
Fig. 1.

Formulas for potential energy of a molecular complex according to the QMPFF potential. The position of the nucleus of atom a is specified by the vector Ra and the offset of the electron cloud by ta; Ba denotes the set of atoms bonded to atom a. The potential is found by minimization of the sum of ES, EX, and IN terms with respect to vectors ta. Summation of the binary atom–atom ES, EX, and DS interactions is performed over all the atoms in the complex, implying the “1–3 rule” with the terms being dropped for atom pairs separated by one or two chemical bonds (along with the interaction of the clouds with their nuclear cores); for “1–4” interactions, a special renormalization is used (see text). These rules are introduced to comply with the next version of QMPFF, which will allow flexible valence interactions. The unary term 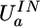 simulates the potential restraining the cloud a to remain close to a reference position. The induction energy can be calculated as the difference between UTOTAL and the energy formally calculated with the clouds fixed at their positions in isolated molecule(s). QMPFF parameters are marked by tilde signs. Parameters for each atom, a: Z̃a, charge;
simulates the potential restraining the cloud a to remain close to a reference position. The induction energy can be calculated as the difference between UTOTAL and the energy formally calculated with the clouds fixed at their positions in isolated molecule(s). QMPFF parameters are marked by tilde signs. Parameters for each atom, a: Z̃a, charge; 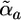 , polarizability; C̃a, exchange strength; w̃a, cloud size; R̃a, dispersion range; Ẽa, dispersion strength. The core charges, Z̃a, are currently fixed at values given in the text; the fixed scale factor R0 is set equal to 1 Å; the parameter t̃max is common for all the atom types. Parameters for each bond, a to b: Q̃ab, bond charge increment; t̃ab, reference cloud shift along bond. (Inset) A schematic representation of a pair of interacting water molecules. (Inset Lower) Atoms drawn as space filling spheres, red for oxygen and gray for hydrogen. (Inset Upper) Nuclei as small spheres with core point charges. The diffuse clouds around (but not necessarily centered on) the nuclei are the electron densities. The arrows illustrate the shifts of the electron cloud centers caused by the external field. It is this movement of diffuse electronic clouds that makes QMPFF polarizable and much more realistic than normal simple point charge force fields. Although the formulae used are much more complicated than for normal force fields, QMPFF is only a factor of 10–20 slower to use; QM would be a million times slower.
, polarizability; C̃a, exchange strength; w̃a, cloud size; R̃a, dispersion range; Ẽa, dispersion strength. The core charges, Z̃a, are currently fixed at values given in the text; the fixed scale factor R0 is set equal to 1 Å; the parameter t̃max is common for all the atom types. Parameters for each bond, a to b: Q̃ab, bond charge increment; t̃ab, reference cloud shift along bond. (Inset) A schematic representation of a pair of interacting water molecules. (Inset Lower) Atoms drawn as space filling spheres, red for oxygen and gray for hydrogen. (Inset Upper) Nuclei as small spheres with core point charges. The diffuse clouds around (but not necessarily centered on) the nuclei are the electron densities. The arrows illustrate the shifts of the electron cloud centers caused by the external field. It is this movement of diffuse electronic clouds that makes QMPFF polarizable and much more realistic than normal simple point charge force fields. Although the formulae used are much more complicated than for normal force fields, QMPFF is only a factor of 10–20 slower to use; QM would be a million times slower.