

Abstract
The metabolomic profile of aging is complex. Here, we analyse 325 nuclear magnetic resonance (NMR) biomarkers from 250,341 UK Biobank participants, identifying 54 representative aging-related biomarkers associated with all-cause mortality. We conduct genome-wide association studies (GWAS) for these 325 biomarkers using whole-genome sequencing (WGS) data from 95,372 individuals and perform multivariable Mendelian randomization (MVMR) analyses, discovering 439 candidate “biomarker - disease” causal pairs at the nominal significance level. We develop a metabolomic aging score that outperforms other aging metrics in predicting short-term mortality risk and exhibits strong potential for discriminating aging-accelerated populations and improving disease risk prediction. A longitudinal analysis of 13,263 individuals enables us to calculate a metabolomic aging rate which provides more refined aging assessments and to identify candidate anti-aging and pro-aging NMR biomarkers. Taken together, our study has presented a comprehensive aging-related metabolomic profile and highlighted its potential for personalized aging monitoring and early disease intervention.
Subject terms: Biomarkers, Diseases, Molecular biology
The metabolomic changes over the course of aging are complex. Here, the authors present a comprehensive metabolomic profile of aging and construct a metabolomic aging score, which has potential for personalized aging monitoring and early disease-risk identification.
Introduction
Aging is a complex biological process1 that leads to impaired physiological functions and may result in frailty2. It is a strong risk factor for multiple morbidities and mortality3,4. The aging-related disease burden accounted for 51.3% of the global health burden among adults in 20174. With advancements in omics technologies, aging research is progressing at an unprecedented pace. Many omics-based biological aging clocks have been developed, ranging from first-generation aging clocks, which were designed to predict chronological age5–9, to second-generation clocks, which were designed to predict aging-related adverse outcomes10–13 and better capture biological aging signals14.
Metabolomics, which integrates intrinsic biological changes with extrinsic exposures15, carries systemic information throughout the body16,17. The advancement in multiple spectroscopy technologies, for example, high-throughput and cost-effective nuclear magnetic resonance (NMR) analysis18,19 and the application of machine learning algorithms, have promoted population-scale metabolomics research, with great potential for disease prediction20. Previous metabolomics-based biological aging scores, such as MetaboHealth18, which was trained on mortality, and MetaboAge19, which was trained on chronological age, performed well in mortality risk prediction and exhibited clinical potential.
The UK Biobank has recently released NMR metabolomics data generated by Nightingale Health for around 275,000 individuals. The large sample size, the broad coverage of NMR biomarkers, and the availability of comprehensive health-related phenotypes make the UK Biobank an ideal repository for metabolomics-based aging research. Here, we aimed to present a metabolomic profile of biological aging and aging-related diseases. We began by identifying aging-related biomarkers from 325 NMR biomarkers (249 biomarkers directly provided and 76 additional biomarker ratios not available in the original data but with potential biological implications21). We then linked these aging-related biomarkers to multiple aging-related adverse health outcomes. Next, we constructed a novel metabolomic aging score and explored its advantages over other aging metrics and its potential clinical applications. Finally, through a longitudinal analysis with follow-up data from 13,263 individuals, we derived a metabolomic aging rate, which provides further insights into participants’ personalized aging status. Based on this rate, we identified potential anti-aging and pro-aging NMR biomarkers that might serve as anti-aging intervention targets (Fig. 1).
Fig. 1. Study overview.

a Identification of 54 representative aging-related metabolomic biomarkers based on a least absolute shrinkage and selection operator (LASSO) Cox regression model developed amongst participants from England and Wales with all-cause mortality as the endpoint. b Links between nuclear magnetic resonance (NMR) biomarkers and aging-related adverse health outcomes. Metabolome-wide association analysis was conducted to explore the association between 54 representative aging-related biomarkers and 50 frailty-related phenotypes. Multivariable Mendelian randomization (MVMR) analysis was performed to identify potential causal relationships between 325 NMR biomarkers and 20 aging-related diseases. c Construction and application of the metabolomic aging score. The metabolomic aging score was integrated as a linear combination of 54 aging-related biomarkers weighted by the estimated coefficients from the model. The metabolomic aging score was compared with other aging metrics among participants recruited from Scotland and was further applied to discriminate future early-onset patients of aging-related diseases and to improve multiple disease-risk prediction. d Longitudinal analysis among 13,263 individuals with revisit metabolomic data. A metabolomic aging rate was calculated and used to identify personalized changing patterns in aging-related metabolomic profiles. 15 pro-aging and 25 anti-aging biomarkers were identified based on distinct changing patterns across different rate groups. COPD chronic obstructive pulmonary disease.
Results
54 aging-related representative metabolomic biomarkers
To identify metabolomic biomarkers representative of biological aging among the 325 highly correlated NMR biomarkers, we developed a least absolute shrinkage and selection operator (LASSO) Cox proportional hazards model22 with all-cause mortality as the predicted outcome. At the individual level, aging is a ubiquitous biological process accompanied by a loss of physiological functions, which ultimately leads to death23. At the populational level, mortality rates may indicate overall health trends24. Thus, all-cause mortality was used as a global aging-related endpoint and a benchmark to compare the predictive performance of different aging metrics5.
Among the 250,341 participants included in our study, 234,553 were recruited from 20 assessment centers in England and Wales, while 15,788 were recruited from two assessment centers in Scotland. Differences in mortality rates and other health-related characteristics between these two subsets have been reported in previous publications25 and were also identified in our study (Supplementary Table 1). Given the differences between participants from these regions, we trained the model in participants from England and Wales and evaluated and validated our model’s performance among participants from Scotland16.
The 325 NMR biomarkers in our study encompassed a wide range of metabolites, including amino acids, ketone bodies, fatty acids, lipoprotein lipids in 14 subclasses, and metabolomic biomarkers involved in glycolysis, fluid balance, and inflammation (Supplementary Fig. 1 and Supplementary Data 1). These biomarkers were highly correlated (Supplementary Data 2, 3). The LASSO Cox model trained with tenfold cross-validation identified a combination of 54 biomarkers (28 in absolute levels and 26 in ratios) with the best predictive performance for all-cause mortality (Supplementary Data 4). These 54 biomarkers were selected as representative aging-related biomarkers and included eight amino acids, three ketone bodies, five polyunsaturated fatty acid-related compositions, 32 lipoprotein-related biomarkers, one inflammation-related biomarker (glycoprotein acetyls, GlycA), two fluid balance biomarkers (creatinine and albumin), and three metabolites involved in glycolysis (glucose, lactate, and citrate). GlycA, a systemic inflammation biomarker and risk factor for cardiovascular and autoimmune diseases26, had the highest hazard ratio (HR) for all-cause mortality (HR = 1.25 per SD), while the linoleic acid to total fatty acids percentage (LA_pct) had the lowest HR (0.82 per SD) (Supplementary Fig. 2).
Next, we investigated the correlations of these biomarkers with different aging metrics, including chronological age, the frailty index (FI), which is a clinical indicator of accumulated health deficits27, and leukocyte telomere length (LTL), a measure of cell division28. Among the 54 aging-related representative biomarkers, 49 were significantly correlated with chronological age, 51 with the FI, and 50 with LTL (BH-adjusted p values <0.05). There were 35 aging-related biomarkers with consistent correlations with all aging metrics (Supplementary Fig. 3).
Compared to previously reported aging or mortality-related NMR biomarkers, four biomarkers associated with all-cause mortality reported by ref. 29 (GlycA, albumin, the average diameter for very-low-density lipoprotein particles, and citrate) were replicated in our study with consistent associations. Ten out of 14 all-cause mortality-related NMR biomarkers included in MetaboHealth18, and 19 out of 56 aging-related NMR biomarkers included in MetaboAge19 were validated in our study (Supplementary Data 5). The overlapping biomarkers included GlycA, creatinine, albumin, glycolysis-related metabolites, ketone bodies, and polyunsaturated fatty acid-related biomarkers. Lipoprotein-related biomarkers were less well replicated.
Associations between aging-related metabolomic biomarkers and frailty
The 54 aging-related representative biomarkers predictive of all-cause mortality were further investigated by analyzing their associations with multiple frailty-related deficits30. Chronological age was included as a covariate in multivariable logistic regression models.
We identified a comprehensive association profile underlying the 54 aging-related representative biomarkers and the 50 frailty-related phenotypes with a total of 1112 statistically significant associations (p values <2E-04) (Supplementary Fig. 4). GlycA, an inflammation-related biomarker, was positively associated with 43 frailty deficits, with odds ratios for pre-frail status of 1.31 and frail status of 1.63 (p values <2.2E-16) compared with non-frail status. Three polyunsaturated fatty acid-related biomarkers (linoleic acid to total fatty acids percentage, omega-3 fatty acids, and polyunsaturated fatty acids to monounsaturated fatty acids ratio) generally exhibited negative associations with multiple frailty deficits and were associated with lower odds of frailty (odds ratios of 0.43, 0.53, and 0.25, respectively, with p values <2.2E-16). Several lipoprotein-related biomarkers, including refined HDL compositions, were negatively associated with prevalent cardiovascular diseases, for example, coronary heart disease, stroke, hypertension, and angina. Several VLDL composition-related biomarkers were potential risk factors for these diseases (Supplementary Data 6,7).
The associations between the 54 aging-related representative NMR biomarkers and multiple aging-related health deficits reflect potential targets for anti-aging interventions or disease prevention through the regulation and monitoring of these metabolomic biomarkers.
Candidate causal relationships between NMR biomarkers and aging-related diseases
Moving beyond cross-sectional associations, we investigated potential causal relationships linking metabolomic biomarkers with aging-related disease onset. It is possible that the NMR biomarkers causally related to aging-related diseases might not be among those selected by the LASSO Cox model due to high collinearity, an inherent feature of metabolomics data31. Therefore, we extended the search for causal biomarkers of aging-related diseases to all 325 NMR biomarkers.
A genome-wide association study (GWAS) for each NMR biomarker was conducted using WGS data from a subset of 95,372 individuals. Variants with a minor allele frequency (MAF) >0.1% were included after quality control (Methods). Based on the GWAS summary statistics of the 325 NMR biomarkers, we calculated pairwise genetic correlations using linkage disequilibrium score regression (LDSC)32. An extensive genetic correlation profile was identified underlying these metabolomic biomarkers, especially for the lipid and lipoprotein-related biomarkers (Supplementary Fig. 5 and Supplementary Data 8, 9).
Given the considerable pleiotropy of significant loci and correlations between the NMR biomarkers, we performed multivariable Mendelian randomization (MVMR) analysis to allow for multiple correlated exposures and pleiotropic instrumental variables33. Index variants marginally associated with each of the 325 NMR biomarkers (p < 1E-09) were selected as candidate instrumental variables (IVs). Twenty chronic non-communicable diseases, including sixteen leading causes of global disability-adjusted life-years (DALYs) among the elderly34, were chosen as outcomes (Supplementary Data 10). After pruning IVs in linkage disequilibrium (LD) and extracting the full column-rank matrix from the original exposure-IV association matrix (Methods), 287 NMR biomarkers were included in the whole set of exposures. A total of 2164 genetic variants, harmonized against the outcome GWAS summary statistics, were used as IVs in the subsequent MVMR analysis (Supplementary Data 11). MVMR analyses were conducted using four different yet complementary methods: MVMR-IVW, MVMR-Egger, MVMR-Lasso, and MVMR-median. The causal estimates from the MVMR-IVW method were considered our main findings, while the other three methods served as sensitivity analyses (Methods).
Out of 5740 possible combinations between the 287 NMR biomarkers and the 20 aging-related diseases, 439 pairs were identified as candidate causal relationships (composed of 213 NMR biomarkers and all 20 diseases) at a nominal statistical significance threshold of 0.05 for both MVMR-IVW and MVMR-Egger methods. Additionally, 14 pairs (involving 13 NMR biomarkers and six diseases) reached a Bonferroni-corrected significance threshold of 5E-04 for both methods (Fig. 2).
Fig. 2. 439 candidate pairs of causal relationships between 213 NMR biomarkers and 20 aging-related diseases identified in multivariable Mendelian randomization (MVMR) analysis.

Biomarker-disease pairs reaching nominal significance in both MVMR-IVW and MVMR-Egger methods were plotted. P values (two-sided, without multiple testing adjustments) and effect sizes are estimated from the MVMR-IVW method. The threshold for significance, adjusted for multiple comparisons, is set at 5E-04 using the Bonferroni-correction (Methods). 14 biomarker-disease pairs reaching the Bonferroni-corrected p value threshold in MVMR-IVW and MVMR-Egger methods are enlarged. NMR biomarkers are represented as dots and aging-related diseases as asterisks. Blue lines indicate negative causal estimates and red lines indicate positive causal estimates. The color intensity of the links is proportional to the corresponding estimated effect size. The transparency and width of the links are proportional to the −log(p value) from MVMR-IVW, which indicates the strength of the association. COPD chronic obstructive pulmonary disease, CKD chronic kidney disease, GERD gastro-esophageal reflux disease. Full names for NMR biomarkers in the plot can be found in Supplementary Data 1. Source data are provided as a Source Data file.
Chronic kidney disease (CKD) had the most candidate causal biomarkers, with 38 NMR biomarkers reaching the nominal p value threshold for both MVMR-IVW and MVMR-Egger methods. Notably, several disease-specific biomarkers emerged as the most significant candidate causal biomarkers for their respective diseases, including glucose for type 2 diabetes (p = 9.1E-09), creatinine for CKD (p = 6.5E-05), glycine for stroke (p = 2.9E-04), and albumin for liver fibrosis and cirrhosis (p = 3.3E-03).
Several NMR biomarkers served as shared risk or protective factors for multiple diseases. For instance, L_HDL_CE was a protective factor for CKD (p = 7.1E-04), asthma (p = 3.8E-03), GERD (p = 2.0E-02), hypertension (p = 2.7E-02), oral diseases (p = 2.8E-02), low back pain (p = 3.0E-02), and senile cataract (p = 3.7E-02). Glycine also showed protective effects for CKD (p = 2.0E-04), stroke (p = 2.9E-04), hypertensive heart disease (p = 2.7E-02), ischemic heart disease (p = 4.0E-02), hypertension (p = 4.0E-02), and type 2 diabetes (p = 4.5E-02). Conversely, M_LDL_L emerged as a risk factor for CKD (p = 2.6E-02), atrial fibrillation (p = 2.8E-02), stroke (p = 3.5E-02), GERD (p = 3.8E-02), asthma (p = 4.1E-02), and ischemic heart disease (p = 4.5E-02). Some biomarkers exhibited dual roles. L_HDL_CE_pct was a risk factor for atrial fibrillation (p = 1.7E-03), hypertension (p = 4.7E-03), CKD (p = 4.4E-02), and type 2 diabetes (p = 4.7E-02), but a protective factor for Parkinson’s disease (p = 5.5E-03). Similarly, total choline was a protective factor against hyperlipidemia (p = 2.1E-02) and ischemic heart disease (p = 2.2E-02), but a risk factor for COPD (p = 9.8E-03) and senile cataract (p = 4.5E-02). The p values reported above were derived from the MVMR-IVW method (Supplementary Data 12). The causal estimates from the other three sensitivity analyses were consistent with the main findings (Supplementary Fig. 6).
The results from the Mendelian randomization analysis might be biased if the exposures and outcomes have distinct but correlated causal variants, such as those in linkage disequilibrium35. Hence, we also conducted colocalization analysis for each pair of the 439 candidate causal relationships that reached nominal significance for both MVMR-IVW and MVMR-Egger methods to explore whether these biomarkers and diseases shared the same causal variants.
Of the 439 candidate causal pairs, 185 pairs (involving 122 NMR biomarkers and 18 diseases) had a posterior probability for hypothesis four (PPH4, which implies that two traits share the same causal variant36) greater than 80% (Supplementary Data 13). For each colocalized NMR biomarker-disease pair, we annotated the colocalized causal SNP with its target gene and associated phenotypes using the Ensembl Variant Effect Predictor37 (Supplementary Data 14).
Overall, for the 185 colocalized pairs, we identified 135 causal variants within 87 target genes, highlighting pleiotropic causal variants that linked multiple NMR biomarkers to aging-related diseases. For instance, rs8176685, a deletion in the intron of the ABO gene, emerged as a highly pleiotropic variant influencing 34 NMR biomarker-disease pairs. This variant linked 23 lipid and lipoprotein-related NMR biomarkers with conditions such as hyperlipidemia, ischemic heart disease, atrial fibrillation, stroke, and asthma. Previous research identified associations of rs8176685 with blood cell traits crucial for immune function, blood clotting, and cardiovascular health indicators like platelet distribution width38, vWF levels39, neutrophil count40, and P-selection levels41. Recent studies underscore the ABO gene’s role in cardiovascular disease risks and lipid metabolism, aligning with rs8176685’s identification in our study as a pleiotropic variant linking multiple lipid and lipoprotein-related biomarkers with cardiovascular diseases42. Additionally, rs11591147 (a missense variant in the PCSK9 gene) and rs7412 (a missense variant in the APOE gene) also exhibited extensive pleiotropic effects, each linking 25 lipid and lipoprotein-related NMR biomarkers with hyperlipidemia, ischemic heart disease, and atrial fibrillation.
Investigation into relevant biological functions and associated phenotypes of colocalized variants also provided insights into potential pathways or mechanisms through which the metabolomic biomarkers may impact disease onset. For example, rs1260326, a missense variant in GCKR, which encodes glucokinase regulators43, was identified as a shared causal variant linking lactate and 12 lipoprotein-related biomarkers with type 2 diabetes. However, rs1260326 was also found to be associated with circulating leptin levels, C-reactive protein levels, fructose-bisphosphate aldolase B levels, gamma-glutamyl transpeptidase (GGT) levels, and serum IGF-1 and IGF-binding protein (IGFBP)−3 levels44. These enzymes and biological molecules associated with rs1260326 might provide hints about the mechanism through which lactate and lipoprotein-related biomarkers are involved in disease onset.
Metabolomic aging score outperforms in short-term mortality risk prediction
Based on the 54 representative aging-related NMR biomarkers, we developed a novel metabolomic aging score as the linear combination of these biomarkers weighted by their estimated coefficients for all-cause mortality (Methods). In our study, the metabolomic aging score was highly correlated with MetaboHealth (Pearson’s r = 0.68), moderately correlated with chronological age (r = 0.29) and the frailty index (r = 0.32), and had the weakest correlation with LTL (r = 0.12) (p values <5E-7, Supplementary Fig. 7).
The predictive performance for all-cause mortality risk across different follow-up intervals, ranging from 1 year to 15 years, was assessed for the metabolomic aging score and compared against other aging metrics (including MetaboHealth, the frailty index, LTL, and chronological age) in an out-of-sample dataset of 15,788 participants from Scotland.
Compared to MetaboHealth, the frailty index, and LTL, the metabolomic aging score had the highest accuracy in mortality risk prediction across all follow-up intervals (Fig. 3a, b). The metabolomic aging score performed best in short-term (1 to 5 years) mortality risk prediction and even outperformed chronological age in these follow-up intervals (1 y, p value = 0.089; 2 y, p value = 0.036; 3 y, p value = 0.044; 4 y, p value = 0.015; 5 y, p value = 0.29). It matched chronological age in 10-year mortality risk prediction (p value = 0.94) but was inferior in 15-year mortality risk prediction (p value = 0.0042) (Supplementary Data 15).
Fig. 3. Predictive performance of different aging metrics for all-cause mortality risk across seven follow-up intervals (from 1 year to 15 years).

a Time-dependent AUCs of five aging metrics’ predictive performance for all-cause mortality risk. b ROC curves for 1-year, 2 years, 3 years, 4 years, 5 years, 10 years, and 15 years mortality risk prediction of five aging metrics. Two-sided P values from Delong’s test comparing the metabolomic aging score and chronological age were 8.9E-02, 3.6E-02, 4.4E-02, 1.5E-02, 2.9E-01, 9.4E-01, 4.2E-03 for 1 y, 2 y, 3 y, 4 y, 5 y, 10 y, and 15 y-prediction, respectively. c Time-dependent AUCs indicating the predictive performance for all-cause mortality risk associated with chronological age (dashed lines) and the residuals of the metabolomic aging score regressed against chronological age (solid lines), stratified by chronological age groups: 40–50 (green), 51–60 (blue), and 61–70 (red). d ROC curves for 1-year, 2 years, 3 years, 4 years, 5 years, 10 years, and 15 years mortality risk prediction of chronological age (dashed lines) and the residuals of the metabolomic aging score (solid lines) stratified by chronological age groups: 40–50 (green), 51–60 (blue), and 61–70 (red). Two-sided P values from Delong’s test comparing the residuals of the metabolomic aging score and chronological age: 40–50 (0.76, 0.67, 0.64, 0.80, 0.43, 0.74, and 1.00; 51–60 (3.0E-04, 3.4E-02, 2.5E-04, 4.2E-06, 7.1E-07, 2.2E-07, and 2.2E-06); 61–70 (6.9E-02, 1.4E-02, 6.3E-03, 2.8E-03, 2.8E-02, 8.0E-05, and 7.6E-04) for 1 y, 2 y, 3 y, 4 y, 5 y, 10 y, and 15 y-prediction, respectively. Met metabolomic aging score; FI frailty index, LTL leukocyte telomere length. Source data are provided as a Source Data file.
Considering the correlations of the four biological aging metrics with chronological age, we regressed each against chronological age and extracted the residuals to investigate whether they retained predictive information independent of chronological age (Supplementary Data 16).
The residuals of the four biological aging metrics regressed against chronological age displayed slightly reduced predictive performance across the seven follow-up intervals (1 y, 2 y, 3 y, 4 y, 5 y, 10 y, and 15 y). Nevertheless, the residuals of the metabolomic aging score outperformed those of the other metrics and had similar prediction accuracy to chronological age across 1-year to 5-year intervals (p values of 0.32, 0.32, 0.67, 0.53, and 0.36, respectively) (Supplementary Fig. 8).
We next conducted age-stratified analyses to explore the predictive performance of the metabolomic aging score across different chronological age groups (40–50, 51–60, and 61–70 years). A 10-year age span was considered sufficient to allow for differences in physiological and aging status between groups. Within the 40–50 age group, the residuals of the metabolomic aging score had a similar predictive performance as chronological age, with no significant difference in AUCs across all follow-up intervals (smallest p value = 0.43). Within the 51–60 and 61–70 age groups, the residuals of the metabolomic aging score outperformed chronological age in mortality risk prediction across all follow-up intervals (p values <0.05 except for p = 0.069 in the 61–70 group with 1-year interval). The best predictive performance was observed for the 51–60 age group with an AUC of 86.8% for 1-year mortality risk, whereas chronological age had an AUC of 56.0% (Fig. 3c, d and Supplementary Data 17).
The residuals of the metabolomic aging score were a significant predictor of short-term all-cause mortality. The age-stratified analysis further confirmed this observation (Supplementary Fig. 9). The residual of the metabolomic aging score exhibited the strongest associations in the 51–60 age group with a 1-year mortality HR per SD of 3.5 (95%CI: 2.6–4.8) and a 15-year mortality HR per SD of 1.9 (95%CI: 1.8–2.1), followed by the 61–70 age group with a 1-year mortality HR per SD of 2.3 (95%CI: 1.8–2.9) and a 15-year mortality risk HR per SD of 1.6 (95%CI: 1.5–1.7) and the 40–50 age group with a 1-year mortality HR per SD of 1.8 (95%CI: 0.6–5.2) and 15-year mortality HR per SD of 1.6 (95%CI: 1.3–1.8).
Metabolomic aging score discriminates future early-onset patients of aging-related diseases
Because the plasma metabolome provides insights into aging-related changes throughout the body45, we investigated whether these metabolomic signals could aid in the discrimination of individuals with accelerated aging. Typically, biologically older individuals are more vulnerable and more susceptible to developing aging-related diseases earlier than their counterparts46. Thus, we examined differences in the average baseline metabolomic aging score between future early-onset, other-onset, and disease-free groups for the 19 aging-related diseases (except for lower-back pain).
The average baseline metabolomic aging score was higher in the early-onset group compared to the other-onset group, followed by the disease-free group for 14 diseases, with chronological age included as a covariate in the analyses (BH-adjusted p values <0.05). These differences were most notable for type 2 diabetes (the early-onset vs the other-onset, BH-adjusted p value = 1.7E-09) and hypertension (the early-onset vs the other-onset, BH-adjusted p value = 1.0E-07) (Fig. 4).
Fig. 4. Metabolomic aging score discriminating future early-onset, other-onset, and disease-free groups among 19 aging-related diseases.

Between-group differences in the average baseline score were compared using the Emmeans test with chronological age-adjusted as a covariate. Red lines represent future early-onset patients (top 10% youngest at disease diagnosis), yellow lines represent other-onset patients (all other patients who did not fall within the youngest 10%), and blue lines represent disease-free participants (controls). Two-sided P values from Emmeans tests are adjusted for multiple comparisons using the Benjamini–Hochberg (BH) procedure. Data were presented as estimated marginal means with 95% confidence intervals. *, BH-adjusted P value <5E-02; **, BH-adjusted P value <5E-03; ***, BH-adjusted P value <5E-04; ****, BH-adjusted P value <5E-05; ns non-significant difference (Exact P values are available in the corresponding Source Data file). The sample size for each disease group as: type 2 diabetes, 226,761, 1675, 15,063; hyperlipidemia, 185,110, 2688, 24,192; hypertension, 148,546, 3449, 31,032; Ischemic heart disease, 222,053, 2008, 18,067; hypertensive heart disease, 249,794, 49, 435; atrial fibrillation and flutter, 228,799, 1734, 15,601; stroke, 245,035, 170, 1522; Parkinson’s disease, 248,047, 183, 1643; Alzheimer’s disease, 248,124, 221, 1985; hearing loss, 245,821, 248, 2224; COPD, 235,335, 1016, 9138; asthma, 213,092, 759, 6831; fibrosis and cirrhosis of liver, 248,910, 108, 965; diseases of oral cavity, 227,986, 1193, 10,729; gastro-esophageal reflux disease, 208,125, 2340, 21,059; CKD, 234,434, 1283, 115,47; osteoporosis, 236,103, 899, 8086; polyarthritis, 241,791, 681, 6123; cataract, 231,365, 1753, 15,773 (control, early-onset, other-onset groups respectively). COPD chronic obstructive pulmonary disease, CKD chronic kidney disease, Met baseline metabolomic aging score. Source data are provided as a Source Data file.
While metabolomic biomarkers circulate throughout the body, various organ systems contribute differently to the systemic metabolomic profile47. Specifically, in the cases of Parkinson’s disease and sensorineural hearing loss, there was no discernible difference in the average baseline metabolomic aging score between the early-onset group and the other-onset groups. This observation might suggest that parts of the nervous system, relative to other organ systems, are not as well reflected in systemic metabolomic profiles, potentially due to the blood–brain barrier48.
Between-group differences in the distribution of several basic physiological and socioeconomic characteristics (including chronological age, self-reported sex, BMI, systolic blood pressure, Townsend deprivation index, alcohol intake frequency, and smoking status) existed for each disease (Supplementary Tables 2–20). To address potential confounding effects derived from these differences, we conducted a multinomial logistic regression with each disease status (disease-free, early-onset, and other-onset) as the dependent variable, the above-mentioned confounders as covariates, and the metabolomic aging score as the independent variable. The baseline metabolomic aging score remained a significant factor distinguishing the future early-onset, other-onset, and disease-free groups (Supplementary Data 18).
Metabolomic aging score improves aging-related disease-risk prediction
We further explored the potential application of the metabolomic aging score for aging-related disease-risk prediction.
First, we investigated the predictive performance of different aging metrics as independent predictors of 19 aging-related diseases. Chronological age generally displayed the broadest applicability in disease-risk prediction, surpassing the metabolomic aging score, the frailty index, and LTL, with the highest AUC for eight diseases (Supplementary Fig. 10). The metabolomic aging score demonstrated good performance in cases where the pathogenesis underlying the disease involved dysregulated metabolic pathways. It outperformed the other aging metrics in the prediction of type 2 diabetes, hypertensive heart disease, fibrosis and cirrhosis of the liver, and CKD. Additionally, it demonstrated comparable performance to chronological age in predicting the risk of ischemic heart disease (DeLong test p value = 0.50). The frailty index, which quantifies accumulated health deficits, exhibited the strongest prediction of COPD, asthma, diseases of the oral cavity, GERD, and polyarthritis. One plausible explanation might be the inclusion of relevant clinical manifestations in the calculation of the frailty index. For example, emphysema or chronic bronchitis are hallmarks of COPD49, while knee pain and long-standing infirmity are common features of polyarthritis50. These health deficits were included in the calculation of the frailty index. LTL generally demonstrated weaker predictive performance.
Additionally, we explored whether the metabolomic aging score improves disease-risk prediction beyond traditional risk factors and other aging measures (Methods). We found that for 17 of the 19 diseases, there was an improvement in risk prediction, as indicated by Harrell’s C-index (p values <0.05), except for Parkinson’s disease and sensorineural hearing loss. However, when the frailty index was also incorporated into the model (Model 4), the improvement due to the metabolomic aging score was attenuated. In line with our earlier findings, type 2 diabetes, stroke, fibrosis, and cirrhosis of the liver, and CKD benefitted most from the inclusion of the metabolomic aging score (Fig. 5).
Fig. 5. Harrell’s C-index of four models for 19 aging-related disease-risk prediction.

Model 1 (light orange), traditional risk factors (self-reported sex, body mass index, systolic blood pressure, Townsend deprivation index, alcohol intake frequency, and smoking status) + chronological age; Model 2 (orange), traditional risk factors + chronological age + metabolomic aging score; Model 3 (blue), traditional risk factors + chronological age + frailty index; Model 4 (red), traditional risk factors + chronological age + frailty index + metabolomic aging score. Two-sided P values indicating the differences in two Harrell’s C-indexes are calculated using Z-score tests without adjustment for multiple comparisons. Data were presented as Harrell’s C-indexes with 95% confidence intervals estimated in the testing dataset (a random 30% subset for each disease). *P value <0.05; **P value <5E-03; ***P value <5E-04; ns non-significant difference (Exact P values are available in the corresponding Source Data file). The sample size for each disease: type 2 diabetes, 73,047; hyperlipidemia, 63,597; hypertension, 54,906; ischemic heart disease, 72,636; hypertensive heart disease, 75,081; atrial fibrillation and flutter, 73,839; stroke, 74,016; Parkinson’s disease, 74,961; Alzheimer’s disease, 75,099; hearing loss, 74,487; COPD, 73,644; asthma, 66,204; fibrosis and cirrhosis of liver, 74,994; diseases of oral cavity, 71,970; gastro-esophageal reflux disease, 69,456; CKD, 74,178; osteoporosis, 73,524; polyarthritis, 74,577; cataract, 74,667. COPD chronic obstructive pulmonary disease, CKD chronic kidney disease. Source data are provided as a Source Data file.
Metabolomic aging rate calculated from longitudinal data
A subset of 13,263 participants had available revisit metabolomics data after a median follow-up duration of 4.4 years. This enabled us to calculate a metabolomic aging rate reflecting the rate of change in the metabolomic aging score.
Based on the change in the metabolomic aging score between the baseline assessment and the revisit, we defined the metabolomic aging rate as Δ metabolomic aging score divided by the follow-up time. There was a negative correlation between the metabolomic aging rate and the baseline metabolomic aging score (r = −0.38, p < 2.2E-16), indicating that individuals with higher metabolomic aging scores at baseline exhibited a more modest rate of change compared to those with lower scores at baseline (Supplementary Fig. 11). Subsequently, we regressed the metabolomic aging rate against the baseline score to obtain residuals that were independent of the baseline score. The residuals of the rate were positively correlated with chronological age, suggesting that chronologically older individuals experienced higher rates of change in their aging-related metabolomic profile (r = 0.18, p < 2.2E-16) (Supplementary Fig. 12).
The residuals of the metabolomic aging rate, regressed against the baseline score, were a significant risk factor (HR = 1.46 per SD, 95%CI: 1.37–1.55, p value < 2.2E-16) for all-cause mortality after adjusting for chronological age. Next, we conducted age-stratified analyses to explore potentially varied associations between the metabolomic aging rate and all-cause mortality risk across three chronological age groups (40–50, 51–60, and 61–70 years) while adjusting for chronological age. Compared with those in the 40–50 age group, the residuals of the metabolomic aging rate exhibited stronger associations with mortality risk among those in the 51–60 and 61–70 age groups (p values of 1.07E-02, 2.79E-15, and 8.06E-19, with HRs per SD of 1.42, 1.53, and 1.43, respectively) (Supplementary Fig. 13).
Furthermore, we stratified individuals into three aging-rate groups based on the distribution of the residuals of the metabolomic aging rate (top 25%, middle 50%, and bottom 25%) to examine differences in their mortality risk. Compared to the middle 50% rate group, the all-cause mortality hazard was higher in the top 25% rate group (HR = 2.31, 95%CI: 1.97–2.70, p value = 3.98E-25) and lower in the bottom 25% rate group (HR = 0.77, 95%CI: 0.62–0.96, p value = 1.84E-02) (Fig. 6). The age-stratified analyses revealed that across the three groups (40–50, 51–60, and 61–70 years), those with a top 25% rate had approximately twice the mortality hazard compared to those with a middle 50% rate. There was no significant difference between the bottom 25% rate group and the middle 50% rate group (Supplementary Fig. 14). Findings were similar after adjustment for chronological age (Supplementary Data 19).
Fig. 6. Survival curves of different metabolomic aging rate groups.
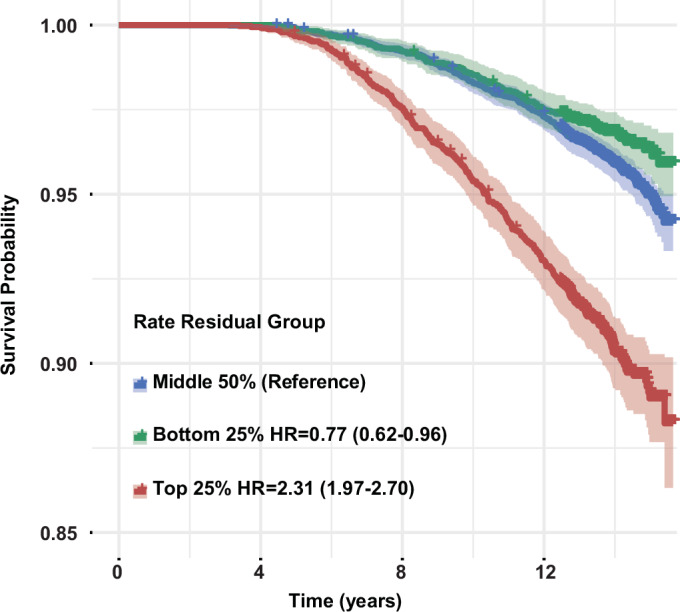
Three aging rate groups were defined based on the distribution of residuals of the metabolomic aging rate regressed against baseline metabolomic aging score: top 25% (red), middle 50% (blue), and bottom 25% (green). Data were presented as Kaplan–Meier estimates with 95% confidence intervals. Hazard ratios (HR) for all-cause mortality with 95% CIs were estimated using Cox proportional hazards regression with the middle rate group as the reference. Source data are provided as a Source Data file.
Identification of potential anti-aging and pro-aging metabolomic biomarkers
Beyond the general exploration of metabolomic aging score changes, we also tested individual changes in each aging-related representative biomarker relative to its baseline level. We discovered distinct changing patterns in the aging-related metabolomic profiles between those with a top 5% rate residual regressed against the baseline metabolomic aging score and those with a bottom 5% rate residual (Supplementary Fig. 15).
To identify potential anti-aging and pro-aging metabolomic biomarkers exhibiting distinct changing patterns across different rate residual groups (top 25%, middle 50%, and bottom 25%), we compared alterations in each biomarker’s level from the baseline (referred to as Δvalue) and included its baseline level as a covariate in the analysis of covariance (Fig. 7).
Fig. 7. Analysis of covariance comparing changes in each biomarker level against its baseline among the different metabolomic aging rate groups.

Changes in each biomarker level against the baseline (Δvalue) were compared between three aging rate groups using the Emmeans test with the baseline biomarker level adjusted as a covariate: top 25% (Fast, colored in red), middle 50% (Middle, colored in blue), and bottom 25% (Slow, colored in green) based on the distribution of the rate residuals regressed against the baseline metabolomic aging score. Two-sided P values from Emmeans tests are adjusted for multiple comparisons using the Benjamini–Hochberg (BH) procedure. Data were presented as estimated marginal means with 95% confidence intervals. *BH-adjusted P value <5E-02; **BH-adjusted P value <5E-03; ***BH-adjusted P value <5E-04; ****BH-adjusted P value <5E-05 (Exact P values are available in the corresponding Source Data file). The full name of each metabolomic biomarker is provided in Supplementary Data 1. The sample size for each NMR biomarker in the plot is 13,263. Source data are provided as a Source Data file.
Fifteen aging-related biomarkers with progressively increasing Δvalue from the bottom to the middle and to the top rate residual group (BH-adjusted p value <0.05) were considered pro-aging because of their greater increase in faster agers. These pro-aging NMR biomarkers included GlycA, tyrosine, creatinine, three glycolysis-related metabolites (glucose, lactate, and citrate), two ketone bodies (acetone and 3-hydroxybutyrate) and seven lipoprotein-related compositions. We observed the opposite pattern for 25 NMR biomarkers for which the Δvalue decreased progressively from the bottom to the middle and to the top rate residual group (BH-adjusted p value <0.05). These 25 biomarkers were considered anti-aging and included four amino acids (valine, histidine, glycine, and leucine), albumin, five polyunsaturated fatty acids-related biomarkers, and 15 lipoprotein-related biomarkers.
Out of the 40 pro-aging and anti-aging biomarkers identified here, we manually retrieved relevant biological functions and disease associations for 13 non-lipid biomarkers. For the remaining 27 lipid-related biomarkers that were highly correlated and had uncertain biological roles, we examined their overlap with the candidate causal biomarkers of aging-related diseases identified in the MVMR analysis. Among the 15 pro-aging biomarkers, GlycA marks the level of inflammatory cytokines in circulation and predicts cardiovascular and severe infection risk51. Impaired and dysregulated glycolysis was identified as a cardiovascular disease mechanism52 and a relevant biological aging process53, which might account for the pro-aging effects of glucose, lactate, and citrate. Acetone and 3-hydroxybutyrate, two ketone bodies whose elevated concentrations in circulation have been linked with ketoacidosis, a complication of uncontrolled diabetes and a significant risk factor for mortality, might exert their pro-aging effects via inducing oxidative stress54. Creatinine serves as a marker of renal damage and was recognized as a risk factor for cerebrovascular diseases55. In addition, XS_VLDL_PL_pct, a lipoprotein-related biomarker, was identified as a shared causal biomarker for multiple aging-related diseases in the MVMR analysis, including senile cataract, atrial fibrillation and flutter, and CKD. Among the 25 anti-aging biomarkers, albumin plays multiple crucial roles, such as inhibiting endothelial apoptosis and protecting against inflammation and oxidative stress56. Several amino acids, including glycine, histidine, leucine, and valine, were associated with a reduced risk of cardiovascular diseases57,58. Dietary supplementation of these amino acids has been recommended to mitigate various health issues59. Among the lipoprotein-related biomarkers recognized as anti-aging factors, LDL_size emerged as a candidate protective causal biomarker for stroke, Alzheimer’s disease, and liver fibrosis and cirrhosis. S_HDL_CE was identified as a candidate protective causal biomarker for Alzheimer’s disease and ischemic heart disease, while S_VLDL_PL_pct was a candidate protective biomarker for COPD and sensorineural hearing loss. Detailed biological functions and candidate causal associations with aging-related diseases for each pro and anti-aging biomarker are listed in Supplementary Data 20.
Discussion
The plasma metabolome carries dynamic biological signals reflecting personal health status60. Previous studies have demonstrated the potential of metabolomic biomarkers for disease16,17 and mortality risk prediction18. With the availability of low-cost, standardized, high-throughput NMR metabolomic profiling61 and the promotion of blood tests during medical checkups62, the identification and quantification of aging-related metabolomic biomarkers hold potential for personalized health monitoring and anti-aging interventions63.
Here, we present the largest aging-related metabolomic profile to date based on 325 NMR biomarkers from 250,341 individuals from the UK Biobank. A subset of 54 aging-related representative metabolomic biomarkers were identified based on their ability to predict all-cause mortality. These aging-related biomarkers are involved in diverse biological functions and metabolic pathways64, which might serve as potential anti-aging intervention targets and facilitate further exploration of the mechanism of aging-related diseases. High-resolution analysis of the refined composition and structure of multiple lipoprotein-related biomarkers, enabled by NMR profiling65, contributes greatly to unraveling the roles of lipid metabolism in the process of aging66.
In contrast to previous metabolomics-based studies that focused narrowly on the associations of selected biomarkers with specific diseases or their contribution to predictive performance16,17, our study harnessed WGS data from 95,372 individuals alongside comprehensive NMR metabolomic profiles. This enabled us to characterize the genetic architecture of the plasma metabolome67, yielding 325 NMR GWAS summary statistics for downstream analyses. Using MVMR analyses, we identified 439 candidate biomarker-disease causal pairs achieving nominal significance, with 14 pairs reaching Bonferroni-corrected significance. Colocalization analysis further supported 185 out of the 439 candidate causal pairs, providing insights into how these risk or protective biomarkers may affect disease onset. Moreover, the GWAS summary statistics for 325 NMR biomarkers provide a repository for future exploration68.
Among the 54 aging-related biomarkers, a significant portion of the non-lipoprotein-related biomarkers had been identified in previous studies18,19,29. These included several amino acids, glycolysis-related metabolites, two kidney function-related biomarkers (albumin and creatinine), and one inflammation-related biomarker (GlycA). There were also several differences from previous studies, particularly related to lipoproteins, which might be attributed to the following factors: (1) different sampling sources: NMR metabolomic biomarkers in the UK Biobank were measured from EDTA plasma samples, whereas MetaboAge utilized serum metabolomics19, and MetaboHealth employed a mixture of EDTA plasma and serum samples from various cohorts18. Previous studies have indicated that the source of sampling, whether it is plasma or serum, can affect metabolomic profiling69; (2) different cohort characteristics: individuals included in our study were between 40 and 70 years old, whereas the studies used for MetaboHealth and MetaboAge had a broader baseline age range spanning from 18 to 109 years. Additionally, differences in other health or socioeconomic features might affect the results; (3) different profiling backgrounds and noise in model training: although the NMR biomarkers in MetaboHealth and MetaboAge were measured using the same platform as in the UK Biobank, there were disparities in the number and variety of measured biomarkers. Moreover, due to the high degree of collinearity among the NMR biomarkers, the selection of aging or mortality-related biomarkers during model training may be influenced by the unique metabolomic background of each study70. Non-replicated aging-related biomarkers across different studies could, in fact, provide concordant and shared predictive information due to their inherent correlation70.
By integrating 54 aging-related biomarkers, we developed a novel metabolomic aging score with superior predictive performance for all-cause mortality compared to alternative biological aging metrics across various follow-up intervals in an out-of-sample dataset, both before and after adjusting for chronological age.
Notably, the metabolomic aging score demonstrated optimal predictive capability for short-term (1 to 5 years) mortality, surpassing chronological age. This superiority might stem from the dynamic nature of metabolomic profiles, which reflect immediate influences from intrinsic and extrinsic factors on health status71. Cellular metabolic reactions undergo rapid changes, while the resulting products or waste build up in the body due to delayed or impaired clearance over the course of aging72. However, the predictive signals carried by these biomarkers may decay due to the numerous changes that occur over longer follow-up periods73.
The characteristics of the plasma metabolome provide this score with prospective clinical value, particularly in monitoring high-risk populations such as frail seniors for whom complex physiological examinations may be challenging74. Additionally, it serves as a tool for detecting subtle metabolomic changes indicative of pathological patterns, thereby facilitating early interventions75. Our subsequent analyses revealed the metabolomic aging score as a significant factor in discriminating future early-onset patients of multiple aging-related diseases who exhibited an accelerated pace of aging compared to their peers76, even after adjustment for chronological age and other potential confounders. Importantly, this score offered additional and complementary predictive signals beyond traditional risk factors indicative of aging-related disease risks.
We propose the application of the metabolomic aging score as a complementary tool in various clinical scenarios, where it can be used alongside chronological age and other clinical parameters to provide a more comprehensive assessment. These include monitoring short-term mortality risk, identifying aging-accelerated populations, and enhancing disease-risk prediction when used in combination with traditional risk factors.
In a longitudinal analysis involving 13,263 individuals with revisit metabolomics data, we further defined a metabolomic aging rate which reflects the rate of change in the aging-related metabolomic profile and identified aging-accelerated individuals with higher mortality risk. After regressing this rate against the baseline score and obtaining the orthogonal residuals, we observed a gradual increase in rate residuals with advancing age, suggesting that the metabolomic profile of chronologically older individuals underwent more rapid changes toward age-related pathology77. While the metabolomic aging score helped discern biologically older individuals within a peer group, the metabolomic aging rate detected subtler differences in the rate of change in their aging-related metabolomic profiles. Drawing an analogy to the concepts of distance and speed in physics, the combination of both can predict how far a person will reach. We believed that combining the metabolomic aging score (reflecting the current distance along the route of biological aging) with the metabolomic aging rate (reflecting the current speed of biological aging) would yield greater predictive power and insights into personal aging status and future disease or mortality risk78. Based on the distinct changing patterns across different aging rate groups, we identified 15 pro-aging and 25 anti-aging biomarkers. These findings not only illuminate potential metabolic dysregulation accelerating the course of aging, but also highlight promising targets for anti-aging interventions79. Aging is a gradual and dynamic biological process. Longitudinal studies are crucial for capturing indicative changes in bio-signals over time and providing insights into the evolving nature of aging pathology80,81. The metabolomic aging rate reflects the rate of change toward aging pathology. With more samples from follow-up visits, increased reassessment frequency, and the development of large-scale longitudinal cohorts82, this rate can be further refined and improved. Such advancements would contribute to more precise and personalized aging assessments. Future research should focus more on the analysis of biological aging rates, as exemplified in our study, which offers a higher resolution of the pace of aging.
However, there are certain limitations to our current study. First, participants included in our study were aged between 40 and 70 years at the baseline assessment, with the majority falling within the 50–70 age group. The limited age range and imbalanced distribution among age groups might introduce potential bias in model training83 and diminish the generalizability of the metabolomic aging score and the metabolomic aging rate to broader populations. Second, the underrepresentation of more deprived and less healthy individuals in the UK Biobank is well documented84. Thus, applying our findings outside the studied population warrants caution. We used a sub-cohort recruited from Scotland as an out-of-sample dataset for validation, given the distinct demographic and health-related characteristics across different regions25. Nevertheless, external validation in other cohorts is warranted. Third, it is crucial to interpret candidate causal relationships identified in the MVMR analysis with caution, as only rigorous randomized controlled trials are the gold standard for testing cause-effect relationships85. Moreover, the MVMR analysis detected the direct effect of an exposure on the outcome, independent of other correlated exposures, rather than the overall effect on that outcome. This is because the exposure might influence the outcome indirectly via other related exposures in a multivariable scenario33. Importantly, similar to multivariable regression, multicollinearity due to the inclusion of correlated exposures could lead to unstable estimates and reduced statistical power in MVMR. Therefore, careful consideration is needed when determining which specific biomarkers influence which diseases86. Lastly, aligning with earlier research into the strengths and limitations of metabolomics for disease-risk prediction, our findings underscored that the predictive power of the plasma metabolome exhibits some degree of disease specificity16. It is plausible that the metabolic profile varies in its contribution to different disease mechanisms47, highlighting the challenge of applying a one-size-fits-all approach in metabolomics-based risk prediction5.
In conclusion, our study presents the most extensive metabolomic profile related to biological aging and highlights the potential of our metabolomic aging score in predicting mortality and disease-risk. However, our intention in devising this score was not to recommend it as a singular authoritative metric of biological aging. Instead, this score captures the aging-related signal at the metabolome level. Given the multifaceted nature of aging1, future research should integrate diverse aging-related metrics from multiple dimensions, for example, combining proteomic aging scores87 and epigenetic aging scores88 with the metabolomic aging score to unveil a more comprehensive profile of aging5.
Methods
Ethical compliance
UK Biobank has approval from the North West Multi-centre Research Ethics Committee as a Research Tissue Bank (RTB) approval. This approval means that researchers do not require separate ethical clearance and can operate under the RTB approval. Details on the ethics and governance framework of the UK Biobank are provided on the website (https://www.ukbiobank.ac.uk/media/0xsbmfmw/egf.pdf). This study has been approved under the UK Biobank application ID 103082.
Data processing and quality control
The UK Biobank is one of the largest biomedical databases89,90 and includes data from more than 500,000 participants recruited between 2006 and 2010 at 22 centers across England, Scotland, and Wales.
Nuclear magnetic resonance (NMR) metabolomics data available in the UK Biobank (updated in July 2023) included 249 metabolomic biomarkers (168 in absolute concentrations and 81 in derived ratios) from EDTA plasma samples of ~275,000 participants. Technical variation in these data was removed using the “ukbnmr” R package21. Seventy-six additional biomarker ratios with potential biological significance, but not available in the original data, were also computed and included in our analysis, resulting in a total of 325 NMR biomarkers21. The inclusion criteria and rationale behind these additional 76 biomarker ratios can be summarized as follows: (1) supplementing 20 additional lipoprotein fractions for three lipoprotein classes (low-density lipoprotein, very-low-density lipoprotein, and high-density lipoprotein) and for total serum lipids based on the original 14 lipoprotein subclasses; (2) decomposing total cholesterol into free cholesterol and esterified cholesterol and subsequently deriving more refined ratios for each lipoprotein class and subclass; (3) decomposing polyunsaturated fatty acids into omega-3 fatty acids and omega-6 fatty acids and subsequently deriving more refined ratios composed of omega-3 and omega-6 fatty acids.
After quality control, the 168 NMR biomarkers in absolute concentrations were log1p-transformed to better approximate a normal distribution.
Sample inclusion and model construction
To select representative NMR biomarkers associated with biological aging and predictive of all-cause mortality from the 325 highly correlated biomarkers, we adopted a LASSO Cox regression model with all-cause mortality as the endpoint. Participants recruited from the 20 assessment centers in England and Wales (n = 234,553) were included in the training dataset, while participants recruited from the two assessment centers in Scotland (n = 15,788) were included in an out-of-sample validation dataset. The date of attending the assessment center (UKB field ID: 53) was established as the baseline time point, while the date of death (up to December 8, 2022, for participants in England and Wales; December 19, 2022, for participants in Scotland) was designated as the endpoint. Participants who died of external accidents were censored (UKB field ID: 40001, deduced from the primary cause of death ICD-10 codes).
The LASSO Cox regression model was trained using the “glmnet” R package91, with 325 NMR biomarkers as independent variables and all-cause mortality as the dependent variable. We performed tenfold cross-validation to identify the optimal hyperparameter λ, which controls the magnitude of the penalty applied. The algorithm searched across 1000 possible values for λ and, for each specific λ, calculated the cross-validation partial likelihood deviance. The λ (0.00088527) that resulted in the lowest cross-validation partial likelihood deviance was chosen to fit the final model. A total of 54 out of 325 metabolomic biomarkers were assigned non-zero β coefficients after L1 regularization. The hazard ratios for all-cause mortality for each selected NMR biomarker were computed by standardizing the processed metabolomic data and exponentiating the corresponding estimated coefficients. A metabolomic aging score was integrated as the linear combination of the 54 aging-related biomarkers weighted by their respective coefficients assigned by the model.
Genome-wide association study of 325 NMR biomarkers
A subset of about 200,000 individuals with available whole-genome sequencing data were selected. Samples that failed to pass quality requirements (UKB field ID: 23093), samples with sex chromosome aneuploidy (UKB field ID: 22019), and samples with discordant genetic sex (UKB field ID: 22001) and self-reported sex (UKB field ID: 31) were excluded. Only samples whose genetic ethnic group was White (UKB field ID: 22006) were included, resulting in a final sample size of 95,372 individuals.
We used the whole-genome sequencing data from the UKB 200k release in GraphTyper joint call pVCF format on the UKB RAP92. Multiallelic variants were decomposed into biallelic variants using bcftools (v1.15.1). Quality control of SNPs and indels was performed based on the following criteria93: (1) alternative alleles with AAscore >0.5; (2) variant sites with the tag “FILTER = PASS”; (3) Hardy–Weinberg P value >10E-15; (4) genotype missing rate <10%. Further, only common variant sites (MAF >0.1%) were included in the GWAS. NMR data processed after quality control was inverse rank normalized with age, , sex, age*sex, *sex, BMI, medication status, smoking status, alcohol intake frequency, fasting time, assessment centre and genetic PCs 1–10 included as covariates. GWAS analyses were conducted using the STAAR framework (individual variant analysis provided within), which was suitable for biobank-scale WGS studies with abundant functional annotations to promote the power of association analysis94,95. The statistical significance threshold was defined as 5E-09 for GWAS analysis, including low-frequency variants (MAF >0.1%)96 divided by five principal components, which together accounted for >80% of the variation in the 325 biomarker levels, instead of dividing the total number of metabolomic biomarkers included in the study, which would be too conservative given the high collinearity between these biomarkers68. Thus, we chose 1E-09 as the appropriate p value threshold to claim statistical significance.
GWAS summary statistics for each biomarker were further clumped to identify independent loci accounting for linkage disequilibrium between variants, with a clumping window size of 500 kb around the index variant (p value <1E-09) and a linkage disequilibrium r2 threshold of 0.1.
Phenotypic and genetic correlations among 325 metabolomic biomarkers
Pairwise phenotypic correlations among the 325 NMR biomarkers were estimated using Pearson’s correlation coefficient. Pairwise genetic correlations were calculated based on the 325 GWAS summary statistics from our study using LDSC32, with the European ancestry population in the 1000 Genomes Project as the LD score reference panel. The p value threshold to claim statistically significant correlations was Bonferroni-corrected to 0.01 (0.05/5), as five principal components accounted for more than 80% of the variation in the metabolomic biomarker data.
Calculation and inclusion of different aging metrics
To compare the metabolomic aging score from our study to previous metabolomics-based aging metrics, we calculated the MetaboHealth score, which was composed of 14 all-cause mortality-related NMR biomarkers18,97, in the Scottish sub-cohort. The frailty index in the UK Biobank cohort was calculated following a previous study and included 49 health deficits30. These deficits met the following criteria: indicators of poor health; more prevalent in older individuals; neither rare nor universal; covering multiple areas of functioning; available for ≥80% of participants. The sum of deficits was divided by the total number of possible deficits, resulting in frailty index scores between zero and one, with higher scores indicating greater levels of frailty. Participants with missing data for ≥10/49 deficits were excluded30. Relative leukocyte telomere length (LTL) was measured by quantitative PCR, calculated as T/S ratio (telomere repeat copy number to single copy gene number), and adjusted for technical and operational parameters98 (UKB field ID: 22191). Chronological age was included as the participant’s age at recruitment (UKB field ID: 21022).
Associations between 54 aging-related metabolomic biomarkers and 50 frailty-related phenotypes
The 54 aging-related NMR biomarkers were standardized before calculating their associations with each of the 49 health deficits included in the frailty index and the overall frailty status. The overall frailty status was defined as “non-frail” (frailty index ≤0.08), “pre-frail” (frailty index: between 0.08 and 0.25), and “frail” (frailty index ≥0.25). The 54 aging-related representative biomarkers were included as independent variables, and each frailty-related phenotype was included as the dependent variable, with chronological age included as a covariate in multivariable logistic regression models. Frailty-related phenotypes with more than two categories were treated as categorical dependent variables with multiple levels, and associations were estimated with multinomial logistic regression models instead. The p value threshold was corrected for multiple testing using the Bonferroni procedure: 0.05/5/50 = 2E-04 (0.05 refers to the alpha level, 5 refers to the number of principal components, and 50 refers to the number of frailty-related phenotypes).
Multivariable Mendelian randomization (MVMR) analysis
We performed two-sample MVMR to identify potential causal relationships between the 325 metabolomic biomarkers, based on our GWAS summary statistics calculated from WGS data in a subset of 95,372 individuals, and 20 aging-related diseases, based on available GWAS summary statistics from the FinnGen consortium (data release R9)99.
MVMR is an extension of traditional Mendelian randomization. It tests for direct causal effects of multiple exposures on an outcome of interest and provides unbiased estimates, allowing for highly correlated exposures and pleiotropic instrumental variables when certain assumptions are satisfied33,100–102. Those assumptions are derived and modified from three critical assumptions for instrumental variables in univariable Mendelian randomization33,100,101: (i) the “relevance” assumption requires the genetic variants to be associated with at least one of the exposures; (ii) the “independence” assumption requires the genetic variants to be independent of all confounders of each exposure-outcome association; (iii) the “exclusion restriction” requires the genetic variants to not affect the outcome except through their effects on the exposures included in the analyses.
Considering the comprehensive genetic correlations between the 325 NMR biomarkers and notable pleiotropy in multiple loci, the 325 NMR biomarkers were initially taken together as a set of exposures, and each aging-related disease was treated as the outcome to minimize unmeasured pleiotropy of the instrumental variables.
Each NMR biomarker’s GWAS summary statistics underwent clumping to identify independently significant variants (using a window size of 500 kb around the lead variant, LD r2 threshold of 0.1 and p value threshold of 1E-09), resulting in 5680 index variants marginally associated with at least one of the NMR biomarkers. To ensure the independence of the instrumental variables (IVs) included in the MVMR analyses, further pruning among these 5680 variants was performed with a 200 kb window size and pairwise r2 threshold of 0.5. This process yielded 3171 independent variants, which were retained as candidate IVs.
Contrary to univariable Mendelian randomization, MVMR requires the marginal association matrix between the IVs and exposures to be of full column-rank. Multicollinearity within the exposure-IV association matrix can lead to unstable estimates and inflated type-I errors, thereby reducing statistical power86. To meet this requirement, we extracted a full-rank matrix (with a tolerance for determining a rank set of 1E-07) and eliminated redundant vectors from the exposure-IV marginal association matrix ( matrix). We retained a total of 3167 candidate IVs and 287 exposures in the full-rank association matrix for further analysis. After harmonizing allele effects between the exposures and outcomes, a total of 2164 IVs were included in the MVMR analysis (Supplementary Data 11).
Four MVMR analysis methods were utilized: MVMR-IVW, MVMR-Egger, MVMR-Median, and MVMR-Lasso. Each method provides valid estimates of causal effects under varying sets of relaxed assumptions103: MVMR-IVW provides unbiased estimates when all genetic variants are valid IVs or, in the presence of invalid IVs, if the pleiotropy is balanced and the InSIDE (Instrument strength independent of direct effect) assumption is met; MVMR-Egger is robust to directional pleiotropy and provides unbiased estimates even when all IVs are invalid, provided that the InSIDE assumption is met; MVMR-Median provides unbiased estimates when at least 50% of the weights come from valid IVs, allowing for the IV assumptions to be violated in a more general manner than MVMR-Egger; MVMR-Lasso identifies valid IVs and accounts for pleiotropy caused by invalid IVs without loss of power and without the requirement for the InSIDE assumption. Each method offers unique advantages tailored to specific scenarios. When combined, they provide complementary and supportive evidence for a candidate causal estimate, thereby strengthening the overall findings.
Results from MVMR-IVW represented our primary findings, while the results from the other three methods represented sensitivity analyses. Two p value thresholds were considered to claim statistical significance: a multiple-testing corrected p value using the Bonferroni-correction set to 0.05/ (5 PCs × 20 diseases) = 5E-04, and a nominal p value without correction for multiple testing set to 0.05.
Colocalization analysis
To further investigate whether the potential causal relationships between the NMR biomarkers and diseases were due to the same causal variant and not from different variants in linkage disequilibrium (which might lead to false-positive results in Mendelian randomization due to horizontal pleiotropy)35, we performed colocalization analysis for 439 candidate biomarker-disease causal pairs (involving 213 NMR biomarkers and 20 aging-related diseases) with nominal significance in both MVMR-IVW and MVMR-Egger analyses (p value <0.05).
Target loci to include in colocalization analysis were defined as a window size of 1 Mb around the independently significant index variants of each NMR biomarker included in colocalization analysis. Bayesian posterior probabilities of different causal variant configurations were calculated using the “coloc” R package36, with prior probabilities of SNP causality and colocalization set to default (p1 = 1E-04, p2 = 1E-04, and p12 = 1E-5). A posterior probability for hypothesis 4 (PPH4, association with both traits at a shared causal variant35) of ≥80% was considered suggestive evidence for colocalization. The most likely colocalized causal variant or variant sets were extracted and annotated for target genes and associated phenotypes using the Ensembl Variant Effect Predictor (VEP)37.
Comparisons of the predictive performance of different aging metrics for all-cause mortality across different follow-up intervals
First, pairwise correlations between different aging measures (the metabolomic aging score, MetaboHealth, the frailty index, LTL, and chronological age) were estimated using Pearson’s correlation. Then, the predictive performance of each aging measure for all-cause mortality across different follow-up intervals (1 year, 2 years, 3 years, 4 years, 5 years, 10 years, and 15 years) were estimated by the area under the receiver operating characteristic curve (AUC) using the “timeROC” R package104 among 15,788 participants from Scotland as an out-of-sample testing dataset. We reverse-coded LTL to ensure that the interpretation of effect sizes for all aging indicators was consistent. Differences in AUCs were tested using DeLong’s test. Next, four biological aging measures (the metabolomic aging score, MetaboHealth, the frailty index, and LTL) were regressed against chronological age, and their residuals were used for downstream analyses to validate their predictive performance independent of chronological age. Age-stratified analyses were conducted in three chronological age groups: 40–50, 51–60, and 61–70 years. Within each chronological age group, the predictive performance of each biomarker’s residual independent of chronological age was investigated in the same manner, and hazard ratios for all-cause mortality across different follow-up intervals were estimated using Cox proportional hazards models both within each age group and among all individuals in the Scottish dataset.
Differences in the metabolomic aging score discriminate future early-onset, other-onset, and disease-free groups for aging-related diseases
We included 19 out of 20 aging-related diseases, except for lower-back pain, in the following analyses. The first disease report dates (UKB field ID: 1712) were compared with the dates of attending the assessment center to calculate time-to-event. Individuals with a prevalent disease diagnosis before the baseline assessment were excluded from each disease-specific analysis.
The “early-onset” group of future patients for each disease was defined as those whose age at disease diagnosis was within the 10% youngest age among all incident cases. The age-at-diagnosis thresholds to define early-onset patients were: 57 for ischemic heart disease, 58 for stroke, 59 for COPD, 68.1 for Alzheimer’s disease, 54.6 for type 2 diabetes, 60 for CKD, 53.8 for sensorineural hearing loss, 58.2 for hypertensive heart disease, 63 for cataract, 60.3 for atrial fibrillation and flutter, 55.7 for fibrosis and cirrhosis of the liver, 63.6 for Parkinson’s disease, 57.4 for polyarthritis, 50.1 for diseases of the oral cavity, 52.2 for asthma, 54.6 for hypertension, 55.2 for hyperlipidemia, 59.2 for osteoporosis, and 53.7 for gastro-esophageal reflux disease. The remaining incident cases were labeled as other-onset and those without the disease as disease-free. Differences in the average metabolomic aging score at baseline among the three groups were compared using pairwise comparisons of estimated marginal means (“emmeans test”) with adjustment for baseline chronological age as a covariate. BH-adjusted p values <0.05 were considered statistically significant.
Moreover, for each disease cohort, differences in baseline distributions of basic health and demographic characteristics (including chronological age, self-reported sex, BMI, systolic blood pressure, Townsend deprivation index, alcohol intake frequency, and smoking status) were compared among future early-onset, other-onset, and disease-free groups using the Kruskal–Wallis rank-sum test and Pearson’s chi-squared test. Multinomial logistic regression models were fitted with the baseline metabolomic aging score as an independent variable, differentially distributed participant characteristics as covariates, and the disease status (defined as early-onset, other-onset, and disease-free) as the dependent variable.
Disease-risk prediction with different aging metrics
For disease-risk prediction, prevalent cases were excluded as previously described. First, four aging measures, including chronological age, the metabolomic aging score, the frailty index, and LTL, were used independently for disease-risk prediction. Performance was quantified by AUCs, and differences between them were tested using DeLong’s test. Then, four multivariable prediction models with different combinations of these aging measures and traditional risk factors (self-reported sex, body mass index, systolic blood pressure, Townsend deprivation index, alcohol intake frequency, and smoking status) were built as follows: Model 1 (traditional risk factors + chronological age), Model 2 (traditional risk factors + chronological age + metabolomic aging score), Model 3 (traditional risk factors + chronological age + frailty index) and Model 4 (traditional risk factors + chronological age + frailty index + metabolomic aging score). The full sample included in our study was randomly divided into a training and test set with a 7:3 ratio. Four multivariable prediction models were built using Cox proportional hazards regression in the training set for each disease. The time-to-event was determined with the baseline date as the starting point and the first disease-report date or the censoring date (the latest disease-report date among all incident cases), whichever came first, as the endpoint. Prediction performance was evaluated in the test set using Harrell’s C-index. The differences in Harrell’s C-indexes of different models were compared using Z-score tests105.
Calculation of metabolomic aging rate with revisit metabolomics data
There were 13,263 individuals with both baseline and revisit metabolomic data (median follow-up interval: 4.4 years), enabling us to calculate their rate of change in the aging-related metabolomic profile.
The metabolomic aging rate in our study was defined as “Δ metabolomic aging score/follow-up time”, corresponding to the average rate of change in score during the follow-up period. The residuals of the metabolomic aging rate were regressed against the baseline metabolomic aging score to exclude the effect of the baseline score on the rate of change. Cox proportional hazard models were fitted to estimate the hazard ratio (per SD) of the residuals of this rate among all the samples as well as within three chronological age groups (40–50, 51–60, and 61–70 years), with chronological age included as a covariate. Subsequently, three aging rate groups were defined based on the interquartile range of the residuals of the metabolomic aging rate: top 25%, middle 50%, and bottom 25%. Differences in the mortality hazards among the different rate residual groups were compared using Cox proportional hazards models. Age-stratified analyses across the three chronological age groups (40–50, 51–60, and 61–70 years), adjusted for chronological age, were also performed.
Identification of anti-aging and pro-aging biomarkers
We calculated the difference between the baseline level and the revisit level for each of the 54 representative aging-related NMR biomarkers and obtained their residuals regressed against the baseline level for downstream analysis. Metabolomic changing profiles for individuals in the top 5% aging rate residual group and the bottom 5% rate residual group were plotted, with each row representing an individual and each column representing a biomarker. Rows and columns were hierarchically clustered using the Ward.D2 method based on their Euclidean distances.
Further, the differences in the average change of each biomarker among different rate residuals groups (top 25%, middle 50% and bottom 25%) were compared, with the baseline level adjusted for as a covariate using the “emmeans test”. BH-adjusted p values <0.05 were considered statistically significant differences in average change.
Statistics and reproducibility
Our study is designed as an observational study with samples recruited from the UK Biobank cohort. No statistical methods were used to predetermine the sample size. Sample inclusion and exclusion criteria have been provided in the above “Sample inclusion and model construction” section. Traditional experimental design elements such as randomization and blinding are not applicable to our study.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Source data
Acknowledgements
This research analysed data provided by the UK Biobank via application 103082. We express our gratitude to all participants and their families, as well as all the investigators and members of the UK Biobank. We appreciate the resources and the platform provided by the High-Performance Computing Center of Central South University. Jinchen Li is funded by the National Key R&D Program of China (2021YFC2502100), the Hunan Innovative Province Construction Project (2021SK1010), and The Central South University Research Program of Advanced Interdisciplinary Study (2023QYJC010). Bin Li is funded by the National Natural Science Foundation of China (82001362), the Natural Science Foundation of Hunan province in China (2021JJ31070), Hunan Youth Science and Technology Innovation Talent Project (2022RC1070), The Scientific Research Program of FuRong Laboratory (No. 2023SK2093-1). Julian Mutz is funded by the King’s Prize Fellowship and the National Institute for Health and Care Research (NIHR) Maudsley Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. Zheng Wang is funded by Fundamental Research Funds for the Central Universities of Central South University (Grants No. 2023ZZTS0831).
Author contributions
J.L. and B.L. conceived the project. J.M. provided methodological support and revised the manuscript. S.Z. and Z.W. performed the data analysis and wrote the manuscript. Y.W., Y.Z., and Q.Z. visualized and organized the main results. X.J. and G.Z. revised the manuscript. J.Q., K.X., and B.T. contributed to the discussion of the results. All the authors have reviewed, revised, and approved the manuscript.
Peer review
Peer review information
Nature Communications thanks Nicola Pirastu, Kun Qian and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Data availability
The data for this research are obtained from the UK Biobank and are publicly available to approved researchers for health-related research (https://www.ukbiobank.ac.uk/enable-your-research/apply-for-access). The NMR metabolomic data in the UK Biobank are generated by Nightingale Health and are provided in Category 220 (https://biobank.ndph.ox.ac.uk/showcase/label.cgi?id=220). The GWAS summary statistics for 325 NMR biomarkers have been deposited in the NHGRI-EBI GWAS Catalog database with study accession IDs ranging from GCST90445833 to GCST90446157. Detailed GWAS Catalog assession IDs for each NMR biomarker are provided in Supplementary Data 21. For example, GWAS summary statistics of acetate has been deposited at http://ftp.ebi.ac.uk/pub/databases/gwas/summary_statistics/GCST90445001-GCST90446000/GCST90445833/. The data for figures in this study are provided in the Source Data file. Also, the data that support the findings of this study are available from the corresponding authors upon request. Source data are provided with this paper.
Code availability
Relevant analyses in this study were conducted using R version 4.2.0 (https://www.r-project.org), PLINK 2.00 alpha (https://www.cog-genomics.org/plink/2.0/), Bcftools (https://samtools.github.io/bcftools/), and LDSC (LD score) v1.0.1 (https://github.com/bulik/ldsc). No customized code was developed.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Shiyu Zhang, Zheng Wang.
Contributor Information
Julian Mutz, Email: julian.mutz@gmail.com.
Jinchen Li, Email: lijinchen@csu.edu.cn.
Bin Li, Email: lebin001@csu.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-52310-9.
References
- 1.López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. Hallmarks of aging: an expanding universe. Cell186, 243–278 (2023). 10.1016/j.cell.2022.11.001 [DOI] [PubMed] [Google Scholar]
- 2.Fedarko, N. S. The biology of aging and frailty. Clin. Geriatr. Med.27, 27–37 (2011). 10.1016/j.cger.2010.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mutz, J., Roscoe, C. J. & Lewis, C. M. Exploring health in the UK Biobank: associations with sociodemographic characteristics, psychosocial factors, lifestyle and environmental exposures. BMC Med.19, 240 (2021). 10.1186/s12916-021-02097-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang, A. Y., Skirbekk, V. F., Tyrovolas, S., Kassebaum, N. J. & Dieleman, J. L. Measuring population ageing: an analysis of the Global Burden of Disease Study 2017. Lancet Public Health4, e159–e167 (2019). 10.1016/S2468-2667(19)30019-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rutledge, J., Oh, H. & Wyss-Coray, T. Measuring biological age using omics data. Nat. Rev. Genet.23, 715–727 (2022). 10.1038/s41576-022-00511-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol.14, 3156 (2013). 10.1186/gb-2013-14-10-r115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hannum, G. et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell49, 359–367 (2013). 10.1016/j.molcel.2012.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin, Q. et al. DNA methylation levels at individual age-associated CpG sites can be indicative for life expectancy. Aging8, 394–401 (2016). 10.18632/aging.100908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mutz, J., Iniesta, R. & Lewis, C. M. Metabolomic age (MileAge) predicts health and lifespan: a comparison of multiple machine learning algorithms. Preprint at medRxiv10.1101/2024.02.10.24302617 (2024).
- 10.Lu, A. T. et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging11, 303–327 (2019). 10.18632/aging.101684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levine, M. E. et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging10, 573–591 (2018). 10.18632/aging.101414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belsky, D. W. et al. Quantification of the pace of biological aging in humans through a blood test, the DunedinPoAm DNA methylation algorithm. eLife9, e54870 (2020). 10.7554/eLife.54870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang, Y. et al. DNA methylation signatures in peripheral blood strongly predict all-cause mortality. Nat. Commun.8, 14617 (2017). 10.1038/ncomms14617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuiper, L. M. et al. Epigenetic and metabolomic biomarkers for biological age: a comparative analysis of mortality and frailty risk. J. Gerontol.78, 1753–1762 (2023). 10.1093/gerona/glad137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bentley, A. R. et al. Multi-ancestry genome-wide gene–smoking interaction study of 387,272 individuals identifies new loci associated with serum lipids. Nat. Genet.51, 636–648 (2019). 10.1038/s41588-019-0378-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buergel, T. et al. Metabolomic profiles predict individual multidisease outcomes. Nat. Med.28, 2309–2320 (2022). 10.1038/s41591-022-01980-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Talmor-Barkan, Y. et al. Metabolomic and microbiome profiling reveals personalized risk factors for coronary artery disease. Nat. Med.28, 295–302 (2022). 10.1038/s41591-022-01686-6 [DOI] [PubMed] [Google Scholar]
- 18.Deelen, J. et al. A metabolic profile of all-cause mortality risk identified in an observational study of 44,168 individuals. Nat. Commun.10, 3346 (2019). 10.1038/s41467-019-11311-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van den Akker, E. B. et al. Metabolic age based on the BBMRI-NL 1H-NMR metabolomics repository as biomarker of age-related disease. Circ. Genom. Precis. Med.13, 541–547 (2020). 10.1161/CIRCGEN.119.002610 [DOI] [PubMed] [Google Scholar]
- 20.Chen, X., Shu, W., Zhao, L. & Wan, J. Advanced mass spectrometric and spectroscopic methods coupled with machine learning for in vitro diagnosis. VIEW4, 20220038 (2023). 10.1002/VIW.20220038 [DOI] [Google Scholar]
- 21.Ritchie, S. C. et al. Quality control and removal of technical variation of NMR metabolic biomarker data in ~120,000 UK Biobank participants. Sci. Data10, 64 (2023). 10.1038/s41597-023-01949-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tibshirani, R. The lasso method for variable selection in the Cox model. Stat. Med.16, 385–395 (1997). [DOI] [PubMed] [Google Scholar]
- 23.Guo, J. et al. Aging and aging-related diseases: from molecular mechanisms to interventions and treatments. Signal Transduct. Target. Ther.7, 391 (2022). 10.1038/s41392-022-01251-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Naghavi, M. et al. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet390, 1151–1210 (2017). 10.1016/S0140-6736(17)32152-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCartney, G. et al. Explaining the excess mortality in Scotland compared with England: pooling of 18 cohort studies. J. Epidemiol. Community Health69, 20–27 (2015). 10.1136/jech-2014-204185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Connelly, M. A., Otvos, J. D., Shalaurova, I., Playford, M. P. & Mehta, N. N. GlycA, a novel biomarker of systemic inflammation and cardiovascular disease risk. J. Transl. Med.15, 219 (2017). 10.1186/s12967-017-1321-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rockwood, K. & Mitnitski, A. Frailty in relation to the accumulation of deficits. J. Gerontol.62, 722–727 (2007). 10.1093/gerona/62.7.722 [DOI] [PubMed] [Google Scholar]
- 28.Harley, C. B., Futcher, A. B. & Greider, C. W. Telomeres shorten during ageing of human fibroblasts. Nature345, 458–460 (1990). 10.1038/345458a0 [DOI] [PubMed] [Google Scholar]
- 29.Fischer, K. et al. Biomarker profiling by nuclear magnetic resonance spectroscopy for the prediction of all-cause mortality: an observational study of 17,345 Persons. PLoS Med.11, e1001606 (2014). 10.1371/journal.pmed.1001606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mutz, J., Choudhury, U., Zhao, J. & Dregan, A. Frailty in individuals with depression, bipolar disorder and anxiety disorders: longitudinal analyses of all-cause mortality. BMC Med.20, 274 (2022). 10.1186/s12916-022-02474-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson, C. H., Ivanisevic, J. & Siuzdak, G. Metabolomics: beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol.17, 451–459 (2016). 10.1038/nrm.2016.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet.47, 291–295 (2015). 10.1038/ng.3211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burgess, S. & Thompson, S. G. Multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am. J. Epidemiol.181, 251–260 (2015). 10.1093/aje/kwu283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vos, T. et al. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet396, 1204–1222 (2020). 10.1016/S0140-6736(20)30925-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zuber, V. et al. Combining evidence from Mendelian randomization and colocalization: review and comparison of approaches. Am. J. Hum. Genet.109, 767–782 (2022). 10.1016/j.ajhg.2022.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet.10, e1004383 (2014). 10.1371/journal.pgen.1004383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McLaren, W. et al. The Ensembl variant effect predictor. Genome Biol.17, 122 (2016). 10.1186/s13059-016-0974-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Astle, W. J. et al. The allelic landscape of human blood cell trait variation and links to common complex disease. Cell167, 1415–1429.e1419 (2016). 10.1016/j.cell.2016.10.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sabater-Lleal, M. et al. Genome-wide association transethnic meta-analyses identifies novel associations regulating coagulation factor VIII and von Willebrand factor plasma levels. Circulation139, 620–635 (2019). 10.1161/CIRCULATIONAHA.118.034532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vuckovic, D. et al. The polygenic and monogenic basis of blood traits and diseases. Cell182, 1214–1231.e1211 (2020). 10.1016/j.cell.2020.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Png, G. et al. Identifying causal serum protein-cardiometabolic trait relationships using whole genome sequencing. Hum. Mol. Genet.32, 1266–1275 (2023). 10.1093/hmg/ddac275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vargas-Alarcón, G. et al. ABO gene polymorphisms are associated with acute coronary syndrome and with plasma concentration of HDL-cholesterol and triglycerides. Biomol. Biomed.23, 1125–1135 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shen, M. et al. Interaction between the GCKR rs1260326 variant and serum HDL cholesterol contributes to HOMA-β and ISI(Matusda) in the middle-aged T2D individuals. J. Hum. Genet.68, 835–842 (2023). 10.1038/s10038-023-01191-9 [DOI] [PubMed] [Google Scholar]
- 44.Yuan, F. et al. The association between rs1260326 with the risk of NAFLD and the mediation effect of triglyceride on NAFLD in the elderly Chinese Han population. Aging14, 2736–2747 (2022). 10.18632/aging.203970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rinschen, M. M., Ivanisevic, J., Giera, M. & Siuzdak, G. Identification of bioactive metabolites using activity metabolomics. Nat. Rev. Mol. Cell Biol.20, 353–367 (2019). 10.1038/s41580-019-0108-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tan, Q. Epigenetic age acceleration as an effective predictor of diseases and mortality in the elderly. eBioMedicine63, 103174 (2021). 10.1016/j.ebiom.2020.103174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tian, Y. E. et al. Heterogeneous aging across multiple organ systems and prediction of chronic disease and mortality. Nat. Med.29, 1221–1231 (2023). 10.1038/s41591-023-02296-6 [DOI] [PubMed] [Google Scholar]
- 48.Knox, E. G., Aburto, M. R., Clarke, G., Cryan, J. F. & O’Driscoll, C. M. The blood-brain barrier in aging and neurodegeneration. Mol. Psychiatry27, 2659–2673 (2022). 10.1038/s41380-022-01511-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barnes, P. J. & Celli, B. R. Systemic manifestations and comorbidities of COPD. Eur. Respir. J.33, 1165–1185 (2009). 10.1183/09031936.00128008 [DOI] [PubMed] [Google Scholar]
- 50.Alpay-Kanıtez, N., Çelik, S. & Bes, C. Polyarthritis and its differential diagnosis. Eur. J. Rheumatol.6, 167–173 (2019). 10.5152/eurjrheum.2019.19145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ritchie, ScottC. et al. The biomarker GlycA is associated with chronic inflammation and predicts long-term risk of severe infection. Cell Syst.1, 293–301 (2015). 10.1016/j.cels.2015.09.007 [DOI] [PubMed] [Google Scholar]
- 52.Ouyang, J., Wang, H. & Huang, J. The role of lactate in cardiovascular diseases. Cell Commun. Signal21, 317 (2023). 10.1186/s12964-023-01350-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Feng, Z., Hanson, R. W., Berger, N. A. & Trubitsyn, A. Reprogramming of energy metabolism as a driver of aging. Oncotarget7, 15410–15420 (2016). 10.18632/oncotarget.7645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kanikarla-Marie, P. & Jain, S. K. Hyperketonemia and ketosis increase the risk of complications in type 1 diabetes. Free Radic. Biol. Med.95, 268–277 (2016). 10.1016/j.freeradbiomed.2016.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wannamethee, S. G., Shaper, A. G. & Perry, I. J. Serum creatinine concentration and risk of cardiovascular disease. Stroke28, 557–563 (1997). 10.1161/01.STR.28.3.557 [DOI] [PubMed] [Google Scholar]
- 56.Bihari, S., Bannard-Smith, J. & Bellomo, R. Albumin as a drug: its biological effects beyond volume expansion. Crit.l Care Resusc.22, 257–265 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jauhiainen, R. et al. The association of 9 amino acids with cardiovascular events in Finnish men in a 12-year follow-up study. J. Clin. Endocrinol. Metab.106, 3448–3454 (2021). 10.1210/clinem/dgab562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ding, Y. et al. Plasma glycine and risk of acute myocardial infarction in patients with suspected stable angina pectoris. J. Am. Heart Assoc.5, e002621 (2016). 10.1161/JAHA.115.002621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu, G. Amino acids: metabolism, functions, and nutrition. Amino Acids37, 1–17 (2009). 10.1007/s00726-009-0269-0 [DOI] [PubMed] [Google Scholar]
- 60.Chen, L. et al. Influence of the microbiome, diet and genetics on inter-individual variation in the human plasma metabolome. Nat. Med.28, 2333–2343 (2022). 10.1038/s41591-022-02014-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wishart, D. S. et al. NMR and metabolomics—a roadmap for the future. Metabolites12, 678 (2022). 10.3390/metabo12080678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schrag, D. et al. Blood-based tests for multicancer early detection (PATHFINDER): a prospective cohort study. Lancet402, 1251–1260 (2023). 10.1016/S0140-6736(23)01700-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang, Y. et al. Polyamine metabolite spermidine rejuvenates oocyte quality by enhancing mitophagy during female reproductive aging. Nat. Aging3, 1372–1386 (2023). 10.1038/s43587-023-00498-8 [DOI] [PubMed] [Google Scholar]
- 64.Julkunen, H. et al. Atlas of plasma NMR biomarkers for health and disease in 118,461 individuals from the UK Biobank. Nat. Commun.14, 604 (2023). 10.1038/s41467-023-36231-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aru, V. et al. Quantification of lipoprotein profiles by nuclear magnetic resonance spectroscopy and multivariate data analysis. Trends Anal. Chem.94, 210–219 (2017). 10.1016/j.trac.2017.07.009 [DOI] [Google Scholar]
- 66.Tsugawa, H. et al. A lipidome landscape of aging in mice. Nat. Aging4, 709–726 (2024). 10.1038/s43587-024-00610-6 [DOI] [PubMed] [Google Scholar]
- 67.Surendran, P. et al. Rare and common genetic determinants of metabolic individuality and their effects on human health. Nat. Med.28, 2321–2332 (2022). 10.1038/s41591-022-02046-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen, Y. et al. Genomic atlas of the plasma metabolome prioritizes metabolites implicated in human diseases. Nat. Genet.55, 44–53 (2023). 10.1038/s41588-022-01270-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu, Z. et al. Differences between human plasma and serum metabolite profiles. PLoS ONE6, e21230 (2011). 10.1371/journal.pone.0021230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Takahashi, Y. et al. Improved metabolomic data-based prediction of depressive symptoms using nonlinear machine learning with feature selection. Transl. Psychiatry10, 157 (2020). 10.1038/s41398-020-0831-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Buchweitz, L. F. et al. Visualizing metabolic network dynamics through time-series metabolomic data. BMC Bioinformatics21, 130 (2020). 10.1186/s12859-020-3415-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell153, 1194–1217 (2013). 10.1016/j.cell.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Agueusop, I., Musholt, P. B., Klaus, B., Hightower, K. & Kannt, A. Short-term variability of the human serum metabolome depending on nutritional and metabolic health status. Sci. Rep.10, 16310 (2020). 10.1038/s41598-020-72914-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu, X. et al. Illness severity assessment of older adults in critical illness using machine learning (ELDER-ICU): an international multicentre study with subgroup bias evaluation. Lancet Digit. Health5, e657–e667 (2023). 10.1016/S2589-7500(23)00128-0 [DOI] [PubMed] [Google Scholar]
- 75.Zhao, Y. et al. NMR and MS reveal characteristic metabolome atlas and optimize esophageal squamous cell carcinoma early detection. Nat. Commun.15, 2463 (2024). 10.1038/s41467-024-46837-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Guida, J. L. et al. Associations of seven measures of biological age acceleration with frailty and all-cause mortality among adult survivors of childhood cancer in the St. Jude Lifetime Cohort. Nat.e Cancer5, 731–741 (2024). 10.1038/s43018-024-00745-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Belikov, A. V. Age-related diseases as vicious cycles. Ageing Res. Rev.49, 11–26 (2019). 10.1016/j.arr.2018.11.002 [DOI] [PubMed] [Google Scholar]
- 78.Kuo, P.-L. et al. Longitudinal phenotypic aging metrics in the Baltimore longitudinal study of aging. Nat. Aging2, 635–643 (2022). 10.1038/s43587-022-00243-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rosoff, D. B. et al. Multivariate genome-wide analysis of aging-related traits identifies novel loci and new drug targets for healthy aging. Nat. Aging3, 1020–1035 (2023). 10.1038/s43587-023-00455-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Özalay, Ö. et al. Longitudinal monitoring of the mouse brain reveals heterogenous network trajectories during aging. Commun. Biol.7, 210 (2024). 10.1038/s42003-024-05873-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee, S. W. et al. Longitudinal modeling of human neuronal aging reveals the contribution of the RCAN1–TFEB pathway to Huntington’s disease neurodegeneration. Nat. Aging4, 95–109 (2024). 10.1038/s43587-023-00538-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang, Y. & Zhao, Y. Cohort studies have great potential in healthy ageing research. Lancet Healthy Longev.4, e450–e451 (2023). 10.1016/S2666-7568(23)00163-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang, J., Soltan, A. A. S., Eyre, D. W. & Clifton, D. A. Algorithmic fairness and bias mitigation for clinical machine learning with deep reinforcement learning. Nat. Mach. Intell.5, 884–894 (2023). 10.1038/s42256-023-00697-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brayne, C. & Moffitt, T. E. The limitations of large-scale volunteer databases to address inequalities and global challenges in health and aging. Nat. Aging2, 775–783 (2022). 10.1038/s43587-022-00277-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hariton, E. & Locascio, J. J. Randomised controlled trials - the gold standard for effectiveness research: Study design: randomised controlled trials. BJOG125, 1716 (2018). 10.1111/1471-0528.15199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lin, Z., Xue, H. & Pan, W. Robust multivariable Mendelian randomization based on constrained maximum likelihood. Am. J. Hum. Genet.110, 592–605 (2023). 10.1016/j.ajhg.2023.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Oh, H. S.-H. et al. Organ aging signatures in the plasma proteome track health and disease. Nature624, 164–172 (2023). 10.1038/s41586-023-06802-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bell, C. G. et al. DNA methylation aging clocks: challenges and recommendations. Genome Biol.20, 249 (2019). 10.1186/s13059-019-1824-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature562, 203–209 (2018). 10.1038/s41586-018-0579-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sudlow, C. et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med.12, e1001779 (2015). 10.1371/journal.pmed.1001779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Simon, N., Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for Cox’s proportional hazards model via coordinate descent. J. Stat. Softw.39, 1–13 (2011). 10.18637/jss.v039.i05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Halldorsson, B. V. et al. The sequences of 150,119 genomes in the UK Biobank. Nature607, 732–740 (2022). 10.1038/s41586-022-04965-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hofmeister, R. J., Ribeiro, D. M., Rubinacci, S. & Delaneau, O. Accurate rare variant phasing of whole-genome and whole-exome sequencing data in the UK Biobank. Nat. Genet.55, 1243–1249 (2023). 10.1038/s41588-023-01415-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li, X. et al. Dynamic incorporation of multiple in silico functional annotations empowers rare variant association analysis of large whole-genome sequencing studies at scale. Nat. Genet.52, 969–983 (2020). 10.1038/s41588-020-0676-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li, Z. et al. A framework for detecting noncoding rare-variant associations of large-scale whole-genome sequencing studies. Nat. Methods19, 1599–1611 (2022). 10.1038/s41592-022-01640-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pulit, S. L., de With, S. A. & de Bakker, P. I. Resetting the bar: statistical significance in whole-genome sequencing-based association studies of global populations. Genet. Epidemiol.41, 145–151 (2017). 10.1002/gepi.22032 [DOI] [PubMed] [Google Scholar]
- 97.Bizzarri, D., Reinders, M. J. T., Beekman, M., Slagboom, P. E. & van den Akker, E. B. MiMIR: R-shiny application to infer risk factors and endpoints from Nightingale Health’s 1H-NMR metabolomics data. Bioinformatics38, 3847–3849 (2022). 10.1093/bioinformatics/btac388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Codd, V. et al. Measurement and initial characterization of leukocyte telomere length in 474,074 participants in UK Biobank. Nat. Aging2, 170–179 (2022). 10.1038/s43587-021-00166-9 [DOI] [PubMed] [Google Scholar]
- 99.Kurki, M. I. et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature613, 508–518 (2023). 10.1038/s41586-022-05473-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sanderson, E., Davey Smith, G., Windmeijer, F. & Bowden, J. An examination of multivariable Mendelian randomization in the single-sample and two-sample summary data settings. Int. J. Epidemiol.48, 713–727 (2018). 10.1093/ije/dyy262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rees, J. M. B., Wood, A. M. & Burgess, S. Extending the MR-Egger method for multivariable Mendelian randomization to correct for both measured and unmeasured pleiotropy. Stat. Med.36, 4705–4718 (2017). 10.1002/sim.7492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sanderson, E., Spiller, W. & Bowden, J. Testing and correcting for weak and pleiotropic instruments in two-sample multivariable Mendelian randomization. Stat. Med.40, 5434–5452 (2021). 10.1002/sim.9133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Grant, A. J. & Burgess, S. Pleiotropy robust methods for multivariable Mendelian randomization. Stat. Med.40, 5813–5830 (2021). 10.1002/sim.9156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kamarudin, A. N., Cox, T. & Kolamunnage-Dona, R. Time-dependent ROC curve analysis in medical research: current methods and applications. BMC Med. Res. Methodol.17, 53 (2017). 10.1186/s12874-017-0332-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kang, L., Chen, W., Petrick, N. A. & Gallas, B. D. Comparing two correlated C indices with right-censored survival outcome: a one-shot nonparametric approach. Stat. Med.34, 685–703 (2015). 10.1002/sim.6370 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The data for this research are obtained from the UK Biobank and are publicly available to approved researchers for health-related research (https://www.ukbiobank.ac.uk/enable-your-research/apply-for-access). The NMR metabolomic data in the UK Biobank are generated by Nightingale Health and are provided in Category 220 (https://biobank.ndph.ox.ac.uk/showcase/label.cgi?id=220). The GWAS summary statistics for 325 NMR biomarkers have been deposited in the NHGRI-EBI GWAS Catalog database with study accession IDs ranging from GCST90445833 to GCST90446157. Detailed GWAS Catalog assession IDs for each NMR biomarker are provided in Supplementary Data 21. For example, GWAS summary statistics of acetate has been deposited at http://ftp.ebi.ac.uk/pub/databases/gwas/summary_statistics/GCST90445001-GCST90446000/GCST90445833/. The data for figures in this study are provided in the Source Data file. Also, the data that support the findings of this study are available from the corresponding authors upon request. Source data are provided with this paper.
Relevant analyses in this study were conducted using R version 4.2.0 (https://www.r-project.org), PLINK 2.00 alpha (https://www.cog-genomics.org/plink/2.0/), Bcftools (https://samtools.github.io/bcftools/), and LDSC (LD score) v1.0.1 (https://github.com/bulik/ldsc). No customized code was developed.