

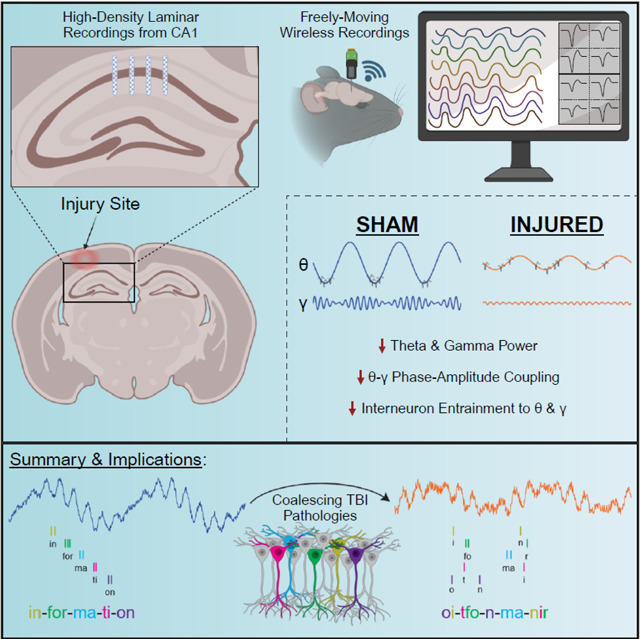
SUMMARY
Traumatic brain injury (TBI) often results in persistent learning and memory deficits, likely due to disrupted hippocampal circuitry underlying these processes. Precise temporal control of hippocampal neuronal activity is important for memory encoding and retrieval and is supported by oscillations that dynamically organize single unit firing. Using high-density laminar electrophysiology, we discovered a loss of oscillatory power across CA1 lamina, with a profound, layer-specific reduction in theta-gamma phase amplitude coupling in injured rats. Interneurons from injured animals were less strongly entrained to theta and gamma oscillations, suggesting a mechanism for the loss of coupling, while pyramidal cells were entrained to a later phase of theta. During quiet immobility, we report decreased ripple amplitudes from injured animals during sharp-wave ripple events. These results reveal deficits in information encoding and retrieval schemes essential to cognition that likely underlie TBI-associated learning and memory impairments, and elucidate potential targets for future neuromodulation therapies.
Graphical abstract
INTRODUCTION
Cognitive deficits, including learning and memory impairments, are one of the most common consequences of TBI1–3. These deficits occur when complex, heterogeneous TBI pathologies affect circuits that support these processes. Local circuits within the hippocampus are especially important for learning and memory, as are interactions between the hippocampus and connected brain regions. Because these networks are dispersed, it is not surprising that TBI-associated learning and memory deficits have been reported both in the presence and absence of hippocampal pathology4–7. Neuronal pathology in hippocampal afferents such as the medial septum and para-hippocampal cortical regions can affect hippocampal processing supporting cognition, as can axonal damage along connecting pathways8–12. Additionally, other coalescing pathological changes such as neurotransmitter dysregulation13–15, inflammation16,17, blood brain barrier breakdown18,19, and metabolic dysfunction20–23, as well as changes in intrinsic neuronal and circuit excitability and synaptic plasticity16,24–34 are all thought to contribute to hippocampal dysfunction following TBI. However, it is unclear how these pathological changes collectively affect information processing in the intact hippocampus. Studying this process in awake subjects is important since network level physiological processes in the hippocampus have been shown to support learning and memory functions, and these electrophysiological processes can be predictive of animal behavior35–39.
Despite our understanding that hippocampal neuronal activity underlies many aspects of learning and memory, surprisingly few studies have investigated how TBI affects these processes in awake animals. Prior studies have reported a loss of oscillatory power in CA1 or broad changes in the firing properties of pyramidal cells post injury5,40–45. However, no studies have examined how TBI affects the timing of single unit activity in relation to local oscillations, a process called spike-field coherence or entrainment. This process is important as hippocampal oscillations dynamically organize the firing of populations of neurons into cell ensembles, and the precise temporal control of cell ensemble firing is important for plasticity mechanisms underlying learning and memory46–49. Hippocampal oscillations coordinate ensembles in a complex manner, nesting high frequency gamma oscillations within a dominant lower frequency theta oscillation during active exploration50,51. Theta oscillations are important for synchronizing neuronal activity both within and across brain structures52,53, while gamma oscillations are thought to reflect local processing within a brain region and are supported by local interneurons54. Importantly the amplitude of gamma oscillations changes as a function of theta phase, a process known as theta-gamma phase amplitude coupling (PAC), which provides a mechanism whereby local processing across distributed networks can be temporally coordinated on a theta timescale55–60. Appropriate PAC and single unit entrainment to hippocampal oscillations are thought to be essential for cognition and have been directly linked to memory performance in humans60–62. Additionally, higher frequency sharp-wave ripples (SWRs) are known to support working memory processes and memory consolidation63–67, however, the effects of TBI on these processes have not been investigated.
We therefore utilized high-density laminar electrodes to characterize layer-specific pathophysiological changes to hippocampal oscillations and single unit activity from freely moving rats following the well-characterized lateral fluid percussion injury (LFPI) model of TBI. We found that injured rats had decreased oscillatory power, PAC, and ripple amplitudes in CA1 compared to controls. Additionally, we report changes in single unit entrainment to theta and gamma oscillations post-injury. These results reveal TBI-associated coding disruptions to processes that are fundamental to cognitive processing in the hippocampus. These disruptions likely underlie learning and memory deficits following TBI and can provide physiological targets for future neuromodulation therapies.
RESULTS
LFPI decreases theta and gamma power across CA1 lamina
Hippocampal recordings were obtained from freely moving rats to characterize differences in CA1 physiology in freely moving sham (n=4) and injured (n=5) animals. Rats were subjected to LFPI (1.68–1.78 atm) or sham surgery then chronically implanted with multi-shank, high-density laminar electrodes in the CA1 region of the hippocampus ipsilateral to the injury/sham site. Recordings were obtained on three separate days within 6–11 days post-injury. On the first day of recording rats freely explored a familiar environment, followed by a novel environment. On the subsequent two recording days, electrodes were slowly advanced, then recordings were obtained in the familiar environment. The use of laminar electrodes allowed us to localize electrode contacts across different CA1 lamina specifically stratum oriens, pyramidale (st. pyr), and radiatum (st. rad). To maximize single unit yields, most electrodes were localized to st. oriens and st. pyr on the first day thus driving on subsequent days allowed us to reach st. rad.
For each recording, electrodes from individual shanks were independently assessed to determine if they were localized to st. pyr or st. rad. The st. pyr channel was defined as the channel with maximum power in the ripple frequency range (100–250 Hz; Fig 1A). As expected, individual pyramidal cells were localized around this channel (Fig 1B). Figure 1C shows the mean SWR waveform and associated current source density (CSD) recorded from this shank. The st. rad channel was defined as the local maximum of the CSD sink (not present in Fig 1C but visible in Fig 1E). Due to the curvature of the hippocampus and position of our electrode array, each shank sampled a slightly different depth of the CA1 lamina, but recordings could be localized based on physiological properties such as ripple frequency power, SWR CSD, and single unit locations (Fig 1D).
Figure 1: LFPI decreases theta and gamma power across CA1 lamina.

(A) Silicon probe recording from dorsal CA1. Schematic of electrode shank widened for visualization but accurately scaled along CA1 lamina. The peak of ripple frequency (100–250 Hz) power across channels was used to define the st. pyr channel (marked with *). Channels within ±80 μm of the defined st. pyr channel were assigned to the pyramidal cell layer (denoted by bracket).
(B) Mean waveforms from 6 single units identified as putative pyramidal cells recorded on the shank shown in A across all electrode contacts.
(C) Mean SWR waveform from the shank shown in A and its associated current source density (CSD)
(D) Schematic of an electrode array with shanks positioned along the proximal-distal axis of CA1. Heatmap contours show the mean SWR CSD for each shank. Normalized ripple frequency power and edges of the pyramidal cell layer are shown as solid and dashed lines respectively for each shank. Individual recorded units are shown at the depth of the channel containing their max amplitude (putative cell types: interneuron=magenta circle, pyramidal cell=green triangle, unclassified=yellow square). Shank 2 shown in A-C.
(E) Example recording from a shank spanning both st. pyr and st rad. The peak of ripple frequency power occurs at channel 3 (identified as pyr.). The st. rad channel was defined as the channel closest to the peak sink in the CSD of the mean SWR waveform (channel 13 denoted by rad.). Note theta (5–10 Hz) and gamma (30–59 Hz) oscillations especially apparent in st. rad.
(F) Power spectra (mean±SEM) from st. pyr (top) and st. rad (bottom) for sham and injured rats moving (>10 cm/sec) in the familiar environment. Theta (5–10 Hz) and gamma (30–59 Hz) bands denoted by dashed lines. Statistically significant differences between sham and injured are outlined in gray (p<0.01 for multiple comparisons; t-test).
(G) Integral of power in the theta and gamma frequency bands (mean±SEM; individual animals labeled by points) from sham and injured rats in st. pyr (top; theta: sham=−7.65±0.09 log10V2, injured=−8.38±0.10 log10V2, p=0.001; gamma: sham=−7.64±0.05 log10V2, injured=−7.90±0.04 log10V2, p=0.009; t-test) and st. rad (bottom; theta: sham=−7.15±0.04 log10V2, injured=−7.51±0.07 log10V2, p=0.001; gamma: sham=−7.20±0.05 log10V2, injured=−7.42±0.07 log10V2, p=0.022; t-test).
After determining electrode positions, we sought to compare hippocampal oscillatory power between sham and injured rats. Theta and gamma oscillations have been well-characterized in the rat hippocampus and can be readily observed while rats are actively moving and exploring the environment50,55. Importantly, these oscillations change in power and phase along the CA1 lamina (Fig 1E), thus it is essential to localize electrodes to specific layers before comparing across animals or groups. To compare oscillatory power between sham and injured rats, power spectra were computed from channels localized to st. pyr or st. rad while rats were actively moving (>10 cm/sec). Power spectra were extracted in the familiar environment, averaged across shanks and recording days to obtain a mean for each animal, then averaged across sham and TBI conditions (Fig 1F). In st. pyr, injured rats had decreased power from 3.5–85.1 Hz, and in st. rad, they had decreased power from 7.1–12.7 Hz, 13.7–19.7 Hz, 30.1–33.8 Hz, and 35.1–86.8 Hz (t-test; p<0.01 for multiple comparisons).
Data were then separated into well-described, physiologically relevant theta (5–10 Hz) and low-gamma (30–59 Hz) bands and power in these bands was compared across sham and TBI rats in both st. pyr and st. rad (Fig 1G). In st. pyr of TBI rats, both theta and low-gamma power were significantly decreased compared to shams (sham theta=−7.65±0.09 log10V2, injured theta=−8.38±0.10 log10V2, p=0.001; sham gamma=−7.64±0.05 log10V2, injured gamma=−7.90±0.04 log10V2, p=0.009; mean±SEM; t-test). In st. rad, both theta and low-gamma power were also significantly decreased in TBI rats compared to shams (sham theta=−7.15±0.04 log10V2, injured theta=−7.51±0.07 log10V2, p=0.001; sham gamma=−7.20±0.05 log10V2, injured gamma=−7.42±0.07 log10V2, p=0.022; mean±SEM; t-test). Overall, injured rats have decreased oscillatory power spanning a range of frequencies that includes theta and gamma, and this loss of power is more pronounced in st. pyr than in st. rad.
LFPI decreases theta-gamma PAC in st. pyr but not st. rad
Theta-gamma PAC (Fig 2A) is a well-characterized phenomenon important for the selection and coordination of cell ensembles that support spatial coding and memory processes56,59,60. To assess the effects of TBI on theta-gamma PAC, we computed the modulation index68 across discrete frequency bands during epochs in which the rat was moving (>10cm/sec) in the familiar environment, then constructed PAC heatmaps for channels localized to st. pyr and st rad (Fig 2B). Sham rats showed strong theta-gamma PAC in the pyramidal cell layer which was drastically reduced in injured rats. This reduction was specific to st. pyr, as the strength of PAC in st. rad was similar between sham and injured rats, though there was a slight shift in coupling to a lower frequency phase and amplitude. Both the loss of PAC strength in st. pyr and the frequency shift in st. rad can be visualized by subtracting PAC heatmaps of injured animals from sham animals (Fig 2C).
Figure 2: LFPI decreases theta-gamma phase-amplitude coupling (PAC) in st. pyr but not st. rad.

(A) Visualization of theta-gamma PAC. Raw 3 sec trace recorded from st. pyr (top), the same trace filtered for theta (5–10 Hz; middle) and for gamma (30–59 Hz; bottom). Envelope amplitude of gamma is plotted in red and peaks in gamma amplitude >1SD above mean are marked with a dashed line. Note that most peaks align to a similar phase of theta (~90°).
(B) Averaged PAC heatmap contours from sham (left) and injured (right) animals in st. pyr (top) and st. rad (bottom).
(C) Difference between sham and injured PAC heatmap contours in both st. pyr (top) and st. rad (bottom).
(D) PAC modulation index values from broadband theta and gamma filtered signals (mean±SEM; individual animals labeled by points) in st. pyr (top; sham=0.00130±0.00016, injured=0.00051±0.00012, p=0.006, t-test) and st. rad (bottom; sham=0.00121±0.00032, injured=0.00145±0.00032, p=0.623, t-test).
(E) Normalized gamma amplitudes across theta phase bins (2 cycles of theta shown in black for reference). Peak gamma in st. pyr (sham=89.2°, injured=115.2°, Δ=26°) and st. rad (sham=200.0°, injured=255.4°, Δ=55.4°).
To quantify differences in theta-gamma PAC between sham and injured rats, we extracted the phase and amplitude from broadband theta (5–10 Hz) and low-gamma (30–59 Hz) filtered signals respectively and compared the magnitude and phase preference of PAC. Similar to the heatmaps in Figures 2B and C, the magnitude of theta-gamma PAC in injured rats was reduced in st. pyr (sham=0.00130±0.00016, injured=0.00051±0.00012, p=0.006; mean±SEM; t-test) but not in st. rad (sham=0.00121±0.00032, injured=0.00145±0.00032, p=0.623; mean±SEM; t-test; Fig 2D). Additionally, the maximal amplitude of gamma was coupled to a later phase of theta in injured rats compared to shams in both st. pyr (sham=89.2°, injured=115.2°, Δ=26.0°) and st. rad (sham=200.0°, injured=255.4°, Δ=55.4deg; Fig 2E). Taken together, injured rats show a decrease in the strength of theta-gamma PAC that is specific to st. pyr, and a shift in peak gamma amplitude to a later phase of theta in both st. pyr and st. rad.
LFPI minimally affects single unit firing rates but increases recruitment of pyramidal cells
To compare neuronal firing properties between sham and injured animals we isolated single units (see methods) then clustered them into one of 3 categories: putative pyramidal cells, putative interneurons, and unclassified (putative identifiers henceforth removed for writing clarity). Cells within ±80 μm of the defined st. pyr channel were manually clustered based on each unit’s spike width, firing rate, and 1st moment of the autocorrelogram (a measure of burstiness) as previously described69 (Fig 3A,B). Pyramidal cells typically have larger spike widths, lower firing rates and are more bursty (lower 1st moment of the autocorrelogram) compared to interneurons (Fig 3A,B). Importantly, there was a 95.47% overlap between manual clustering and automated k-means consensus clustering of cells within ±80 μm of the defined st. pyr channel when unclassified cells were excluded from the comparison (Sup Fig 1). We chose to use manual clustering because the inclusion of the unclassified group allowed for a more conservative classification, and manual clustering is more robust to outliers than automated clustering (see Sup Fig 1). Cells located >80 μm above the defined st. pyr channel were classified as interneurons as anatomically these cells would be located in st. oriens. When the location of each shank was centered to the defined st. pyr channel, cells are visibly distributed into the distinct st. oriens and st. pyr cell layers (Fig 3C). Importantly, cells classified as interneurons based on their anatomical location had firing properties that clustered with other interneurons further supporting their classification (Sup Fig 2).
Figure 3: LFPI minimally affects single unit firing rates but increases recruitment of pyramidal cells.

(A) Waveforms (mean±SD) from an example putative pyramidal cell (top) and interneuron (bottom) and their associated autocorrelograms. W=width, FR=firing rate
(B) Scatterplot of spike width, first moment of the autocorrelogram, and firing rate of all cells located within the pyramidal cell layer from both sham and injured animals. Note separation of putative pyramidal cells and interneurons with unclassified cells typically lying between these 2 populations.
(C) Location of all cells relative to the defined st. pyr channel (based on peak ripple frequency power). Note the peak of the pyramidal cell distribution is located on the defined st. pyr channel, and the bimodal distribution of interneurons reflects populations localized to st. oriens and st. pyr.
(D) Left: firing rates (mean±SEM) of putative interneurons in the familiar and novel environments across sham and injured animals (familiar: sham=20.3±3.8 Hz, n=20, injured=13.0±1.3 Hz, n=88, p=0.116; novel: sham=17.4±3.3 Hz, n=19, injured=12.6±1.4 Hz, n=89, p=0.185; ks-test). Right: cumulative distributions of interneuron firing rates.
(E) Left: firing rates (mean±SEM) of putative pyramidal cells in the familiar and novel environments across sham and injured animals (familiar: sham=1.63±0.23 Hz, n=51, injured=2.11±0.20 Hz, n=141, p=0.446; novel sham=1.84±0.18 Hz, n=55, injured=2.23±0.21 Hz, n=134, p=0.170, ks-test). Right: cumulative distributions of pyramidal cell firing rates.
(F) Recruitment of putative pyramidal cells across environments (sham=74%, injured=87%, p=0.025, Fisher’s exact test).
After clustering individual units, we compared the firing rates of active (firing rate >0.1Hz) pyramidal cells and interneurons in the familiar and novel environment. There were no significant differences in the firing rate of interneurons (sham familiar=20.3±3.8 Hz, n=20, injured familiar=13.0±1.3 Hz, n=88, p=0.116; sham novel=17.4±3.3 Hz, n=19, injured novel=12.6±1.4 Hz, n=89, p=0.185; mean±SEM; ks-test; Fig 3D) or pyramidal cells (sham familiar=1.63±0.23 Hz, n=51, injured familiar=2.11±0.20 Hz, n=141, p=0.446; sham novel=1.84±0.18 Hz, n=55, injured novel=2.23±0.21 Hz, n=134, p=0.170; mean±SEM; ks-test; Fig 3E) between sham and injured groups, but a higher percentage of pyramidal cells were active (firing rate >0.1Hz) in both the familiar and novel environment in injured rats compared to shams (sham=74%, injured=87%, p=0.025, Fisher’s exact test; Fig 3F). While the firing rates of each cell type were similar across groups, the higher percentage of pyramidal cells active in both environments in injured animals suggests a possible over-recruitment of pyramidal cells across distinct conditions.
LFPI disrupts spike field coherence and impairs single unit entrainment to theta oscillations
To assess potential mechanisms for the loss of oscillatory power and PAC in injured rats, we investigated whether there were changes in single unit entrainment to local oscillations. Many CA1 neurons are entrained to local oscillations and are more likely to fire around a specific phase of the oscillation (example of an interneuron entrained to theta shown in Fig 4A). Proper entrainment of cells is essential for temporal coding supporting learning and memory functions of the hippocampus. We first assessed entrainment strength by calculating the mean vector length (MVL) of spike times across a range of discrete frequency bands in st. pyr for interneurons (Fig 4B) and pyramidal cells (Fig 4D) while the rat was either still (<10 cm/sec) or moving (>10 cm/sec). Clear peaks in entrainment can be seen in the theta frequency range for both neuron subtypes.
Figure 4: LFPI disrupts spike field coherence to hippocampal oscillations.

(A) Example 2 sec recording trace containing a large amplitude interneuron entrained to theta (all spikes are from the same unit). Right: probability of spike times across theta phase bins (line shows 2 cycles of theta for reference).
(B) Entrainment strength of all interneurons across frequencies in both environments while the rat was still <10 cm/sec; top) or moving (>10 cm/sec; bottom).
(C) Percent of interneurons significantly entrained to theta (5–10 Hz) and gamma (30–59 Hz) while animals were still or moving (theta still: sham=97.44%, injured=91.53%, p=0.315; theta moving: sham=97.44%, injured=91.53%, p=0.315; gamma still: sham=89.74%, injured=67.80%, p=0.006; gamma moving: sham=89.74%, injured=65.54%, p=0.002; Fisher’s exact test).
(D) Entrainment strength of all pyramidal cells across frequencies in both environments while the rat was still (top) or moving (bottom).
(E) Percent of pyramidal cells significantly entrained to theta (5–10 Hz) and gamma (30–59 Hz) while animals were still or moving (theta still: sham=71.57%, injured=67.17%, p=0.454; theta moving: sham=58.82%, injured=65.28%, p=0.277; gamma still: sham=25.49%, injured=37.74%, p=0.028; gamma moving: sham=29.41%, injured=30.19%, p>0.999; Fisher’s exact test).
To investigate single unit entrainment to physiologically relevant hippocampal oscillations, we calculated the percentage of cells that were significantly entrained to broadband filtered theta (5–10 Hz) and gamma (30–59 Hz) oscillations in st. pyr using circular shuffling (see methods). There was no difference in the percentage of interneurons significantly entrained to theta between sham and injured rats when animals were still (sham=97.44%, injured=91.53%, p=0.315, Fisher’s exact test) or moving (sham=97.44%, injured=91.53%, p=0.315, Fisher’s exact test), but a lower percentage of interneurons were entrained to gamma in injured rats compared to shams both while animals were still (sham=89.74%, injured=67.80%, p=0.006, Fisher’s exact test) and moving (sham=89.74%, injured=65.54%, p=0.002, Fisher’s exact test; Fig 4C). For pyramidal cells, there was no difference in the percentage of cells significantly entrained to theta while rats were still (sham=71.57%, injured=67.17%, p=0.454, Fisher’s exact test) or moving (sham=58.82%, injured=65.28%, p=0.277, Fisher’s exact test); however, a larger percentage of pyramidal cells were significantly entrained to gamma in injured rats compared to shams while animals were still (sham=25.49%, injured=37.74%, p=0.028, Fisher’s exact test), but not while they were moving (sham=29.41%, injured=30.19%, p>0.999, Fisher’s exact test; Fig 4E).
Because a similar percentage of interneurons were significantly entrained to theta across sham and injured animals, we next investigated the apparent reduction in entrainment strength of interneurons in the theta frequency band seen in Fig 4B. To quantify this, we extracted only cells that were significantly entrained to theta and compared their entrainment strength and phase preference across all conditions. Figure 5A shows entrainment strength cumulative distributions for interneurons significantly entrained to theta. Interneurons were less entrained to theta in injured rats compared to shams in both the familiar (sham=0.31±0.03, n=19, injured=0.21±0.01, n=83, p=0.004; mean±SEM; ks-test) and novel (sham=0.27±0.03, n=19, injured=0.20±0.01, n=79, p=0.026; mean±SEM; ks-test) environment while rats were moving (Fig 5B). Additionally, the MVL of interneurons drastically increased when injured rats were moving compared to still (familiar: still=0.14±0.01, n=79, moving=0.21±0.01, n=83, p<0.001; novel: still=0.13±0.01, n=83, moving=0.20±0.01, n=79, p<0.001; mean±SEM; ks-test), which did not occur in sham rats (familiar: still=0.27±0.02, n=19, moving=0.31±0.03, n=19, p=0.462; novel: still=0.23±0.02, n=19, moving=0.27±0.03, n=19, p=0.462; mean±SEM; ks-test; Fig 5B). The mean angle of entrainment for interneurons significantly entrained to theta was not different between sham and injured rats in either environment while the rat was moving (familiar: sham=173.7°, injured=196.6°, p=0.446; novel: sham=167.6°, injured=190.2°, p=0.443; circular Kruskal-Wallis test; Fig 5C).
Figure 5: LFPI impairs single unit entrainment to theta oscillations.

(A) Cumulative distributions of all interneurons significantly entrained to theta across all conditions.
(B) Left: average entrainment (mean±SEM) of interneurons significantly entrained to theta while rats were moving in the familiar (sham=0.31±0.03, n=19, injured=0.21±0.01, n=83, p=0.004, ks-test) and novel (sham=0.27±0.03, n=19, injured=0.20±0.01, n=79, p=0.026, ks-test) environment. Right: average entrainment of interneurons while rats were still or moving in each environment (sham familiar: still=0.27±0.02, n=19, moving=0.31±0.03, n=19, p=0.462; sham novel: still=0.23±0.02, n=19, moving=0.27±0.03, n=19, p=0.462; injured familiar: still=0.14±0.01, n=79, moving=0.21±0.01, n=83, p<0.001; injured novel: still=0.13±0.01, n=83, moving=0.20±0.01, n=79, p<0.001; ks-test).
(C) Polar histograms showing the mean angle of entrainment for all significantly entrained interneurons while animals were moving in the familiar or novel environment (familiar: sham=173.7°, injured=196.6°, p=0.446; novel: sham=167.6°, injured=190.2°, p=0.443; circular Kruskal-Wallis test).
(D) Cumulative distributions of pyramidal cells significantly entrained to theta across all conditions.
(E) Left: average entrainment (mean±SEM) of pyramidal cells significantly entrained to theta while rats were moving in the familiar (sham=0.19±0.02, n=25, injured=0.20±0.01, n=89, p=0.518, ks-test) and novel
(sham=0.16±0.01, n=35, injured=0.19±0.01, n=84, p=0.191, ks-test) environment. Right: average entrainment of pyramidal cells while rats were still or moving in each environment (sham familiar: still=0.18± 0.02, n=35, moving=0.19±0.02, n=25, p=0.780; sham novel: still=0.15±0.01, n=38, moving=0.16±0.01, n=35, p=0.908; injured familiar: still=0.12±0.01, n=85, moving=0.2±0.01, n=89, p<0.001; injured novel: still=0.14±0.01, n=93, moving=0.19±0.01, n=84, p<0.001; ks-test).
(F) Polar histograms showing the mean angle of entrainment for all significantly entrained pyramidal cells while animals were moving in the familiar or novel environment (familiar: sham=197.4, injured=250.1, p=0.003; novel: sham=214.5, injured=267.9, p<0.001: circular Kruskal-Wallis test).
Figure 5D shows entrainment strength cumulative distributions for pyramidal cells significantly entrained to theta. There were no differences in pyramidal cell MVL between sham and injured groups in either the familiar (sham=0.19±0.02, n=25, injured=0.20±0.01, n=89, p=0.518; mean±SEM; ks-test) or novel (sham=0.16±0.01, n=35, injured=0.19±0.01, n=84, p=0.191; mean±SEM; ks-test) environment while rats were moving (Fig 5E). Like interneurons, pyramidal cells in injured animals had drastically increased MVLs when rats were moving compared to when they were still (familiar: still=0.12±0.01, n=85, moving=0.2±0.01, n=89, p<0.001; novel: still=0.14±0.01, n=93, moving=0.19±0.01, n=84, p<0.001; mean±SEM; ks-test) which again did not occur in sham rats (familiar: still=0.18±0.02, n=35, moving=0.19±0.02, n=25, p=0.780; novel: still=0.15±0.01, n=38, moving=0.16±0.01, n=35, p=0.908; mean±SEM; ks-test; Fig 5E). Additionally, the mean angle of entrainment for pyramidal cells significantly entrained to theta was shifted to a later phase in injured rats compared to shams in both environments while the rat was moving (familiar: sham=197.4, injured=250.1, p=0.003; novel: sham=214.5, injured=267.9, p<0.001; circular Kruskal-Wallis test; Fig 5F).
Overall, the percentage of interneurons significantly entrained to theta was similar across sham and injured animals, but the entrainment strength of significantly entrained interneurons was decreased in injured rats. Additionally, a lower percentage of interneurons were significantly entrained to gamma in injured animals compared to shams. For pyramidal cells, the percentage of cells significantly entrained to theta was similar between sham and injured animals as was the entrainment strength of significantly entrained pyramidal cells. However, pyramidal cells were entrained to a later phase of theta in injured rats. In both interneurons and pyramidal cells, theta entrainment strength drastically increased when rats began moving in injured but not sham animals. Together, these results reveal disruptions to temporal coding in the hippocampus presumed to underlie information encoding and retrieval.
LFPI decreases SWR amplitude
CA1 SWRs are highly coordinated events implicated in memory consolidation and planning behaviors64. These events require precise synchronization of distributed cell populations resulting in a sharp-wave sink in st. rad with an associated ripple oscillation (150–250 Hz) in st. pyr63. The decrease in power, PAC, and entrainment in injured animals suggests an overall desynchronization of hippocampal activity in the active state, thus we evaluated whether TBI also disrupts highly coordinated SWRs in CA1. We used time-frequency decomposition as described previously70 to detect ripple events, and manually verified all candidate events that coincided with a sharp-wave (see methods). Figure 6A shows an example SWR event along with its time-frequency map. We found that the vast majority of detected SWR events occurred while animals were still (sham: familiar=96.7±0.7%, novel=97.5±0.3%; injured: familiar=97.9±0.7%, novel=96.9±0.7%; mean±SEM; Fig 6B), which is expected, as SWRs are known to preferentially occur during awake immobility63,64,70.
Figure 6: LFPI decreases SWR amplitude.

(A) Example SWR event recorded across all electrode contacts from a single shank (st. pyr channel labeled and colored blue), the associated time-frequency map from the st. pyr channel (red contour outlines where ripple event exceeds amplitude threshold; white dashed lines mark edges of ripple event in time), and the frequency-amplitude spectrum of the event averaged over time between white dashed lines
(B) Percentage of SWRs that were detected while the animal was still (sham: familiar=96.7±0.7%, novel=97.5±0.3%, injured: familiar=97.9±0.7%, novel=96.9±0.7%; mean±SEM; points represent individual animals).
(C) SWR event rate normalized to the amount of time animals were still (sham: familiar=0.26±0.03 Hz, novel=0.56±0.05 Hz, p=0.011; injured: familiar=0.36±0.03 Hz, novel=0.52±0.03 Hz, p=0.052; t-test; mean±SEM; points represent individual animals).
(D) Average (mean±SEM) frequency-amplitude spectrum of all ripples across conditions.
(E) Left: cumulative distributions of ripple amplitudes across conditions (familiar: sham=91.2±1.7 mV, n=490, injured=73.3±0.9 mV, n=1082, p<0.001; novel: sham=126.2±1.6 mV, n=1285, injured=98.4±1.3 mV, n=1464, p<0.001; sham still vs moving: p<0.001; injured still vs moving: p<0.001; mean±SEM; ks-test). Right: cumulative distributions of ripple durations across conditions (familiar: sham=34.3±0.9 ms, injured=34.7±0.6 ms, p=0.019; novel: sham=36.9±0.5 ms, injured=38.5±0.5 ms, p=0.062; sham still vs moving: p<0.001; injured still vs moving: p<0.001; mean±SEM; ks-test).
To evaluate whether TBI disrupts ripple generation, we assessed SWR event frequency between sham and injured rats across environments normalized to the total amount of time the rats were still (Fig 6C). While there were no significant differences between sham and injured animals, the rate of SWR events was significantly higher in the novel compared to the familiar environment in sham but not injured animals (sham: familiar=0.26±0.03 Hz, novel=0.56±0.05 Hz, p=0.011; injured: familiar=0.36±0.03 Hz, novel=0.52±0.03 Hz, p=0.052; mean±SEM; t-test), likely due to the slightly higher frequency of SWR events in injured animals in the familiar environment compared to shams.
Figure 6D shows the spectral profile of ripples detected in st. pyr across conditions. Ripple amplitudes were significantly larger in sham compared to injured rats across both environments (familiar: sham=91.2±1.7 mV, n=490, injured=73.3±0.9 mV, n=1082, p<0.001; novel: sham=126.2±1.6 mV, n=1285, injured=98.4±1.3 mV, n=1464, p<0.001; mean±SEM; ks-test; Fig 6E). Additionally, ripple amplitudes were significantly larger in the novel compared to the familiar environment in both sham and injured animals (sham: p<0.001; injured: p<0.001, ks-test). Ripple durations were slightly longer in injured rats compared to shams in the familiar but not the novel environment (familiar: sham=34.3±0.9 ms, injured=34.7±0.6 ms, p=0.019; novel: sham=36.9±0.5 ms, injured=38.5±0.5 ms, p=0.062; mean±SEM; ks-test; Fig 6E); however, ripple durations were significantly longer in the novel environment compared to the familiar in both sham and injured rats (sham: p<0.001; injured: p<0.001, ks-test). Overall, ripples were more frequent and had larger amplitudes and durations in the novel environment compared to familiar in both groups; however, ripples in injured rats were smaller compared to shams.
LFPI disrupts spatial memory and induces stereotypic pathologies
The LFPI model of TBI in rats is known to produce spatial memory deficits and stereotypic patterns of pathology71–75, however, LFPIs can vary significantly across studies. Injury severity can be titrated based on the intensity of the fluid pulse generated, but other factors such as the precise placement of the craniectomy as well as differences in surgical technique and injury devices used can all contribute to LFPI heterogeneity76,77. Because LFPIs are not always standardized, we assessed whether LFPIs from this study produced pathology in line with previously published results from this model and reproduced spatial memory deficits43,74.
Spatial memory following TBI was assessed in a separate cohort of rats using the Morris Water Maze (MWM) as described previously74 (see methods). Animals were trained to find a submerged hidden platform in a pool then were subjected to a LFPI (1.34–1.80 atm) or sham surgery. Two days later, rats were placed back into the pool to test their memory of the platform location. On test day, injured rats had a lower memory score than shams (sham=114.8±21.8, n=9; injured=51.5±6.8, n=14; p=0.020; mean±SEM; Welch’s t-test) indicating poor spatial memory (Sup Fig 3A).
A subset of 1 sham and 2 injured rats were sacrificed immediately after testing in the MWM (at 48hrs post LFPI/sham surgery) to verify that pathology generated in this study was in line with previously published results. Injured animals demonstrated multifocal hemorrhagic lesions including within the corpus callosum directly underlying the impact site, the angular bundle, fimbria-fornix and the lateral temporoparietal cortex (Sup Fig 3B). In one animal the temporoparietal hemorrhage extended into the lateral aspect of the ipsilateral hippocampus. APP immunoreactive axonal pathology was also observed consistently post-injury. Injured axons displayed abnormal morphologies including varicose swellings and axon bulbs indicative of transport interruption78–81 (Sup Fig 3C,D). Consistent with prior descriptions of the model43, axonal pathology was observed in a multifocal distribution post-injury including within the perilesional cortex, corpus callosum, thalamus, angular bundle (Sup Fig 3C), and fimbria-fornix (Sup Fig 3D). Notably, the fimbria-fornix and angular bundle serve as two major input/output pathways connecting the hippocampus to other structures. Degenerating cells, identified via Fluoro-Jade C staining were observed within the peri-lesional cortex and thalamus (Sup Fig 3F). While cell degeneration was also identified in the dentate granule cell layer in both animals, no cell degeneration was observed in CA1 (Sup Fig 3G) or hilus of the hippocampus, or entorhinal cortex. In one injured animal, minimal neuronal cell death was observed in CA2–3. No axonal pathology, cell death or hemorrhage was observed in the sham animal in any region examined (Sup Fig 3E,H).
DISCUSSION
Learning and memory deficits are common following TBI, however it remains poorly understood how TBI-associated pathological changes disrupt information processing in brain regions important for these cognitive processes. Therefore, we utilized high-density electrophysiology to record from the hippocampus in freely moving animals following a well-characterized rat model of TBI. We took advantage of laminar probes to characterize layer-specific pathophysiology across CA1 lamina following TBI and found a loss of theta and gamma power across st. pyr and st. rad in injured animals, along with a drastic reduction in PAC that was specific to st. pyr. Interneurons from TBI rats were less entrained to theta and gamma, providing a potential mechanism for the loss of oscillatory power and coupling. Pyramidal cells from injured animals were more strongly recruited to fire across different environments and were entrained to a later phase of theta. Additionally, sharp wave ripples (SW-Rs) in injured rats had decreased amplitudes compared to shams, suggesting a desynchronization of the CA1 network even in the non-theta state. Taken together, these results reveal disruptions to electrophysiological processes essential for learning and memory, potentially providing a mechanism through which TBI-associated pathological changes collectively disrupt cognition.
Layer Specific Changes
A loss of hippocampal oscillatory power in awake animals following TBI has previously been reported40,42,45, however, these studies used either monopolar electrodes or twisted wires which are difficult to localize to specific lamina. Localization of electrodes within lamina is important when examining oscillations, as power varies greatly across layers. The use of high-density laminar probes in this study allowed us to precisely localize electrodes to both st. pyr and st. rad in CA1 revealing a loss of oscillatory power in injured animals from both layers that was more pronounced in st. pyr, and a dramatic reduction in PAC in injured rats that was specific to st. pyr. The more pronounced loss of power and PAC from st. pyr suggests TBI-associated disruptions to cells and circuits specifically targeting this layer.
One cell type implicated in these disruptions are parvalbumin positive (PV+) basket cells, which provide strong peri-somatic inhibition to CA1 pyramidal cells that can tightly control the timing of pyramidal cell firing through shunting inhibition82,83. PV+ interneurons as a class are entrained to theta and gamma oscillations and can entrain pyramidal cells to these oscillations84–86. Here we found that interneurons from injured animals were less entrained to theta and gamma oscillations, and pyramidal cells from TBI rats were entrained to a different angle of theta. These combined results support a model in which TBI reduces the entrainment strength of basket cells, which then results in a change in the angle of entrainment of pyramidal cells. Because basket cells target the soma of pyramidal cells, this model would predict a loss of theta and gamma power and PAC in st. pyr matching our observed results. Additionally, PV+ basket cells are strongly recruited to fire during SWRs and support ripple generation64,87–89, thus the decrease in ripple power during SWR events may also suggest PV+ basket cell dysfunction. While hippocampal PV+ interneuron loss and dysfunction has previously been reported in a variety of injury models41,90–92, it is likely that TBI also disrupts other interneuron populations which could contribute to the observed deficits especially the loss of oscillatory power in st. rad. Future studies recording from specifically identified interneuron classes would be needed to address whether TBI preferentially disrupts distinct interneuron subpopulations.
Loss of Afferent Input
Disruptions to afferent input from CA3 and EC to CA1 can also contribute to electrophysiological deficits observed following TBI. CA3 pyramidal neurons are prone to excitotoxicity and are susceptible to cell loss in the LFPI model, and human cases of TBI43,93–96. Additionally, axonal injury has been demonstrated in pathways connecting CA3 and EC to CA143,97. Axonal injury is known to cause action potential conduction delays and may therefore lead to desynchronization of CA1 afferent input98,99. This deafferentation of CA1 may also contribute to circuit hyperexcitability in TBI which been reported in both DG and CA1 in brain slices across different TBI models25,31,33,34. Compensatory mechanisms following TBI and/or deafferentation can alter the intrinsic properties of cells in CA1 and also disrupt long term potentiation16,24,27–30,32,33. Additionally, TBI has been shown to disrupt hyperpolarization-activated cation nonselective (HCN) channels responsible for Ih100–102 which can affect cell excitability and could disrupt theta generation especially in the apical dendrites where these channels are expressed in large numbers.
TBI-associated changes in oscillatory power, PAC, and entrainment may also arise from desynchronization between the hippocampus and medial septum and diagonal band of Broca (MSDB). Reciprocal connections between MSDB and hippocampus lie within the fimbria and fornix where axonal pathology is reliably seen in the LFPI model43,103. Importantly, the extent of fimbria/fornix disruption has been correlated with poor learning and memory performance in human TBI10–12. In rodents, MSDB is especially important for theta generation and sets the pace of theta through theta frequency firing of MSDB interneurons50,104–106. Using the LFPI model, we previously demonstrated hippocampal-MSDB desynchronization in injured rats under isoflurane anesthesia43. Here, in awake rats, we show that CA1 interneurons are less entrained to theta in injured animals. This further supports hippocampal-MSDB desynchronization as the entrainment of CA1 interneurons can be controlled by MSDB projecting interneurons50,107. MSDB glutamatergic and cholinergic inputs are also affected by fimbria/fornix pathology which may further contribute to theta dysfunction in TBI. MSDB acetylcholine neurons have been demonstrated to be susceptible to injury resulting in cholinergic dysfunction103,108,109. This can disrupt type II theta which occurs when animals are not actively moving and exploring the environment50,110–112. We found that single unit entrainment to theta was substantially decreased in injured rats when they were not moving, a phenomenon not seen in shams, which suggests a disruption in type II theta. This provides further evidence that cholinergic signaling may be dysfunctional following TBI.
Implications of Temporal Coding Deficits
Oscillations provide the temporal framework to coordinate the firing of distinct cell ensembles within and across brain regions46,113–115. Theta oscillations arise from complex interactions between distributed circuits which can temporally link distant brain regions in time, while gamma oscillations reflect local processing important for selecting behaviorally relevant cell ensembles locally49,52,53,113,115. Thus theta-gamma PAC represents a mechanism whereby local processing in many distributed networks can be temporally coordinated on a theta timescale46,58,116. Importantly, LTP and LTD may preferentially occur at specific phases of the theta oscillation creating temporal windows for plasticity and potentially supporting spike-timing dependent plasticity across long distances47. In CA1, theta oscillations can provide temporal windows in which CA3 and EC input is integrated46,117. Additionally, coordinated CA3 and EC inputs recruit subsets of CA1 interneurons that drive layer-specific gamma oscillations46,58,116. CA3 driven low gamma (gammaL; 30–59 Hz) and EC driven mid gamma (gammaM; 60–80Hz) are respectively coupled to the descending phase (~90°) and trough (~180°) of theta measured in st. pyr116. This provides temporal separation of CA3 and EC inputs, which is hypothesized to prevent interference of previously stored associations in the CA3 recurrent network with the formation of new associations driven by EC47. Our findings of reduced theta-gamma PAC in TBI rats and a shift in the peak of gammaL amplitude to a later phase of theta suggests that memory encoding and retrieval may not be sufficiently temporally separated in TBI rats. Further supporting this, we found that a larger percentage of pyramidal cells in injured animals fired across 2 distinct environments, one familiar and one novel. One proposed mechanism of gamma is to select only relevant cell ensembles while inhibiting activity from other ensembles113,115. The loss of gamma power and theta-gamma PAC along with the over recruitment of pyramidal cells across environments suggests that this mechanism is dysfunctional in TBI, which could lead to interference between the encoding and retrieval of memories and disrupt memory updating.
Since information in the hippocampus is thought to be chunked on a theta timescale38,116–120, findings from this study would predict a disruption in theta phase precession and the formation of theta sequences, processes that require precise temporal integration of afferent inputs with local circuits38,118–122. While we did not assess this due to limited occupancy of the largely open environments during recording, previous studies have reported TBI-associated disruptions in place cell specificity and stability5,41. Future studies tying dysfunctional place fields to deficits in phase precession and theta sequences during a spatial navigation task could directly link impaired integration of afferent input to disrupted behavior following TBI. In the non-theta state, SWRs are frequently observed during which cell ensembles are activated in the same or reverse temporal order they fired during behavior but on a compressed timescale66,67. This replay of activity is important for memory consolidation, recall, and updating processes as well as planning behaviors64,65. Here we showed that ripples had decreased amplitudes in TBI animals, which may affect all of the above processes. Since we were unable to assess replay in this dataset, future studies are needed to address whether the replay of behaviorally relevant cells ensembles is affected by TBI.
Future Therapies
Neuromodulation therapies, including theta stimulation of the MSDB, fornix, or hippocampus, have been shown to improve learning and memory deficits in the LFPI model of TBI123–126. While the physiological mechanisms underlying behavioral improvements have not been investigated, results from this study provide outcome measures to target and examine with future therapies. For example, stimulation paradigms that successfully re-entrain cells to theta or boost oscillatory power and theta-gamma PAC may be more successful at restoring TBI-associated cognitive deficits. There is a large parameter space to explore when performing brain stimulation, so having electrophysiological targets may allow for faster optimization of stimulation parameters. Transplantation of stem cells or interneurons have also been shown to restore cognitive deficits in preclinical models of TBI127,128. Understanding how these transplanted cells functionally integrate into the network may help to unveil how pathophysiology correlates to behavior, especially if transplanted interneurons become entrained to local theta and gamma oscillations or if transplantation alters oscillatory power and coupling.
Overall, TBI pathologies can vary across patients and have a wide range of effects on the brain. Fixing a single TBI-associated pathological change is unlikely to improve cognitive outcomes, thus it is important to study how coalescing TBI pathological changes disrupt neural circuit activity underlying cognition in order to develop better targets and treatments129–131. While some of the TBI pathophysiology we report here may be model and timepoint specific, similar electrophysiological deficits have been reported in Alzheimer’s disease, aging, epilepsy, and schizophrenia132–140, all of which can be associated with impaired cognition. Thus, temporal coding disruptions described in this study may be a good readout of hippocampal circuit function and could be predictive of cognitive dysfunction. As we learn more about how coding in the human hippocampus supports learning and memory, and how TBI disrupts these processes, we may be able to optimize deep brain stimulation treatments for TBI patients141–144 and potentially incorporate non-invasive tools such as transcranial magnetic stimulation145–147, transcranial alternating current stimulation148,149, and focused ultrasound150,151 to address cognitive impairments of TBI.
METHODS
Animals
All procedures and animal care related to this study were approved by the University of Pennsylvania Institutional Care and Use Committee at an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) accredited site. Young healthy adult male Long Evans rats (RGD catalog: 2308852, Charles River Laboratories; RRID: RGD_2308852) naïve to any previous procedures or investigations were used in this study. Rats were kept on a strict 12 hr light/dark cycle and had access to standard chow and water ad libitum except when food restricted for electrophysiological experiments (see below). Animals were examined daily for signs of pain, or distress after all surgical procedures.
LFPI Surgery
Lateral fluid percussion injury (LFPI) surgeries were performed as described previously43. Briefly, rats were anesthetized with isoflurane and affixed to a stereotaxic holder. A midline incision was made on the head to visualize the skull, then a 5mm craniectomy was made over the left parietal cortex centered between lambda and bregma in the anterior/posterior direction and between midline and the lateral edge of the skull in the medial/lateral direction. A needle hub (cut from the needle) was affixed to the craniectomy site and secured with glue and dental cement. Rats were then removed from anesthesia, monitored for increased respiration and a response to toe pinch, then immediately attached to the FPI device. To induce injury, a pendulum was dropped to strike a tube filled with sterile saline delivering a fluid pulse to the intact dura within the craniectomy. A transducer was used to measure the force of the pressure wave. The needle hub was then removed and Kwik-Sil was place over the site of the craniectomy. The incision was then sutured up and rats were placed under a warming lamp and administered buprenorphine (0.05 mg/kc s.c.) after demonstrating a righting reflex. Sham rats underwent the same surgical procedure as injured rats, including the craniectomy, but no fluid pulse was delivered. Animals were excluded from further study if a dural breach was present or if the craniectomy was not properly centered between lambda/bregma and midline/lateral edge. Four rats (3 injured and 1 sham) were excluded from the MWM based on these criteria.
Electrode Implantation Surgery
Rats underwent electrode implantation surgery 3–4 days after LFPI/sham surgery. During this procedure, rats were anesthetized with isoflurane and affixed to a stereotaxic holder. Sutures from the FPI procedure were removed, the midline incision was re-opened to expose the skull surface, and the Kwik-Sil cover over the LFPI craniectomy was removed. A stainless-steel screw was placed on the surface of the dura over the cerebellum and was used as ground. A small tungsten wire (~10 kohms) was placed in the lateral ventricle (0.7 mm posterior to bregma, 2.8 mm lateral (assuming angle), 3.8 mm DV, at a 10° angle lateral to medial) and was used as a reference. A durotomy was then performed over the LFPI craniectomy and electrodes mounted to drives (nano-Drive; Cambridge NeuroTech; Cambridge, United Kingdom) were slowly advanced into cortex (AP: −4.2 mm, ML: 2.8 mm, DV: ~1.3 mm). Dura-Gel (Cambridge NeuroTech) was then applied to the brain surface and Vaseline was melted onto the electrode and cable using a low-temp cauterizer to allow them to move when advancing the electrode post-implant. Anchor screws were inserted bilaterally into the lateral ridge of the skull, and dental cement was applied to secure the implant. Rats were either implanted with a Cambridge NeuroTech E1 (4 shank) electrode (n=2 sham, n=2 injured) or a Cambridge NeuroTech H2 electrode (n=2 sham, n=3 injured).
Behavior During Electrophysiological Recordings
Animals used for electrophysiological recordings were handled for 5–7 days then were food restricted (up to 85% of body weight) and introduced to a 1 m2 open field environment containing small pieces of Fruit Loops to encourage exploration. Rats were exposed to the environment for 15 mins/day for a minimum of 3 days before injury to become familiarized to it. Rats were then returned to normal ad libitum food access for at least 24 hrs then subjected to LFPI/sham surgery. Rats were chronically implanted with recording electrodes 3–4 days after sham/injury surgery. 24 hrs after implantation, rats were again food restricted (up to 85% of body weight) and habituated to the wireless recording transmitter (FreeLynx; Neuralynx; Bozeman, MT) while exploring the same open field environment for food rewards. The following days, electrodes were advanced deeper into the brain, and the rat was again placed in the open field environment to find food rewards. This process was repeated until electrodes reached st. pyr in CA1 (6–9 days post-injury) which was determined based on real-time observation of multi-unit activity and the presence of sharp-wave ripples. Once in st. pyr, rats were placed in their home cage for at least 20mins to allow electrodes to settle then recordings were obtained in the familiar open field environment. Rats were then placed back in their home cage for 5 mins while the battery for the wireless transmitter was replaced, then were placed into a novel environment for an additional recording. The novel environment was an 8-arm radial arm maze with Fruit Loops scattered throughout to encourage exploration (no behavioral task was performed in the radial arm maze). On 2 subsequent days, electrodes were advanced further into the CA1 lamina, allowed to settle, then rats were recorded in the familiar open field environment.
Data Acquisition
Electrical signals were sampled at 30 kHz, amplified, digitized, then wirelessly transmitted using the FreeLynx (Neuralynx). Real-time signals were visualized in the Cheetah recording software (Neuralynx) as local field potentials (LFPs) and filtered signals (600–6000 Hz) were organized into tetrodes and thresholded for real-time spike detection.
Selection of St. Pyramidale and St. Radiatum Channels
On each shank, only a single st. pyr and st. rad channel was chosen. The channel containing the local maximum of power in the ripple frequency (100–250 Hz) range was chosen as the st. pyr channel. As electrodes were advanced on later recording days, st. pyr was not always present on every electrode shank thus only shanks with a local maximum of ripple frequency power were included. The st. rad channel was chosen as the local maximum of the CSD sink for the mean sharp-wave ripple (SWR) waveform. A local maximum of the SWR CSD sink was not observed on all shanks especially on the first recording day when most electrodes were localized to st. pyr, thus, shanks without a local maximum in the SWR CSD sink were not included.
Velocity Calculations
The position of the animal was obtained by tracking a light on the wireless transmitter affixed to the rat’s head. Tracking was curated by manually removing artifacts (sudden large displacements), interpolating between removed positions, and smoothing pixel locations with a gaussian kernel. Pixels were then converted to real-world distances (cm) using the size of the environments for scaling. Instantaneous velocity (Δ position / Δ time) was computed between each frame then smoothed with a gaussian kernel.
Power Analyses
Raw signals were down sampled to 3 kHz using an anti-aliasing filter (resample function in MATLAB; Mathworks, MA). Power was then computed using a continuous wavelet transform using the cwt function in MATLAB. First a filter bank of complex bump wavelets was created from 1–1500 Hz using 36 wavelets per octave. Bump wavelets were selected because they have narrow variance in frequency. Wavelets were then convolved with the down sampled signal and power was computed as the magnitude of the analytic signal squared for each convolution. For each recording, the mean power for each frequency was computed specifically for times when the rat was moving (>10 cm/sec). These values were then averaged across shanks and days to get an average per animal for the defined st. pyr and st. rad channels respectively, and sham and injured animals were averaged with each other to create the power spectra shown in Fig 1F. Theta and gamma power was extracted from each animal’s mean power spectrum by computing the area under the curve in the 5–10 and 30–59 Hz bands respectively. These individual values are shown in Fig 1G.
Current Source Density
The current source density (CSD) was calculated across electrode channels on a shank using the spline inverse CSD method from152 in the freely available MATLAB CSDplotter (v0.1.1) package.
PAC Analyses
The strength of theta-gamma PAC was assessed independently in st. pyr and st. rad by calculating the modulation index (MI)68 while rats were moving (>10 cm/sec) in the familiar environment. Data was down sampled to 600Hz, separately filtered into theta (5–10 Hz) and gamma (30–59Hz) frequency bands using a 2nd order Butterworth filter, and the instantaneous theta phase and gamma amplitude was extracted for each sample using the Hilbert transform. The mean gamma amplitude () was calculated across 18 theta phase bins (; 20° width) for epochs when the rat was moving then converted to a probability distribution (equation 1).
| Eq. 1 |
Shannon Entropy () was calculated (equation 2) for the distribution, then the modulation index () was computed (equation 3) and averaged across shanks and recording days to get a mean MI per rat (individual points in Fig 2D).
| Eq. 2 |
| Eq. 3 |
Figure 2E depicts the mean gamma amplitude across theta phase bins averaged across shanks and days to get a mean distribution for each rat, then averaged across animals in the sham and injured conditions. This distribution was then up sampled by a factor of 100 and the theta phase angle corresponding to the peak in gamma amplitude was extracted to compare between sham and injured animals. PAC heatmaps in Fig 2B were constructed by calculating the MI across a range of low-frequency bins for phase (40 logarithmically spaced bins spanning 1–15 Hz each with a width of 1 Hz) and high-frequency bins for amplitude (20 logarithmically spaced bins spanning 20–180 Hz each with a width of 20 Hz). Heatmaps were averaged across shanks and recording days to get an average for each rat, then averaged across sham and injured animals.
Single Unit Isolation and Clustering
Automated spike sorting was done using Klusta (RRID: SCR_014480) and manually curated in Phy (https://github.com/kwikteam/phy) by a reviewer blind to sham/injured condition. Each shank was assessed independently as the inter-shank spacing of 250 uM ensured that units were not visible on multiple shanks. Familiar and novel environment recordings on day 1 were spliced together, and spike sorting was performed on this joined data to retain unit identity across recordings. After single units were extracted, they were clustered into 3 different groups: putative pyramidal cells, interneurons, or unclassified. All units >80 μm above the defined st. pyr channel were automatically clustered as interneurons. Cells within ±80 μm of the defined st. pyr channel were manually classified by a reviewer blind to animal ID and sham/injured condition based on firing rate, spike shape, and autocorrelogram.
Automated k-means consensus clustering was performed to further validate manual clustering. Firing rates, spike width (width of mean action potential waveform at half-max spike height), and the first moment of the autocorrelogram (mean of autocorrelogram values at lags from 0–50 ms) were normalized from 0–1 for each cell within ±80 μm of the defined st. pyr channel. K-means clustering (with 2 groups) was performed on these normalized features 1000 times, and the consensus of all 1000 permutations was used to identify cells as pyramidal cells or interneurons. Classification of cells that were manually identified as pyramidal cells or interneurons (excluding unclassified cells) were compared to automated clustering results and there was >95% agreement between the two methods.
Entrainment Analyses
Entrainment was assessed by filtering the st. pyr LFP into discrete frequency bands using a 2nd order Butterworth filter, finding the instantaneous angle associated with each spike using the Hilbert transform, then computing the mean vector length (MVL) and mean angle of entrainment using circular statistics (CircStat toolbox in MATLAB; RRID:SCR_016651)153. Entrainment strength was visualized across a range of frequency bins (24 bins spanning 1–15 Hz with a width of 1 Hz, and 30 bins spanning 15–180 Hz with a width of 20 Hz) separately for epochs when the rat was moving (>10 cm/sec) and still (<10 cm/sec) (Figs 4B&D). Units were excluded if their firing rate was below 0.1 Hz in the environment or if they did not have at least 25 spikes in both moving and still epochs.
Theta and gamma entrainment was assessed by filtering the st. pyr LFP from 5–10 Hz or 30–59 Hz respectively and calculating the MVL and mean angle of entrainment. To determine if cells were significantly entrained to theta or gamma oscillations (Fig 4C&E), we used a circular shuffling procedure with 500 permutations. For each permutation, the vector of st. pyr theta and gamma phase angles was cut at a random point (minimum 10 second offset from the original data), and the phase angles after the cut point were placed in front of the phase angles before the cut point to create a circularly shuffled distribution of phase angles. This maintains the spiking profile of all units as well as the temporal structure of the data except at the cut point. The MVL was computed for each permutation, and significantly entrained cells were defined as cells that had a MVL greater than 95% of shuffled permutations. Any units not significantly entrained to theta oscillations were excluded from analysis in Figure 5.
Ripple Detection and Analysis
Raw signals were down sampled to 3000 Hz using an anti-aliasing filter (resample function in MATLAB). Then a continuous wavelet transform was used to construct time-frequency maps of signals using an open-source algorithm (fCWT)154. Morlet wavelets with 12 were used for higher frequency resolution. High frequency events were isolated by thresholding the time-frequency maps using empirical cumulative distribution function of amplitudes. The thresholding value was calculated for 80–250 Hz and 250–500 Hz separately to minimize the effect of decrease in the amplitude of oscillations with frequency. These isolated high frequency blobs were accepted if they passed several criteria such as their duration (at least 3 cycles based on the frequency of oscillation), or their amplitudes compared to background activity. The hyperparameters of this algorithm were chosen by evaluating the accuracy and precision of this method using synthetic data70.
Sharp wave events were detected separately on st. rad (Note that for some animals, the first day of recording did not include any channels located at st. rad because most electrodes were localized to st. pyr). First, the down sampled raw signals were filtered between 8 and 40 Hz using a 2nd order Butterworth filter. Then, local minimums of the filtered signal were found using islocalmin function in MATLAB as candidates for the center of sharp waves. Next, islocalmax function in MATLAB was used to find the local maximums of the filtered signal for the starting and stopping point of a sharp wave. A sharp wave was identified if a center (local minimum) was located between to local maximums. Finally, sharp waves were only accepted if their amplitude of the local minimum was larger than 98% of all local minimums. The described method for sharp wave detection was generous by design (it can be stricter by applying a harsher criterion for amplitudes).
Sharp-wave ripples were identified if the detected ripples events (accepted contiguous blobs in time-frequency maps) overlapped with detected sharp wave events. These putative SWRs were then manually curated by an observer blind to animal ID, sham/injured condition, and animal behavior during event. The percentage of accepted SWR events that occurred while the animals were not moving was calculated (>95% in all conditions) and SWR event frequency was normalized to the amount of time animals were not moving to compare SWR event frequency across conditions while controlling for any potential differences is the amount of time animals spent moving. Frequency amplitude spectra were computed for each event by averaging amplitudes at each frequency bin over times coinciding with the blob edges (see white lines in Fig 6A), then averaged across conditions. Event durations and peak amplitudes (the maximum value in the time-frequency map) were then computed to create the cumulative distributions shown in Fig 6E.
Morris Water Maze
The MWM apparatus was a circular pool (2 m in diameter, 50 cm tall, filled to a depth of 25 cm with 18° C water) containing a hidden platform (11.5 cm x 11.5 cm x 24 cm tall) just below the water’s surface. Both the interior of the pool and the platform were painted black making the platform invisible from the surface. External visual cues could be easily seen from the pool for navigation purposes. Rats received 10 trials of training in the MWM ~20 hrs before TBI/sham surgery followed by another 10 trials ~2 hrs before surgery. For each trial, the rat was placed in one of 4 randomly selected starting locations located 90° apart and given 2 mins to freely swim until they found the platform. If the rat was unable to find the platform after 2 mins, they were placed there for 15–30 sec. Rats were tested ~42 hrs after FPI or sham surgery to assess memory of the platform location. During testing, the platform was removed, and rats freely swam for 1 minute. A memory score74 was calculated based on the amount of time the rat spent in 5 distinct zones with zones closer to the platform’s previous location being weighted higher. All trials were video recorded from a camera placed above the pool, and the rat’s position was obtained using automated video tracking (Ethovision XT, Noduls; RRID: SCR_000441).
Histological Examinations
To ensure the nature and severity of injury were consistent with prior characterizations of the model, a separate set of animals underwent FPI without electrode implantation (n=2; 1.8 atm) or sham surgery (n=1) for histopathological analysis. All animals were survived for 48 hours. At the study endpoint, ketamine / xylazine / acepromazine (75/15/2 mg/kg, i.p.) was administered and following adequate anesthesia, animals were transcardially perfused with chilled 1X phosphate buffered saline (PBS) followed immediately with chilled 10% neutral buffered formalin. Brains were extracted and post-fixed for 24 hours at 4°C before being blocked in the coronal plane at 2 mm intervals and processed to paraffin using standard techniques. 8 μm thick whole brain coronal sections were obtained at the level of the anterior hippocampus, posterior hippocampus and posterior angular bundle using a rotary microtome.
All regions were stained using hematoxylin and eosin (H&E). In addition, to determine the presence and distribution of axonal pathology, immunohistochemical (IHC) techniques were performed as previously described79,155. Briefly, following deparaffinization and rehydration of tissue sections, endogenous peroxidase activity tissue was quenched using 3% aqueous hydrogen peroxide. Antigen retrieval was performed using a pressure cooker with sections immersed in Tris EDTA buffer (pH 8.0). Subsequent blocking was performed for 30 minutes in 1% normal horse serum (Vector Labs, Burlingame, CA, USA) in Optimax buffer (BioGenex, San Ramon, CA, USA). An antibody reactive for the N-terminal amino acids 66–81 of the amyloid precursor protein (APP) (Clone 22C11: Millipore, Billerica, MA) was applied at 1:40K and incubated overnight at 4°C. After rinsing, sections were incubated with the appropriate biotinylated secondary antibody for 30 minutes followed by avidin-biotin complex and visualization achieved using DAB (all reagents: Vector Labs, Burlingame, CA, USA). Sections were rinsed and dehydrated in graded alcohols and cleared in xylenes before being coverslipped. Positive control tissue for APP IHC included sections of contused rat brain tissue with previously established axonal pathology. Omission of the primary antibody was performed on positive control tissue to control for non-specific binding.
Fluoro-Jade C staining was performed to assess for neuronal degeneration using the Biosensis Fluoro-Jade C staining Reagent Kit per the manufacturer’s instructions (Biosensis, Thebarton, Australia). Briefly, following dewaxing and rehydration to water as above, tissue was the immersed in potassium permanganate solution for 25 minutes at room temperature. After being rinsed in gently flowing dH2O, tissue was then incubated in Fluoro-Jade C solution at room temperature for 30 minutes. After rinsing, sections were dried at 37° C in an oven for 90 minutes before being immersed in xylenes and coverslipped.
Analysis of Histological Findings
All sections were reviewed for the presence of hemorrhage. The presence or absence of axonal pathology was determined in all sections including in the fimbria fornix, angular bundle, hippocampus, corpus callosum and thalamus. In addition, cell death was assessed by examination for Fluoro-Jade C positive neurons in all sections, including all hippocampal subfields, entorhinal cortex and thalamus. Two independent observers reviewed all sections for the presence or absence of axonal pathology and cell degeneration with excellent interrater reliability (Cohen’s Kappa 0.91). Notably, histological observations were performed at 48 hours to identify pathologies which are readily visualized at this timepoint including acute axonal and neuronal degeneration. However, due to the complex temporal evolution of post-traumatic pathologies, observations at 48 hrs may differ from the pathologies at 7 days post-injury, when electrophysiological recordings were performed.
Supplementary Material
Funding
This work was supported by a VA RR&D MERIT (RX002705) and an NINDS R01 (NS101108).
REFERENCES
- 1.Draper K. & Ponsford J. Cognitive Functioning Ten Years Following Traumatic Brain Injury and Rehabilitation. Neuropsychology 22, 618–625, doi: 10.1037/0894-4105.22.5.618 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Monti J. M. et al. History of mild traumatic brain injury is associated with deficits in relational memory, reduced hippocampal volume, and less neural activity later in life. Front. Aging Neurosci. 5, 41, doi: 10.3389/fnagi.2013.00041 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vakil E., Greenstein Y., Weiss I. & Shtein S. The Effects of Moderate-to-Severe Traumatic Brain Injury on Episodic Memory: a Meta-Analysis. Neuropsychol Rev 29, 270–287, doi: 10.1007/s11065-019-09413-8 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Bigler E. D. et al. Traumatic Brain Injury and Memory: The Role of Hippocampal Atrophy. Neuropsychology 10, 333–342, doi: 10.1037/0894-4105.10.3.333 (1996). [DOI] [Google Scholar]
- 5.Eakin K. & Miller J. P. Mild Traumatic Brain Injury Is Associated with Impaired Hippocampal Spatiotemporal Representation in the Absence of Histological Changes. Journal of Neurotrauma 29, 1180–1187, doi: 10.1089/neu.2011.2192 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Lyeth B. G. et al. Prolonged memory impairment in the absence of hippocampal cell death following traumatic brain injury in the rat. Brain Research 526, 249–258, doi: 10.1016/0006-8993(90)91229-a (1990). [DOI] [PubMed] [Google Scholar]
- 7.Tate D. F. & Bigler E. D. Fornix and Hippocampal Atrophy in Traumatic Brain Injury. Learn. Mem. 7, 442–446, doi: 10.1101/lm.33000 (2000). [DOI] [PubMed] [Google Scholar]
- 8.Adnan A. et al. Moderate–severe traumatic brain injury causes delayed loss of white matter integrity: Evidence of fornix deterioration in the chronic stage of injury. Brain Inj. 27, 1415–1422, doi: 10.3109/02699052.2013.823659 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Blumbergs P. C. et al. Stalning af amyloid percursor protein to study axonal damage in mild head Injury. Lancet 344, 1055–1056, doi: 10.1016/s0140-6736(94)91712-4 (1994). [DOI] [PubMed] [Google Scholar]
- 10.Kinnunen K. M. et al. White matter damage and cognitive impairment after traumatic brain injury. Brain 134, 449–463, doi: 10.1093/brain/awq347 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palacios E. M. et al. Diffusion tensor imaging differences relate to memory deficits in diffuse traumatic brain injury. BMC Neurol. 11, 24, doi: 10.1186/1471-2377-11-24 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tomaiuolo F. et al. Gross morphology and morphometric sequelae in the hippocampus, fornix, and corpus callosum of patients with severe non-missile traumatic brain injury without macroscopically detectable lesions: a T1 weighted MRI study. J. Neurol., Neurosurg. Psychiatry 75, 1314, doi: 10.1136/jnnp.2003.017046 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Faden A. I., Demediuk P., Panter S. S. & Vink R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science 244, 798–800, doi: 10.1126/science.2567056 (1989). [DOI] [PubMed] [Google Scholar]
- 14.Guerriero R. M., Giza C. C. & Rotenberg A. Glutamate and GABA Imbalance Following Traumatic Brain Injury. Curr. Neurol. Neurosci. Rep. 15, 27, doi: 10.1007/s11910-015-0545-1 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McGuire J. L., Ngwenya L. B. & McCullumsmith R. E. Neurotransmitter changes after traumatic brain injury: an update for new treatment strategies. Mol. Psychiatry 24, 995–1012, doi: 10.1038/s41380-018-0239-6 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Aungst S. L., Kabadi S. V., Thompson S. M., Stoica B. A. & Faden A. I. Repeated Mild Traumatic Brain Injury Causes Chronic Neuroinflammation, Changes in Hippocampal Synaptic Plasticity, and Associated Cognitive Deficits. J. Cereb. Blood Flow Metab. 34, 1223–1232, doi: 10.1038/jcbfm.2014.75 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muccigrosso M. M. et al. Cognitive deficits develop 1month after diffuse brain injury and are exaggerated by microglia-associated reactivity to peripheral immune challenge. Brain, Behav., Immun. 54, 95–109, doi: 10.1016/j.bbi.2016.01.009 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shetty A. K., Mishra V., Kodali M. & Hattiangady B. Blood brain barrier dysfunction and delayed neurological deficits in mild traumatic brain injury induced by blast shock waves. Front. Cell. Neurosci. 8, 232, doi: 10.3389/fncel.2014.00232 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sulhan S., Lyon K. A., Shapiro L. A. & Huang J. H. Neuroinflammation and blood–brain barrier disruption following traumatic brain injury: Pathophysiology and potential therapeutic targets. J. Neurosci. Res. 98, 19–28, doi: 10.1002/jnr.24331 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lai J. q., Shi Y.-C., Lin S. & Chen X.-R. Metabolic disorders on cognitive dysfunction after traumatic brain injury. Trends Endocrinol. Metab. 33, 451–462, doi: 10.1016/j.tem.2022.04.003 (2022). [DOI] [PubMed] [Google Scholar]
- 21.Li J. et al. Exploring Temporospatial Changes in Glucose Metabolic Disorder, Learning, and Memory Dysfunction in a Rat Model of Diffuse Axonal Injury. Journal of Neurotrauma 29, 2635–2646, doi: 10.1089/neu.2012.2411 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weil Z. M., Gaier K. R. & Karelina K. Injury timing alters metabolic, inflammatory and functional outcomes following repeated mild traumatic brain injury. Neurobiology of Disease 70, 108–116, doi: 10.1016/j.nbd.2014.06.016 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Yi L. et al. Serum Metabolic Profiling Reveals Altered Metabolic Pathways in Patients with Post-traumatic Cognitive Impairments. Sci. Rep. 6, 21320, doi: 10.1038/srep21320 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.D’Ambrosio R., Maris D. O., Grady M. S., Winn H. R. & Janigro D. Selective loss of hippocampal long-term potentiation, but not depression, following fluid percussion injury. Brain Research 786, 64–79, doi: 10.1016/s0006-8993(97)01412-1 (1998). [DOI] [PubMed] [Google Scholar]
- 25.Folweiler K. A., Samuel S., Metheny H. E. & Cohen A. S. Diminished Dentate Gyrus Filtering of Cortical Input Leads to Enhanced Area Ca3 Excitability after Mild Traumatic Brain Injury. Journal of Neurotrauma 35, 1304–1317, doi: 10.1089/neu.2017.5350 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Katayama Y., Becker D. P., Tamura T. & Hovda D. A. Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. Journal of Neurosurgery 73, 889–900, doi: 10.3171/jns.1990.73.6.0889 (1990). [DOI] [PubMed] [Google Scholar]
- 27.Lei Z., Deng P., Li J. & Xu Z. C. Alterations of A-Type Potassium Channels in Hippocampal Neurons after Traumatic Brain Injury. Journal of Neurotrauma 29, 235–245, doi: 10.1089/neu.2010.1537 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyazaki S. et al. Enduring suppression of hippocampal long-term potentiation following traumatic brain injury in rat. Brain Research 585, 335–339, doi: 10.1016/0006-8993(92)91232-4 (1992). [DOI] [PubMed] [Google Scholar]
- 29.Reeves T. M., Lyeth B. G. & Povlishock J. T. Long-term potentiation deficits and excitability changes following traumatic brain injury. Exp Brain Res 106, 248–256, doi: 10.1007/bf00241120 (1995). [DOI] [PubMed] [Google Scholar]
- 30.Sanders M. J., Sick T. J., Perez-Pinzon M. A., Dietrich W. D. & Green E. J. Chronic failure in the maintenance of long-term potentiation following fluid percussion injury in the rat. Brain Research 861, 69–76, doi: 10.1016/s0006-8993(00)01986-7 (2000). [DOI] [PubMed] [Google Scholar]
- 31.Santhakumar V., Ratzliff A. D. H., Jeng J., Toth Z. & Soltesz I. Long-term hyperexcitability in the hippocampus after experimental head trauma. Ann Neurol 50, 708–717, doi: 10.1002/ana.1230 (2001). [DOI] [PubMed] [Google Scholar]
- 32.Schwarzbach E., Bonislawski D. P., Xiong G. & Cohen A. S. Mechanisms underlying the inability to induce area CA1 LTP in the mouse after traumatic brain injury. Hippocampus 16, 541–550, doi: 10.1002/hipo.20183 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Witgen B. M. et al. Regional hippocampal alteration associated with cognitive deficit following experimental brain injury: A systems, network and cellular evaluation. Neuroscience 133, 1–15, doi: 10.1016/j.neuroscience.2005.01.052 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Wolf J. A. et al. Concussion Induces Hippocampal Circuitry Disruption in Swine. Journal of Neurotrauma 34, 2303–2314, doi: 10.1089/neu.2016.4848 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang Y., Brandon M. P., Griffin A. L., Hasselmo M. E. & Eden U. T. Decoding Movement Trajectories Through a T-Maze Using Point Process Filters Applied to Place Field Data from Rat Hippocampal Region CA1. Neural Comput. 21, 3305–3334, doi: 10.1162/neco.2009.10-08-893 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Lisman J. & Redish A. D. Prediction, sequences and the hippocampus. Philos. Trans. R. Soc. B: Biol. Sci. 364, 1193–1201, doi: 10.1098/rstb.2008.0316 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pastalkova E., Itskov V., Amarasingham A. & Buzsáki G. r. Internally Generated Cell Assembly Sequences in the Rat Hippocampus. Science 321, 1322–1327, doi: 10.1126/science.1159775 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wikenheiser A. M. & Redish A. D. Hippocampal theta sequences reflect current goals. Nat Neurosci 18, 289–294, doi: 10.1038/nn.3909 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu H., Baracskay P., O’Neill J. & Csicsvari J. Assembly Responses of Hippocampal CA1 Place Cells Predict Learned Behavior in Goal-Directed Spatial Tasks on the Radial Eight-Arm Maze. Neuron 101, 119–132.e114, doi: 10.1016/j.neuron.2018.11.015 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Biswas C., Marković D. & Giza C. C. Alterations in Mesoscopic Oscillations affecting Episodic Memory following Developmental Traumatic Brain Injury. Experimental Neurology 300, 259–273, doi: 10.1016/j.expneurol.2017.10.021 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Broussard J. I. et al. Mild Traumatic Brain Injury Decreases Spatial Information Content and Reduces Place Field Stability of Hippocampal CA1 Neurons. Journal of Neurotrauma 37, 227–235, doi: 10.1089/neu.2019.6766 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fedor M., Berman R. F., Muizelaar J. P. & Lyeth B. G. Hippocampal Theta Dysfunction after Lateral Fluid Percussion Injury. Journal of Neurotrauma 27, 1605–1615, doi: 10.1089/neu.2010.1370 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koch P. F. et al. Traumatic Brain Injury Preserves Firing Rates but Disrupts Laminar Oscillatory Coupling and Neuronal Entrainment in Hippocampal CA1. Eneuro 7, ENEURO.0495–0419.2020, doi: 10.1523/eneuro.0495-19.2020 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Munyon C., Eakin K. C., Sweet J. A. & Miller J. P. Decreased bursting and novel object-specific cell firing in the hippocampus after mild traumatic brain injury. Brain Research 1582, 220–226, doi: 10.1016/j.brainres.2014.07.036 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Paterno R., Metheny H., Xiong G., Elkind J. & Cohen A. S. Mild Traumatic Brain Injury Decreases Broadband Power in Area CA1. Journal of Neurotrauma 33, 1645–1649, doi: 10.1089/neu.2015.4107 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fernández-Ruiz A. et al. Entorhinal-CA3 Dual-Input Control of Spike Timing in the Hippocampus by Theta-Gamma Coupling. Neuron 93, 1213–1226.e1215, doi: 10.1016/j.neuron.2017.02.017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hasselmo M. E., Bodeln C. & Wyble B. P. A Proposed Function for Hippocampal Theta Rhythm: Separate Phases of Encoding and Retrieval Enhance Reversal of Prior Learning. Neural Comput. 14, 793–817, doi: 10.1162/089976602317318965 (2002). [DOI] [PubMed] [Google Scholar]
- 48.Feldman, Daniel E. The Spike-Timing Dependence of Plasticity. Neuron 75, 556–571, doi: 10.1016/j.neuron.2012.08.001 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sirota A. et al. Entrainment of Neocortical Neurons and Gamma Oscillations by the Hippocampal Theta Rhythm. Neuron 60, 683–697, doi: 10.1016/j.neuron.2008.09.014 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Buzsáki G. Theta Oscillations in the Hippocampus. Neuron 33, 325–340, doi: 10.1016/s0896-6273(02)00586-x (2002). [DOI] [PubMed] [Google Scholar]
- 51.Vanderwolf C. H. Hippocampal electrical activity and voluntary movement in the rat. Electroencephalogr. Clin. Neurophysiol. 26, 407–418, doi: 10.1016/0013-4694(69)90092-3 (1969). [DOI] [PubMed] [Google Scholar]
- 52.Solomon E. A. et al. Widespread theta synchrony and high-frequency desynchronization underlies enhanced cognition. Nature Communications 8, 1704, doi: 10.1038/s41467-017-01763-2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Summerfield C. & Mangels J. A. Coherent theta-band EEG activity predicts item-context binding during encoding. NeuroImage 24, 692–703, doi: 10.1016/j.neuroimage.2004.09.012 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Csicsvari J., Jamieson B., Wise K. D. & Buzsáki G. Mechanisms of Gamma Oscillations in the Hippocampus of the Behaving Rat. Neuron 37, 311–322, doi: 10.1016/s0896-6273(02)01169-8 (2003). [DOI] [PubMed] [Google Scholar]
- 55.Bragin A. et al. Gamma (40–100 Hz) oscillation in the hippocampus of the behaving rat. The Journal of Neuroscience 15, 47–60, doi: 10.1523/jneurosci.15-01-00047.1995 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Buzsáki G., S, L. L.-W. & Vanderwolf, C. H. Cellular bases of hippocampal EEG in the behaving rat. Brain Res. Rev. 6, 139–171, doi: 10.1016/0165-0173(83)90037-1 (1983). [DOI] [PubMed] [Google Scholar]
- 57.Canolty R. T. et al. High Gamma Power Is Phase-Locked to Theta Oscillations in Human Neocortex. Science 313, 1626–1628, doi: 10.1126/science.1128115 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Colgin L. L. Theta–gamma coupling in the entorhinal–hippocampal system. Current Opinion in Neurobiology 31, 45–50, doi: 10.1016/j.conb.2014.08.001 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jensen O. & Colgin L. L. Cross-frequency coupling between neuronal oscillations. Trends Cogn Sci 11, 267–269, doi: 10.1016/j.tics.2007.05.003 (2007). [DOI] [PubMed] [Google Scholar]
- 60.Lega B., Burke J., Jacobs J. & Kahana M. J. Slow-Theta-to-Gamma Phase–Amplitude Coupling in Human Hippocampus Supports the Formation of New Episodic Memories. Cereb. Cortex 26, 268–278, doi: 10.1093/cercor/bhu232 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Axmacher N. et al. Cross-frequency coupling supports multi-item working memory in the human hippocampus. Proceedings of the National Academy of Sciences 107, 3228–3233, doi: 10.1073/pnas.0911531107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Daume J. et al. Control of working memory by phase–amplitude coupling of human hippocampal neurons. Nature, 1–9, doi: 10.1038/s41586-024-07309-z (2024). [DOI] [PMC free article] [PubMed]
- 63.Buzsáki G. Hippocampal sharp waves: Their origin and significance. Brain Research 398, 242–252, doi: 10.1016/0006-8993(86)91483-6 (1986). [DOI] [PubMed] [Google Scholar]
- 64.Buzsáki G. Hippocampal sharp wave-ripple: A cognitive biomarker for episodic memory and planning. Hippocampus 25, 1073–1188, doi: 10.1002/hipo.22488 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carr M. F., Jadhav S. P. & Frank L. M. Hippocampal replay in the awake state: a potential substrate for memory consolidation and retrieval. Nat Neurosci 14, 147–153, doi: 10.1038/nn.2732 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Davidson T. J., Kloosterman F. & Wilson M. A. Hippocampal Replay of Extended Experience. Neuron 63, 497–507, doi: 10.1016/j.neuron.2009.07.027 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wilson M. A. & McNaughton B. L. Reactivation of Hippocampal Ensemble Memories During Sleep. Science 265, 676–679, doi: 10.1126/science.8036517 (1994). [DOI] [PubMed] [Google Scholar]
- 68.Tort A. B. L. et al. Dynamic cross-frequency couplings of local field potential oscillations in rat striatum and hippocampus during performance of a T-maze task. Proceedings of the National Academy of Sciences 105, 20517–20522, doi: 10.1073/pnas.0810524105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Csicsvari J., Hirase H., Czurko A. & Buzsáki G. Reliability and State Dependence of Pyramidal Cell–Interneuron Synapses in the Hippocampus an Ensemble Approach in the Behaving Rat. Neuron 21, 179–189, doi: 10.1016/s0896-6273(00)80525-5 (1998). [DOI] [PubMed] [Google Scholar]
- 70.Mirzakhalili E., Adam C. D., Ulyanova A. V., Johnson V. E. & Wolf J. A. Automatic High-Frequency Oscillations Detection Using Time-Frequency Analysis. 2023 11th Int Ieee Embs Conf Neural Eng Ner 00, 1–6, doi: 10.1109/ner52421.2023.10123882 (2023). [DOI]
- 71.Bramlett H. M. & Dietrich D. W. Quantitative structural changes in white and gray matter 1 year following traumatic brain injury in rats. Acta Neuropathol. 103, 607–614, doi: 10.1007/s00401-001-0510-8 (2002). [DOI] [PubMed] [Google Scholar]
- 72.McIntosh T. K. et al. Traumatic brain injury in the rat: Characterization of a lateral fluid-percussion model. Neuroscience 28, 233–244, doi: 10.1016/0306-4522(89)90247-9 (1989). [DOI] [PubMed] [Google Scholar]
- 73.Saatman K. E., Graham D. I. & McIntosh T. K. The Neuronal Cytoskeleton Is at Risk After Mild and Moderate Brain Injury. Journal of Neurotrauma 15, 1047–1058, doi: 10.1089/neu.1998.15.1047 (1998). [DOI] [PubMed] [Google Scholar]
- 74.Smith D. H., Okiyama K., Thomas M. J., Claussen B. & McIntosh T. K. Evaluation of Memory Dysfunction Following Experimental Brain Injury Using the Morris Water Maze. Journal of Neurotrauma 8, 259–269, doi: 10.1089/neu.1991.8.259 (1991). [DOI] [PubMed] [Google Scholar]
- 75.Thompson H. J. et al. Lateral Fluid Percussion Brain Injury: A 15-Year Review and Evaluation. Journal of Neurotrauma 22, 42–75, doi: 10.1089/neu.2005.22.42 (2005). [DOI] [PubMed] [Google Scholar]
- 76.Ndode-Ekane X. E. et al. Harmonization of lateral fluid-percussion injury model production and post-injury monitoring in a preclinical multicenter biomarker discovery study on post-traumatic epileptogenesis. Epilepsy Res. 151, 7–16, doi: 10.1016/j.eplepsyres.2019.01.006 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vink R., Mullins P. G. M., Temple M. D., Bao W. & Faden A. I. Small Shifts in Craniotomy Position in the Lateral Fluid Percussion Injury Model Are Associated with Differential Lesion Development. Journal of Neurotrauma 18, 839–847, doi: 10.1089/089771501316919201 (2001). [DOI] [PubMed] [Google Scholar]
- 78.Gentleman S. M., Nash M. J., Sweeting C. J., Graham D. I. & Roberts G. W. β-Amyloid precursor protein (βAPP) as a marker for axonal injury after head injury. Neurosci. Lett. 160, 139–144, doi: 10.1016/0304-3940(93)90398-5 (1993). [DOI] [PubMed] [Google Scholar]
- 79.Johnson V. E. et al. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 136, 28–42, doi: 10.1093/brain/aws322 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Johnson V. E., Stewart W. & Smith D. H. Axonal pathology in traumatic brain injury. Experimental Neurology 246, 35–43, doi: 10.1016/j.expneurol.2012.01.013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Johnson V. E. et al. SNTF immunostaining reveals previously undetected axonal pathology in traumatic brain injury. Acta Neuropathol. 131, 115–135, doi: 10.1007/s00401-015-1506-0 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Freund T. F. & Buzsáki G. Interneurons of the hippocampus. Hippocampus 6, 347–470, doi: (1996). [DOI] [PubMed] [Google Scholar]
- 83.Pelkey K. A. et al. Hippocampal GABAergic Inhibitory Interneurons. Physiol Rev 97, 1619–1747, doi: 10.1152/physrev.00007.2017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lasztóczi B. & Klausberger T. Layer-Specific GABAergic Control of Distinct Gamma Oscillations in the CA1 Hippocampus. Neuron 81, 1126–1139, doi: 10.1016/j.neuron.2014.01.021 (2014). [DOI] [PubMed] [Google Scholar]
- 85.Royer S. et al. Control of timing, rate and bursts of hippocampal place cells by dendritic and somatic inhibition. Nat Neurosci 15, 769–775, doi: 10.1038/nn.3077 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vida I., Bartos M. & Jonas P. Shunting Inhibition Improves Robustness of Gamma Oscillations in Hippocampal Interneuron Networks by Homogenizing Firing Rates. Neuron 49, 107–117, doi: 10.1016/j.neuron.2005.11.036 (2006). [DOI] [PubMed] [Google Scholar]
- 87.Schlingloff D., Káli S., Freund T. F., Hájos N. & Gulyás A. I. Mechanisms of Sharp Wave Initiation and Ripple Generation. The Journal of Neuroscience 34, 11385–11398, doi: 10.1523/jneurosci.0867-14.2014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stark E. et al. Pyramidal Cell-Interneuron Interactions Underlie Hippocampal Ripple Oscillations. Neuron 83, 467–480, doi: 10.1016/j.neuron.2014.06.023 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ylinen A. et al. Sharp wave-associated high-frequency oscillation (200 Hz) in the intact hippocampus: network and intracellular mechanisms. Journal of Neuroscience 15, 30–46, doi: 10.1523/jneurosci.15-01-00030.1995 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Almeida-Suhett C. P. et al. GABAergic interneuronal loss and reduced inhibitory synaptic transmission in the hippocampal CA1 region after mild traumatic brain injury. Experimental Neurology 273, 11–23, doi: 10.1016/j.expneurol.2015.07.028 (2015). [DOI] [PubMed] [Google Scholar]
- 91.Frankowski J. C., Kim Y. J. & Hunt R. F. Selective vulnerability of hippocampal interneurons to graded traumatic brain injury. Neurobiology of Disease 129, 208–216, doi: 10.1016/j.nbd.2018.07.022 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ulyanova A. V. et al. Hippocampal interneuronal dysfunction and hyperexcitability in a porcine model of concussion. Commun. Biol. 6, 1136, doi: 10.1038/s42003-023-05491-w (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cortez S. C., McIntosh T. K. & Noble L. J. Experimental fluid percussion brain injury: vascular disruption and neuronal and glial alterations. Brain Research 482, 271–282, doi: 10.1016/0006-8993(89)91190-6 (1989). [DOI] [PubMed] [Google Scholar]
- 94.Hicks R. R., Smith D. H., Lowenstein D. H., Marie R. S. & McIntosh T. K. Mild Experimental Brain Injury in the Rat Induces Cognitive Deficits Associated with Regional Neuronal Loss in the Hippocampus. Journal of Neurotrauma 10, 405–414, doi: 10.1089/neu.1993.10.405 (1993). [DOI] [PubMed] [Google Scholar]
- 95.Maxwell W. L. et al. There Is Differential Loss of Pyramidal Cells from the Human Hippocampus with Survival after Blunt Head Injury. J. Neuropathol. Exp. Neurol. 62, 272–279, doi: 10.1093/jnen/62.3.272 (2003). [DOI] [PubMed] [Google Scholar]
- 96.Swartz B. E. et al. Hippocampal Cell Loss in Posttraumatic Human Epilepsy. Epilepsia 47, 1373–1382, doi: 10.1111/j.1528-1167.2006.00602.x (2006). [DOI] [PubMed] [Google Scholar]
- 97.Christidi F. et al. Diffusion Tensor Imaging of the Perforant Pathway Zone and Its Relation to Memory Function in Patients with Severe Traumatic Brain Injury. Journal of Neurotrauma 28, 711–725, doi: 10.1089/neu.2010.1644 (2011). [DOI] [PubMed] [Google Scholar]
- 98.Colley B. S., Phillips L. L. & Reeves T. M. The effects of cyclosporin-A on axonal conduction deficits following traumatic brain injury in adult rats. Experimental Neurology 224, 241–251, doi: 10.1016/j.expneurol.2010.03.026 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Reeves T. M., Phillips L. L. & Povlishock J. T. Myelinated and unmyelinated axons of the corpus callosum differ in vulnerability and functional recovery following traumatic brain injury. Experimental Neurology 196, 126–137, doi: 10.1016/j.expneurol.2005.07.014 (2005). [DOI] [PubMed] [Google Scholar]
- 100.Deng P. & Xu Z. C. Contribution of Ih to Neuronal Damage in the Hippocampus after Traumatic Brain Injury in Rats. Journal of Neurotrauma 28, 1173–1183, doi: 10.1089/neu.2010.1683 (2011). [DOI] [PubMed] [Google Scholar]
- 101.Karimi S. A., Hosseinmardi N., Sayyah M., Hajisoltani R. & Janahmadi M. Enhancement of intrinsic neuronal excitability-mediated by a reduction in hyperpolarization-activated cation current (Ih) in hippocampal CA1 neurons in a rat model of traumatic brain injury. Hippocampus 31, 156–169, doi: 10.1002/hipo.23270 (2021). [DOI] [PubMed] [Google Scholar]
- 102.Sizemore G. et al. Temporal Lobe Epilepsy, Stroke, and Traumatic Brain Injury: Mechanisms of Hyperpolarized, Depolarized, and Flow-Through Ion Channels Utilized as Tri-Coordinate Biomarkers of Electrophysiologic Dysfunction. OBM Neurobiol. 2, 1-1, doi: 10.21926/obm.neurobiol.1802009 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Leonard J. R., Grady M. S., Lee M. E., Paz J. C. & Westrum L. E. Fluid Percussion Injury Causes Disruption of the Septohippocampal Pathway in the Rat. Experimental Neurology 143, 177–187, doi: 10.1006/exnr.1996.6366 (1997). [DOI] [PubMed] [Google Scholar]
- 104.Borhegyi Z., Varga V., Szilágyi N., Fabo D. & Freund T. F. Phase Segregation of Medial Septal GABAergic Neurons during Hippocampal Theta Activity. The Journal of Neuroscience 24, 8470–8479, doi: 10.1523/jneurosci.1413-04.2004 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hangya B., Borhegyi Z., Szilagyi N., Freund T. F. & Varga V. GABAergic Neurons of the Medial Septum Lead the Hippocampal Network during Theta Activity. Journal of Neuroscience 29, 8094–8102, doi: 10.1523/jneurosci.5665-08.2009 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Varga V. et al. The presence of pacemaker HCN channels identifies theta rhythmic GABAergic neurons in the medial septum. J. Physiol. 586, 3893–3915, doi: 10.1113/jphysiol.2008.155242 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Freund T. F. & Antal M. GABA-containing neurons in the septum control inhibitory interneurons in the hippocampus. Nature 336, 170–173, doi: 10.1038/336170a0 (1988). [DOI] [PubMed] [Google Scholar]
- 108.Arciniegas D. B. The cholinergic hypothesis of cognitive impairment caused by traumatic brain injury. Curr Psychiat Rep 5, 391–399, doi: 10.1007/s11920-003-0074-5 (2003). [DOI] [PubMed] [Google Scholar]
- 109.Shin S. S. & Dixon C. E. Alterations in Cholinergic Pathways and Therapeutic Strategies Targeting Cholinergic System after Traumatic Brain Injury. Journal of Neurotrauma 32, 1429–1440, doi: 10.1089/neu.2014.3445 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kramis R., Vanderwolf C. H. & Bland B. H. Two types of hippocampal rhythmical slow activity in both the rabbit and the rat: Relations to behavior and effects of atropine, diethyl ether, urethane, and pentobarbital. Experimental Neurology 49, 58–85, doi: 10.1016/0014-4886(75)90195-8 (1975). [DOI] [PubMed] [Google Scholar]
- 111.Vandecasteele M. et al. Optogenetic activation of septal cholinergic neurons suppresses sharp wave ripples and enhances theta oscillations in the hippocampus. Proceedings of the National Academy of Sciences 111, 13535–13540, doi: 10.1073/pnas.1411233111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vanderwolf C. H. Neocortical and hippocampal activation in relation to behavior: Effects of atropine, eserine, phenothiazines, and amphetamine. J. Comp. Physiol. Psychol. 88, 300–323, doi: 10.1037/h0076211 (1975). [DOI] [PubMed] [Google Scholar]
- 113.Almeida L. d., Idiart M. & Lisman J. E. A Second Function of Gamma Frequency Oscillations: An E%-Max Winner-Take-All Mechanism Selects Which Cells Fire. The Journal of Neuroscience 29, 7497–7503, doi: 10.1523/jneurosci.6044-08.2009 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dragoi G. Cell assemblies, sequences and temporal coding in the hippocampus. Current Opinion in Neurobiology 64, 111–118, doi: 10.1016/j.conb.2020.03.003 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Senior T. J., Huxter J. R., Allen K., O’Neill J. & Csicsvari J. Gamma Oscillatory Firing Reveals Distinct Populations of Pyramidal Cells in the CA1 Region of the Hippocampus. The Journal of Neuroscience 28, 2274–2286, doi: 10.1523/jneurosci.4669-07.2008 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Colgin L. L. et al. Frequency of gamma oscillations routes flow of information in the hippocampus. Nature 462, 353–357, doi: 10.1038/nature08573 (2009). [DOI] [PubMed] [Google Scholar]
- 117.Jezek K., Henriksen E. J., Treves A., Moser E. I. & Moser M.-B. Theta-paced flickering between place-cell maps in the hippocampus. Nature 478, 246–249, doi: 10.1038/nature10439 (2011). [DOI] [PubMed] [Google Scholar]
- 118.Foster D. J. & Wilson M. A. Hippocampal theta sequences. Hippocampus 17, 1093–1099, doi: 10.1002/hipo.20345 (2007). [DOI] [PubMed] [Google Scholar]
- 119.Gupta A. S., Meer M. A. A. v. d., Touretzky D. S. & Redish A. D. Segmentation of spatial experience by hippocampal theta sequences. Nat Neurosci 15, 1032–1039, doi: 10.1038/nn.3138 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Skaggs W. E., McNaughton B. L., Wilson M. A. & Barnes C. A. Theta phase precession in hippocampal neuronal populations and the compression of temporal sequences. Hippocampus 6, 149–172, doi: (1996). [DOI] [PubMed] [Google Scholar]
- 121.Dragoi G. & Buzsáki G. Temporal Encoding of Place Sequences by Hippocampal Cell Assemblies. Neuron 50, 145–157, doi: 10.1016/j.neuron.2006.02.023 (2006). [DOI] [PubMed] [Google Scholar]
- 122.O’Keefe J. & Recce M. L. Phase relationship between hippocampal place units and the EEG theta rhythm. Hippocampus 3, 317–330, doi: 10.1002/hipo.450030307 (1993). [DOI] [PubMed] [Google Scholar]
- 123.Broussard J. I. et al. Optogenetic Stimulation of CA1 Pyramidal Neurons at Theta Enhances Recognition Memory in Brain Injured Animals. Journal of Neurotrauma 40, 2442–2448, doi: 10.1089/neu.2023.0078 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lee D. J. et al. Medial Septal Nucleus Theta Frequency Deep Brain Stimulation Improves Spatial Working Memory after Traumatic Brain Injury. Journal of Neurotrauma 30, 131–139, doi: 10.1089/neu.2012.2646 (2013). [DOI] [PubMed] [Google Scholar]
- 125.Lee D. J. et al. Septohippocampal Neuromodulation Improves Cognition after Traumatic Brain Injury. Journal of Neurotrauma 32, 1822–1832, doi: 10.1089/neu.2014.3744 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sweet J. A., Eakin K. C., Munyon C. N. & Miller J. P. Improved learning and memory with theta-burst stimulation of the fornix in rat model of traumatic brain injury. Hippocampus 24, 1592–1600, doi: 10.1002/hipo.22338 (2014). [DOI] [PubMed] [Google Scholar]
- 127.Chang J., Phelan M. & Cummings B. J. A meta-analysis of efficacy in pre-clinical human stem cell therapies for traumatic brain injury. Experimental Neurology 273, 225–233, doi: 10.1016/j.expneurol.2015.08.020 (2015). [DOI] [PubMed] [Google Scholar]
- 128.Zhu B., Eom J. & Hunt R. F. Transplanted interneurons improve memory precision after traumatic brain injury. Nature Communications 10, 5156, doi: 10.1038/s41467-019-13170-w (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Girgis F., Pace J., Sweet J. & Miller J. P. Hippocampal Neurophysiologic Changes after Mild Traumatic Brain Injury and Potential Neuromodulation Treatment Approaches. Frontiers in Systems Neuroscience 10, 8, doi: 10.3389/fnsys.2016.00008 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pevzner A., Izadi A., Lee D. J., Shahlaie K. & Gurkoff G. G. Making Waves in the Brain: What Are Oscillations, and Why Modulating Them Makes Sense for Brain Injury. Frontiers in Systems Neuroscience 10, 30, doi: 10.3389/fnsys.2016.00030 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wolf J. A. & Koch P. F. Disruption of Network Synchrony and Cognitive Dysfunction After Traumatic Brain Injury. Frontiers in Systems Neuroscience 10, 43, doi: 10.3389/fnsys.2016.00043 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Babiloni C. et al. Brain neural synchronization and functional coupling in Alzheimer’s disease as revealed by resting state EEG rhythms. Int. J. Psychophysiol. 103, 88–102, doi: 10.1016/j.ijpsycho.2015.02.008 (2016). [DOI] [PubMed] [Google Scholar]
- 133.Bazzigaluppi P. et al. Early-stage attenuation of phase-amplitude coupling in the hippocampus and medial prefrontal cortex in a transgenic rat model of Alzheimer’s disease. J. Neurochem. 144, 669–679, doi: 10.1111/jnc.14136 (2018). [DOI] [PubMed] [Google Scholar]
- 134.Caixeta F. V., Cornélio A. M., Scheffer-Teixeira R., Ribeiro S. & Tort A. B. L. Ketamine alters oscillatory coupling in the hippocampus. Sci. Rep. 3, 2348, doi: 10.1038/srep02348 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chauvière L. et al. Early Deficits in Spatial Memory and Theta Rhythm in Experimental Temporal Lobe Epilepsy. The Journal of Neuroscience 29, 5402–5410, doi: 10.1523/jneurosci.4699-08.2009 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Dugladze T. et al. Impaired hippocampal rhythmogenesis in a mouse model of mesial temporal lobe epilepsy. Proceedings of the National Academy of Sciences 104, 17530–17535, doi: 10.1073/pnas.0708301104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Jacobson T. K. et al. Hippocampal theta, gamma, and theta-gamma coupling: effects of aging, environmental change, and cholinergic activation. J. Neurophysiol. 109, 1852–1865, doi: 10.1152/jn.00409.2012 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Moran L. V. & Hong L. E. High vs Low Frequency Neural Oscillations in Schizophrenia. Schizophr. Bull. 37, 659–663, doi: 10.1093/schbul/sbr056 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Shuman T. et al. Breakdown of spatial coding and interneuron synchronization in epileptic mice. Nat Neurosci 23, 229–238, doi: 10.1038/s41593-019-0559-0 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Shuman T., Amendolara B. & Golshani P. Theta Rhythmopathy as a Cause of Cognitive Disability in TLE. Epilepsy Curr. 17, 107–111, doi: 10.5698/1535-7511.17.2.107 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kundu B., Brock A. A., Englot D. J., Butson C. R. & Rolston J. D. Deep brain stimulation for the treatment of disorders of consciousness and cognition in traumatic brain injury patients: a review. Neurosurg. Focus 45, E14, doi: 10.3171/2018.5.focus18168 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mankin E. A. & Fried I. Modulation of Human Memory by Deep Brain Stimulation of the Entorhinal-Hippocampal Circuitry. Neuron 106, 218–235, doi: 10.1016/j.neuron.2020.02.024 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Miller J. P. et al. Visual-spatial memory may be enhanced with theta burst deep brain stimulation of the fornix: a preliminary investigation with four cases. Brain 138, 1833–1842, doi: 10.1093/brain/awv095 (2015). [DOI] [PubMed] [Google Scholar]
- 144.Ponce F. A. et al. Bilateral deep brain stimulation of the fornix for Alzheimer’s disease: surgical safety in the ADvance trial. Journal of Neurosurgery 125, 75–84, doi: 10.3171/2015.6.jns15716 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Brignani D., Manganotti P., Rossini P. M. & Miniussi C. Modulation of cortical oscillatory activity during transcranial magnetic stimulation. Hum. Brain Mapp. 29, 603–612, doi: 10.1002/hbm.20423 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hallett M. Transcranial Magnetic Stimulation: A Primer. Neuron 55, 187–199, doi: 10.1016/j.neuron.2007.06.026 (2007). [DOI] [PubMed] [Google Scholar]
- 147.Huerta P. T. & Volpe B. T. Transcranial magnetic stimulation, synaptic plasticity and network oscillations. J. Neuroeng. Rehabilitation 6, 7, doi: 10.1186/1743-0003-6-7 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Helfrich Randolph F. et al. Entrainment of Brain Oscillations by Transcranial Alternating Current Stimulation. Curr. Biol. 24, 333–339, doi: 10.1016/j.cub.2013.12.041 (2014). [DOI] [PubMed] [Google Scholar]
- 149.Vogeti S., Boetzel C. & Herrmann C. S. Entrainment and Spike-Timing Dependent Plasticity – A Review of Proposed Mechanisms of Transcranial Alternating Current Stimulation. Frontiers in Systems Neuroscience 16, 827353, doi: 10.3389/fnsys.2022.827353 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mueller J., Legon W., Opitz A., Sato T. F. & Tyler W. J. Transcranial Focused Ultrasound Modulates Intrinsic and Evoked EEG Dynamics. Brain Stimul. 7, 900–908, doi: 10.1016/j.brs.2014.08.008 (2014). [DOI] [PubMed] [Google Scholar]
- 151.Yuan Y., Yan J., Ma Z. & Li X. Noninvasive Focused Ultrasound Stimulation Can Modulate Phase-Amplitude Coupling between Neuronal Oscillations in the Rat Hippocampus. Front. Neurosci. 10, 348, doi: 10.3389/fnins.2016.00348 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Pettersen K. H., Devor A., Ulbert I., Dale A. M. & Einevoll G. T. Current-source density estimation based on inversion of electrostatic forward solution: Effects of finite extent of neuronal activity and conductivity discontinuities. J Neurosci Meth 154, 116–133, doi: 10.1016/j.jneumeth.2005.12.005 (2006). [DOI] [PubMed] [Google Scholar]
- 153.Berens P. CircStat : A MATLAB Toolbox for Circular Statistics. J. Stat. Softw. 31, doi: 10.18637/jss.v031.i10 (2009). [DOI] [Google Scholar]
- 154.Arts L. P. A. & Broek E. L. v. d. The fast continuous wavelet transformation (fCWT) for real-time, high-quality, noise-resistant time–frequency analysis. Nat. Comput. Sci. 2, 47–58, doi: 10.1038/s43588-021-00183-z (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Weber M. T., Arena J. D., Xiao R., Wolf J. A. & Johnson V. E. CLARITY reveals a more protracted temporal course of axon swelling and disconnection than previously described following traumatic brain injury. Brain Pathol. 29, 437–450, doi: 10.1111/bpa.12677 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.