
Fig. 3 ∣. Flowcharts of the SCcaller-pipeline.
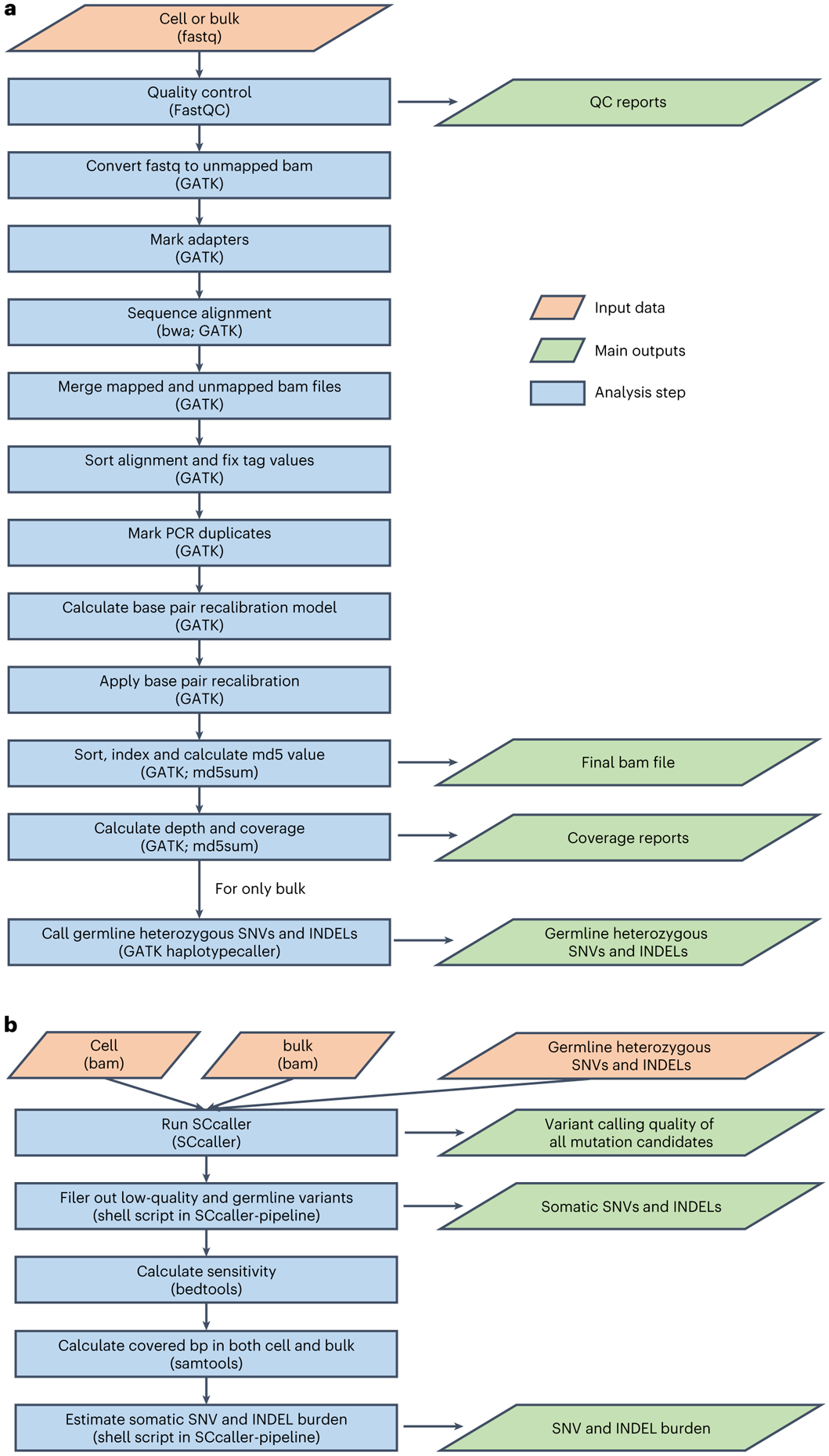
The pipeline includes two major stages (i.e., commands). a, QC, sequence alignment and germline variant calling. This command should be performed for single-cell sequencing data and bulk sequencing data separately. For single-cell sequencing data, it takes fastq files of a sample (either bulk or single cell) as input and outputs aligned bam file as well as reports of QC and genome coverage. For bulk sequencing data, in addition to the above, it outputs a list of germline heterozygous SNPs and INDELs. b, Somatic mutation calling and mutation burden estimation. This command takes bam files of a single cell and its corresponding bulk DNA, and the list of germline heterozygous SNPs and INDELs (all are outputs from a) as input, and outputs somatic mutations, their variant calling qualities, and estimated mutation burden (i.e., the number of mutations per cell after adjusting variant calling accuracy and genomic coverage).