

Abstract
Dry eye disease (DED) is a multifactorial aging disorder leading to tear film insufficiency and instability. Yet, an important knowledge gap lingers in understanding senescence-associated ocular pathogenesis, due to limited in vitro translational lacrimal gland (LG) models. Consequently, this remains a major roadblock to discover effective therapies for the restoration of tear film secretion. Herein, the authors reported the magnetic bioassembly of two LG organoid platforms to recapitulate functional and aging states. Using a proof-of-concept approach, porcine primary LG cells were assembled into organoids via a magnetic 3D bioprinting (M3DB) platform. This platform could form reproducible LG organoids with epithelial hallmarks (AQP5+) and exhibit epithelial secretory functions (lysozyme activity). DNA damage-induced senescence and cell death was induced with etoposide, and LG organoid hypofunction and senescence-associated pathogenesis were observed. To confer DNA protection against aging, a novel gene therapy with Box A domain of high-mobility group box-1 (HMGB1-Box A) previously established by our group, was applied here to prevent LG cellular senescence for the first time. HMGB1-Box A transfection prevented LG organoids from senescence-associated pathogenesis at the transcriptomic, metabolomic and proteomic levels. Thus, M3DB platforms could generate functional and DNA damage-induced senescence LG organoids, and this latter damage could be prevented with HMGB1-Box A gene therapy.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-73101-8.
Keywords: Dry eye disease, Lacrimal gland, Cellular Senescence, Bioprinting, Organoid, Gene therapy
Subject terms: Eye diseases, Biotechnology, Cell biology, Biomedical engineering
Introduction
Cellular senescence is a major risk factor for common eye disorders affecting the lacrimal gland (LG) including dry eye disease (DED) also known as xerophthalmia1–3. This condition results from tear deficiency or/and excessive evaporation of the tear film. Epidemiological studies and meta-analysis have demonstrated that individuals aged 50–60 years and older are more prone to DED due to an imbalance in the produced tear film3,4. The underlying mechanisms of senescence-related DED remain poorly understood and effective treatments are limited. Earlier studies have reported reduced tear flow and increased levels of late peroxidation markers in the tear film of elderly aqueous-deficient DED patients5,6.
Our researchers have successfully tested gene therapy strategies when fluid secretion is severely diminished in other craniofacial exocrine glands alike the LG (i.e. salivary glands)7. More recently, other members of our research group have unveiled new mechanisms on how DNA is damaged during senescence-associated pathogenesis and offered strategies to reverse the cellular senescence phenomena8,9. Moreover, we’ve shown evidence that Box A from high mobility group box 1 (HMGB1) produced DNA gaps via its molecular scissoring activity. Transfer of Box A-producing plasmid increases the DNA gaps and enhances DNA durability, by protecting the DNA from all types of damage including base changes and losses, and single-strand and double-strand DNA breaks. This Box A-induced cascade of events ultimately leads to the prevention of DNA damage and suppression of senescence-associated pathogenesis. When new Youth-DNA‐GAPs by HMGB1-Box A are supplemented, the complete rejuvenation is observed of in vitro senescent cells and in two aging rat models. Hence, HMGB1-Box A may constitute a promising candidate for future anti-senescent gene therapies for many tissues and exocrine organs including the LG.
However, complex ethical challenges make clinical studies on LG senescence-associated pathogenesis difficult to undertake. Alternative pre-clinical animal models (rabbits or rodents) are widely utilized, but the LG in these models lack physiological resemblance with the human LG organ10,11. Recently, to address this gap, researchers have developed functional LG organoids from human or porcine primary LG cells using either Matrigel as a scaffold to support the viability of the three-dimensional (3D) organoids12,13 or scaffold-free constructs generated via bioprinting platforms developed by our research team14,15. Since Matrigel has limited translation towards clinical applications, our previous work on magnetic 3D bioprinting (M3DB) offers a superior strategy for assembling exocrine gland organoids (i.e. lacrimal and salivary glands) that can be used as functional and senescent models for studying novel therapies for glandular hypofunction14–18. These platforms provide user-friendly, step-by-step short procedures for manufacturing reproducible and consistent LG organoids. As per limited availability of healthy LG tissues, primary cells from porcine LG can be used as a proof-of-concept14. These primary cells can be promptly assembled to fabricate a functional LG organoid with a M3DB protocol. To establish senescence-associated pathogenesis, an in vitro LG organoid model was developed upon DNA damage induced by etoposide, which can lead to cell death in vitro8,14. Therefore, developing senescent LG organoid models mimicking DED is of ultimate importance to evaluate potential anti-senescent gene therapies, alike with HMGB1-Box A gene transfer, to prevent DNA damage.
Thus, the main goal of this study was to determine whether HMGB1-BoxA gene therapy can prevent cellular senescence in bioprinted LG organoids exposed to etoposide. First, functional LG organoids were developed by magnetic 3D bioprinting and characterized using multi-omics approaches. Next, a LG organoid model with DNA damage-induced senescence was generated upon the induction with etoposide. Finally, a plasmid with the HMGB1-Box A gene was transfected using a standard lipid-based nanoparticle technology to assess the prevention of cellular senescence in the LG organoid and functional preservation of fluid secretion.
Materials and methods
Primary LG cell isolation and differentiation
Primary LG cells were isolated from LG of a 3- to 5-month-old porcine head and cultured as published previously14, and all reagents are listed in Supplementary Tables S1 and S2 in Supplementary File. Briefly, the gland was mechanically disrupted, and the tissue fragments were enzymatically digested in 1x PBS buffer with collagenase type II 1 mg/mL (Gibco, Thermo Fisher Scientific, Waltham, MA, USA), Hyaluronidase 1 mg/mL (Sigma-Aldrich, Merck KGaA, Darmstadt, Germany), and 1.25 mM CaCl2. A 2 mL (for 200 mg of tissue) of this enzymatic dissociation was used and place inside a 15 mL tube at 37 °C water beaker with magnetic stirring at 500 rpm and incubated for 30 min (vortex the tube every 15 min). The resulting cell suspensions were filtered through 100 μm and 40 μm cell strainers (SPL, Pocheon-si, Gyeonggi-do, South Korea) and plated on a T75 flask coated with Cultrex basement membrane extract (Bio-Techne, Minneapolis, MN, USA) at a density of 1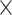 106 cells per flask. Cells were then cultured in 10 mL expansion media in a 5% CO2 incubator at 37 °C until they reached a confluency of 70–80%, which typically took 6 culture days. The expansion media was composed of DMEM/F12 (Sigma-Aldrich), 1 mM l-Glutamine, 1% Penicillin/Streptomycin, 20 ng/mL EGF (Thermo Fisher Scientific), 5% FBS (HyClone, Cytiva, Marlborough, MA, USA). To assess the viability of primary cells, the Trypan blue exclusion reagent and methodology was utilized (Thermo Fisher Scientific) and cell viability was determined with a Countess 3 FL Automated Cell Counter (Thermo Fisher Scientific). To induce differentiation of the epithelial cells, the expansion medium was replaced with 10 mL of fresh epithelial enrichment media, and the cells were maintained in culture for an additional 8 days. The epithelial enrichment media contained Defined Keratinocyte Serum-Free basal media (DKSFM, Gibco, Thermo Fisher Scientific) with 20 ng/mL EGF, 50 ng/mL FGF-10, 50 ng/mL FGF-7 (ImmunoTools GmbH, Friesoythe, Germany).
106 cells per flask. Cells were then cultured in 10 mL expansion media in a 5% CO2 incubator at 37 °C until they reached a confluency of 70–80%, which typically took 6 culture days. The expansion media was composed of DMEM/F12 (Sigma-Aldrich), 1 mM l-Glutamine, 1% Penicillin/Streptomycin, 20 ng/mL EGF (Thermo Fisher Scientific), 5% FBS (HyClone, Cytiva, Marlborough, MA, USA). To assess the viability of primary cells, the Trypan blue exclusion reagent and methodology was utilized (Thermo Fisher Scientific) and cell viability was determined with a Countess 3 FL Automated Cell Counter (Thermo Fisher Scientific). To induce differentiation of the epithelial cells, the expansion medium was replaced with 10 mL of fresh epithelial enrichment media, and the cells were maintained in culture for an additional 8 days. The epithelial enrichment media contained Defined Keratinocyte Serum-Free basal media (DKSFM, Gibco, Thermo Fisher Scientific) with 20 ng/mL EGF, 50 ng/mL FGF-10, 50 ng/mL FGF-7 (ImmunoTools GmbH, Friesoythe, Germany).
Plasmid construction
Box A-GFP plasmid with the HMGB1 sequence and the Scramble-GFP plasmid were developed as described previously8. The plasmids were transformed into competent Escherichia coli (DH5α, Invitrogen, Carlsbad, CA, USA) and the transformants were selected on LB agar. The colonies were then cultured in LB broth with 34 µg/mL chloramphenicol at 37 °C for 16 h with shaking. The plasmids were then extracted using the GeneJET Plasmid Miniprep Kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions.
Plasmid transfection
The differentiated epithelial-enriched porcine LG cells at confluency of 50–60% were maintained in each well of a 6-well plate containing 2 mL of differentiated culture medium. Then, 2.0 µg of each plasmid was transfected for 24 h using Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Organoid biofabrication and cellular senescence induction
The biofabrication of LG organoids was conducted by using M3DB platform as per previous studies14,15, and their size and morphology was assessed for 6 culture days using a flatbed scanner Epson Perfection V600 (Epson, Japan) and ImageJ (NIH, Bethesda, MD, USA). To fabricate the organoids in the M3DB, a monolayer of differentiated LG cells with a confluency of 70–80% was dissociated and resuspended in a culture medium at a density 1 × 106 cells/mL. The cells were then magnetized with magnetic nanoparticle solution (MNP) at the ratio 10,000 cells/µL and incubated for 2 h at 37 °C with shaking (250 rpm). Then, cells were resuspended in culture medium to a density of 3.33 × 105 cells/mL and transferred into an ultra-low attachment flat bottom 96-well plate, with 150 µL of cell suspension in each well and a magnetic dot drive underneath to gain the spheroid-like shape. The LG organoids were cultured in a 5% CO2 incubator at 37 °C for 24 h on top of the drive before being treated with 10 µM etoposide for another 24 h. The vehicle group was treated with 0.1% DMSO. Cytotoxicity or late apoptosis was evaluated with propidium iodide (PI) and nuclear Hoechst 33,342 staining (Thermo Fisher Scientific). These organoids were imaged with a fluorescence microscope (EVOS FL auto) through the z-axis to generate a z-stack (at 10 μm distance between each scan), and whole images were annotated for late apoptotic cells (PI-stained) and ImageJ software was used to determine the fluorescent intensity signal (as per measurement of “integrated density”).
Luciferase-based ATP 3D assays
Cell viability in the LG organoids was evaluated by determining their ATP activity using CellTiter-Glo 3D Cell Viability Assay (Promega Corporation, Madison, WI, USA) according to the manufacturer’s protocol. Briefly, reagents and plates were allowed to equilibrate at RT prior to the addition of 20 µL of reagent to each well of a 96-well plate containing organoid in 100 µL of DMEM. Next, plates were vigorously mixed for 5 min to induce cell lysis, followed by incubation at 37 °C for 25 min. Subsequently, 100 µL of the resulting solution was transferred to a white opaque-walled 96-well plate and the relative luminescence units (RLU) were measured on a microplate reader (GloMax Explorer Multimode Microplate Reader, Promega Corporation).
Reactive oxygen species assessment
Reactive oxygen species (ROS) generation was determined by ROS-Glo H2O2 assay kit (Promega Corporation) as per manufacturer’s instructions. This assay was performed 48 h after cellular senescence was induced in the LG organoids while these were cultured in 5% CO2 at 37 °C. To run the ROS-Glo H2O2 assay, the culture medium was replaced with 100 µL of medium containing H2O2 substrate and incubated for 6 h. Next, 100 µL of working ROS-Glo detection reagent was added to each well, and the plate was incubated for 40 min at RT. The relative luminescence units (RLU) were then measured on a microplate reader (GloMax Explorer Multimode Microplate Reader).
Calcium influx assay
To determine the functionality of LG organoid upon neurostimulation, the intracellular Ca2+ mobilization in the organoid was determined with a Fluo-4 Calcium Imaging Kit (Thermo Fisher Scientific) according to the manufacturer’s protocol. The organoids were treated with Fluo-4AM for 25 min at 37 °C, followed by incubation at RT for 25 min. After rinsing with 1× PBS, organoids were stimulated with 10 µg/mL Carbachol (Sigma, MA, USA) and Ca2+ intracellular influx was observed using a fluorescence and confocal microscopes (EVOS FL auto and Zeiss LSM 980, respectively).
Histology and Immunofluorescence
LG organoids were initially washed with PBS and then fixed in 4% paraformaldehyde (Sigma, MA, USA) for 20 min and embedded in paraffin. Paraffin Sect. (5 μm thick) were deparaffinized in xylene and performed histological processes of hematoxylin and eosin (H&E) staining. As for immunofluorescence staining, the sections were pre-blocked with 5% normal horse serum and 10% bovine serum albumin (BSA) in PBS for 2 h at RT. The sections were then incubated with primary antibodies against Aquaporin 5 (AQP5), Cytokeratin 5 (K5), Cytokeratin 14 (K14), Cytokeratin 19 (K19), the ion co-transporter NKCC1 at 4 °C overnight. After washing with PBS, sections were incubated with secondary antibodies for 1 h at RT. Nuclear counterstaining was performed using Hoechst 33,342 (Thermo Fisher Scientific) for 20 min. All primary and secondary antibodies and respective dilutions are listed in supplementary data and were validated (Supplementary Table S1 and Supplementary Fig. S1).
Senescence-associated β-galactosidase detection
LG organoids were washed with 1× PBS and fixed with 4% paraformaldehyde solution for 20 min. Next, organoids were stained using the Senescence-associated β-galactosidase (SA-β-gal) staining kit (Cell Signaling Technology, Danvers, MA, USA) according to the manufacturer’s instructions. The SA-β-gal in organoids was visualized under a light microscope (DMi1 Inverted Microscope, Leica Microsystems, Wetzlar, Germany) and quantified the results from five random fields using Image J software (NIH, Bethesda, MD, USA).
Lysozyme activity
The EnzChek™ Lysozyme Assay kit (cat. no. E-22013, Thermo Fisher Scientific) was utilized to measure lysozyme activity in the conditioned media solution where LG organoids were cultured. The protocol followed the manufacturer instructions. Briefly, a 10 mg/mL stock suspension of the DQTM lysozyme substrate (Component A) was prepared in 2 mL deionized water. Then, the Component C reagent was dissolved in 1 mL of deionized water. A lysozyme standard curve was generated in a total volume of 100 µL per well, whereas 50 µL of 1× of Component B was added, followed by 50 µL of the 1000 U/mL stock solution of lysozyme (prepared earlier). These were mixed by pipetting in the first well, then transferred 50 µL to the second well, and so forth until the final one is discarded. Next, lysozyme-containing experimental samples were diluted in 1× Component B to prepate a volume of 50 µL for each reaction. Samples were serially diluted to determine the optimal sample concentration. To start the reaction, 50 µL of the 50 µg/mL DQ lysozyme substrate working suspension was added to each well containing the experimental samples. Then, we incubate the reaction mixtures for 30 min or longer at 37 °C and always protecting samples from light. Since the assay is continuous (does not terminate), the fluorescence can be measured at multiple time points to follow the reaction kinetics. We measured the fluorescence intensity of each reaction in a fluorescence microplate reader (GloMax Explorer Multimode). Digestion products from the DQ lysozyme substrate have a maximum absorption at 494 nm and maximum fluorescence emission at 518 nm, hence, these wavelengths were used to measure fluorescence intensity.
Transcriptome arrays
RNA total extraction was performed by using manufacturer instructions with a total RNA extraction kit (FFPET RNA Isolation Kit version 13, ref. no. 06 483 852 001, Roche, Germany). The RNA isolation kit protocol comprised of 5 steps: (1) Formalin-fixed paraffin embedded organoid samples were disrupted in Tissue Lysis Buffer and homogenized during an incubation with Proteinase K. In the presence of a chaotropic salt, nucleic acids (NA) bound specifically to the surface of glass fibers pre-packed in the purification filter tube. Next, bound nucleic acids were washed, and thereby purified of salts, proteins, and other impurities. Then, to eliminate residual DNA, an on-column DNase I digestion was performed. For the final step, a low salt elution released the RNA from the glass fibers. RNA was quantified and assessed for quality using NanoDrop spectrophotometer. A total of 100 ng of RNA from native tissue, LG organoid, etoposide-treated LG organoid with or without gene therapy in 10 µL of RNase-free water was subjected to NanoString nCounter Elements workflow, according to the nCounter SPRINT Profiler User Manual (NanoString Technologies, Seattle, WA, USA). All raw values generated by the nCounter Analysis System (NanoString Technologies) were based on the digital detection of molecular barcodes present in reporter probes that hybridize with the target RNA. These reporter probes are associated with target-probe complexes that have also a capture probe which hybridizes with the native RNA transcript (your target of interest). Hence, the nCounter assay technology and their gene expression output for each gene panel generates data with high specificity and sensitivity that can be promptly replicated with negligible variations in gene expression (< 1 copy per cell). Due to the high precision of the nCounter technology, the statistics are often not necessary simply because researchers are really getting the precise counts or RNA transcript copies. Both the nSolver 4.0 and Rosalind software were used to generate heatmaps based on Z-scores and these scores are always positive since the barcode detection output for each gene has a positive target count. RNA transcripts of the 394 targeted genes from 760 genes of the stem cell panel (Supplementary Table S1) was assessed using nSolver 4.0 software (NanoString Technologies) by normalizing with 6 reference genes (ACTB, CLTC, GAPDH, PPIA, RPLP0, and TBP). Three hundred sixty-six genes that have percent identity to human genes lower than 85% were excluded, and briefly, an analysis was started by running the analysis function section on the nSolver 4.0 software. The Agglomerative Cluster analysis was run and a Z-score transformation function on genes was generated by the software. This dictates how the heatmap was centered and scaled. Heatmaps were generated using the R studio and GraphPad Prism version 9 (GraphPad, San Diego, CA, USA).
Western blot
To study proteins expressed by LG organoids after aging induction and gene therapy, 20–30 organoids were harvested 48 h after etoposide treatment for each experimental group and analyzed through Western blot analysis. In brief, 250 µg of protein were extracted by using 250 µL RIPA lysis buffer containing protease inhibitors (Thermo Fisher Scientific). Then, 20 µg of denatured total proteins were resolved on 10% SDS-PAGE gels and transferred to a polyvinylidene difluoride membrane (Millipore, Merck) by semi-dry electroblotting. Non-specific protein bindings were blocked in blocking solution, 5% non-fat dry milk in 1× Tris-Buffered Saline with 0.1% Tween-20 (TBST), for 2 h at RT. Next, membranes were incubated with monoclonal antibodies against AQP5 (AB92320, Abcam, UK) and NKCC1 (AB59791, Abcam, UK) at 4 °C, overnight. After washing with TBST, they were incubated with horseradish peroxidase-conjugated secondary antibodies that corresponded to each primary antibody before subjecting to enhance chemiluminescence detection (Bio-Rad, Hercules, CA, USA).
Statistical analysis
All experiments were performed in triplicate at minimum. Data was plotted as the mean ± standard deviation (SD). The number of biological replicates is indicated in the figure captions as well as whether fold change was plotted or normalization was done. D’Agostino-Pearson normality test was performed to confirm normal shape distribution of all data sets. Since all data exhibited a normal distribution, we used the Student’s t-test for comparison between two groups. A one-way or two-way ANOVA was used for comparison of multiple groups, followed by Tukey’s or Dunnett’s post-hoc tests. GraphPad Prism 9 (GraphPad, San Diego, CA, USA) was used for all statistical analysis and significance was defined at 5%.
Results
Establishment of LG organoids by M3DB
To generate LG-like organoids, a specific biofabrication workflow was followed using the M3DB platform (Fig. 1A). After differentiated primary LG cells were magnetically assembled into 3D spheroids, consistent 3D sphere-like cell clusters within 24 h but this was not the case for MNP-free (-MNP) cells grown in basement membrane extract (Fig. 1B). Moreover, consistent 3D sphere-like cell clusters within 24 h with a stable ATP cellular production and viability (Fig. 1C, Supplementary Fig. S2). These spheroids exhibited a very consistent and uniform size over 3 days of culture (250 ± 10 μm) and started to slightly enlarge thereafter (Fig. 1D). Histological examination revealed that LG spheroids had cellular complexity with lumens and acinar-like structures mimicking the native LG tissue (Fig. 1E).
Fig. 1.
Epithelial morphology and viability of a M3DB-assembled LG organoid. (A) LG organoid biofabrication workflow steps. EEM epithelial-enriched media, MNP magnetic nanoparticles, M3DB magnetic 3D bioprinting/bioassembly. Created with BioRender.com. (B) Brightfield micrographs with phase contrast showing consistent spheroid formation in MNP-tagged LG primary cells. Scale bars: 100 μm. (C) Fold change in total ATP of each organoid was determined by a CellTiter-Glo 3D assay (n = 4–5) and all values were normalized to day 1 (baseline). The total ATP was deemed stable through 6 days of culture (n = 5). (D) After biofabrication, organoid diameter was determined and consistent up to day 3, after which there was wide variation in organoid size (n = 10). *p < 0.05 when compared to baseline or day 1, with one-way ANOVA with Dunnett’s post-hoc test. (E) H&E micrographs displayed a structural integrity and epithelial parenchyma with acini-like clusters (gray arrowheads, top white framed inset at higher magnification) and lumen-like regions (black arrowhead, bottom black framed inset at higher magnification) of the LG organoid at day 3 of culture resembling that of native LG fresh biopsy tissue. Inset is a focused area arising from black frame box. Scale bars: 50 μm.
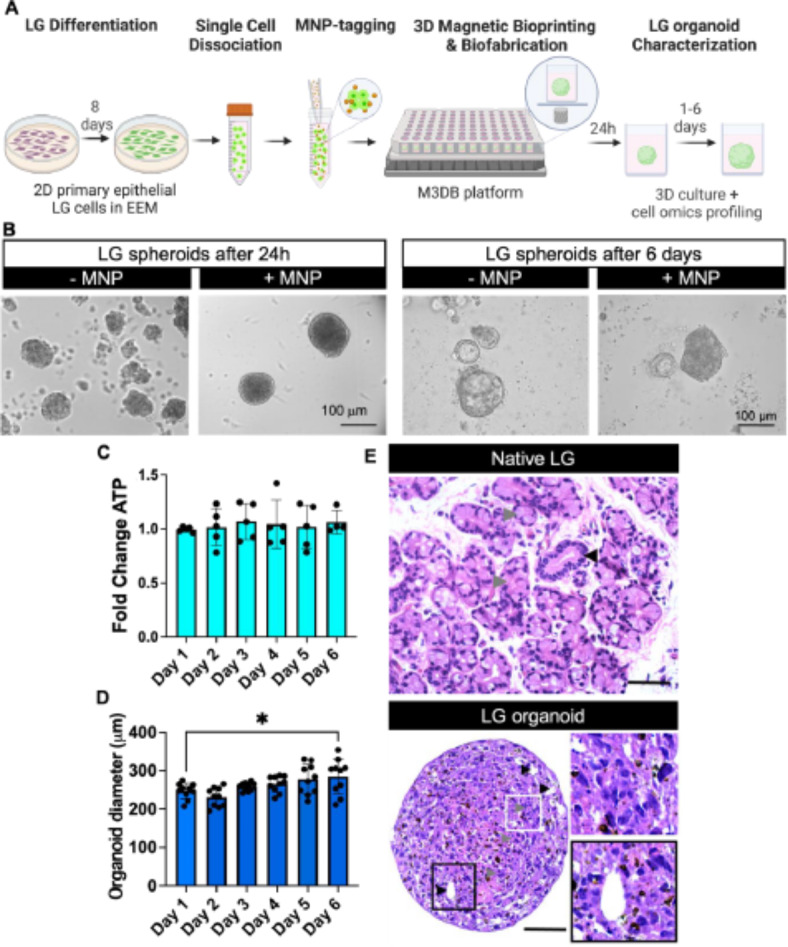
LG spheroids expressed pro-acinar AQP5+ (39.55 ± 7.05%) and NKCC1 + cells (4.83 ± 1.65%), ductal basal progenitors K14+ (76.03 ± 10.10%), and ductal luminal progenitors K19+ (3.24 ± 0.99%), indicating the presence of an epithelial LG-like organoid with acinar and ductal cellular compartments alike the native LG (Fig. 2A,B). Lacrimal secretory function was assessed upon stimulation with 10 µM Carbachol, a cholinergic agonist (Fig. 2C,D). After 1 min of Carbachol treatment, intracellular Ca2+ was actively mobilized in the cells within the LG-like organoid.
Fig. 2.
Epithelial phenotypic, functional and transcriptomic mimicry of a M3DB-assembled LG organoids (LGO). (A) Whole mount immunofluorescence staining confirmed the presence of secretory pro-acinar (AQP5 and NKCC1), progenitor ductal/myoepithelial (K14), ductal epithelial (K19) protein markers, as well as Live/Dead staining with propidium iodide (PI) in the LG, alike the native LG control (NGL CTL). All nuclei were counterstained with Hoechst 33,342. Scale bars: 200 μm. (B) Expression of LG epithelial markers was quantified by ImageJ. The fluorescent signal for each marker was assessed using “integrated density” output measurement in ImageJ at each confocal microscopy scan and summed for the entire z-stack and normalized to the total nuclear fluorescence signal (n = 3–4). (C) Secretory epithelial function was evaluated by a fluorescently labeled Ca2+ assay after parasympathetic stimulation with 10 µM Carbachol from baseline (t0 just before adding this stimuli) and 1 min. Scale bar: 200 μm. (D) Ca2+ fluorescence signal was assessed to determine the mobilization of this cation (n = 5) and plotted data is normalized to baseline signal. ****p < 0.0001, while using a Student t-test. (E) Heatmap to compare the Z-scores in gene expression (color scale) of pluripotent and stemness-related genes between native LG tissue biopsies (NLG) and the LG organoid (LGO) samples using the nCounter stem cell transcriptome panel and ROSALIND analysis software, which follows the nCounter advanced analysis protocol of dividing counts within a lane by the geometric mean of the normalizer probes from the same lane. The set of housekeeping genes was selected based on the standard geNorm algorithm as implemented in the NormqPCR R library (n = 3). The Agglomerative Cluster analysis was run using the nSolver 4.0 software, and a Z-score transformation function on genes was performed by the software. This dictated how the heatmap was centered and scaled. All adjusted p-values are listed on Supplementary Table S3.
Next, transcriptome profiling showed many similarities in the gene expression patterns between native LG tissues (NLG1-3) and LG organoids (LGO1-3) in the nCounter panel in terms of stemness, multipotency and epithelial markers (Fig. 2E). In summary, LG organoids assembled by M3DB nanotechnology exhibited phenotypic such as AQP5+ (~ 39%), NKCC1+ (~ 5%), K14+ (~ 75%) and K19 + cells (~ 4%) and the presence of calcium mobilization upon cholinergic stimulation confirmed their functional properties. These particular features are somewhat comparable to the native LG. This prompted us to move to next experimental stage, and develop a senescent organoid model for the LG.
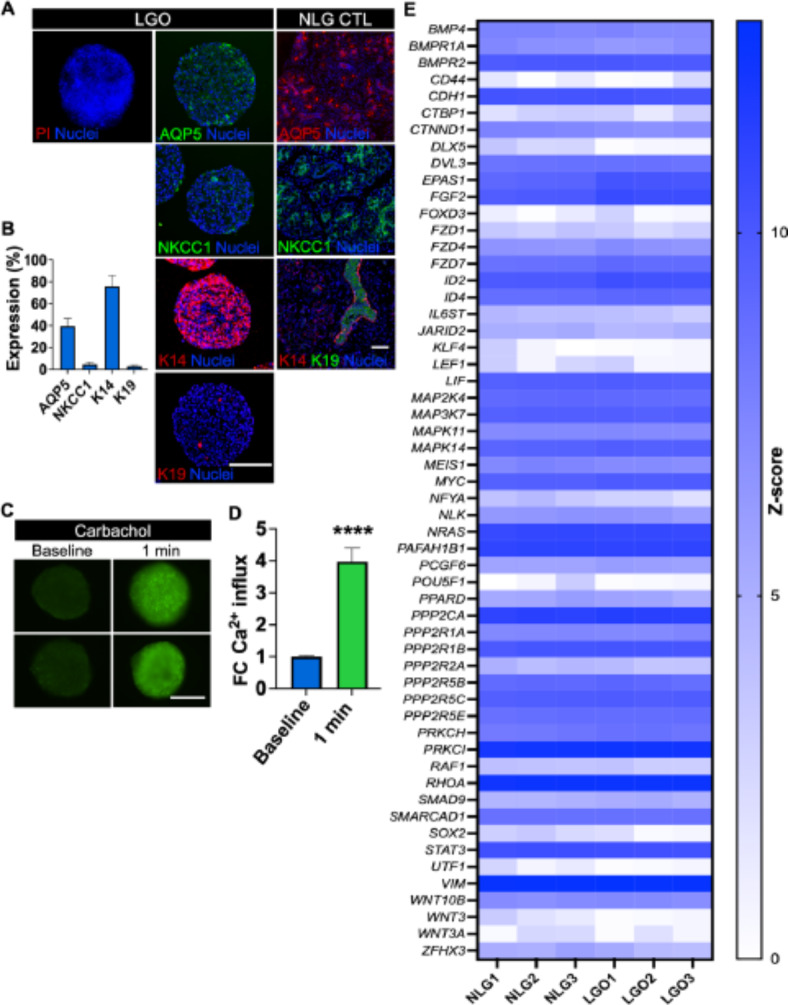
Induction of cellular senescence in lacrimal gland organoids
To create an organoid with cellular senescence hallmarks, LG organoids were assembled for 24 h in the magnetic drive and then exposed to etoposide (Fig. 3A). This reagent was previously demonstrated to induce cellular senescence8. Upon etoposide exposure, there was an increase in cell death in the organoids at 24 h and 48 h (Fig. 3B). This was further confirmed by a reduction in cellular ATP activity (Fig. 3C). Histological examination of the senescence-induced organoids revealed epithelial atrophy and cell death, as evidenced by nuclear pyknosis and fragmentation, eosinophilic cytoplasm, and disrupted cellular architecture (Fig. 3D). At the transcriptome level, the apoptosis-related cysteine protease CASP3 was overexpressed (Fig. 3E), but the apoptosis was not mediated by tumor suppressor gene TP53 or downstream TP53 via SFN-mediated genotoxicity19. The cellular senescence induction was followed by a decrease in the expression of the regulator of mitosis progression CDC20. Ubiquitin conjugating enzymes (UBE2C, UBE2D1) involved in the degradation of mitotic cyclins were also significantly decreased together with cyclins that tightly regulate the transition phases of the cell cycle (G1/S and G2/M) like CCNA2 and CCNB1. The induction of apoptosis was confirmed by the robust increase in the number of cleaved Caspase 3-positive cells in the organoid (Fig. 3F). The average ratio of apoptotic cells was approximately 40% for the etoposide-treated LGO group (LGO + Eto) and 10% for the control LGO group (LGO) but one needs to account for a substantial decrease in size and number of cells in the senescent organoid (LGO + Eto).
Fig. 3.
LG organoid displayed senescence-associated pathogenesis features at the proteomic, metabolomic and transcriptomic levels after Etoposide (Eto) exposure. (A) Biofabrication and experimental steps to produce a senescent LG organoid (LGO). MNP magnetic nanoparticles. Created with BioRender.com. (B) Fluorescence micrographs of PI staining of LG organoids treated with 10 µM Eto. (C) Fold change (FC) in total ATP was determined by CellTiter-Glo 3D assay. **p < 0.01, ****p < 0.0001 using a paired Student t-test. (D) H&E micrographs showed abundant apoptotic-like cells in Eto-treated LG organoid (LGO + Eto) when compared to non-treated LG organoid (LGO). Mag.: 40×, scale bar: 100 μm. (E) Expression of genes related to apoptosis (CASP3, CASP9), DNA repair (SFN, TP53), autophagy via ubiquitination (CDC20, UBE2C, UBE2D1), cell cycle progression (CCNA2, CCNB1) assessed by nCounter transcriptome panel. Z-score function was calculated by transforming the mean counts after normalization to the nCounter panel of a predefined set of 6 reference genes via nSolver software analysis, where *p < 0.05 using multiple Student t-tests (n = 3). All adjusted p-values are listed on Supplementary Table S4. (F) Fold change in late apoptosis was calculated after normalization to control LG organoids (LGO). Both organoid groups were stained with PI and the fluorescence signal was quantified for each z-stack. Student t-test was performed (n = 3–5): ***p < 0.001. Senescence markers were detected by: (G) ROS-Glo assay, ****p < 0.0001 using a Student t-test (n = 3–5) and (H) Senescence-associated β-galactosidase activity. ***p < 0.001 using a Student t-test (n = 3–5). (I,J) After carbachol stimulation (1 min), the function was evaluated with a Ca2+ influx assay (I), ****p < 0.0001 using a Student t-test (n = 3–5) and lysozyme activity assay (J). ****p < 0.0001 using a Student t-test (n = 3–5). Data on graph (H) is plotted as mean ± SD, and data for (C), (F), (G), (I) and (J) graphs are plotted as fold change (FC) after normalization to the control LGO group (C,F,G) or to the LGO + Eto group (I,J).
Furthermore, senescence biomarkers such as reactive oxygen species (ROS) were increased in senescence-induced LG organoids (Fig. 3G) as well as β-galactosidase activity (Fig. 3H). Next, lacrimal secretory function was assessed, and senescence-induced LG organoids displayed LG functional impairment upon cholinergic (carbachol) stimulation as per the reduction of both Ca2+ influx (Fig. 3I) and lysozyme activity (Fig. 3J). Overall, our findings demonstrated that in the presence of etoposide, LG organoids exhibited cell cycle arrest and DNA damage-induced senescence phenotypic features leading to cell death.
HMGB1 Box A gene can be successfully transferred and prevent cell death
Genetically engineered HMGB1 Box A (Box A) can stabilize DNA strands and was shown to protect DNA and prevent aging in our previous reports8. To test this hypothesis in our LG organoids with cellular senescence signatures, a GFP-tagged Box A plasmid was utilized to assess its transfection efficiency in primary LG cells and in the organoids, while a GFP-Scramble plasmid served as a transfection control (Fig. 4A). To increase the transfection efficiency of the plasmids into epithelial LG cells, the lipofectamine: DNA ratio was initially optimized, and 1–2 µg of DNA plasmid with either 0.75 µL or 1.5 µL of Lipofectamine increased the GFP expression, indicating higher transfection efficiency in the LG cell monolayer when compared with other conditions (Supplementary Figs. S3 and S4). Next, the transfected LG primary cells were successfully assembled into 3D LG organoids expressing GFP (Fig. 4B) and the organoids were exposed to etoposide for induction of cellular senescence as per previous experiments. After 48 h of exposure, Box A gene transfection significantly improved the cellular ATP levels in the LG organoids (Fig. 4C) and reduced both cell death (Fig. 4D), cellular vacuolization and apoptosis by a third as compared to the scramble plasmid (Fig. 4E, F). In all groups, organoids were expected to slightly undergo cell compaction and fluctuations in their sizes14,15. Moreover, Box A transfected organoids maintained their epithelial integrity and architecture (Supplementary Figs. S5 and S6). Furthermore, transcriptome profiling showed the overexpression of HMGB1 and cell cycle regulatory genes (TP53, SFN, CDC20, CCNA2, CCNB1) following the Box A gene therapy, as well as the downregulation of apoptotic markers like CASP3 (Fig. 4G).
Fig. 4.
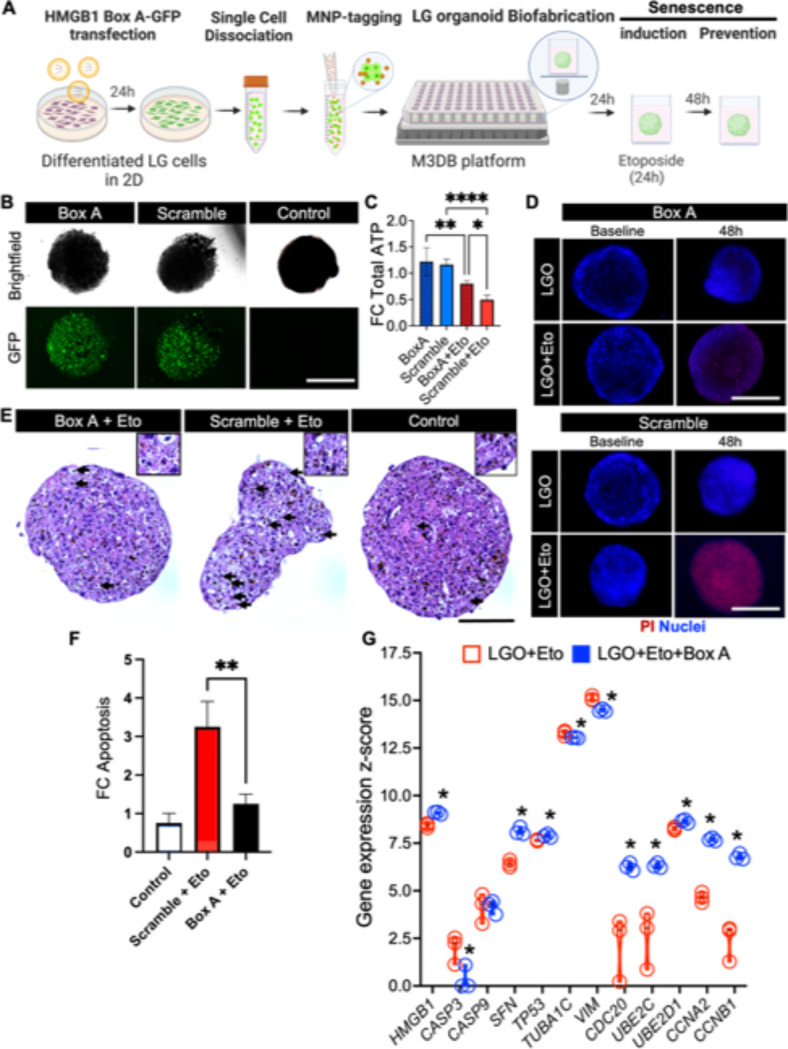
Protection effect of HMGB1 Box A gene therapy against senescence-associated pathogenesis in LG organoids. (A) Biofabrication and experimental steps to test Box A gene therapy to protect a LG organoid against cellular senescence. MNP magnetic nanoparticles. Created with BioRender.com. (B) Fluorescence micrographs of GFP-conjugated High Mobility Group A Box-1 domain plasmid (Box A), GFP-scramble plasmid (scramble) transfected organoid and LGO-treated vehicle control to confirm transfection protocol for subsequent gene therapy. (C) Total ATP was detected by CellTiter-Glo 3D assay in normal (Eto-free) LG organoids transfected groups Box A and Scramble, and the Eto senescence-induced transfected groups, Box A + Eto and Scramble + Eto. *p < 0.05, **p < 0.01, ****p < 0.0001 using a one-way ANOVA with Tukey post-hoc test (n = 3–4). (D) Cell death in all four LG organoid transfected groups was determined by PI staining counterstained with nuclear Hoechst. (E) H&E micrographs of senescence-induced LGO with Box A and scramble plasmid (Box A + Eto and Scramble + Eto, respectively), and LGO-treated vehicle control (control) are performed to identify cellular changes caused by apoptosis (arrows). Insets are displayed on top with higher magnification and focus. Mag.: 40×, scale bar: 100 μm. (F) Apoptosis (late stage) was quantified after PI staining and fluorescent microscopy imaging and plotted as fold change in fluorescent signal relative to control. Average apoptotic cell values were: 10% for Control group, 32% for Scramble + Eto, and 13% for Box A + Eto). **p < 0.01 using a one-way ANOVA with Tukey post-hoc test (n = 3–4). (G) Expression of genes related to gene therapy target (HMGB1), apoptosis (CASP3, CASP9), DNA repair (SFN, TP53), autophagy via cytoskeletal reorganization (TUBA1C, VIM), autophagy via ubiquitination (CDC20, UBE2C, UBE2D1), cell cycle progression (CCNA2, CCNB1) assessed by nCounter transcriptome panel. Z-score was determined via nSolver software analysis by a comparison with a predefined set of 6 reference genes, where *p < 0.05 using multiple Student t-tests (n = 3). All adjusted p-values are listed on Supplementary Table S5.
HMGB1 Box A gene therapy can prevent senescence-associated pathogenesis and hypofunction in LG organoids
Whole-mount β-galactosidase (β-gal) staining had a stronger signal in the scramble-transfected LG organoid (after etoposide exposure); meanwhile, the β-gal enzymatic activity was significantly decreased in the Box A-transfected organoids (Fig. 5A, B). In addition, PAS staining revealed a few areas where mucin was produced and deposited inside the LG organoid after Box A treatment, although such areas were larger and more robust in the untreated controls. These observations were also confirmed with H&E stained micrographs (Supplementary Fig. S6). Next, to determine whether Box A gene therapy could restore the physiological function of the epithelial cells in the senescence-induced LG organoids, the intracellular Ca2+ influx was assessed following stimulation with Carbachol. The levels of Ca2+ influx in Box A-transfected organoids increased approximately 3-fold when compared to the scramble-transfected controls upon etoposide exposure (Fig. 5C), and this was confirmed via real-time imaging (Fig. 5D). Oxidative stress measured via ROS was also decreased by more than 2-fold with Box A gene therapy (Fig. 5E). Next, protein expression of pro-acinar markers AQP5 and NKCC1 in LG organoids were evaluated by Western blot (Fig. 5F). In the Scramble-transfected LG organoids (after etoposide exposure), both AQP5 and NKCC1 expression patterns were decreased after 24 and 48 h when compared to control organoids at their naive baseline state. However, upon Box A gene therapy, the expression of both pro-acinar markers increased to levels comparable to the control group after 48 h. Regarding the transcriptome analysis of the etoposide-exposed organoids (Fig. 5G), Box A gene therapy upregulated Wnt signaling (WNT5A), and markers regulating DNA stabilization and cellular senescence (HMGA1, FOXA2, KLF5) and energy metabolism (LEPR, PPARGC1A), when compared to Scramble-treated organoids.
Fig. 5.
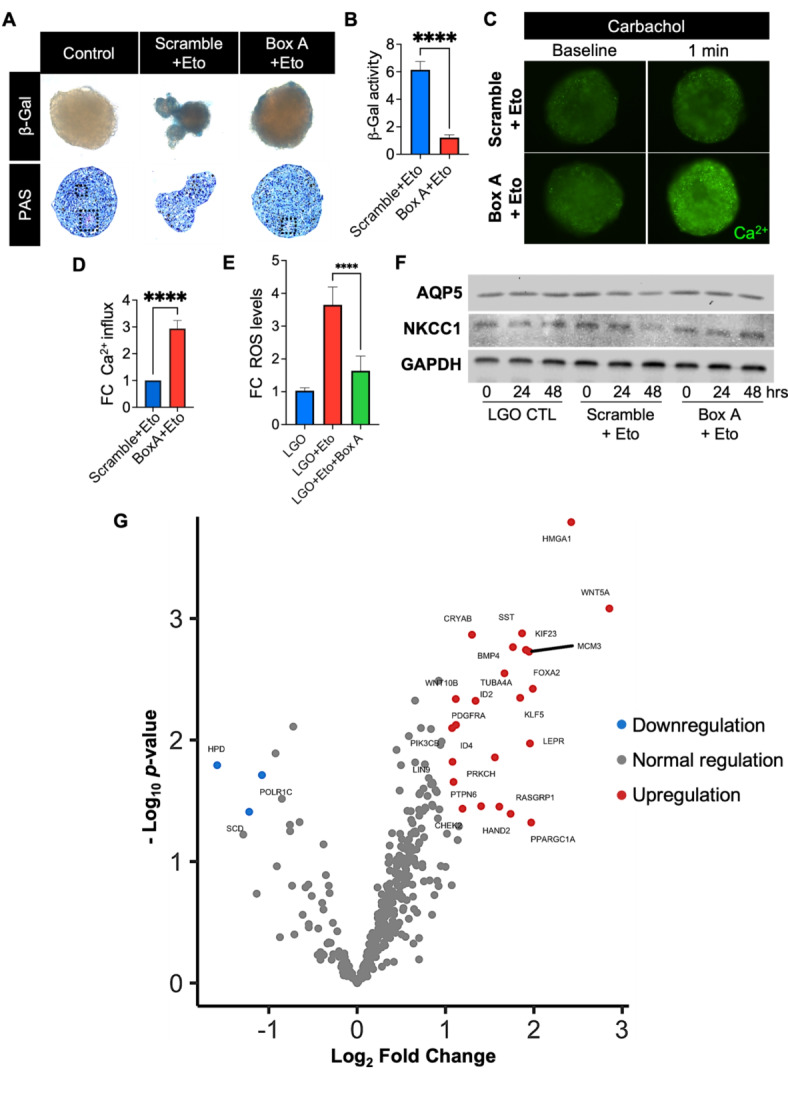
Protection against cellular senescence after Box A gene therapy in LG organoids. (A) Brightfield micrographs of whole-mount staining of senescence-associated β-galactosidase (β-gal) activity and Period acid-Schiff (PAS) staining in healthy LG organoids (LGO) and senescent-associated organoids exposed to Etoposide (Eto) after scramble or Box A gene therapy. Dashed black lines represent bright magenta regions with mucin deposition in vacuoles. (B) The intensity of β-gal-stained cells was quantified by ImageJ, and data plotted as mean ± SD. ****p < 0.0001 while using Student t-test (n = 3–4). (C) Fluorescence micrographs confirmed the Ca2+ influx and secretory function before and upon cholinergic stimulation with carbachol (1 min). (D) Ca2+-labeled fluorescence signal plotted as a fold change (FC) relative Scramble + Eto group, ****p < 0.0001 while using Student t-test (n = 3–4). (E) Senescence-associated ROS levels were determined by ROS-Glo assay kit, normalized to control naïve LG organoids (LGO) and plotted as fold change (FC). ****p < 0.0001 while using Student t-test (n = 3–4). (F) Western blot displaying expression of AQP5 and NKCC1 pro-acinar proteins at 0 h, 24 h, and 48 h after Box A gene transfer (Box A), scrambled plasmid transfer (Scramble) in etoposide-exposed LG organoids and control naïve LG organoids (LGO). GAPDH was used as a loading control. The grouping of blots was cropped from 3 different parts of the same gel for each protein and exposures are explicit by using white spaces for a clear delineation. The protein content was extracted from 20–30 organoids/group, and which were combined at each group level and loaded into each lane. The blot was run one time for each protein of interest. (G) Differential gene expression between Box A HMGB1 and Scramble plasmid gene therapy in LG organoids was quantified using a nCounter transcriptome panel and nSolver software and plotted with Rosalind software. A list of significant differentially expressed genes was determined by a calculated cut-off filter as dictated by the software. Default settings for the filter are at a fold change of 1.5 for upregulated and 1.5 for downregulated with a p-adjusted value of 0.05.
Overall, we report herein a bioprinted etoposide-exposed LG organoid that can be utilized for investigating anti-aging gene therapy for the LG.
Discussion
There is a significant lack of in vitro LG disease models for researchers and clinicians to grasp a clear understanding of senescence-related ocular pathogenesis. This study reported magnetically assembled LG organoids with cellular senescence hallmarks and investigated therapies to prevent senescence-associated pathogenesis of the LG. Consequently, this LG organoid holds potential as a pre-clinical model to discover new gene therapies to prevent senescence. To our knowledge, this is the first attempt to generate senescent-associated LG organoids from primary cells by magnetic 3D bioassembly. Recently, Bannier-Helaouet et al. from Hans Clevers group12,13 and Jeong et al.20 have successfully developed LG organoids from mouse and human tissue biopsies, and Hayashi and colleagues21 have generated LG organoids from human induced pluripotent stem cells (iPSC) and embryonic stem cells (ESC). However, these studies only fabricated functional models with secretory features, but no disease modeling or functional impairment was induced. Interestingly, our group unveiled herein that HMGB1-Box A gene therapy can prevent cellular senescence in the assembled LG organoid by suppressing Caspase 3-mediated apoptosis and oxidative stress via the reduction of ROS. This organoid biofabrication and modeling strategy was achieved utilizing porcine LG tissue biopsies as a proof-of-concept since the retrieval of healthy human LG tissue biopsies was limited at the time this research was initiated; nonetheless, porcine LG biopsies were available and deemed feasible for the biofabrication process since these are comparable to their human counterparts14,15,22. Despite such endeavors, the modeling in M3DB platforms needs further validation with LG primary cells from human biopsies.
Alike previous efforts to fabricate LG organoids12,21, we reported herein LG organoids displaying similarities to native LG in terms of their morphology, immunolabelling features and transcriptomic patterns. It is relevant to note that these reports have used biofabrication methodologies with high variability and poor clinical translation potential since organoids were grown and embedded in Matrigel12,13,20,21, which is an ill-defined heterogenous matrix also known to have batch-to-batch and single batch variations23,24. Efforts have been made to improve the secretory function in LG organoids by replacing Matrigel with a porcine decellularized LG hydrogel, but the high biodegradation rates of this hydrogel and its suboptimal rheological properties are a concern for bioprinting applications22. The high variability of self-organized organoid growth observed with Matrigel can be replaced with magnetic-based bioengineering approaches that allow spatiotemporal 3D control of organoid growth, morphology and shape14,16,18. Hence, these were implemented in our workflow for the LG organoid biofabrication, and thus, the organoids reached a robust, consistent, and reproducible diameter size of 250 ± 10 μm after 3 culture days (Fig. 1D), which gave us enough time to carry on with our in vitro protocols for cellular senescence induction towards the modeling of senescence-associated pathogenesis (Fig. 2). Longer organoid culture times are possible but those can lead to larger sizes (> 300 μm) and generate less reproducible shapes due to poorly controlled nutrient mass transfer to the central core of the organoids. To tackle these challenges, microfluidic or milifluidic approaches can eventually support the integration of mechano-physiological parameters in the organoid biofabrication process and enhances viability and functional readouts23. The application of bioengineering principles to organoid processing platforms improves reproducibility and provides experimental control, which will be required to ultimately enable clinical translation. Due to the limited cell density and organoid size, the number of lumenized areas was reduced and the apical location of the water channels (AQP5) could not be accurately determined (Fig. 1); hence, epithelial acinar polarity should be assessed with large organoids in integrated fluid flow systems.
In previous studies, LG organoids derived from human iPSC and embedded in Matrigel had limited secretory functions in vitro and can only functionally mature into myoepithelial-like cells when transplanted adjacent to the rat eye21. In our static M3DB in vitro platforms, the LG organoids secreted lysozyme, produced mucin vacuoles, and were amenable to calcium mobilization after neurotransmitter stimulation with a cholinergic agent. As expected, these secretory functions were diminished in the presence of etoposide mimicking the LG hypofunction often seen in aqueous-deficient DED3. In addition, etoposide increased oxidative stress (i.e. ROS), senescence-associated β-galactosidase activity and apoptosis (i.e. number of apoptotic bodies, Caspase 3 expression levels), indicating the development of a LG model with senescence-associated pathogenesis. Thus, this is the first report showing the development of a senescence-associated LG organoid with impaired function (pathogenesis) and cellular senescence features. Despite such endeavor, the need for other senescence induction approaches triggering other mechanisms would be relevant to better understand the complex aging phenomena. In addition, human DED cannot be fully assessed in porcine organoids because of limited homology in certain DED-related peroxidation genes (< 90%) plus our organoids have limited size (< 300 μm), therefore, these cannot produce tear film on a robust manner for proteomic studies.
Next, to prevent this cellular senescence phenomenon in the LG organoids, a plasmid with the HMGB1-Box A gene was successfully transfected using a well-established lipid-based nanoparticle technology. The prevention of senescence-associated pathogenesis and the functional preservation of fluid secretion in the LG organoid was observed after plasmid transfection of Box A, which protected the organoids against DNA damage and apoptosis. As expected, plasmid transfection efficiency was limited, and viral non-pathogenic gene therapy strategies with more stable and long-term gene expression profiles may constitute a better alternative7. Nevertheless, one can hypothesize that these events possibly occurred due to the generation of DNA gaps and the increase in DNA durability as previously observed by our research team in aging rat models8,9. Thus, the HMGB1-Box A gene therapy may constitute a promising anti-senescence strategy for DED prompted by a hypofunctional LG and may have potential applications in aging-associated chronic diseases affecting other exocrine glands.
M3DB in vitro platforms can generate functional LG organoids as well as hypofunctional LG organoids after DNA-damage induced by etoposide exposure. Such senescence-associated pathogenesis phenotype can be prevented by novel gene therapies with HMGB1-Box A. Future studies with human LG organoids are necessary to confirm these findings when non-pathological LG biopsies become available.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
This research is funded by Thailand Science research and Innovation Fund Chulalongkorn University to JNF, RC and AM. This project is also supported by the National Research Council of Thailand (NRCT) and Chulalongkorn University (Project number: N42A670176) to JNF (main PI), RC (Co-I) and PJ (Co-I), and in part from the Faculty Research Grant (Grant number: DRF66033) from Faculty of Dentistry, Chulalongkorn University. Center of Excellence and Innovation for Oral Health and Healthy Longevity is funded by the Ratchadaphiseksomphot Endowment Fund, Chulalongkorn University. National Science and Technology Development Agency, Thailand (Research Chair Grant, P-19-50189) to AM. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We would like to give a special thanks to Dr. Teerapat Rodboon for performing many of the 3D cell culture experiments and Mr. Somchai Yodsanga from Department of Oral Pathology for the assistance to prepare and stain the organoid sections for histology. Dr. Teerapat Rodboon expressed his wish not to be included in the authorship since he was not able to approve the initial, final and revised version of the manuscript.
Author contributions
J.N.F.: conception and design, data collection and assembly, data curation, analysis and interpretation, validation, visualization, grant funding, project administration, supervision, validation, visualization, original manuscript draft writing, editing and revision of final manuscript. N.B.: Conception and design, performed preliminary transfection studies, data curation, analysis and interpretation, manuscript revision and editing. R.C.: Conception and design of histological studies, analysis and interpretation of micrographs after histological staining, funding acquisition, validation, visualization, manuscript editing; T.V.P., and Y.O.: Performing porcine LG surgical dissections, extraction and isolation of LG primary cells and 2D cell culture for expansion, manuscript editing and revision. P.J.: Funding acquisition, providing access and resources for LG biopsies, manuscript revision and editing. O.M.: Providing resources for plasmid transfection experiments, manuscript revision and editing. A.M.: conception and design, data curation, grant funding, methodology, resources, supervision, original manuscript draft writing, editing and revision of final manuscript. All authors understood the data and provided important contribution to the manuscript. All authors read and approved the final manuscript.
Data availability
Datasets and figures/tables supporting the conclusions of this article are available in the OSF repository: [https://osf.io/8fdx7/?view_only=c0b1d2e859544d26af2491d7ae9d0f52]. The organoid culture protocol described in this article is published on protocols.io [DOI 10.17504/protocols.io.b5ttq6nn].
Declarations
Competing interests
The authors declare no competing interests.
Study approval statement
All methods were carried out in accordance with relevant guidelines and regulations. Only in vitro studies were conducted and all in vitro experimental work was approved by the Institutional Biosafety Committee at Chulalongkorn University Faculty of Dentistry (certificate of notification number: DENT CU-IBC 022/2021, approved on July 19, 2021). No in vivo experiments in live vertebrates were conducted and this work did not involve the use of human specimens. According to the Thai law, tissues that are obtained from postmortem animal pig carcasses can be freely used for research, education or for other means. Hence, neither an ethical application nor a IACUC animal protocol needs to be approved for the extraction of porcine cells. This study is reported in accordance with ARRIVE guidelines (https://arriveguidelines.org).
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.de Paiva, C. S. Effects of aging in dry eye. Int. Ophthalmol. Clin.57, 47–64. 10.1097/iio.0000000000000170 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dogru, M., Kojima, T., Simsek, C. & Tsubota, K. Potential role of oxidative stress in ocular surface inflammation and dry eye disease. Investig Ophthalmol. Vis. Sci.59, DES163–DES168. 10.1167/iovs.17-23402 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Kitazawa, K. et al. Impact of aging on the pathophysiology of dry eye disease: a systematic review and meta-analysis. Ocul Surf.25, 108–118. 10.1016/j.jtos.2022.06.004 (2022). [DOI] [PubMed] [Google Scholar]
- 4.Listed, N. A. The epidemiology of dry eye disease: report of the Epidemiology Subcommittee of the International Dry Eye WorkShop (2007). Ocul Surf.5, 93–107. 10.1016/s1542-0124(12)70082-4 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Choi, W. et al. Expression of lipid peroxidation markers in the tear film and ocular surface of patients with non-sjogren syndrome: potential biomarkers for dry eye disease. Curr. Eye Res.41, 1143–1149. 10.3109/02713683.2015.1098707 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Macri, A. et al. Evaluation of oxidative stress levels in the conjunctival epithelium of patients with or without dry eye, and dry eye patients treated with preservative-free hyaluronic acid 0.15% and vitamin B12 eye drops. Graefes Arch. Clin. Exp. Ophthalmol.253, 425–430. 10.1007/s00417-014-2853-6 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Ferreira, J. N. A. et al. Neurturin gene therapy protects parasympathetic function to prevent irradiation-induced murine salivary gland hypofunction. Mol. Ther. Methods Clin. Dev.9, 172–180. 10.1016/j.omtm.2018.02.008 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yasom, S. et al. The roles of HMGB1-produced DNA gaps in DNA protection and aging biomarker reversal. FASEB Bioadv. 4, 408–434. 10.1096/fba.2021-00131 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watcharanurak, P. & Mutirangura, A. Genome wide hypomethylation and youth-associated DNA gap reduction promoting DNA damage and senescence-associated pathogenesis. Med. Res. Arch.. 11. 10.18103/mra.v11i12.4952 (2023).
- 10.Hu, J. L., Todhunter, M. E., LaBarge, M. A. & Gartner, Z. J. Opportunities for organoids as new models of aging. J. Cell. Biol.217, 39–50. 10.1083/jcb.201709054 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bucolo, C. et al. Antioxidant and osmoprotecting activity of taurine in dry eye models. J. Ocul Pharmacol. Ther.34, 188–194. 10.1089/jop.2017.0008 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Bannier-Helaouet, M. et al. Exploring the human lacrimal gland using organoids and single-cell sequencing. Cell Stem Cell 28, 1221–1232 e1227 (2021). 10.1016/j.stem.2021.02.024 [DOI] [PubMed]
- 13.Bannier-Helaouet, M., Geurts, M. H., Korving, J., Begthel, H. & Clevers, H. Establishment, maintenance, differentiation, genetic manipulation, and transplantation of mouse and human lacrimal gland organoids. J. Vis. Exp.10.3791/65040 (2023). [DOI] [PubMed] [Google Scholar]
- 14.Rodboon, T., Souza, G. R., Mutirangura, A. & Ferreira, J. N. Magnetic bioassembly platforms for establishing craniofacial exocrine gland organoids as aging in vitro models. PLoS One. 17, e0272644. 10.1371/journal.pone.0272644 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodboon, T., Yodmuang, S., Chaisuparat, R. & Ferreira, J. N. Development of high-throughput lacrimal gland organoid platforms for drug discovery in dry eye disease. SLAS Discov. 27, 151–158. 10.1016/j.slasd.2021.11.002 (2022). [DOI] [PubMed] [Google Scholar]
- 16.Adine, C., Ng, K. K., Rungarunlert, S., Souza, G. R. & Ferreira, J. N. Engineering innervated secretory epithelial organoids by magnetic three-dimensional bioprinting for stimulating epithelial growth in salivary glands. Biomaterials. 180, 52–66. 10.1016/j.biomaterials.2018.06.011 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Urkasemsin, G., Rungarunlert, S. & Ferreira, J. N. Bioprinting strategies for secretory epithelial organoids. Methods Mol. Biol.2140, 243–249. 10.1007/978-1-0716-0520-2_16 (2020). [DOI] [PubMed] [Google Scholar]
- 18.Chansaenroj, A. et al. Magnetic bioassembly platforms towards the generation of extracellular vesicles from human salivary gland functional organoids for epithelial repair. Bioact Mater.18, 151–163. 10.1016/j.bioactmat.2022.02.007 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sheekey, E. & Narita, M. p53 in senescence - it’s a marathon, not a sprint. FEBS J.290, 1212–1220. 10.1111/febs.16325 (2023). [DOI] [PubMed] [Google Scholar]
- 20.Jeong, S. Y. et al. Establishment of functional epithelial organoids from human lacrimal glands. Stem Cell. Res. Ther.12, 247. 10.1186/s13287-021-02133-y (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayashi, R. et al. Generation of 3D lacrimal gland organoids from human pluripotent stem cells. Nature. 605, 126–131. 10.1038/s41586-022-04613-4 (2022). [DOI] [PubMed] [Google Scholar]
- 22.Wiebe-Ben Zakour, K. E. et al. Enhancement of lacrimal gland cell function by decellularized lacrimal gland derived hydrogel. Biofabrication. 10.1088/1758-5090/ad2082 (2024). [DOI] [PubMed] [Google Scholar]
- 23.Zhao, Z. et al. Organoids. Nat. Rev. Methods Primers. 2. 10.1038/s43586-022-00174-y (2022). [DOI] [PMC free article] [PubMed]
- 24.Hofer, M. & Lutolf, M. P. Engineering organoids. Nat. Rev. Mater.6, 402–420. 10.1038/s41578-021-00279-y (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets and figures/tables supporting the conclusions of this article are available in the OSF repository: [https://osf.io/8fdx7/?view_only=c0b1d2e859544d26af2491d7ae9d0f52]. The organoid culture protocol described in this article is published on protocols.io [DOI 10.17504/protocols.io.b5ttq6nn].