
Fig 1.
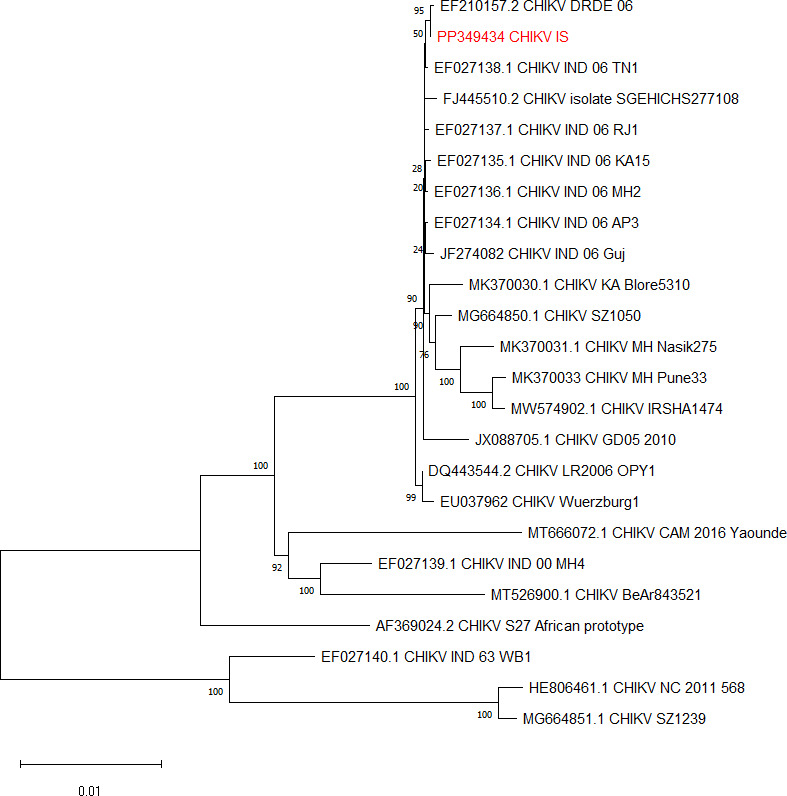
Phylogeny of chikungunya virus strains. Evolutionary history was inferred using the neighbor-joining method with default parameters. The optimal tree is shown and drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the maximum composite likelihood method and are in units of the number of base substitutions per site. MUSCLE tool was used to build the alignment and cleaned using the “pairwise deletion” option. Neighbor-joining method was the statistical method of choice with 1,000 bootstrap replicates. MEGA11 (v.11.0.13) was used to build and analyze the tree.