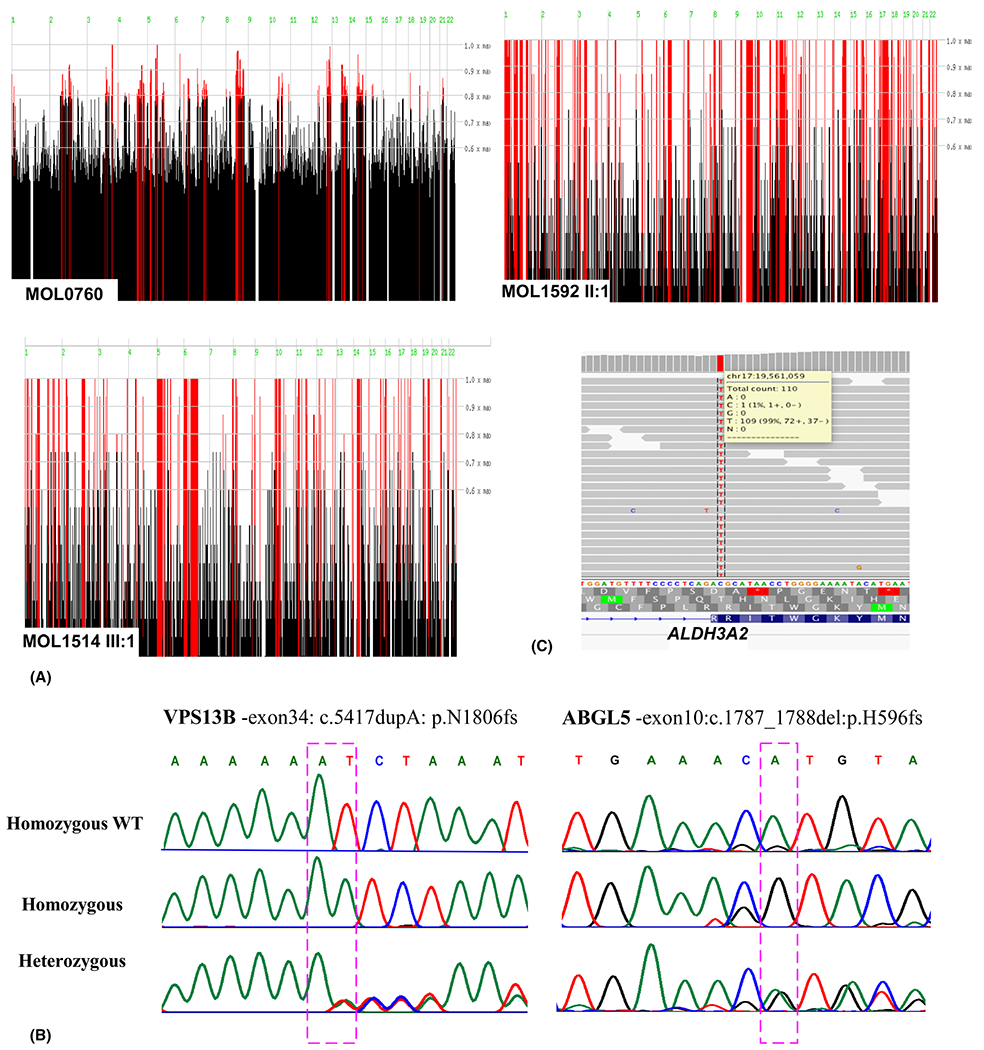
Homozygosity mapping and Mutation Identification. (A) Homozygosity analysis of the recruited families. Homozygosity mapping of MOL0760 (HM-WES-MOL0760 IV:1, -IV:4 and -III:2 and SNP array HM-MOL0760 IV:1), MOL1592 II:1 and MOL1514 III:1. Data were analysed using the HomozygosityMapper server (Berlin, Germany [03/2016]): the x-axis represents the genomic location and y-axis represents the threshold for the Homozygosity score as a ratio over the maximum score. (B) Chromatograms of a homozygous affected individual (top), a heterozygous unaffected family member (bottom) in the families MOL760 (left) and MOL1514 (right). (C) Exome sequence alignment of the ALDH3A2 c.682C>T, p.(Arg228Cys) mutation region in MOL1592II:1. Only a representative set of reads is depicted.
Table 1.
Data on variants identified by the whole-exome sequencing analysis.
| Sample | Total no. of variants | No. of homozygous missense, nonsense and splicing variants | Variants in 1000 genome project < 0.001 | Variants with gnomAD MAF < 0.1% | Variants with CG46 MAF < 0.1% | No. of variants fitting the disease mode | No. of variants within linked region |
|---|---|---|---|---|---|---|---|
| MOL0760 IV:1 | 19865 | 3741 | 143 | 85 | 26 | ||
| MOL0760 IV:4 | 19023 | 3800 | 125 | 80 | 18 | 7 | 1 |
| MOL1514 III:1 | 45509 | 3772 | 111 | 34 | 31 | 2 | |
| MOL1592 II:1 | 44652 | 4204 | 158 | 48 | 44 | 1 | |
| SJ002 II:1 | 154086 | 4670 | 215 | 70 | 67 | 1 |
Table 2.
Presumed diagnoses of families included in this report, identified mutated gene leading to precise clinical diagnosis.
| Family number | Presumed diagnosis | Identified gene and mutation (c./p.) | Precise clinical diagnosis based on genetics |
|---|---|---|---|
| MOL0760 | Kabuki syndrome |
VPS13B c.5492dup p.(Asn1831Lysfs*8) |
Cohen syndrome |
| MOL1514 | Syndromic RP |
AGBL5 c.1787_1788del p.(His596Argfs*47) |
|
| MOL1592 | Crystalline retinopathy, ichthyosis, short stature, congenital adrenal hypoplasia |
ALDH3A2 c.682C>T p.(Arg228Cys) |
Sjögren-Larsson syndrome (SLS) |
| SJ002 | Nystagmus, low VA, hypotony, global severe psychomotor delay and dysmorphic facial features |
VARS2 c.1691C>T p.(Ala564Val) |
Combined oxidative phosphorylation deficiency-20 (COXPD20) |
Table 3.
Population frequency and predicted pathogenicity of the identified mutations.
| Gene | Mutation c./p. | gnomAD Allele Frequency | MutationTaster Prediction Disease causing | PolyPhen-2 | GVGD | SIFT | Conservation |
|---|---|---|---|---|---|---|---|
| VPS13B | c.5492dup p.(Asn1831Lysfs*8) g. 100654235dup |
Not found | 1 | High | |||
| AGBL5 | c.1787_1788del p.(His596Argfs*47) g.27281383_27281384del |
0.000004061 Never homo |
1 | High | |||
| ALDH3A2 | c.682C>T p.(Arg228Cys) g.19561059C>T |
0.00001625 Never Homo |
1 | Probably damaging (1) | C65 | Deleterious (0) | High |
| VARS2 | c.1691C>T p.(Ala564Val) g.30921277C>T |
Not found | 1 | Probably damaging (0.998) | C0 | Tolerated (0.2) | High |
Genome Reference—GRCh38 (hg38); Genome Aggregation Database (gnomAD): http://gnomad.broadinstitute.org/; MutationTaster: http://www.mutationtaster.org/; Polymorphism Phenotyping v2 (PolyPhen-2): http://genetics.bwh.harvard.edu/pph2/; GVGD: http://agvgd.hci.utah.edu/; Sorting intolerant from tolerant (SIFT): http://sift.jcvi.org/; Conservation: http://genome.ucsc.edu/index.html; VPS13B- NM_017890.4, AGBL5- NM_021831.5, ALDH3A2- NM_001031806.1
Of note, no pathogenic variants were identified in the KMT2D (MLL2) and KDM6A genes reported previously to cause Kabuki syndrome (in up to 80% of cases), which strengthened our findings regarding the VPS13B homozygous frameshift as the disease-causing variant.
Family MOL1514
MOL1514 is a consanguineous Arab-Christian family from the Jerusalem area (Fig. 1), consisting of one affected female (MOL1514 III:1) and three unaffected siblings. The index case was examined at 5 YO. She was found to have a BCVA of 0.50 Snellen and refraction of +2.00(−1.50)180° with unremarkable anterior and posterior segments in BE. Systemically, blood tests showed elevated liver transaminases from 9 months of age including: alkaline phosphatase (ALP), aspartate aminotransferase (AST), alanine aminotransferase (ALT) and gamma-glutamyl transferase (GGT), along with slightly elevated inflammatory markers, including erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP). Kidney function tests were within normal limits. A liver echography showed enlarged fatty liver. She was examined again at 12 years old and presented with a BCVA of 0.40 Snellen in BE, with normal anterior segments, while fundoscopy showed attenuated retinal vessels, peripheral atrophy combined with patches of pigmentation and CME in BE. This was treated with systemic and topical carbonic anhydrase inhibitors, resulting in a partial response. Farnsworth D-15 colour test was within normal limits, ffERG showed extinct rod-cone responses in RE, while LE was not examined due to technical difficulties. The visual field was mildly constricted (central 40 degrees in BE) tested by Goldmann perimetry (target 4IVe). The co-presentation of RP and liver dysfunction was suspected to be associated with a distinctive syndromic disease. Homozygosity mapping of the index case showed a large number of homozygous regions, which was expected due to the consanguineous marriage (Fig. 3A). Whole-exome sequencing (WES) analyses of the index case revealed a homozygous 2-bp deletion (c.1787_1788del) leading to a premature stop codon c.1787_1788del [p.(His596Argfs*47)] in the AGBL5 (CCP5) gene (NM_021831.5) (Tables 2 and 3, Fig. 3B), which was reported to cause non-syndromic ARRP (Kastner et al. 2015; Astuti et al. 2016). None of the genes causing liver dysfunction was found to be mutated via the WES analysis.
Family MOL1592
MOL1592 II:1 is the index case, descending from a first-degree Arab Muslim family from the Jerusalem area. She was 20 years old when referred to our IRD centre due to low visual acuity and photophobia and was suspected to be suffering from Stargardt disease. She had been diagnosed previously with developmental delay, short stature, ambiguous genitalia, congenital adrenal hypoplasia (CAH) and 21-hydroxylase deficiency treated with systemic corticosteroids (Dexamethasone 0.4 mg/day and fludrocortisone 0.1 mg/day); and skin ichthyosis treated with Neotigason (acitretin). Family history included one brother who had died at 6 months of age with no obvious clinical data. Another brother, who was known to suffer from CAH, presented with hearing impairment, skin disease and visual impairment. Her aunt was also diagnosed with the same phenotype except for CAH. Unfortunately, the family provided limited co-operation and refused further investigation. BCVA was counting fingers (CF) 3 metres RE and 0.10 LE. The anterior segments were unremarkable including clear lens. Via fundoscopy, normal looking optic nerve and retinal vessels were encountered, while in the fovea, small yellowish deposits surrounded with subtle pigmentary changes were seen, with a normal peripheral retina (Fig. 2C). Hyperreflective dots along the inner nuclear and inner plexiform layers were observed by SD-OCT, with normal retinal thickness and an intact ellipsoid zone compatible with crystalline retinopathy. Infrared reflectance showed white small macular dots that stained in fluorescein angiography (FA) (data not shown). Ishihara and Farnsworth D-15 colour tests were normal. Full-field electroretinography (ffERG) using a bright white stimulus after 30-min-long dark adaptation demonstrated mild decrease of b-wave in each eye and normal a-wave of the mixed cone-rod responses and slightly low responses of rods and cones in the LE. The EOG was within normal limits (RE-303%, LE-280%; normal-250%). Genetic analysis included a normal karyotype (46XX). Whole-exome sequencing (WES) analysis performed on the DNA sample of the index case revealed a missense variant c.682C>T [p.(Arg228Cys)] in exon 5 of ALDH3A2 (NM_001031806.1), encoding the fatty aldehyde dehydrogenase (FALDH) protein (Tables 2 and 3, Fig. 3C). Fibroblasts harbouring this mutation showed residual activity of the FALDH protein (Gloerich et al. 2006). Mutations in this gene were reported to cause Sjögren-Larsson syndrome (SLS), an autosomal recessive disorder of fatty alcohol metabolism, characterized by short stature, seizures, ichthyosis, spastic di- or tetraplegia, crystalline retinopathy, mental retardation and other minor features (OMIM #270200) (Jagell et al. 1981).
Family SJ002
The index case SJ002 II:1 was a 7-year-old male whose parents are first-degree cousins in a Palestinian Arab Muslim family. He was examined in the St. John’s Eye Hospital clinic due to nystagmus and poor visual acuity since birth. General medical examination revealed global hypotony, severe psychomotor delay and dysmorphic facial features. Visual acuity of light perception and cycloplegic refraction −12 in BE. He also demonstrated upper lid ptosis and progressive external ophthalmoplegia, but the anterior segments were unremarkable. Fundoscopy revealed waxy optic discs and diffuse retinal atrophy with RPE changes compatible with Leber congenital amaurosis (LCA). ffERG showed extinct cone and rod responses in both eyes. Imaging of the patient was not possible due to his young age, mental and general status. Per anamnesis, his younger brother (3 YO) has similar external dysmorphism but was not examined. The multiple pathologies were suspected to be associated with syndromic retinal degeneration; hence, WES was performed for both affected subjects and their parents using TruSeq Capture Exome kit (Illumina) with 45 Mb coverage of exonic content. The WES analysis revealed a homozygous variant c.1691C>T [p.(Ala564-Val)] in the Valyl-tRNA Synthetase 2 (VARS2) gene (NM_001167734.1) (Tables 2 and 3) in both affected subjects, while the parents were found to be heterozygous. Valyl-tRNA Synthetase 2 (VARS2) encodes for the mitochondrial valyl-tRNA (Val-tRNA) synthetase protein reported to be involved in mitochondrial translation and to cause combined oxidative Phosphorylation deficiency-20 (COXPD20) when mutated as reported previously (Taylor et al. 2014). Ala565 is within the alpha-helical structure of class I aminoacyl-tRNA synthetases, which is coupled to the 2′-hydroxyl of the tRNA. The reported patients presented with major hypotony, microcephaly and poor eye contact at age 4 months, nystagmus, global psychomotor development delay and failure to thrive. This mutation was predicted to be disease causing and highly conserved among 100 species indicating the pathogenic potential of this variant.
Discussion
The recent progress of clinical diagnostic tests utilizing newer imaging and laboratory modalities combined with the revolution of high capacity sequencing techniques such as whole-genome (WGS) and whole-exome sequencing (WES) significantly have improved our molecular diagnostic abilities and extended the precise clinical spectrum of rare diseases considered previously as one entity. Next-generation sequencing (NGS) is considered a paradigm-shifting technology translated into clinical diagnostic laboratories. It allows diagnosis of genetic disorders considered previously as cost prohibitive or difficult in various fields of medicine including: inherited cardiomyopathies (Gowrisankar et al. 2010; Voelkerding K et al. 2010), metabolic diseases (Jones et al. 2011) and mitochondrial diseases (Vasta et al. 2009). It is estimated to identify 30–70% of the causative mutations/genes in total (Robinson et al. 2011; Singh et al. 2016). Inherited retinal diseases (IRDs) are considered genetically highly heterogeneous, and using high-throughput sequencing techniques, such as NGS, have contributed largely to the identification of the causative mutation in already known genes (Khateb et al. 2012, 2016; Beryozkin et al. 2015) as well as novel disease-causing genes (El Shamieh et al. 2014; Corton et al. 2016; Namburi et al. 2016; Khateb et al. 2018). Inherited retinal diseases (IRDs) frequently have a large spectrum of clinical phenotypes, even when caused by the same causative mutation/gene. Syndromic phenotypes that include retinal dystrophies are even more difficult to investigate, mainly due to disease complexity and variability even among siblings who share the same causative mutations. Such phenotypes tend to show variable expressivity leading to unusual cases in which a specific organ, known to be affected in the vast majority of cases, appears normal or asymptomatic in some individuals. Since the number of affected organs in a specific syndrome can be extensive, such variable expressivity can easily lead to a wrong clinical diagnosis, or to cases in which no clinical diagnosis can be determined. Therefore, during the last decade, an increasing number of syndromic cases are referred to genetic analysis with the aim of more accurately defining the phenotype.
Here, we report four unrelated cases who suffer from different, distinct and rare inherited diseases including various retinal dystrophies, which were misdiagnosed or lacked precise diagnosis based on clinical findings, but which, via genetic analysis, including WES, enabled us to provide a more accurate clinical diagnosis. All the identified mutations completely co-segregated and were found to be pathogenic based on the different prediction tools.
In MOL0760, two affected subjects suffering from short stature, developmental delay, congenital mental retardation, microcephaly, facial dysmorphism and RP had been incorrectly diagnosed with Kabuki syndrome [KABUK1 (Niikawa-Kuroki syndrome) OMIM #147920; KABUK2 OMIM #300867, which has been reported to affect multiple organs as well as distinctive facial features (a flat, broadened tip of the nose and large protruding earlobes)]. Moreover, it leads to mild to severe developmental delay and intellectual disability, seizures, microcephaly, hypotonia and many other structural pathologies. Previously reported ocular findings of Kabuki patients include the following: arched eyebrows, long eyelashes, long palpebral fissures, everted lower lids, nystagmus, strabismus, colobomatous microphthalmos; dysplastic and elevated disc without central cupping (Chen et al. 2014), and myelinated nerve fibre layer.(Duval et al. 2010) Retinal abnormalities include prepapillary gliosis, tortuous retinal vessels, foveal irregular pigmentation (Chuah et al. 2009) and macular dystrophy (Lindfield et al. 2011), but no previous reports of RP. Whole-exome sequencing (WES) analysis revealed a pathogenic nonsense mutation in VPS13B that is known to cause Cohen syndrome in various populations (OMIM #216550) (Kolehmainen et al. 2003; Mochida et al. 2004) consisting of variable combination of clinical manifestations including psychomotor retardation, microcephaly, dysmorphic facial features, progressive pigmentary retinopathy appearing at mid-childhood, early onset and severe myopia; and intermittent neutropenia (Michael et al. 1973; Norio et al. 1984; Kivitie-Kallio & Norio 2001). This combination of the clinical phenotype and genetic findings together with the absence of Kabuki syndrome causing mutations established the probably correct diagnosis of Cohen syndrome in this family.
The index case of MOL1514 was previously clinically diagnosed with RP and liver dysfunction reflected by elevated transaminases and an enlarged fatty liver. This combination of pathologies was suspected to be related and caused by a syndromic disease. Whole-exome sequencing (WES) identified a nonsense mutation in the AGBL5 gene, which had been reported to cause nonsyndromic RP. Initially, a homozygous missense mutation p.Asp295Asn (c.883G>A) was reported to segregate with recessive RP in a consanguineous Turkish family (Kastner et al. 2015). Detailed clinical information was missing, but one patient was reported with a diagnosis of mental retardation in addition to RP. Using WES, Patel and colleagues (Patel et al. 2016) reported a p.Arg276Trp (c.826C>T) homozygous variant segregating with recessive RP in a consanguineous Saudi Arabian pedigree absent of additional information regarding other ocular or extraocular pathologies. Branham reported two affected subjects descending from European ancestry suffering from adolescent-onset recessive retinitis pigmentosa (arRP), who harboured compound heterozygous mutations p.Arg281Cys and p.Arg487* in the AGBL5 gene identified using WGS.(Branham et al. 2016) One of the two patients had had petit mal seizures as a child controlled with medications, while the other older affected sibling did not have seizures. No other extraocular pathologies were reported. The AGBL5 gene encodes an 886-amino acid metallocarboxypeptidase that is a member of the peptidase M14 family of proteins.(Berezniuk et al. 2013) It cleaves branching point glutamates (Rogowski et al. 2010), and recently, it was found to remove longer α-linked glutamate chains from different substrates (Berezniuk et al. 2013). It is expressed in the human retina, specifically in the inner nerve fibre- and inner ganglion cell layer (GCL), as well as in the inner (IPL) and outer (OPL) plexiform layers, and in the brain.(Kastner et al. 2015) CCP5 zebrafish morphants show smaller eye size (Lyons et al. 2013), and knockdown of CCP5 resulted in increased cilia tubulin glutamylation which led to multiple ciliopathies including: acutely curved body axis, hydrocephalus and pronephric cysts. This was due to inhibition of motility-associated functions of microtubules (Pathak et al. 2014). Up to date, genes involved in ciliogenesis are known to cause a wide variety of syndromic and nonsyndromic disorders (Waters & Beales 2011). Moreover, ciliopathy genes may be implicated in either syndromic or non-syndromic retinal degeneration (Waters & Beales 2011; Wheway et al. 2014; Hull et al. 2016; Dharmat et al. 2017). In addition, other ciliary genes were reported to cause RP in combination with liver disease as in Bardet-Biedl syndrome (Aldahmesh et al. 2014; Bujakowska et al. 2015; Ylldlz Bölükbasi et al. 2018). Our affected subject presented with fatty liver and elevated transaminases which may be related to the AGBL5 mutation, but additional analysis is needed to clarify the gene function and explain the extraocular pathologies related to this gene. None of the genes associated with liver dysfunction were found to be mutated.
The index case MOL1592 suffered from a very distinctive combination of crystalline retinopathy, developmental delay, short stature, ambiguous genitalia, congenital adrenal hypoplasia (CAH), 21-hydroxylase deficiency and skin ichthyosis. Multiple affected subjects were reported in this family, but unfortunately due to patients’ low compliance, clinical data are lacking for the other family members. Initial clinical diagnosis failed to associate accurately between both the ocular and extraocular pathologies in one specific diagnosis. Whole-exome sequencing (WES) analysis revealed a missense mutation p.(Arg228Cys) in the ALDH3A2 gene that encodes fatty aldehyde dehydrogenase (FALDH) protein and was reported to cause Sjögren-Larsson syndrome (SLS) (Rizzo et al. 1988). Most cases are born preterm, but the index case that we present here was born full term, as far as we know. Systemically, they presented with generalized pruritic ichthyosis, which is prominent in flexural areas, neck and lower abdomen. Neurological deficits include bilateral spastic paresis involving the legs more than the arms, cognitive impairment; dysarthria combined with periventricular white matter abnormalities as shown by magnetic resonance imaging (MRI). Other signs include short stature, peripheral neuropathy and dental abnormalities. Congenital adrenal hypoplasia has not been reported previously in SLS cases, as in our case, which association requires further confirmation. Ocular abnormalities include reduced visual acuity and photophobia, glistening intraretinal crystalline deposits on fundoscopy compatible with hyperreflective dots along the INL and OPL in OCT within the fovea (Burgueño-Montañés et al. 2014; Theelen et al. 2014). Most patients show cystic foveal degeneration (Fuijkschot et al. 2008) and reduced central macular pigmentation (van der Veen et al. 2010). Most reported mutations result in a complete loss of catalytic activity of FALDH, although there are several (missense) mutations associated with residual FALDH activity. Gloerich et al. (2006) reported that Bezafibrate induces FALDH activity in SLS fibroblasts harbouring the same mutation with residual enzyme activity (Gloerich et al. 2006).
Family SJ002 included two affected subjects suffering from serious hypotony, global severe psychomotor delay and dysmorphic facial features. Ophthalmologically, they presented with pathologic myopia, progressive external ophthalmoplegia and diffuse retinal atrophy. This phenotype combination was suspected to be a primary mitochondrial disorder such as an atypical presentation of Kearns-Sayre syndrome (KSS). Of note, the inheritance pattern was compatible with AR rather than mitochondrial. Whole-exome sequencing (WES) analysis results confirmed our suspicion, revealing a homozygous mutation in VARS2 gene encoding a mitochondrial aminoacyl-tRNA synthetase. Valyl-tRNA Synthetase 2 (VARS2) protein is encoded by the nuclear DNA and imported to the mitochondria; hence, it is inherited following the classic Mendelian rules. A deficiency in VARS2 protein has been reported in several cases recently, causing mitochondrial encephalopathy (Diodato et al. 2014; Taylor et al. 2014; Pronicka et al. 2016). Diodato et al. 2014 reported a homozygous mutation (c.1100C>T, p.Thr367Ile) in a 8-year-old patient, characterized by psychomotor delay and epilepsia partialis continua associated with a mitochondrial Complex I defect (Diodato et al. 2014). Baertling et al. 2017 reported compound heterozygous mutations (c.1100C>T, p.Thr367Ile and c.601C>T, p.Arg201Trp) causing structural brain abnormalities, hypertrophic cardiomyopathy and severe lactic acidosis but no ocular defects in a Greek male (Baertling et al. 2017). Bruni et al. 2018; described nine unrelated families with VARS2-associated mitochondrial diseases including encephalomyopathy and cardiomyopathy, hypotonia, psychomotor delay, seizures, feeding difficulty, abnormal brain MRI and elevated lactate. One patient showed prominent eyes with shallow orbits; one was found to have poor eye contact; and one demonstrated restricted range of eye movements in all directions with some sparing of downgaze and bilateral ptosis (Bruni et al. 2018).
None of these cases were reported to have retinal degeneration as in our family, thus expanding the ocular phenotypes of the disease. Of note, however, the nystagmus and poor eye contact described in some of the cases could be due to primary retinal degeneration rather than to a neurological defect.
In conclusion, the cases that we present here demonstrate the efficacy of WES analysis combined with clinical characteristics in establishing a precise diagnosis of rare inherited diseases involving retinal dystrophies. We, therefore, recommend using WES in the genetic workup of syndromic cases with an unclear or an ambiguous clinical phenotype.
Acknowledgments
We thank the family members reported here and Michelle Grunin, Ann and Colin Marks for editing the manuscript.
This study was financially supported by the Foundation Fighting Blindness USA (BR-GE-0214-0639 to DS and EB), Israel Science Foundation (#2154/15 to SK), Chief Scientist Office of the Israeli Ministry of Health and the Lirot association (#300009177 to SK) and the Yedidut Research grant (to EB)
References
- Aldahmesh MA, Li Y, Alhashem A et al. (2014): IFT27, encoding a small GTPase component of IFT particles, is mutated in a consanguineous family with bardet-biedl syndrome. Hum Mol Genet 23: 3307–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astuti GDN, Arno G, Hull S et al. (2016): Mutations in AGBL5, encoding a-tubulin deglutamylase, are associated with autosomal recessive retinitis pigmentosa. Investig Ophthalmol Vis Sci 57: 6180–6187. [DOI] [PubMed] [Google Scholar]
- Bacchelli C & Williams HJ (2016): Opportunities and technical challenges in next-generation sequencing for diagnosis of rare pediatric diseases. Expert Rev Mol Diagn 16: 1073–1082. [DOI] [PubMed] [Google Scholar]
- Baertling F, Alhaddad B, Seibt A et al. (2017): Neonatal encephalocardiomyopathy caused by mutations in VARS2. Metab Brain Dis 32: 267–270. [DOI] [PubMed] [Google Scholar]
- Berezniuk I, Lyons PJ, Sironi JJ, Xiao H, Setou M, Angeletti RH, Ikegami K & Fricker LD (2013): Cytosolic carboxypeptidase 5 removes α- And γ-linked glutamates from tubulin. J Biol Chem 288: 30445–30453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beryozkin A, Shevah E, Kimchi A et al. (2015): Whole exome sequencing reveals mutations in known retinal disease genes in 33 out of 68 Israeli families with inherited retinopathies. Sci Rep 5: 13187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branham K, Matsui H, Biswas XP et al. (2016): Establishing the involvement of the novel gene AGBL5 in retinitis pigmentosa by whole genome sequencing. Physiol Genomics 48: 922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruni F, Di Meo I, Bellacchio E et al. (2018): Clinical, biochemical and genetic features associated with VARS2 -related mitochondrial disease. Hum Mutat 39: 563–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujakowska KM, Zhang Q, Siemiatkowska AM et al. (2015): Mutations in IFT172 cause isolated retinal degeneration and Bardet-Biedl syndrome. Hum Mol Genet 24: 230–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bundey S & Crews SJ (1984): A study of retinitis pigmentosa in the City of Birmingham. II Clinical and genetic heterogeneity. J Med Genet 21: 421–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgueño-Montañés C, García-Fernández M, Colunga-Cueva M & García-López A (2014): Sjögren-Larsson syndrome: optical coherence tomography and a novel mutation. Arch Soc Esp Oftalmol 89: 504–507. [DOI] [PubMed] [Google Scholar]
- Chen YH, Sun MH, Hsia SH, Lai CC & Wu WC (2014): Rare ocular features in a case of Kabuki syndrome (Niikawa-Kuroki syndrome). BMC Ophthalmol 14: 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuah JL, Chuah JK & Brown R (2009): New fundus findings in a case of Kabuki syndrome. Eye 23: 1483–1485. [DOI] [PubMed] [Google Scholar]
- Corton M, Avila-Fernández A, Campello L et al. (2016): Identification of the photoreceptor transcriptional Co-Repressor SAMD11 as novel cause of autosomal recessive retinitis pigmentosa. Sci Rep 6: 35370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharmat R, Liu W, Ge Z et al. (2017): IFT81 as a candidate gene for nonsyndromic retinal degeneration. Investig Ophthalmol Vis Sci 58: 2483–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diodato D, Melchionda L, Haack TB et al. (2014): VARS2 and TARS2 mutations in patients with mitochondrial encephalomy-opathies. Hum Mutat 35: 983–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolled-Filhart MP, Lordemann A, Dahl W, Haraksingh RR, Ou-Yang CW & Lin JCH (2012): Personalizing rare disease research: How genomics is revolutionizing the diagnosis and treatment of rare disease. Per Med 9: 805–819. [DOI] [PubMed] [Google Scholar]
- Duval R, Hammamji K, Aroichane M, Michaud JL & Ospina LH (2010): Acquired myelinated nerve fibers in association with optic disk drusen. J AAPOS 14: 544–547. [DOI] [PubMed] [Google Scholar]
- El Shamieh S, Neuillé M, Terray A et al. (2014): Whole-exome sequencing identifies kiz as a ciliary gene associated with autosomal-recessive rod-cone dystrophy. Am J Hum Genet 94: 625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Marmiesse A, Gouveia S & Couce ML (2018): NGS technologies as a turning point in rare disease research, diagnosis and treatment. Curr Med Chem 25: 404–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuijkschot J, Cruysberg JRM, Willemsen MAAP, Keunen JEE & Theelen T (2008): Subclinical changes in the juvenile crystalline macular dystrophy in Sjögren-Larsson syndrome detected by optical coherence tomography. Ophthalmology 115: 870–875. [DOI] [PubMed] [Google Scholar]
- Gilissen C, Hoischen A, Brunner HG & Veltman JA (2011): Unlocking Mendelian disease using exome sequencing. Genome Biol 12: 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloerich J, IJlst L, Wanders RJA & Ferdinandusse S (2006): Bezafibrate induces FALDH in human fibroblasts; implications for Sjögren-Larsson syndrome. Mol Genet Metab 89: 111–115. [DOI] [PubMed] [Google Scholar]
- Gowrisankar S, Lerner-Ellis JP, Cox S et al. (2010): Evaluation of second-generation sequencing of 19 dilated cardiomyopathy genes for clinical applications. J Mol Diagnostics 12: 818–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haim M (2002): Epidemiology of retinitis pigmentosa in Denmark. Acta Ophthalmol 80: 1–34. [DOI] [PubMed] [Google Scholar]
- Hartong DT, Berson EL & Dryja TP (2006): Retinitis pigmentosa. Lancet 368: 1795–809. [DOI] [PubMed] [Google Scholar]
- Hull S, Owen N, Islam F et al. (2016): Nonsyndromic retinal dystrophy due to biallelic mutations in the ciliary transport gene IFT140. Investig Ophthalmol Vis Sci 57: 1053–1062. [DOI] [PubMed] [Google Scholar]
- Jagell S, Gustavson KH & Holmgren G (1981): Sjögren-Larsson syndrome in Sweden. A clinical, genetic and epidemiological study. Clin Genet 19: 233–256. [DOI] [PubMed] [Google Scholar]
- Jones MA, Bhide S, Chin E et al. (2011): Targeted polymerase chain reaction-based enrichment and next generation sequencing for diagnostic testing of congenital disorders of glycosylation. Genet Med 13: 921–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastner S, Thiemann IJ, Dekomien G et al. (2015): Exome sequencing reveals AGBL5 as novel candidate gene and additional variants for retinitis pigmentosa in five Turkish families. Investig Ophthalmol Vis Sci 56: 8045–8053. [DOI] [PubMed] [Google Scholar]
- Khateb S, Zelinger L, Ben-Yosef T, Merin S, Crystal-Shalit O, Gross M, Banin E & Sharon D (2012): Exome sequencing identifies a founder Frameshift mutation in an alternative exon of USH1C as the cause of autosomal recessive retinitis pigmentosa with late-onset hearing loss. PLoS One 56: 8045–8053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khateb S, Zelinger L, Mizrahi-Meissonnier L et al. (2014): A homozygous nonsense CEP250 mutation combined with a heterozygous nonsense C2orf71 mutation is associated with atypical Usher syndrome. J Med Genet 51: 460–469. [DOI] [PubMed] [Google Scholar]
- Khateb S, Hanany M, Khalailah A et al. (2016): Identification of genomic deletions causing inherited retinal degenerations by coverage analysis of whole exome sequencing data. J Med Genet 53: 1–8. [DOI] [PubMed] [Google Scholar]
- Khateb S, Kowalewski B, Bedoni N et al. (2018): A homozygous founder missense variant in arylsulfatase G abolishes its enzymatic activity causing atypical Usher syndrome in humans. Genet Med 20: 1004–1012. [DOI] [PubMed] [Google Scholar]
- Kivitie-Kallio S & Norio R (2001): Cohen syndrome: essential features, natural history, and heterogeneity. Am J Med Genet 102: 125–135. [DOI] [PubMed] [Google Scholar]
- Kolehmainen J, Black GCM, Saarinen A et al. (2003): Cohen syndrome is caused by mutations in a novel gene, COH1, encoding a transmembrane protein with a presumed role in vesicle-mediated sorting and intracellular protein transport. Am J Hum Genet 72: 1359–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindfield D, Griffiths MFP, Thompson DA & Moore AT (2011): Macular dystrophy in kabuki syndrome: a new clinical feature? J Pediatr Ophthalmol Strabismus 48: E40–E42. [DOI] [PubMed] [Google Scholar]
- Lyons PJ, Sapio MR & Fricker LD (2013): Zebrafish cytosolic carboxypeptidases 1 and 5 are essential for embryonic development. J Biol Chem 288: 30454–30462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado TMB, Bomfim TF, Souza LV, Soares N, Santos FL, Acosta AX & Abe-Sandes K (2013): Types of marriages, population structure and genetic disease. J Biosoc Sci 45: 461–470. [DOI] [PubMed] [Google Scholar]
- Michael Cohen M, Hall BD, Smith DW, Benjamin Graham C & Lampert KJ (1973): A new syndrome with hypotonia, obesity, mental deficiency, and facial, oral, ocular, and limb anomalies. J Pediatr 83: 280–284. [DOI] [PubMed] [Google Scholar]
- Mochida GH, Rajab A, Eyaid W et al. (2004): Broader geographical spectrum of Cohen syndrome due to COH1 mutations. J Med Genet 41: e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namburi P, Ratnapriya R, Khateb S et al. (2016): Bi-allelic truncating mutations in CEP78, encoding centrosomal protein 78, cause cone-rod degeneration with sensorineural hearing loss. Am J Hum Genet 99: 777–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norio R, Raitta C & Lindahl E (1984): Further delineation of the Cohen syndrome; report on chorioretinal dystrophy, leukopenia and consanguinity. Clin Genet 25: 1–14. [DOI] [PubMed] [Google Scholar]
- Özgül RK, Siemiatkowska AM, Yücel D et al. (2011): Exome sequencing and cis-regulatory mapping identify mutations in MAK, a gene encoding a regulator of ciliary length, as a cause of retinitis pigmentosa. Am J Hum Genet 89: 253–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel N, Aldahmesh MA, Alkuraya H et al. (2016): Expanding the clinical, allelic, and locus heterogeneity of retinal dystrophies. Genet Med 18: 554–562. [DOI] [PubMed] [Google Scholar]
- Pathak N, Austin-Tse CA, Liu Y, Vasilyev A & Drummond IA (2014): Cytoplasmic carboxypeptidase 5 regulates tubulin glutamylation and zebrafish cilia formation and function. Mol Biol Cell 25: 1836–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronicka E, Piekutowska-Abramczuk D, Ciara E et al. (2016): New perspective in diagnostics of mitochondrial disorders: two years’ experience with whole-exome sequencing at a national paediatric centre. J Transl Med 14: 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo WB, Dammann AL & Craft DA (1988): Sjogren-Larsson syndrome. Impaired fatty alcohol oxidation in cultured fibroblasts due to deficient fatty alcohol:nicotinamide adenine dinucleotide oxidoreductase activity. J Clin Invest 81: 738–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson P, Krawitz P & Mundlos S (2011): Strategies for exome and genome sequence data analysis in disease-gene discovery projects. Clin Genet 80: 127–132. [DOI] [PubMed] [Google Scholar]
- Rogowski K, van Dijk J, Magiera MM et al. (2010): A family of protein-deglutamylating enzymes associated with neurodegeneration. Cell 143: 564–578. [DOI] [PubMed] [Google Scholar]
- Shen T, Lee A, Shen C & Lin CJ (2015): The long tail and rare disease research: the impact of next-generation sequencing for rare Mendelian disorders. Genet Res (Camb) 97: e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Mishra A, Pandian AJ et al. (2016): Next-generation sequencing-based method shows increased mutation detection sensitivity in an Indian retinoblastoma cohort. Mol Vis 22: 1036–1047. [PMC free article] [PubMed] [Google Scholar]
- Taylor RW, Pyle A, Griffin H et al. (2014): Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA – J Am Med Assoc 312: 68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theelen T, Berendschot TTJM, Klevering BJ, Fuijkschot J, Hoyng CB & Willemsen MAAP (2014): Multimodal imaging of the macula in hereditary and acquired lack of macular pigment. Acta Ophthalmol 92: 138–142. [DOI] [PubMed] [Google Scholar]
- Vasta V, Ng SB, Turner EH, Shendure J & Hahn SH (2009): Next generation sequence analysis for mitochondrial disorders. Genome Med 1: 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Veen RLP, Fuijkschot J, Willemsen MAAP, Cruysberg JRM, Berendschot TTJM & Theelen T (2010): Patients with Sjögren-Larsson syndrome lack macular pigment. Ophthalmology 117: 966–971. [DOI] [PubMed] [Google Scholar]
- Voelkerding KV, Dames S & Durtschi JD (2010): Next generation sequencing for clinical diagnostics-principles and application to targeted resequencing for hypertrophic cardiomyopathy: a paper from the 2009 William Beaumont Hospital Symposium on Molecular Pathology. J Mol Diagnostics 12: 539–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters AM & Beales PL (2011): Ciliopathies: an expanding disease spectrum. Pediatr Nephrol 26: 1039–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheway G, Parry DA & Johnson CA (2014): The role of primary cilia in the development and disease of the retina. Organogenesis 10: 69–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylldlz Bölükbasi EE, Mumtaz S, Afzal M, Woehlbier U, Malik S & Tolun A (2018): Homozygous mutation in CEP19, a gene mutated in morbid obesity, in Bardet-Biedl syndrome with predominant postaxial polydactyly. J Med Genet 55: 189–197 [DOI] [PubMed] [Google Scholar]
- Zeitz C, Jacobson SG, Hamel CP et al. (2013): Whole-exome sequencing identifies LRIT3 mutations as a cause of autosomal-recessive complete congenital stationary night blindness. Am J Hum Genet 92: 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Züchner S, Dallman J, Wen R et al. (2011): Whole-exome sequencing links a variant in DHDDS to retinitis pigmentosa. Am J Hum Genet 88: 201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]