

Abstract
Antibody–oligonucleotide conjugates (AOCs) are promising treatments for Duchenne muscular dystrophy (DMD). They work via induction of exon skipping and restoration of dystrophin protein in skeletal and heart muscles. The structure–activity relationships (SARs) of AOCs comprising antibody–phosphorodiamidate morpholino oligomers (PMOs) depend on several aspects of their component parts. We evaluate the SAR of antimouse transferrin receptor 1 antibody (αmTfR1)–PMO conjugates: cleavable and noncleavable linkers, linker location on the PMO, and the impact of drug-to-antibody ratios (DARs) on plasma pharmacokinetics (PK), oligonucleotide delivery to tissues, and exon skipping. AOCs containing a stable linker with a DAR9.7 were the most effective PMO delivery vehicles in preclinical studies. We demonstrate that αmTfR1-PMO conjugates induce dystrophin protein restoration in the skeletal and heart muscles of mdx mice. Our results show that αmTfR1-PMO conjugates are a potentially effective approach for the treatment of DMD.
Introduction
Duchenne muscular dystrophy (DMD) is an X-linked, progressive neuromuscular disease, predominantly caused by out-of-frame mutations in the dystrophin gene.1,2 Dystrophin forms a glycoprotein-associated complex with other signaling and scaffolding proteins, which is integral to the development and function of the musculoskeletal system.3 In the absence of functional dystrophin, there is increased fragility in muscle fibers and muscle degeneration occurs.4 Affecting 1 in 5000 males globally, DMD results in loss of ambulation prior to adolescence and eventual death in the third to fourth decade of life due to respiratory complications and heart failure.5,6 No cure is available for DMD, but exon-skipping therapies aimed at restoring the translational reading frame and, thereby, production of a functional dystrophin protein, have recently been approved as treatments for DMD.7
These approved therapeutics for the treatment of DMD make use of antisense oligonucleotides (ASOs), which are small, synthetic, single-stranded nucleic acids that can act upon mRNA in several ways, including targeting it for degradation or correcting splicing defects that lead to disease.8,9 More specifically, these therapies make use of phosphorodiamidate morpholino oligomers (PMOs), which are short, synthetic single-stranded DNA analogs. PMOs have a unique chemical structure that binds to complementary sequences of target dystrophin pre-mRNA by Watson–Crick base pairing to affect exon skipping and enable expression of a shorter but functional protein.10
PMOs consist of a backbone made up of charge-neutral phosphorodiamidate linkages instead of the typical phosphodiester linkages found in natural DNA. Morpholino rings, containing nitrogen atoms, replace the sugar–phosphate backbone of conventional nucleotides. This modification imparts PMOs with enhanced stability and resistance to enzymatic degradation, making them suitable for in vivo applications without the need for additional chemical modifications.11,32 These PMOs are designed to bind to dystrophin pre-mRNA at sites where the spliceosome would normally bind, thereby skipping over the target exon, bringing the rest of the RNA into frame, and allowing a functional, albeit shorter dystrophin protein to be translated.12,13 Each exon-skipping therapy is designed to skip a particular exon. Patients’ potential responsiveness to the therapy is dependent on the specific mutation they carry, which dictates whether skipping the exon of focus will bring the rest of the RNA into frame and thereby allow production of functional dystrophin.14
While PMOs represent an advancement in DMD treatment, there is limited efficacy afforded by these treatments due to poor delivery.15 The currently available PMOs require high, repeated doses owing to their limited cellular uptake and rapid clearance from the system.16 In addition, delivering PMOs to target organs and tissues such as the heart and large muscle groups poses a significant challenge.5 However, unconjugated PMO-based therapeutics demonstrate good safety and tolerability profile, which has supported their conditional approval by the US Food and Drug Administration (FDA) despite marginal increases in dystrophin levels observed. This highlights the need for improved delivery methods.8
Antibody-drug conjugates (ADCs) use antibodies to deliver small-molecule cytotoxic drugs to specific tissues in the body to improve cellular uptake and have been approved by the FDA and the European Medicines Agency for the treatment of different cancers.17 Antibody–oligonucleotide conjugates (AOCs) are a noncytotoxic subclass of ADCs consisting of an antibody and an oligonucleotide (e.g., small interfering RNA [siRNA] or PMO) bound together by a chemical linker that can deliver oligonucleotides to targeted tissues.18
Work by Sugo et al. has evaluated targeted delivery of oligonucleotides to muscle for the treatment of muscle disorders via the transferrin receptor 1 (TfR1)-mediated approach.5,19 TfR1 is expressed on the surface of several muscle groups including smooth, skeletal, and heart muscles.5 Recent publications, such as the work by Malecova et al. and Desjardins et al., have demonstrated effective delivery of diverse oligonucleotides to muscle tissue, with promising applications for treatments in development for DMD.5,18 AOCs leverage the precision of oligonucleotides with the targetability of antibodies, addressing the challenges associated with oligonucleotide delivery through tissue-specific delivery and receptor-mediated internalization capabilities of antibodies.18,20,21
In our investigation of the structure–activity relationship (SAR) for PMO AOCs, we evaluated several design parameters, one of which was linkers. Linkers are used to covalently tether the antibody to the PMO and play a crucial role in the functionality of AOCs, enhancing stability during circulation and facilitating the specific release of oligonucleotides in target tissues.22 The stability of the linker selected is vital to prevent premature release of PMO prior to reaching the target tissues. There are two types of linkers, cleavable and noncleavable linkers. Cleavable linkers, such as the enzymatically triggered valine-citrulline linker (ValCit), have a para-aminobenzyl carbamate spacer which undergoes a chemical reaction when the ValCit peptide is cleaved, resulting in the release of the payload with no residual components of the antibody or linker attached.23,24 Once internalized into the target cells, the linker needs to be cleaved to ensure quick release of the PMO. This is significant because the presence of a linker could potentially interfere with the PMO’s function. If PMOs cannot tolerate linkers attached after intracellular release, their activity could be compromised. Therefore, the use of cleavable linkers could be advantageous in ensuring the optimal performance of the PMO once it is released inside the cell. On the other hand, noncleavable linkers, like 4-(N-maleimidomethyl) cyclohexane-1-carboxylate (MCC) or bismaleimide (BisMal), lack a structural chemical trigger that could facilitate the release of PMO without attached remnants of antibody or linker.25,26 Noncleavable linkers rely on lysosomal proteolytic degradation of the antibody component to release the PMO after internalization. They typically have increased plasma stability compared with cleavable linkers.25 We expect AOCs with noncleavable linkers to be digested in the lysosomes upon internalization into cells and to release PMO attached to a small fragment or an amino acid of the antibody. Therefore, the choice of linker–cleavable or noncleavable–depends on the specific structure–activity requirements of the PMO and the target tissues. Both types of linkers have their advantages and potential drawbacks, and the selection should be made based on the desired functionality of the AOC.
The drug-to-antibody ratio (DAR) is another important consideration when evaluating AOC efficacy. While a low DAR may reduce payload delivery per antibody, a high DAR could affect antibody structure, stability, PK, delivery, or antibody binding capabilities, all of which may result in loss of AOC activity.27 The DAR of ADCs typically ranges from 2–8 depending upon the conjugation chemistry and hydrophobic nature of the drug.28,29
This study investigated the SAR and bioconjugation strategies of antimouse TfR1 (αmTfR1)-PMO conjugates, including the impact of linker chemistry, PMO conjugation positions, and various DARs on efficacy, as measured by exon skipping and dystrophin restoration in murine models of DMD. The mdx mouse model stands as the predominant murine model in DMD research. It harbors a stop codon mutation in exon 23 of the Dmd gene, leading to the production of a truncated nonfunctional protein. PMOs targeting mouse exon 23 (pmoEx23) obstructs the 5′ splice site of exon 23, thereby interfering with its recognition by the splicing machinery. Consequently, exon 23 is excluded from the dystrophin transcript, resulting in the production of a shorter yet functional mouse dystrophin protein.30,31 To evaluate αmTfR1-PMO conjugates in this model, we conjugated pmoEx23 to αmTfR1 antibodies to create conjugates that we term “αmTfR1-pmoEx23”.
This manuscript describes studies demonstrating that AOCs can achieve robust exon 23 skipping of the dystrophin gene and restore dystrophin protein in mdx mice. Additionally, this manuscript details the implications of the SAR on these results. We first describe a general overview of the synthesis used to create the AOCs we studied, before discussing our investigations on the impact of the conjugation positions (3′ versus 5′) on exon-skipping efficacy and the impact of cleavable versus noncleavable linkers on conjugate performance. We then describe the impact of DAR on exon skipping, including the impact of dose dependencies on exon skipping over time and the effect of conjugate treatment on dystrophin protein restoration in mdx mice.
AOC Naming Convention
In the rapidly evolving field of AOCs, the need for a clear, standardized naming convention is paramount because a multitude of variables exist, such as the type of antibody, its target, the linker used, and the PMO conjugated. Our naming convention, detailed below, offers a systematic approach using specific symbols and abbreviations to denote each component of an AOC.
Take the following conjugate as an example: αmTfR1-MCC-pmoEx23 DAR4. Here, “α” means “anti” to indicate that the antibody binds to transferrin receptor 1 or TfR1. The “h” or “m” denote whether the antibody is human (h) or mouse (m) specific. “MCC” is the linker, 4-(N-maleimidomethyl)cyclohexane-1-carboxyamide. The “pmoEx23” is the PMO targeting exon 23 skipping of the dystrophin gene in the mouse model. DAR4 indicates a drug-to-antibody ratio of four. Thus, “αmTfR1-MCC-pmoEx23 DAR4” would be an anti-TfR1 antibody that is specific to mice, with four MCC linkers connecting PMOs targeting exon 23 skipping.
In our naming convention for linkers, we use the linker name, rather than the reagent used in the reaction. This is because during the reaction, a part of the reagent is removed. For instance, instead of using “SMCC” (the reagent) as the name of the linker, we simply use “MCC”. This approach ensures that the names we use are representative of the actual structure of the AOCs. However, in the Experimental section, we use the full reagent name when discussing the linker reaction with the PMO, for example, SMCC (reagent) versus MCC (linker).
Results
Table S1 describes the antibody target, linker, PMO sequence, purity information, as well as dosing values by PMO and mAb for each antibody–PMO conjugate.
αmTfR1-pmoEx23 Conjugate Generation and SAR Evaluation
We produced the AOCs evaluated in this publication by conjugating PMOs to antibodies via a linker, with the aim of using antibodies to facilitate targeted delivery to certain tissues, such as the heart and skeletal muscle. αmTfR1-pmoEx23 conjugates were produced to conduct a proof-of-concept study in mdx mice.30Figure 1 illustrates the sequence and chemical structure of pmoEx23 with a β-alanine conjugation handle at the end of the 3′ morpholino amine. The β-alanine conjugation handle provides a flexible connection between the PMO and the antibody, which could potentially influence how the PMO interacts with its target.
Figure 1.
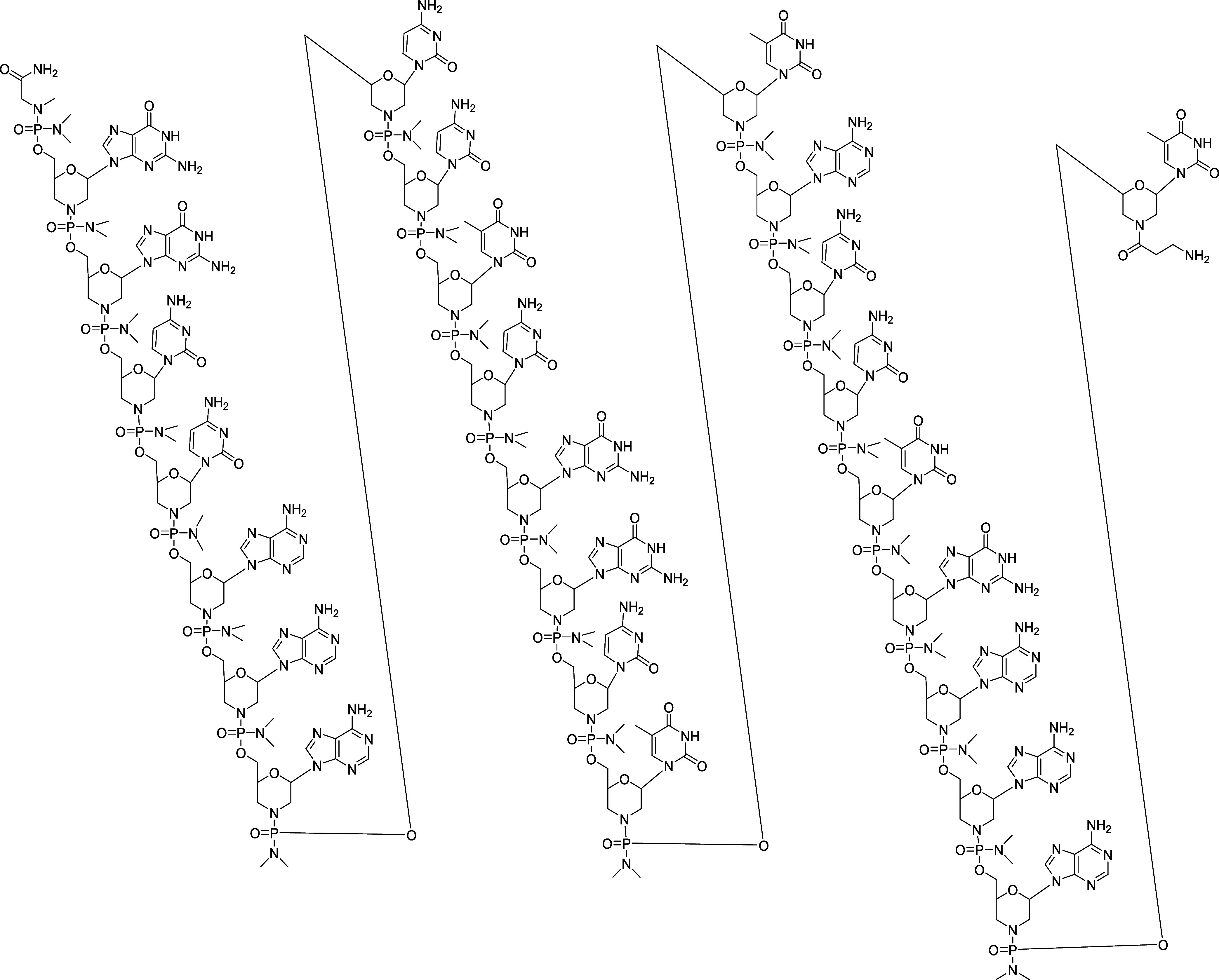
Sequence and chemical structure of pmoEx23 with 3′ amine.
Scheme 1 outlines the general synthetic scheme we used to generate αmTfR1-pmoEx23 conjugates. We procured pmoEx23, equipped with an amino conjugation handle at the 3′ or 5′ end of the PMO, from GeneTools. Subsequently, we conjugated to an N-hydroxysuccinimide (NHS)-linker-maleimide to produce an intermediate PMO-linker-maleimide (Scheme 1a). We reduced the interchain disulfide of the αmTfR1 antibody, meaning the disulfide bonds between the cysteine residues in the antibody were broken, exposing the cysteine residue thiol groups, which were then used for conjugation (Scheme 1b). We then conjugated the antibody to the PMO-linker-maleimide. The resulting intermediate conjugate was processed with N-ethylmaleimide (NEM) to cap any unconjugated thiols, purified with hydrophobic interaction chromatography (HIC) to isolate the desired subset of DAR species, and buffer exchanged to generate the final αmTfR1-PMO conjugate in a mixture of DAR. The ratio of different DARs was controlled by the stoichiometric ratios of tris(2-carboxyethyl)phosphine (TCEP) and PMO to antibody.
Scheme 1. (a) General Synthesis of PMO-linker-maleimide. (b) Synthesis of the αTfR1-PMO Conjugate. (c) A Representative Antibody–PMO Conjugate Structure with a Cleavable ValCit Linker. (d) A Representative Antibody–PMO Conjugate Structure with a Noncleavable MCC Linker.
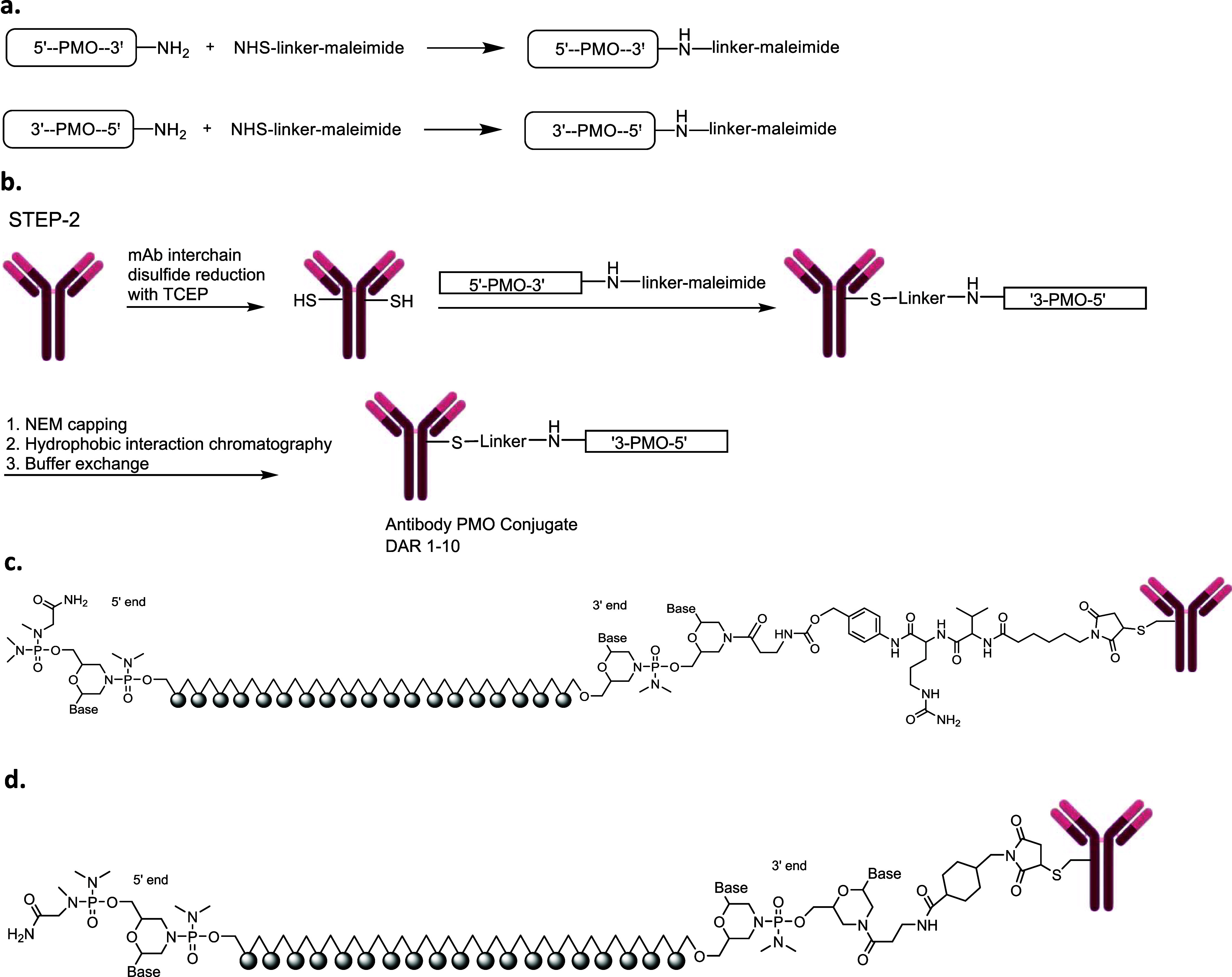
We synthesized αmTfR1-PMO conjugates utilizing both cleavable (ValCit) and noncleavable (BisMal, MCC, maleimidocaproyl [MC] and polyethylene glycol 2-maleimide [PEG2]) linkers (Scheme 1c,d, respectively). The DAR for representative antibody–PMO conjugates in panels 1c and 1d is flexible and can range from DAR1 to DAR8 or higher depending on the antibody subtype. For a human immunoglobulin G1 (IgG1), there are 4 interchain disulfides that can be reduced, leading to as many as 8 conjugation sites for the PMO. The mouse IgG2a antibody has additional interchain disulfides that can be reduced, allowing for average DARs higher than 8.33,34 A compound with DAR3.8, for example, is a mixture of conjugates with variable ratios that average 3.8. To generate DAR9.7 αmTfR1-PMO conjugates, HIC purification was not needed because a stoichiometric excess of PMO-linker was used to ensure that all the available reactive cysteines of the fully reduced monoclonal antibody (mAb) were conjugated by the PMO.
Impact of the Conjugation Positions (3′ versus 5′) on Exon-Skipping Efficacy
Given that PMOs work by the steric blocking of splicing enhancer sites, we were unsure if the linker and antibody fragments, which we expected to remain after the PMO is released from the AOC, would impact the PMO’s ability to bind to mRNA and prevent recruitment of splicing proteins. Therefore, we compared the effect of conjugation at both ends of pmoEx23, i.e., 3′ conjugation (Figure 2a) and 5′ conjugation (Figure 2b) on exon-skipping efficacy in the gastrocnemius of wild-type (WT) C57BL/6 mice using conjugates αmTfR1-MCC-pmoEx23 3′ DAR3.6 and αmTfR1-MCC-pmoEx23 5′ DAR3.7. Our analysis suggests that the 3′ conjugation position achieved marginally better exon skipping than the 5′ conjugation position in WT mice (Figure 2c). While the linker location on the PMO does not impact the melting temperature of the PMO with its target in vitro, it is possible that there are more complex factors that may impact the PMOs access to the target pre-mRNA in vivo.
Figure 2.

Impact of antibody conjugation site on PMO exon-skipping efficacy. (a) 3′ conjugation. (b) 5′ conjugation. (c) Impact of conjugate site on skipping efficiency in gastrocnemius. (d) 3′ PEG2-maleimide conjugation. (e) Impact of conjugation site on gastrocnemius tissue concentration. For (c) and (e), n = 4 per group, error bars represent SEM. DIPEA, N,N-diisopropylethylamine; DMSO, dimethyl sulfoxide; MCC, 4-(N-maleimidomethyl) cyclohexane-1-carboxylate; PEG2, polyethylene glycol 2-maleimide linker; PBS, phosphate buffered saline; SCRAM, scramble PMO control; SEM, standard error of the mean; RT, room temperature.
We also compared the exon skipping of AOCs conjugated to the PMO′s 3′ end via a β-alanine spacer versus PEG2-maleimide conjugated to the 3′ morpholine ring directly (αmTfR1-PEG2-pmoEx23 3′ DAR3.9, Figure 2d). We achieved a similar amount of exon skipping with both conjugates, however, to obtain comparable conjugation efficiency, a linker with less steric bulk (tetrafluorophenyl-PEG2-maleimide linker [TFP-PEG2] versus SMCC) was required (Figure 2d). We also included a nontargeting PMO scramble control (αmTfR1-MCC-SCRAM 3' DAR3.4) in our experiment, i.e., a version of the PMO that does not target any specific sequence. No exon skipping was observed with the PMO scramble control, suggesting that the exon skipping observed with the targeted PMO is a specific effect of that PMO, rather than a nonspecific effect or artifact of the experiment.
Additionally, we evaluated the impact of conjugation position on tissue concentration in gastrocnemius (Figure 2e). We observed slightly higher PMO tissue concentration with the 5′ conjugate, αmTfR1-MCC-pmoEx23 5′ DAR3.7, despite there being lower exon-skipping activity. While these differences were small and not statistically significant, it is possible that the bulkier triazine linker included in the 5′ conjugate slightly improved the PMOs′ retention within the cell and reduced the PMOs′ ability to effectively enter the nucleus and bind to the target pre-mRNA. The tissue concentration of the unconjugated PMO was not assessed in this experiment, given the anticipated rapid clearance from circulation. To confirm the hypothesis of rapid clearance, we directly compared the conjugated antibody–PMO to an unconjugated PMO in a subsequent experiment with PK data (reported later in the Results section, Figure 4c).
Figure 4.

Impact of DAR on AOC binding affinity, PK, muscle tissue delivery of PMO, and exon-skipping efficacy. (a) Impact of DAR on binding affinity. (b) Impact of DAR on PK in plasma. (c) Impact of DAR on PMO concentration in gastrocnemius tissue. (d) Impact of DAR on exon-skipping efficacy. For (b) to (d), n = 4 per group, error bars represent SEM.
Impact of Cleavable versus Noncleavable Linkers on Conjugate Performance
Scheme 2 summarizes the synthetic schemas we used for conjugating MCC, PEG2, BisMal, ValCit, and MC linkers to the 3′ end of a PMO.
Scheme 2. Schematic Diagram Illustrating the Synthetic Schema Conjugating (a) MCC, (b) PEG2, (c) BisMal, (d) ValCit, and (e) MC Linkers to the 3′ End of a PMO.
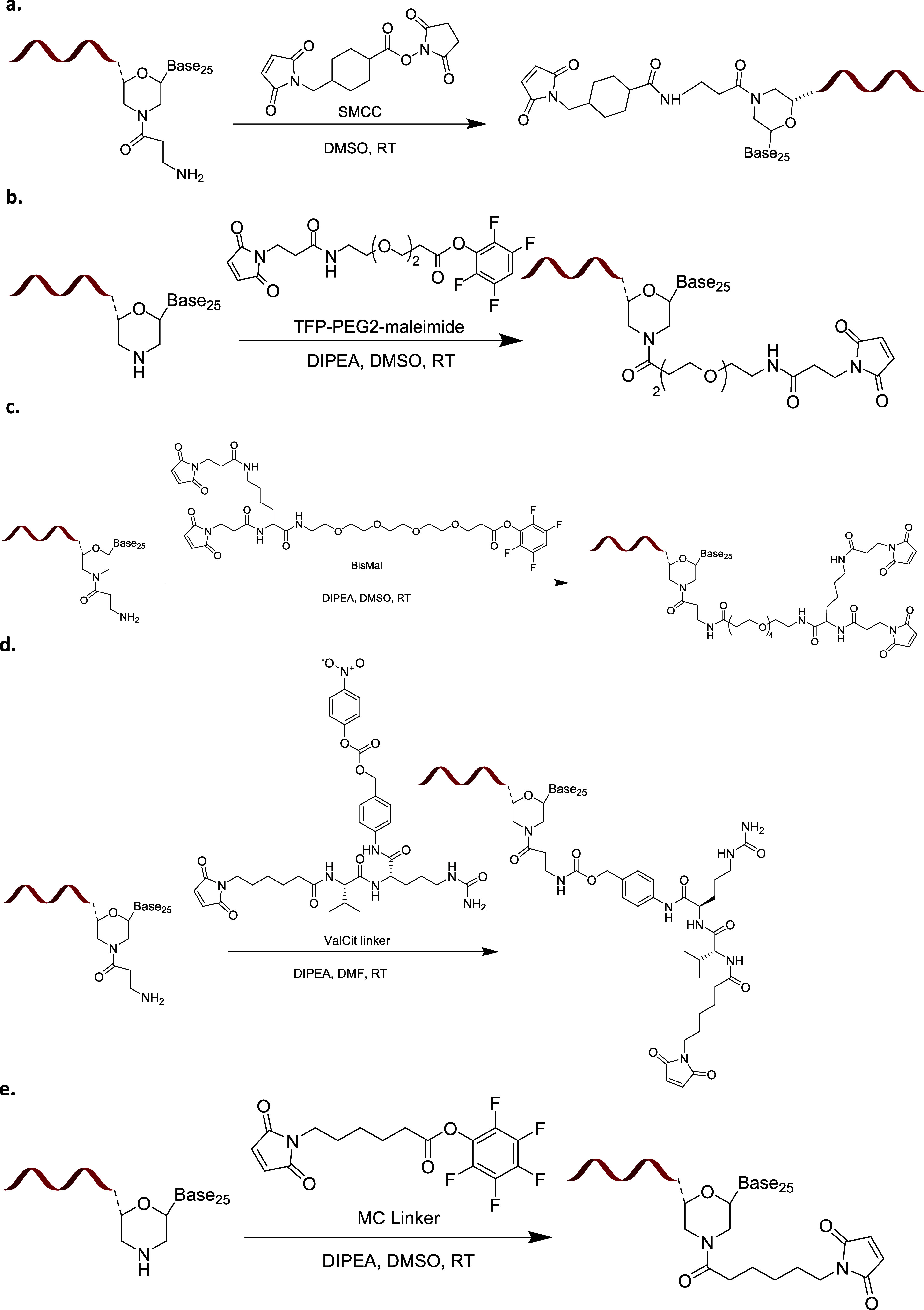
We used cleavable (ValCit) and noncleavable (MCC and BisMal) linkers to construct αmTfR1-pmoEx23 conjugates and investigated their efficacy of exon skipping in gastrocnemius and heart muscles (Figure 3). In previous research, we demonstrated that the stability of cleavable versus noncleavable linkers does not significantly impact the delivery of siRNA oligonucleotides. This was observed in SAR studies where we evaluated the impact of linker stability on oligonucleotide delivery for TfR1-containing AOCs.35 In this current study, we hypothesized that the ValCit cleavable linker would improve exon skipping by detaching more rapidly from the antibody fragments, leaving no linker or antibody remaining. This is in comparison to noncleavable linkers, which are likely to release PMO along with the linker and a fragment of antibody. Of the two noncleavable linkers, we further hypothesized that the larger BisMal linker, which is conjugated to two cysteines on the antibody through its two maleimides, could potentially be released along with a larger linker/remaining peptide. This could result in additional steric hindrance which might obstruct the binding of the PMO to the target mRNA binding and consequently lead to a reduction in exon-skipping activity.
Figure 3.
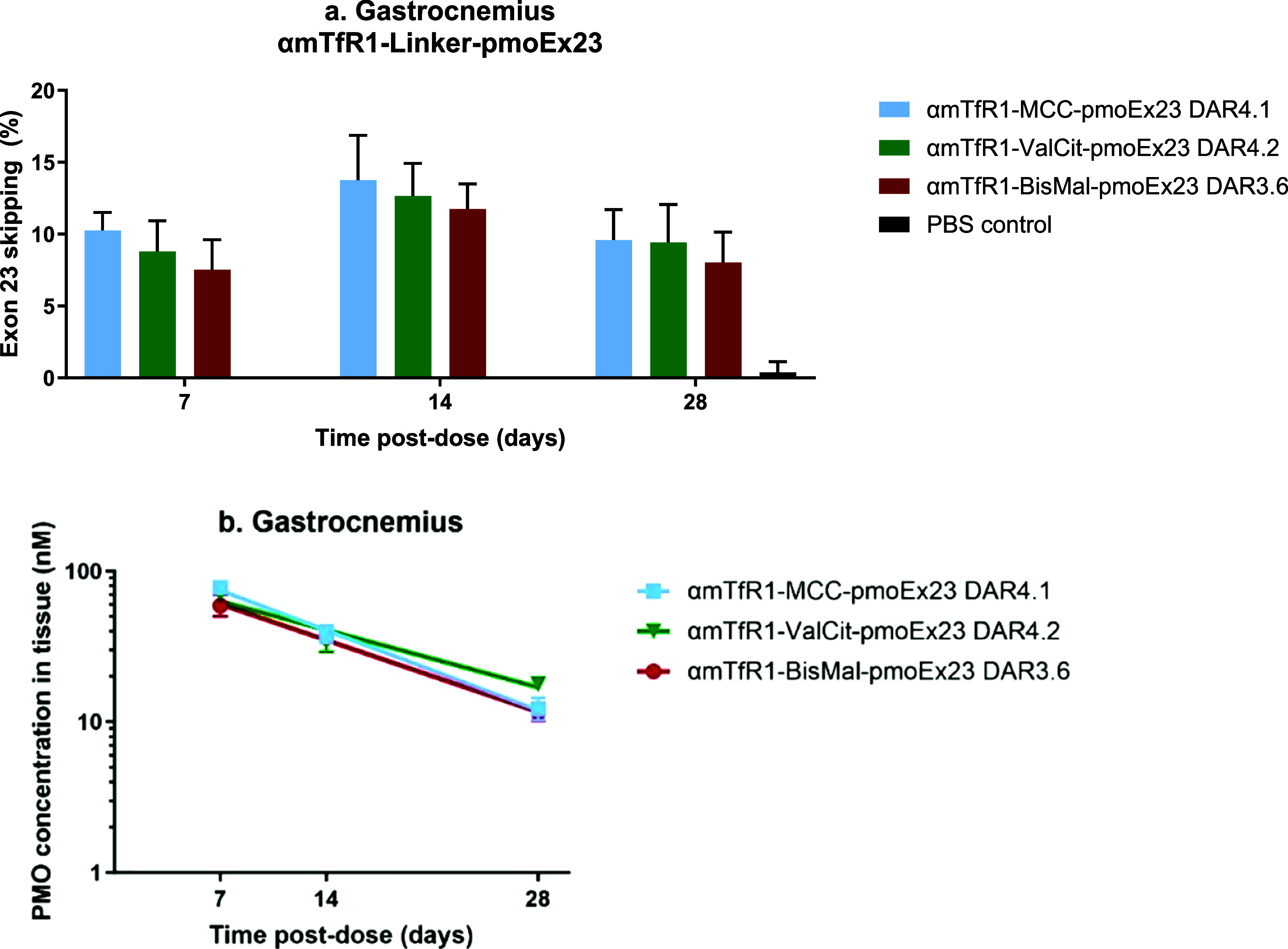
Effects of pmoEx23 linkers MCC (11.6 mg/kg PMO), ValCit (11.9 mg/kg PMO), and BisMal (10.2 mg/kg PMO) at the 3′ position on exon 23 skipping (a) and PMO concentration (b) in gastrocnemius muscle tissue (n = 4 per group, error bars represent SEM).
Our results showed that all three linkers performed equivalently in delivering PMO to muscle tissue and inducing exon skipping. This was consistent across all time points and in both tissue types examined. This indicates that PMOs conjugated at the 3′ termini exhibit a high tolerance for additional structures attached to them, without compromising exon-skipping activity. However, it is possible that the slightly lower activity observed previously when conjugating to the 5′ end of a PMO with a stable linker may have been rescued by using a cleavable linker. Based on these findings, we selected MCC as the linker for additional SAR studies. The selection of MCC was influenced by its proven clinical validation and the analytical simplicity it offers when used in conjugate formation. All linkers evaluated were comparable in terms of delivery, activity, and duration of exon skipping.
Impact of DAR on Exon Skipping
After identifying the optimal linker and its location on the PMO, we then focused on optimizing the DAR. Just as with ADCs, achieving an optimal DAR is critical for the efficient delivery and effectiveness of AOCs. In the context of siRNA AOCs, we determined that a DAR of 1 is ideal for targeting specific tissues while minimizing nonspecific uptake in the liver.35 However, for PMO AOCs, the DAR could be higher, akin to ADCs, because of the neutral charge and limited protein binding characteristics of PMOs. The challenge lies in balancing the DAR; while a higher-DAR AOC could deliver more PMOs per antibody molecule, it could also lead to nonspecific uptake due to the increased size and hydrophobicity of the drug. Additionally, a large payload could inhibit binding to the receptor.27 We conducted a series of experiments to assess the impact of DAR, including performing an enzyme-linked immunosorbent assay (ELISA) to assess binding affinity, PK studies to assess antibody-conjugate clearance from plasma and evaluations of PMO delivery to muscle and exon skipping.
The ELISA showed that while increasing the DAR beyond 5 reduced the binding affinity of the AOC by nearly 5-fold compared with the unconjugated antibody (Figure 4a), the AOC maintained low picomolar affinities that were not expected to inhibit delivery.
The PK studies were conducted with purified AOCs with DAR 2, 3, and 4 at a constant PMO dose of 1 mg/kg (Figure 4b). The AOCs evaluated showed similar PK regardless of the DAR, indicating that all DAR species may have similar clearance. While HIC indicated that increasing the PMO payload increased the AOC hydrophobicity, it did not result in significant nonspecific binding in vivo, as evidenced by the similar delivery profiles. Additionally, the unconjugated PMO was cleared rapidly from plasma, supporting the hypothesis that the PMOs had little-to-no nonspecific protein binding.
To understand the impact of DAR on PMO delivery to muscle (Figure 4c) and its subsequent impact on exon skipping (Figure 4d), we administered conjugates to mdx mice. Surprisingly, the nonbinding antibody control (control.mAb-MCC-pmoEx23 DAR3.9) resulted in nearly 90 nM concentrations of PMO detected in the muscle. However, it is likely that the PMO found in the tissue was not delivered via the same productive pathway that is used by the TfR1 antibody, resulting in significantly reduced exon skipping in gastrocnemius muscle compared with conjugation with the TfR1 antibody (Figures 4d and S1). This underscores the importance of receptor-mediated delivery of oligonucleotides, as evidenced by studies on siRNA delivery.18 Most importantly, higher-DAR PMO AOCs appeared to enable more exon skipping with a lower mAb dose. Our results indicate that exon skipping was primarily driven by PMO dose, with DAR having no significant impact.
We determined that increasing the PMO payload from 2 to 5.5 had no significant impact on PMO delivery or exon skipping when animals were dosed with an equivalent amount of PMO. However, this dramatically improved skipping for an equivalent mAb dose (9.3 to 18% when increasing DAR from 2 to 4). We performed further analyses in mdx mice to assess whether DAR impacted exon skipping in other muscles, including quadriceps, diaphragm, and heart. We also decided to extend our investigations to higher DAR. While purification of individual DAR species higher than 4 by HIC is not efficient, AOCs with average DAR of 4 and 6 were generated by controlling the amount of TCEP used in the antibody reduction step. The mouse IgG2a antibody used in this experiment contains six interchain disulfide bonds. While this could hypothetically produce a DAR12, we found that addition of the sterically bulky PMO resulted in a maximum of DAR9.7, with 2 PMOs attached to the light chains and an additional 4 PMOs on each of the heavy chains. We rationalized that if this approach proved successful in mice using the mIgG2a antibody, the adoption of DAR8 AOCs in the clinic using the human IgG1 antibody (that contain 4 interchain disulfides) would significantly enhance not only PMO delivery but also the manufacturing process and control of AOCs (See Figure S2 for characterization of a αhTfR1-MCC-pmo DAR8 AOC). This advancement would eliminate production of a heterogeneous DAR mixture requiring tedious chromatographic separation for achieving the intended average DAR.
Furthermore, we assessed an alternative noncleavable linker, MC, widely used with ADCs. We considered this option due to its simplified linker addition process, which could potentially reduce the number of steps involved in linker-pmoEx23 synthesis compared to the use of the MCC linker.
We assessed the impact of DAR on plasma clearance in mice by comparing an equal antibody dose of αmTfR1-pmoEx23 AOCs with an average DAR4 or DAR7 made with the MCC linker (Figure 5). Noncompartmental analysis showed comparable clearance parameters for both conjugates (0.136 and 0.106 mL/min/kg for DAR4 and DAR7, respectively), confirming that increasing the PMO payload up to 7 did not increase nonspecific clearance of the AOC. We then compared the delivery of PMO to muscle and percentage of exon 23 skipping in αmTfR1-pmoEx23 AOCs, with comparisons between MCC and MC linkers, and between DARs 3.9, 5.8, and 9.7. Statistical analysis was performed by comparing all groups to αmTfR1-MCC-pmoEx23 DAR4. The results suggest that neither linker nor DAR negatively affected delivery (Figure 6a) or exon skipping in gastrocnemius, quadriceps, or diaphragm (Figure 6b–d). We found that exon skipping increased significantly between αmTfR1-MCC-pmoEx23 DAR3.9 and αmTfR1-MCC-pmoEx23 DAR9.7 in heart muscle, but no significant changes in exon skipping occurred between MCC and MC linkers (Figure 6e). Across all muscles examined, exon skipping was significantly higher for αmTfR1-MCC-pmoEx23 DAR3.9 compared with PBS-treated WT and mdx mice. It is important to note that while increased delivery of PMO to any individual muscle tissue is correlated with increased exon skipping, we see higher PMO concentrations in the heart but less exon skipping compared to skeletal muscle. One limitation of measuring PMO concentrations of homogenized tissue is that it is not possible to identify how much PMO is located within different cell types or intracellular compartments. It is possible that much of the PMO found in the heart has been taken up through unproductive mechanisms. This hypothesis is supported by the high concentrations of PMO found in the heart when conjugated to a nontargeting antibody that do not result in significant exon skipping (Figure S3).
Figure 5.

Impact of DAR upon plasma concentrations of mdx mice treated with αmTfR1-MCC-pmoEx23 DAR4 (10.0 mg/kg PMO) or αmTfR1-MCC-pmoEx23 DAR7 (18.3 mg/kg PMO) via a single IV injection. Test articles were diluted in PBS. Mice (n = 4 per group, error bars represent SEM). PMO concentrations in plasma were taken at 2, 10, 30 min and 1, 4, and 24 h and were measured by PMO ELISA.
Figure 6.

Impact of DAR on levels of (a) tissue concentration and exon skipping in (b) gastrocnemius, (c) quadriceps, (d) diaphragm, and (e) heart muscles. mdx mice were treated with a single IV injection of 10 mg/kg PMO dose for the DAR3.9 and DAR5.8 AOCs, and 12 mg/kg for the DAR9.7 AOCs. Mice (n = 4 per group, error bars represent SEM) were treated with different combinations of linkers (MCC or MC), DARs, and PMO conjugated to αmTfR1. Control groups included B10 WT mice and mdx mice treated with IV PBS. Tissues were harvested at 14 days post-treatment and exon skipping was assessed by ddPCR. Statistical analysis was performed using one-way ANOVA, with all groups compared against αmTfR1-MCC-pmoEx23 DAR4. *: P ≤ 0.05, **: P ≤ 0.01, ***: P ≤ 0.001; ****: P ≤ 0.0001. ANOVA, analysis of variance; ddPCR, droplet digital polymerase chain reaction; IV, intravenous; ns, not significant.
Overall, these data demonstrate that increasing DAR does not compromise antibody-mediated delivery of PMO. Additionally, DAR9.7 PMO AOCs were able to deliver equivalent amounts of the active PMO to target muscles with half the antibody dose and significantly increase exon skipping in the heart. Optimizing the DAR, particularly with DAR9.7, not only enhances the therapeutic potential but also yields tangible benefits in terms of cost-effectiveness and manufacturing efficiency. By simplifying the linker chemistry, we achieved an equivalent level of biological activity between MCC and MC linkers. As shown in Scheme 2, MC linkers require simpler manufacturing as fewer steps are needed in comparison with MCC.
Impact of the Dose Level of αmTfR1-MCC-pmoEx23 on Exon Skipping over Time
Using mdx mice, we administered PMO doses ranging between 10 to 60 mg/kg of αmTfR1-MCC-pmoEx23 DAR3.7 and compared percentages of exon 23 skipping over 84 days (12 weeks) (Figure 7). Our results suggest that exon skipping was dose-dependent, with highest exon skipping occurring in the 60 mg/kg group on Day 14 post-treatment. However, a dosing strategy of 3 × 10 mg/kg (administered on Days 0, 7, and 14) resulted in the highest exon skipping between Days 28 and 84 across all muscle types tested, possibly due to reduced efficiency of TfR1-mediated uptake occurring with higher doses. The nonlinear increase in exon skipping at higher doses may be caused by TfR1 receptor saturation limiting efficient uptake of the AOC, which has been reported with other targeted drugs, including GalNAc-siRNAs and antibody-drug conjugates, this can be overcome with adjusted dosing schedules or subcutaneous dosing.36−38 These data show that a single dose of αmTfR1-MCC-pmoEx23 can sustain 10–20% exon 23 skipping in gastrocnemius and quadriceps muscles for at least 12 weeks. Lower levels of exon skipping were detected in heart muscle compared with gastrocnemius, quadriceps, and diaphragm. Exon skipping was not detected in mdx mice treated with PBS only.
Figure 7.

Analysis of exon 23 skipping in gastrocnemius, quadriceps, diaphragm, and heart muscles of mdx mice treated with varying doses of αTfR1-MCC-pmoEx23 DAR3.7. mdx mice were treated via iv injection on Day 0 with αmTfR1-MCC-pmoEx23 DAR3.7 (10, 30, or 60 mg/kg PMO doses) or 3 doses of 10 mg/kg on Days 0, 7, and 14. Control groups included mdx and B10 WT mice treated with PBS via IV injection on Day 0. Mice (n = 4 per group/time point, error bars represent SEM) receiving a single dose were euthanized for tissue collection on Days 7, 14, 28, 56, and 84 post dose. Mice receiving 3 × 10 mg/kg doses, were euthanized for tissue collection on Days 28, 56, and 84 postfirst dose. Exon skipping was assessed by ddPCR.
Effect of αmTfR1-MCC-pmoEx23 Treatment on Dystrophin Protein Restoration in mdx Mice
As exon skipping was achieved with αmTfR1-MCC-pmoEx23, we examined the subsequent effect on dystrophin protein restoration in mdx mice over 84 days. As seen with exon skipping, dystrophin restoration was found to be dose-dependent, with the highest levels observed for 30 and 60 mg/kg doses of αmTfR1-pmoEx23 DAR3.7 (Figure 8).
Figure 8.

Analysis of dystrophin restoration in gastrocnemius and heart muscles of mdx mice treated with varying doses of αmTfR1-MCC-pmoEx23. mdx mice were treated via IV injection once on Day 0 with αmTfR1-MCC-pmoEx23 DAR3.7 (10 mg/kg, 30 mg/kg, or 60 mg/kg PMO doses) or 3 doses of 10 mg/kg on Days 0, 7, and 14. Control groups included mdx and B10 WT mice treated with PBS via IV injection on Day 0. Mice (n = 4 per group/time point, error bars represent SEM) were euthanized, and tissues harvested on Days 7, 14, 28, 56, and 84 post-treatment. Mice receiving 3 × 10 mg/kg doses, were euthanized for tissue collection on Days 28, 56, and 84 postfirst dose. Dystrophin restoration was assessed by a capillary Western blot analysis using the ProteinSimple Jess Western system with a 66–440 kDaA separation module. Data were analyzed by Compass following a protocol adapted from Beekman et al. presented as percentage of dystrophin levels in WT mice.39
We noted highest dystrophin protein restoration in the gastrocnemius muscle, reaching a maximum of 30%, whereas in heart muscle, a maximum of 8% restoration was observed. As expected, treatment with PBS resulted in no dystrophin restoration.
Discussion
Therapeutic Potential of Oligonucleotide Therapeutics
Of the PMO delivery vehicles investigated in these preclinical studies, αmTfR1-PMO conjugates containing a stable linker with a DAR9.7 are the most effective. We have shown they induce dystrophin protein restoration in both skeletal and heart muscle of mdx mice, thereby presenting a potentially effective approach for the treatment of DMD.
This study focused on the SAR of antibody–PMO conjugate αmTfR1-pmoEx23. It examined the role of different linkers, DARs, and conjugation sites, and how these parameters impact drug targeting to specific tissues, exon skipping, and dystrophin restoration in the mdx mouse, a well-characterized murine model of DMD.
SAR Evaluation of αmTfR1-pmoEX23 AOCs
Linker Comparisons
We compared both cleavable and noncleavable linkers, including MCC, BisMal, and ValCit on exon skipping in mdx mice. The cleavable linker (ValCit) was predicted to improve exon skipping by releasing the PMO from the antibody faster and without any linker or protein attached to it. BisMal, a larger linker with two maleimides that allow it to conjugate to two cysteines on the antibody, was predicted to have additional linker and peptide fragments remaining on the 3′ end of the PMO, potentially impacting PMO delivery to the nucleus and its binding to the pre-mRNA target. Regardless of linker chemistry, the PMO conjugates ultimately achieved similar levels of exon skipping by Day 14, indicating that their mechanistic differences did not play a major role in drug efficacy over 14 days. It is likely that additional linker appendages on the PMO do not have a substantial effect on its binding to pre-mRNA. A previous study screened the efficacy of αmTfR1-siRNA with different linkers. The results demonstrated that the ValCit linker exhibited silencing efficacy (66%) similar to a noncleavable linker (65%). Comparisons of silencing activity over 7 days were found to be dose-dependent and, as with this study, no advantages of cleavable linkers were observed when compared with noncleavable linkers.19
While the linker in a PMO AOC may not significantly impact the activity in mice, its significance in the downstream manufacturing process underscores its crucial role in ensuring the successful development and production of these therapeutic agents. MCC and MC linkers were compared for the efficiency of their respective synthetic routes. MCC does not react efficiently with the 3′ morpholine, resulting in less than 15% of the desired product. This is likely due to steric hindrance presented by the SMCC. The addition of β-alanine to the 3′ end of the PMO efficiently reacts with SMCC. However, this addition necessitates further steps, such as reaction and deprotection, which must be performed post-PMO synthesis and purification. These additional steps prolong the manufacturing process and increase its cost. While enhancing the reaction efficiency is possible by increasing the concentrations and temperature, it leads to the unwanted consequence of superfluous linker attachment to the PMO base unit. Conversely, the MC linker reacts efficiently with the 3′ end of the PMO and maintains in vivo delivery and activity.
DAR Comparisons
There have been numerous studies on the effects of DAR on the pharmacologic properties of ADCs. A key study was performed by Sun et al., whereby ADCs with a higher DAR (mean DAR 10) displayed faster rates of distribution to tissues and blood clearance, along with a decrease in efficacy. However, ADCs with a DAR of less than 6 commonly displayed similar PK properties and overall greater tolerability compared with higher DAR preparations.40 Previous research has also demonstrated the impact of DAR on plasma clearance of ADCs, with DAR8 clearing 3-fold faster than DAR4 and 5-fold faster than DAR2, demonstrating that higher drug loading on the antibody has the potential to lower drug delivery to target tissues.27 Similar SAR studies assessing AOC comprised of antibody-siRNA conjugates also showed improved therapeutic index with a lower DAR of 1.35 However, our findings demonstrate that the charge-neutral PMO-based AOCs behave differently and tolerate higher DARs, achieving higher PMO delivery to target tissues per antibody molecule. Exon skipping in skeletal muscle did not appear to be significantly impacted by DAR and was more likely to be driven by PMO dose. Comparisons between studies are limited as DARs in this study were not as high as those described by Sun et al. We hypothesize that these results could be due to rapid muscle delivery of the transferrin receptor-targeting antibody as well as the hydrophilicity of PMOs compared with highly hydrophobic cytotoxic cancer drugs widely used in ADCs. Higher-DAR AOCs offer several benefits, including improved efficacy, lower cost of goods, product homogeneity, simplified manufacturing processes, and higher yields. The enhancement in the PMO per antibody translates to a more potent and effective treatment. This approach effectively reduces the cost of goods, as a smaller quantity of antibodies is necessary to deliver an equivalent amount of therapeutic payload. Notably for an AOC made with a human IgG1 antibody (αhTfR1-MCC-pmo DAR8 AOC; Figure S2) a DAR of 8 (DAR8) indicates a homogeneous product, unlike mixtures that have lower DARs. Furthermore, DAR8 eliminates the need for chromatography purification. This streamlines the manufacturing process and offers a cost-effective advantage.
In summary, the strategic focus on optimizing higher-DAR AOCs not only enhances the therapeutic potential but also brings about tangible benefits in terms of cost-effectiveness and manufacturing efficiency.
Effect of Antibody-PMOs on Dystrophin Protein Restoration in mdx Mouse Models
Our studies showed that a single dose of 10–60 mg/kg of αmTfR1-MCC-pmoEx23 could induce significant levels of exon skipping and dystrophin protein restoration for up to 12 weeks (84 days) postdose, with maximum dystrophin restoration occurring between Day 28 and Day 56, depending upon the dosing levels and muscle tissue type. This prolonged duration of action is potentially due to the TfR1 antibody-mediated effective delivery of PMO to skeletal muscles and also due to the stability of PMOs in target tissues.5
αmTfR1-pmoEx23 induced exon skipping and dystrophin restoration in the mdx model, and results were comparable with other TfR1-targeting PMO-conjugate studies using mdx mice.5 In addition, differences could be observed between DAR and linker chemistry in a range of muscles, suggesting the model is reliable for assessment of bioconjugation strategies for antibody–PMO conjugates, and is suitable for use in the drug development process.
Using these SAR data generated using αmTfR1-pmoEx23, Avidity has developed multiple clinical candidates for the treatment of DMD. AOC 1044, a clinical candidate aimed at inducing exon 44 skipping, is currently in clinical trials. Results from this study have advanced our understanding of the SAR of antibody–PMO conjugates, which will aid the much-needed development of improved exon-skipping therapies for patients with DMD.
PMO versus siRNA
When comparing siRNA-antibody conjugates with PMO-antibody conjugates, several similarities and differences emerge. Both conjugate types perform well with stable linkers, with no observed benefit from cleavable linkers. Both also tolerate conjugation at either end of the oligonucleotide, although for different underlying reasons. For siRNA, conjugation to the sense strand is well tolerated because it is discarded after the antisense strand is loaded into the RNA-induced silencing complex, while the PMO mechanism of inducing exon skipping through steric blocking appears to be tolerant to significant residual linker fragments.
Key differences lie in the chemistry of the two payloads. siRNAs require careful optimization of the sugar and internucleotide chemistry at each position to maintain potency and activity, while the uniform chemistry of a PMO is sufficiently stable and active in mice. A PMO in muscle has a similar half-life compared to an siRNA with an optimized modification pattern (8.1 vs 11.6 days respectively). The chemistry of the siRNA also limits the DAR of siRNA AOCs to one due to rapid clearance by the liver of DAR2 and higher, while PMO AOCs can be loaded with up to ten PMOs without negatively impacting plasma clearance or muscle delivery. This substantial difference in DAR is useful for PMO AOCs which need to be dosed at higher levels compared to siRNA AOCs. While siRNA AOCs are limited to DAR1, the potency of siRNA allows for prolonged activity with a single dose due to the catalytic mechanism of effect.
Conclusions
The data herein provide a SAR of PMO AOCs. We have shown that, by using a TfR1 antibody, PMO delivery to different skeletal muscles, heart, and diaphragm can be improved. By improving PMO delivery, antibody–PMO conjugates improve PMO efficacy for the treatment of DMD. The studies conducted indicate that a stable linker with a DAR9.7 could be the most effective PMO delivery vehicle in preclinical studies. These data further support the development of this technology for potential therapeutic application in patients with DMD with mutations amenable to exon skipping.
Experimental Section
PMO Synthesis
PMOs were purchased from GeneTools (Philomath, OR) and the identity was confirmed by matrix-assisted laser desorption/ionization–time-of-flight mass spectrometry (MS). Reversed-phase high-performance liquid chromatography (RP-HPLC) using Aeris WIDEPORE XB-C18 (200 Å 3.6 μm 4.6 × 250 mm; Phenomenex, Torrance, CA) analysis was used to measure the purity of the received material (69.2%) with the primary impurity having a mass of +117 Da (N2-phenylacetyl guanosine converted to a diaminopurine derivative with the phenylacetyl group remaining at the N2 position after deprotection) (Figure S4). PMOs were purified by strong anion exchange chromatography; 53 mg PMO was dissolved in 4 mL of 10 mM NaOH and loaded onto a TSKgel SuperQ-5PW column (21.5 mm internal diameter × 150 mm, 13 μm; Tosoh Biosciences, Tokyo, Japan) and eluted with a gradient from 26.5 to 28% over 8 column volumes (CVs) (mobile phase A: 10 mM NaOH + 10% acetonitrile; mobile phase B: 10 mM NaOH, 500 mM NaCl + 10% acetonitrile) (Figure S5). Fractions (10 mL) were collected in tubes containing 1 mL 1 M phosphate buffer pH 7.2 to neutralize the pH and prevent hydrolysis of the amide bond at the 3′ end of the PMO. Fractions with acceptable purity were pooled and buffer exchanged with water followed by lyophilization to achieve PMO purities greater than 90% by RP-HPLC.
Reagents used for (linker) evaluation were maleimidocaproyl-l-valine-L-citrulline-p-aminobenzyl alcohol p-nitrophenyl carbonate (Mc-Val-Cit-PABC-PNP) (Broadpharm, San Diego, CA), 6-maleimidocaproic acid PFP ester (MC) (BroadPharm, San Diego, CA), succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC) (Thermo Fisher, Waltham, MA), bismaleimide-dPEG4-TFP ester (BisMal) (Quanta Biodesign, Plain City OH). The PMO-linker was generated by dissolving PMO at 30 mg/mL in 50 mM phosphate buffer (pH 7.4) followed by the addition of an equal volume of DMSO and 9 mol equiv of linker dissolved in DMSO at 30 mg/mL followed by incubation at room temperature for 40 min. Excess linker was removed by precipitating the PMO in 20 times volume ice-cold acetone followed by centrifugation at 3500g for 10 min. The pellet was dissolved in 10 mM acetate buffer pH 6.0 followed by an additional precipitation step. Linker addition to PMO and free linker removal were confirmed by electrospray ionization MS and RP-HPLC. This was done for pmoEx23 with and without a 3′ β-alanine linker (Figures S6 and S7).
AOC Synthesis
AOCs were generated using a standard random cysteine conjugation method. The interchain disulfide bonds of the αmTfR1 antibody with a mIgG2a backbone (GenScript, Piscataway, NJ) were reduced with TCEP (Thermo Fisher, Waltham, MA) at 37 °C for 4 h in phosphate buffered saline (PBS, pH 7.4) prior to conjugation with a maleimide linker-PMO. PMO-linker in either DMSO or acetate buffer (10 mM sodium acetate, pH 6) was added to the reduced antibody at room temperature and the reaction allowed to proceed for 1 h. Unreacted cysteine thiols were capped by the addition of 10 mol equiv of N-ethylmaleimide (NEM, Sigma-Aldrich, St. Louis, MO) to the reaction mixture from a 50 mg/mL stock of NEM in DMSO. PMO reaction with the reduced antibody was monitored by analytical HIC on an Agilent 1200 with a MAbPac HIC-Butyl column (5 μm 4.6 × 100 mm, Thermo Scientific, Waltham MA).
When indicated, individual DAR species or enriched DAR fractions were isolated by preparative HIC. The antibody-PMO reaction mixture was spiked with 3 M ammonium sulfate to bring the solution to 0.4 M ammonium sulfate, then loaded onto a HiScreen Butyl HP column (4.7 mL; GE Healthcare, Chicago, IL). Each DAR species was eluted with a gradient from 5 to 100% B over 30 CVs (mobile phase A: 50 mM phosphate buffer, 0.8 M ammonium sulfate, pH 7.0; mobile phase B: 80% 50 mM phosphate buffer pH 7.0, 20% isopropanol). The appropriate fractions were then pooled and exchanged into PBS, pH 7.4. Mixed DAR antibody-PMO reaction mixtures were purified to remove the unreacted PMO using preparative strong cation chromatography (SCX). PMO-antibody reaction mixtures were diluted 1:4 (v/v) with acetate buffer (25 mM sodium acetate, 25 mM sodium phosphate, pH 6.0). They were purified with a 20 mL HiPrep SP HP 16/10 column (GE Healthcare, Chicago, IL). Unconjugated PMO was removed with 3 CVs of mobile phase A, then AOCs were eluted using a gradient from 0% B to 60% B over 1 CV (mobile phase A: 25 mM sodium acetate, 25 mM sodium phosphate, pH 6.0 and mobile phase B: 25 mM sodium acetate, 25 mM sodium phosphate, 500 mM sodium chloride, pH 6.0).
AOCs were characterized by HIC, size exclusion chromatography (SEC), and reduced capillary gel electrophoresis. SEC was performed on Agilent 1200 HPLC system with a BioResolve mAb 7.8 × 300 mm SEC column (Waters Corporation, Millford, MA). Reduced capillary gel electrophoresis was conducted using a Maurice CE-SDS instrument (Bio-Techne, Minneapolis, MN) according to manufacturer instructions. Antibody-PMOs concentrations were quantified using the Pierce BCA Protein Assay Kit (Thermo Scientific, Waltham, MA) according to manufacturer specifications. Average DAR was determined by two methods. HIC was utilized to rapidly assess average DAR for up to DAR6 AOCs, and reduced capillary gel electrophoresis was used to provide better resolution for DAR8 or higher. Figures S8–10 show examples of how the DAR was determined by weighted average calculations with HIC and reduced capillary gel electrophoresis for three αmTfR1-MCC-pmoDAR AOCs ranging from DAR3.9 to DAR9.7.
Nine AOCs were ≥95% pure by SEC using a BioResolve mAb column (7.8 × 300 mm 2.5 μm, Waters Corporation, Millford, MA), and seven were between 90 and 95% pure (Table S1). While SEC is not able separate individual DAR species, additional characterization by HIC and confirmation by reduced capillary gel electrophoresis was used to calculate the average DAR as well as the percentage of unconjugated antibody in each conjugate (Table S1).
Murine Models
All animal procedures were approved by the Institutional Animal Care and Use Committee of Explora BioLabs in accordance with guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care. Mice were housed in ventilated cages under a 12-h light–dark cycle and fed ad libitum with standard rodent chow.
WT mouse studies were performed in 5–6-week-old female Hsd:ICR mice (Envigo Indianapolis, IN) or 6–8-week-old male C57BL/6 mice (Envigo). Studies in C57BL/10ScSn-Dmdmdx/J (Jackson) mice were performed in 8–10-week-old males with C57BL/10ScSnJ for control groups. All test articles were administered intravenously at 5 mL/kg. At end of study, mice were euthanized, and tissue collection was performed immediately after euthanasia. Skeletal and cardiac muscles were frozen on dry ice and stored at −80 °C until exon skipping, dystrophin restoration, and PMO tissue concentration analyses were performed.
Measurement of Exon Skipping
Tissue samples were processed for RNA isolation using the Direct-zol-96 RNA purification kit (Zymo Research) according to the manufacturer′s instructions. Total RNA (100–200 ng) was converted to cDNA using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) for exon-skipping analysis by quantitative PCR (qPCR) or droplet digital PCR (ddPCR). For qPCR, cDNA was amplified using a QuantStudio 7 Flex Real-Time PCR System (Applied Biosystems) with TaqMan Fast Advanced Master Mix for qPCR (Applied Biosystems) and TaqMan probes at 95 °C for 20 s, followed by 40 cycles of 95 °C for 1 s and 60 °C for 20 s. Exon 23-skipped and nonskipped dystrophin transcripts were detected by qPCR, individually normalized to PPIB gene expression, and used in ΔΔCt calculations for exon 23 skipping quantification. For ddPCR, cDNA was partitioned into droplets in the QX200 AutoDG Droplet Digital PCR System (Bio-Rad Laboratories, Hercules, CA) in combination with TaqMan probes (Applied Biosystems), 2× ddPCR Supermix for Probes (no dUTP) (Bio-Rad), and BamHI-HF restriction enzyme (New England BioLabs, Ipswich, MA). Following droplet generation, the mixture was loaded into a C1000 Touch Thermal Cycler with 96-Deep Well Reaction Module (Bio-Rad) for PCR amplification. Absolute quantification of the target molecules was measured in the QX200 Droplet Reader (Bio-Rad) using the QX Manager software (Bio-Rad).41,42 Exon 23 skipping percentage was calculated as (Δexon23 Dmd transcript)/(total Dmd transcript) × 100. Primers are listed in Table S2.
Measurement of Dystrophin Restoration
For protein extraction, skeletal and cardiac tissues were mechanically homogenized in RIPA buffer (Thermo Fisher) supplemented with Pierce Protease Inhibitor Mini Tablets (Thermo Fisher) using the Micro Tube Homogenizer System (Wilmad-LabGlass, Vineland, NJ). The suspension was then centrifuged at 14,000g for 15 min at 4 °C, and glycerol (20% of final volume) was added to the supernatant for cryo-protection. Quantification on total protein was measured with BCA Protein Assay Kit (Pierce) with bovine serum albumin as a standard. The Jess Simple Western system (ProteinSimple) was used to quantify dystrophin protein chemiluminescence and normalized to total protein chemiluminescence. Jess Simple Western assay was performed according to the manufacturer’s instructions using a 66–440 kDa Separation Module, Anti-Rabbit Detection Module, Total Protein Detection Module, and the Replex Module (all from ProteinSimple). A rabbit monoclonal antidystrophin antibody was used (Abcam ab154168) and diluted 1:1000 in Antibody Dilution Buffer (ProteinSimple). Using Compass software, the resulting electropherograms were inspected to check whether automatic peak detection required any manual correction, and peaks were quantified by calculation of the area under the curve. The following criteria were used to discriminate low dystrophin signals from background: the peak signal-to-noise ratio given by the software ≥10, and the peak height/baseline ratio ≥3 (protocol adapted from Beekman et al., 2018).39
Statistical Analysis
GraphPad Prism software (version 9.0) was used for all descriptive and statistical analyses. As appropriate, one- or two-way analysis of variance (ANOVA) or t tests were performed for the data sets analyzed. In the case of ANOVA, an appropriate post hoc test was used to determine the differences among the treatment groups. Significant differences were defined as p < 0.05.
Measurement of pmoEx23 Concentrations in Mouse Plasma and Tissues
To evaluate pmoEx23 concentrations in mouse plasma and tissues, an oligonucleotide probe hybridization ELISA was developed following a previously described method.43 The method was subsequently converted to the Meso Scale Discovery platform by using a ruthenium-labeled detector.44 Calibration standards and quality control samples were prepared in the same matrix as unknown study samples with anti-Digoxigenin antibody (Sheep polyclonal IgG, Bio-Rad catalog number 3210-0488), labeled with SULFO-TAG at a molar challenge ratio of 16 with final Tag:Protein of 6.8 using MSD GOLD 96-well streptavidin plate (MSD catalog number L15SA-6) and MSD GOLD SULFO-TAG NHS-Ester (MSD catalog number R91AO-2) and MSD GOLD Read Buffer A (MSD catalog number R92TG-2) using the Meso Sector S 600 instrument (Meso Scale Diagnostics, Rockford, IL). A 5-parameter logistic fit of the calibration standard curve with 1/y2 weighting was used to interpolate unknown samples via GraphPad Prism.
Acknowledgments
The authors would like to thank Yanling Chen and Michael Moon for their help performing animal studies. The authors would like to thank Gemma Hall, DPhil, and Megan Hotard Jarrell, MS, from Lighthouse Medical Communications, New York, NY, USA, for providing medical writing support. Medical writing support was funding by Avidity Biosciences, San Diego, CA, USA, in accordance with Good Publication Practice (GPP3) guidelines.
Glossary
Abbreviations
- αmTfR1
antimouse transferrin receptor 1 antibody
- ADC
antibody-drug conjugate
- ANOVA
analysis of variance
- AOC
antibody–oligonucleotide conjugate
- ASO
antisense oligonucleotide
- BisMal
bismaleimide
- CV
column volume
- DAR
drug-to-antibody ratio
- ddPCR
droplet digital polymerase chain reaction
- DIPEA
N,N-diisopropylethylamine
- DMD
Duchenne muscular dystrophy
- DMSO
dimethyl sulfoxide
- ELISA
enzyme-linked immunosorbent assay
- FDA
US Food and Drug Administration
- GalNAc
N-acetylgalactosamine
- HIC
hydrophobic interaction chromatography
- IgG1
immunoglobulin G1
- IV
intravenous
- mAb
monoclonal antibody
- MC
maleimidocaproyl
- MCC
4-(N-maleimidomethyl) cyclohexane-1-carboxylate
- mdx
X chromosome-linked muscular dystrophy
- MS
mass spectrometry
- NEM
N-ethylmaleimide
- NHS
N-hydroxysuccinimide
- ns
not significant
- PBS
phosphate buffered saline
- PEG2
polyethylene glycol 2-maleimide
- PK
pharmacokinetics
- PMO
phosphorodiamidate morpholino oligomer
- pmoEx23
PMO targeting mouse exon 23
- qPCR
quantitative polymerase chain reaction
- RP-HPLC
reverse-phase high-performance liquid chromatography
- RT
room temperature
- SAR
structure–activity relationship
- SCRAM
scramble PMO control
- SCX
strong cation chromatography
- SEC
size exclusion chromatography
- SEM
standard error of the mean
- siRNA
small interfering RNA
- SMCC
succinimidyl 4-(N-maleimidomethyl) cyclohexane-1-carboxylate
- TCEP
tris(2-carboxyethyl)phosphine
- TFP-PEG2
tetrafluorophenyl-polyethylene glycol 2-maleimide linker
- TfR1
transferrin receptor 1
- ValCit
valine-citrulline linker
- WT
wild type
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c00803.
PMO conjugate structures; primers used for exon-skipping analysis; plasma clearance of targeted versus untargeted AOCs; characterization of an αhTfR1-MCC-pmo DAR8 AOC; PMO concentration and exon skipping in heart; RP-HPLC analysis of pmoEx23 starting material; post strong anion exchange chromatography purification of pmoEx23; electrospray ionization MS and RP-HPLC characterization of pmoEx23 with and without a 3′ β-alanine linker; characterization of αmTfR1-MCC-pmoEx23 DAR4, DAR6, and DAR10 AOCs (PDF)
Author Present Address
§ Seawolf Therapeutics, One Sansome Street Suite 3630, San Francisco, California 94104, United States
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): At the time this research was conducted, all authors were employees of Avidity Biosciences and may have equity in the company.
Supplementary Material
References
- Duan D.; Goemans N.; Takeda S.; Mercuri E.; Aartsma-Rus A. Duchenne Muscular Dystrophy. Nat. Rev. Dis. Primer 2021, 7 (1), 13 10.1038/s41572-021-00248-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake D. J.; Weir A.; Newey S. E.; Davies K. E. Function and Genetics of Dystrophin and Dystrophin-Related Proteins in Muscle. Physiol. Rev. 2002, 82 (2), 291–329. 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- Wilson D. G. S.; Tinker A.; Iskratsch T. The Role of the Dystrophin Glycoprotein Complex in Muscle Cell Mechanotransduction. Commun. Biol. 2022, 5 (1), 1022 10.1038/s42003-022-03980-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro A. F.; Souza L. S.; Almeida C. F.; Ishiba R.; Fernandes S. A.; Guerrieri D. A.; Santos A. L. F.; Onofre-Oliveira P. C. G.; Vainzof M. Muscle Satellite Cells and Impaired Late Stage Regeneration in Different Murine Models for Muscular Dystrophies. Sci. Rep. 2019, 9 (1), 11842 10.1038/s41598-019-48156-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desjardins C. A.; Yao M.; Hall J.; O’Donnell E.; Venkatesan R.; Spring S.; Wen A.; Hsia N.; Shen P.; Russo R.; Lan B.; Picariello T.; Tang K.; Weeden T.; Zanotti S.; Subramanian R.; Ibraghimov-Beskrovnaya O. Enhanced Exon Skipping and Prolonged Dystrophin Restoration Achieved by TfR1-Targeted Delivery of Antisense Oligonucleotide Using FORCE Conjugation in Mdx Mice. Nucleic Acids Res. 2022, 50 (20), 11401–11414. 10.1093/nar/gkac641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariyawasam D.; D’Silva A.; Mowat D.; Russell J.; Sampaio H.; Jones K.; Taylor P.; Farrar M. Incidence of Duchenne Muscular Dystrophy in the Modern Era; an Australian Study. Eur. J. Hum. Genet. 2022, 30 (12), 1398–1404. 10.1038/s41431-022-01138-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilton-Clark H.; Yokota T. Recent Trends in Antisense Therapies for Duchenne Muscular Dystrophy. Pharmaceutics 2023, 15 (3), 778. 10.3390/pharmaceutics15030778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts T. C.; Langer R.; Wood M. J. A. Advances in Oligonucleotide Drug Delivery. Nat. Rev. Drug Discovery 2020, 19 (10), 673–694. 10.1038/s41573-020-0075-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saifullah; Motohashi N.; Tsukahara T.; Aoki Y. Development of Therapeutic RNA Manipulation for Muscular Dystrophy. Front. Genome Ed. 2022, 4, 863651 10.3389/fgeed.2022.863651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan Y.; Zhang Y.-J. Antisense Phosphorodiamidate Morpholino Oligomers as Novel Antiviral Compounds. Front. Microbiol. 2018, 9, 750 10.3389/fmicb.2018.00750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudziak R. M.; Barofsky E.; Barofsky D. F.; Weller D. L.; Huang S.-B.; Weller D. D. Resistance of Morpholino Phosphorodiamidate Oligomers to Enzymatic Degradation. Antisense Nucleic Acid Drug Dev. 1996, 6 (4), 267–272. 10.1089/oli.1.1996.6.267. [DOI] [PubMed] [Google Scholar]
- Summerton J.; Weller D. Morpholino Antisense Oligomers: Design, Preparation, and Properties. Antisense Nucleic Acid Drug Dev. 1997, 7 (3), 187–195. 10.1089/oli.1.1997.7.187. [DOI] [PubMed] [Google Scholar]
- Duan D. Dystrophin Gene Replacement and Gene Repair Therapy for Duchenne Muscular Dystrophy in 2016: An Interview. Hum. Gene Ther.: Clin. Dev. 2016, 27 (1), 9–18. 10.1089/humc.2016.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman E. P.; Bronson A.; Levin A. A.; Takeda S.; Yokota T.; Baudy A. R.; Connor E. M. Restoring Dystrophin Expression in Duchenne Muscular Dystrophy Muscle. Am. J. Pathol. 2011, 179 (1), 12–22. 10.1016/j.ajpath.2011.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim K. R.; Maruyama R.; Yokota T. Eteplirsen in the Treatment of Duchenne Muscular Dystrophy. Drug Des., Dev. Ther. 2017, 11, 533–545. 10.2147/DDDT.S97635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama R.; Yokota T. Molecular Diagnosis and Novel Therapies for Neuromuscular Diseases. J. Pers. Med. 2020, 10 (3), 129. 10.3390/jpm10030129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura A.; Takeda S. Exon-skipping Therapy for Duchenne Muscular Dystrophy. Neuropathology 2009, 29 (4), 494–501. 10.1111/j.1440-1789.2009.01028.x. [DOI] [PubMed] [Google Scholar]
- Riccardi F.; Dal Bo M.; Macor P.; Toffoli G. A Comprehensive Overview on Antibody-Drug Conjugates: From the Conceptualization to Cancer Therapy. Front. Pharmacol. 2023, 14, 1274088 10.3389/fphar.2023.1274088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malecova B.; Burke R. S.; Cochran M.; Hood M. D.; Johns R.; Kovach P. R.; Doppalapudi V. R.; Erdogan G.; Arias J. D.; Darimont B.; Miller C. D.; Huang H.; Geall A.; Younis H. S.; Levin A. A. Targeted Tissue Delivery of RNA Therapeutics Using Antibody–Oligonucleotide Conjugates (AOCs). Nucleic Acids Res. 2023, 51 (12), 5901–5910. 10.1093/nar/gkad415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugo T.; Terada M.; Oikawa T.; Miyata K.; Nishimura S.; Kenjo E.; Ogasawara-Shimizu M.; Makita Y.; Imaichi S.; Murata S.; Otake K.; Kikuchi K.; Teratani M.; Masuda Y.; Kamei T.; Takagahara S.; Ikeda S.; Ohtaki T.; Matsumoto H. Development of Antibody-siRNA Conjugate Targeted to Cardiac and Skeletal Muscles. J. Controlled Release 2016, 237, 1–13. 10.1016/j.jconrel.2016.06.036. [DOI] [PubMed] [Google Scholar]
- Cuellar T. L.; Barnes D.; Nelson C.; Tanguay J.; Yu S.-F.; Wen X.; Scales S. J.; Gesch J.; Davis D.; van Brabant Smith A.; Leake D.; Vandlen R.; Siebel C. W. Systematic Evaluation of Antibody-Mediated siRNA Delivery Using an Industrial Platform of THIOMAB–siRNA Conjugates. Nucleic Acids Res. 2015, 43 (2), 1189–1203. 10.1093/nar/gku1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sela T.; Mansø M.; Siegel M.; Marban-Doran C.; Ducret A.; Niewöhner J.; Ravn J.; Martin R. E.; Sommer A.; Lohmann S.; Krippendorff B.-F.; Ladefoged M.; Indlekofer A.; Quaiser T.; Bueddefeld F.; Koller E.; Mohamed M. Y.; Oelschlaegel T.; Gothelf K. V.; Hofer K.; Schumacher F. F. Diligent Design Enables Antibody-ASO Conjugates with Optimal Pharmacokinetic Properties. Bioconjugate Chem. 2023, 34 (11), 2096–2111. 10.1021/acs.bioconjchem.3c00393. [DOI] [PubMed] [Google Scholar]
- Su D.; Zhang D. Linker Design Impacts Antibody-Drug Conjugate Pharmacokinetics and Efficacy via Modulating the Stability and Payload Release Efficiency. Front. Pharmacol. 2021, 12, 687926 10.3389/fphar.2021.687926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugal-Tessier J.; Thirumalairajan S.; Jain N. Antibody-Oligonucleotide Conjugates: A Twist to Antibody-Drug Conjugates. J. Clin. Med. 2021, 10 (4), 838. 10.3390/jcm10040838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon R. Drawing Lessons from the Clinical Development of Antibody-Drug Conjugates. Drug Discovery Today Technol. 2018, 30, 105–109. 10.1016/j.ddtec.2018.10.001. [DOI] [PubMed] [Google Scholar]
- Lambert J. M.; Chari R. V. J. Ado-Trastuzumab Emtansine (T-DM1): An Antibody–Drug Conjugate (ADC) for HER2-Positive Breast Cancer. J. Med. Chem. 2014, 57 (16), 6949–6964. 10.1021/jm500766w. [DOI] [PubMed] [Google Scholar]
- Dovgan I.; Kolodych S.; Koniev O.; Wagner A. 2-(Maleimidomethyl)-1,3-Dioxanes (MD): A Serum-Stable Self-Hydrolysable Hydrophilic Alternative to Classical Maleimide Conjugation. Sci. Rep. 2016, 6 (1), 30835 10.1038/srep30835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamblett K. J.; Senter P. D.; Chace D. F.; Sun M. M. C.; Lenox J.; Cerveny C. G.; Kissler K. M.; Bernhardt S. X.; Kopcha A. K.; Zabinski R. F.; Meyer D. L.; Francisco J. A. Effects of Drug Loading on the Antitumor Activity of a Monoclonal Antibody Drug Conjugate. Clin. Cancer Res. 2004, 10 (20), 7063–7070. 10.1158/1078-0432.CCR-04-0789. [DOI] [PubMed] [Google Scholar]
- Tang Y.; Tang F.; Yang Y.; Zhao L.; Zhou H.; Dong J.; Huang W. Real-Time Analysis on Drug-Antibody Ratio of Antibody-Drug Conjugates for Synthesis, Process Optimization, and Quality Control. Sci. Rep. 2017, 7 (1), 7763 10.1038/s41598-017-08151-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gébleux R.; Wulhfard S.; Casi G.; Neri D. Antibody Format and Drug Release Rate Determine the Therapeutic Activity of Noninternalizing Antibody–Drug Conjugates. Mol. Cancer Ther. 2015, 14 (11), 2606–2612. 10.1158/1535-7163.MCT-15-0480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann C. J.; Honeyman K.; Cheng A. J.; Ly T.; Lloyd F.; Fletcher S.; Morgan J. E.; Partridge T. A.; Wilton S. D. Antisense-Induced Exon Skipping and Synthesis of Dystrophin in the Mdx Mouse. Proc. Natl. Acad. Sci. U.S.A. 2001, 98 (1), 42–47. 10.1073/pnas.98.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney J. E.; Gurpur P. B.; Burkin D. J. Laminin-111 Protein Therapy Prevents Muscle Disease in the Mdx Mouse Model for Duchenne Muscular Dystrophy. Proc. Natl. Acad. Sci. U.S.A. 2009, 106 (19), 7991–7996. 10.1073/pnas.0811599106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wypych J.; Li M.; Guo A.; Zhang Z.; Martinez T.; Allen M. J.; Fodor S.; Kelner D. N.; Flynn G. C.; Liu Y. D.; Bondarenko P. V.; Ricci M. S.; Dillon T. M.; Balland A. Human IgG2 Antibodies Display Disulfide-Mediated Structural Isoforms. J. Biol. Chem. 2008, 283 (23), 16194–16205. 10.1074/jbc.M709987200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. D.; Wang T.; Chou R.; Chen L.; Kannan G.; Stevenson R.; Goetze A. M.; Jiang X. G.; Huang G.; Dillon T. M.; Flynn G. C. IgG2 Disulfide Isoform Conversion Kinetics. Mol. Immunol. 2013, 54 (2), 217–226. 10.1016/j.molimm.2012.12.005. [DOI] [PubMed] [Google Scholar]
- Cochran M.; Arias D.; Burke R.; Chu D.; Erdogan G.; Hood M.; Kovach P.; Kwon H. W.; Chen Y.; Moon M.; Huang H.; Levin A.; Doppalapudi V. R.. Structure-Activity Relationship of Antibody-Oligonucleotide Conjugates: Evaluating Bioconjugation Strategies for Antibody-siRNA Conjugates for Drug Development. [DOI] [PMC free article] [PubMed]
- Willoughby J. L. S.; Chan A.; Sehgal A.; Butler J. S.; Nair J. K.; Racie T.; Shulga-Morskaya S.; Nguyen T.; Qian K.; Yucius K.; Charisse K.; Van Berkel T. J. C.; Manoharan M.; Rajeev K. G.; Maier M. A.; Jadhav V.; Zimmermann T. S. Evaluation of GalNAc-siRNA Conjugate Activity in Pre-Clinical Animal Models with Reduced Asialoglycoprotein Receptor Expression. Mol. Ther. 2018, 26 (1), 105–114. 10.1016/j.ymthe.2017.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyar V. S.; Song D.; Zheng S.; Carpenter T.; Heald D. L. Minimal Physiologically Based Pharmacokinetic-Pharmacodynamic (mPBPK-PD) Model of N-Acetylgalactosamine–Conjugated Small Interfering RNA Disposition and Gene Silencing in Preclinical Species and Humans. J. Pharmacol. Exp. Ther. 2021, 379 (2), 134–146. 10.1124/jpet.121.000805. [DOI] [PubMed] [Google Scholar]
- Lu G.; Nishio N.; Van Den Berg N. S.; Martin B. A.; Fakurnejad S.; Van Keulen S.; Colevas A. D.; Thurber G. M.; Rosenthal E. L. Co-Administered Antibody Improves Penetration of Antibody–Dye Conjugate into Human Cancers with Implications for Antibody–Drug Conjugates. Nat. Commun. 2020, 11 (1), 5667 10.1038/s41467-020-19498-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beekman C.; Janson A. A.; Baghat A.; Van Deutekom J. C.; Datson N. A. Use of Capillary Western Immunoassay (Wes) for Quantification of Dystrophin Levels in Skeletal Muscle of Healthy Controls and Individuals with Becker and Duchenne Muscular Dystrophy. PLoS One 2018, 13 (4), e0195850 10.1371/journal.pone.0195850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X.; Ponte J. F.; Yoder N. C.; Laleau R.; Coccia J.; Lanieri L.; Qiu Q.; Wu R.; Hong E.; Bogalhas M.; Wang L.; Dong L.; Setiady Y.; Maloney E. K.; Ab O.; Zhang X.; Pinkas J.; Keating T. A.; Chari R.; Erickson H. K.; Lambert J. M. Effects of Drug–Antibody Ratio on Pharmacokinetics, Biodistribution, Efficacy, and Tolerability of Antibody–Maytansinoid Conjugates. Bioconjugate Chem. 2017, 28 (5), 1371–1381. 10.1021/acs.bioconjchem.7b00062. [DOI] [PubMed] [Google Scholar]
- Hiller M.; Falzarano M. S.; Garcia-Jimenez I.; Sardone V.; Verheul R. C.; Popplewell L.; Anthony K.; Ruiz-Del-Yerro E.; Osman H.; Goeman J. J.; Mamchaoui K.; Dickson G.; Ferlini A.; Muntoni F.; Aartsma-Rus A.; Arechavala-Gomeza V.; Datson N. A.; Spitali P. A Multicenter Comparison of Quantification Methods for Antisense Oligonucleotide-Induced DMD Exon 51 Skipping in Duchenne Muscular Dystrophy Cell Cultures. PLoS One 2018, 13 (10), e0204485 10.1371/journal.pone.0204485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheul R. C.; Van Deutekom J. C. T.; Datson N. A. Digital Droplet PCR for the Absolute Quantification of Exon Skipping Induced by Antisense Oligonucleotides in (Pre-)Clinical Development for Duchenne Muscular Dystrophy. PLoS One 2016, 11 (9), e0162467 10.1371/journal.pone.0162467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burki U.; Keane J.; Blain A.; O’Donovan L.; Gait M. J.; Laval S. H.; Straub V. Development and Application of an Ultrasensitive Hybridization-Based ELISA Method for the Determination of Peptide-Conjugated Phosphorodiamidate Morpholino Oligonucleotides. Nucleic Acid Ther. 2015, 25 (5), 275–284. 10.1089/nat.2014.0528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinne A. S.; Tillman E. J.; Abdeen S. J.; Johnson D. E.; Parmer E. S.; Hurst J. P.; De Temple B.; Rinker S.; Rolph T. P.; Bowsher R. R. Noncompetitive Immunoassay Optimized for Pharmacokinetic Assessments of Biologically Active Efruxifermin. J. Pharm. Biomed. Anal. 2023, 232, 115402 10.1016/j.jpba.2023.115402. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.