
Table 1.
Bioinformatics algorithms for sequence/genome searching and their comparisons. S represents a document/genome. N is the total number of documents/genomes. Us∈S |s| represents the total number of terms in N documents/genomes and ∑s∈S |s| is total unique terms/k-mer. MPH is minimal Perfect Hashing. For the Inverted Index size, the extra log (N) comes from the bit precision document IDs. For SBTs, log(N) is the height of the tree, and for Bloom Filters at each level is 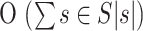 in total. MinHash time and space complexity was based on k hash functions for N sets, each set has d’ non-zero element. Note that the most recent B-bit MinHash with optimal densification can further improve to O(N*(d’+k)). O(vdlog(N)) for GSearch is O(log(N)) in practice since v and d are small and single pair genome comparison is a constant (Supplementary Note 3). τ depends on the user-specified parameter and is generally <0.01 in practice
in total. MinHash time and space complexity was based on k hash functions for N sets, each set has d’ non-zero element. Note that the most recent B-bit MinHash with optimal densification can further improve to O(N*(d’+k)). O(vdlog(N)) for GSearch is O(log(N)) in practice since v and d are small and single pair genome comparison is a constant (Supplementary Note 3). τ depends on the user-specified parameter and is generally <0.01 in practice
| Query time | Space | Recall benchmark (Biologically) | Comments | |
|---|---|---|---|---|
| Inverted Index1 |
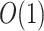
|
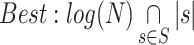
|
Only for document search/retrieval, could be applied to genomic/sequence search | Long construction time; impractical for bigger datasets; best case needs MPH and a known k-mer (term) distribution |
| BIGSCI/COBS2,3 |
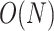
|
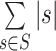
|
A hybrid between an inverted index and Bloom filters (COBS), high false positive rate, no benchmark with mutation rate by ANI/Mash | Query time is linear in N, small index size |
| Sequence Bloom Tree4 |
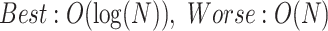
|
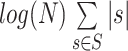
|
Given the k-mers from a query sequence, the task is to determine which of the N documents contain all the k-mers present in the query; no benchmark with mutation rate by ANI/Mash | Sequential query process is bottleneck; designed for sequential implementation |
| RAMBO5 |
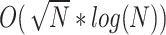
|
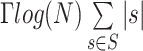
|
Similar to SBT, finding which of the N documents/genomes contain all the k-mers present in the query, no benchmark with ANI/Mash | Only for 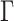 < 1, query time is sub-linear < 1, query time is sub-linear |
| MinHash6 |
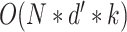
|
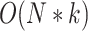
|
Average Nucleotide Identity (ANI) or mutation rate via Mash distance | Query time is linear in N |
| GSearch (MinHash-like + HNSW) |
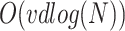
|
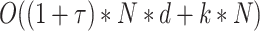
|
Average Nucleotide Identity (ANI) via Mash-like mutation rate/index | Long database construction time 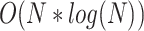 , but users are free from construction. , but users are free from construction. |
| FLINNG7 |
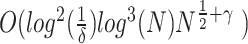
|
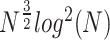
|
No Benchmark with Average Nucleotide Identity (ANI) via Mash-like mutation rate/index, only 15% of RefSeq genome meet the 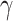 -stable criteria -stable criteria |
The 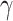 -stable query condition is a relatively strong requirement for the query. Limitation: works for queries for which the neighbors are all above a (relatively high) similarity threshold to the query -stable query condition is a relatively strong requirement for the query. Limitation: works for queries for which the neighbors are all above a (relatively high) similarity threshold to the query |