
Fig. 4 ∣. Flowchart of the SCcaller algorithm.
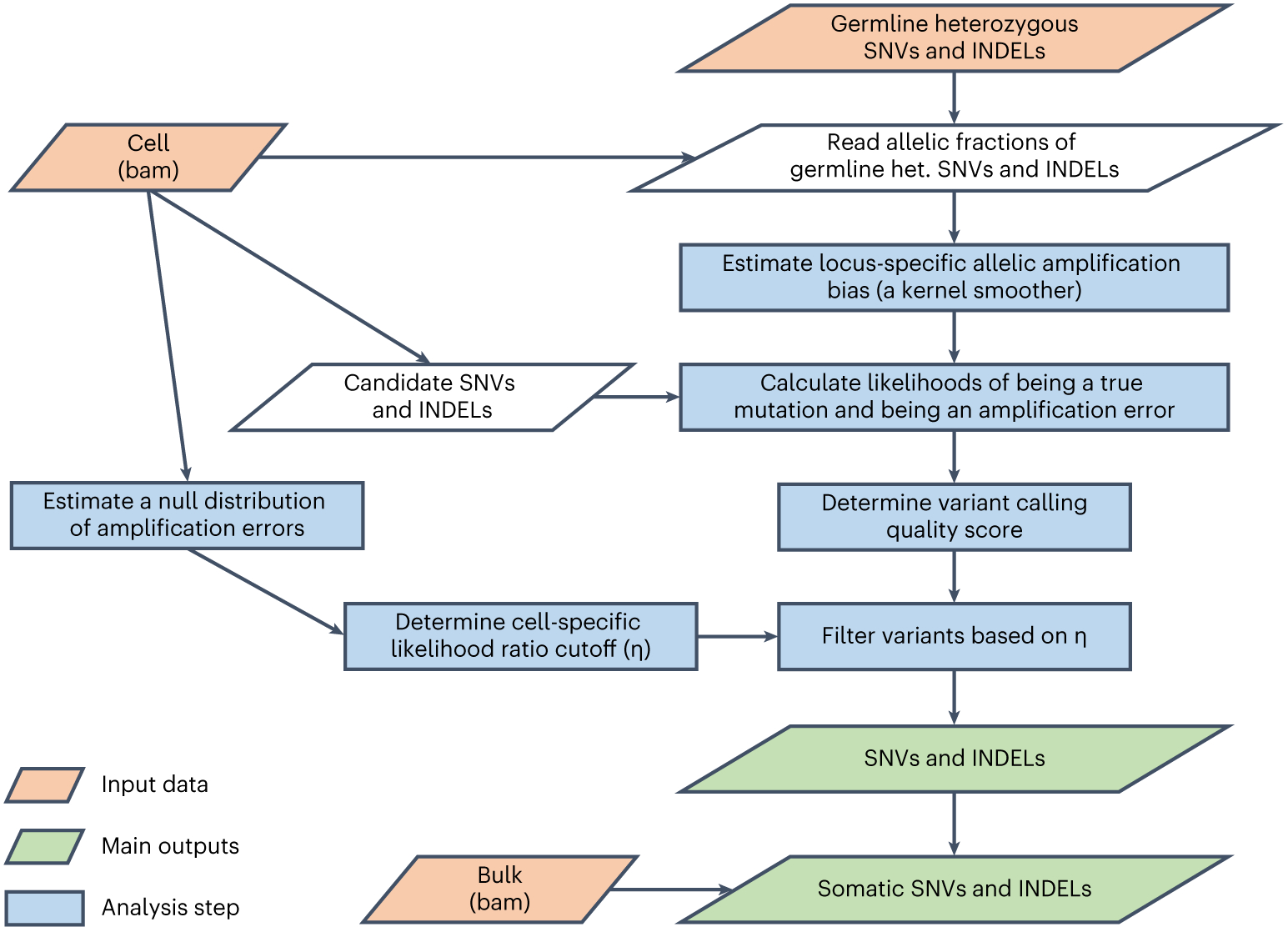
In brief, a heterozygous (het.) mutation should ideally present in 50% of sequencing reads of a diploid cell, but in real single-cell sequencing data, it often deviates from 50% substantially, because the two copies of chromosome are often not amplified to an equivalent amount. In single-cell sequencing data, SCcaller estimates such allelic amplification bias for every position in the genome from the allelic amplification bias observed for known germline heterozygous SNPs and INDELSs close to the position, and adjusts the bias in calling somatic variations: a true somatic mutation should be observed with a similar allelic fraction of sequencing reads as its neighboring germline heterozygous SNPs and INDELs (which may not be 50%). Please see ref. 17 for more descriptions.