

Abstract
Background
PITX2 is a bicoid-related homeodomain transcription factor that plays an important role in asymmetric cardiogenesis. Loss of function experiments in mice cause severe heart malformations, including transposition of the great arteries (TGA). TGA accounts for 5–7% of all congenital heart diseases affecting 0.2 per 1000 live births, thereby representing the most frequent cyanotic heart defect diagnosed in the neonatal period.
Methods
To address whether altered PITX2 function could also contribute to the formation of dTGA in humans, we screened 96 patients with dTGA by means of dHPLC and direct sequencing for mutations within the PITX2 gene.
Results
Several SNPs could be detected, but no stop or frame shift mutation. In particular, we found seven intronic and UTR variants, two silent mutations and two polymorphisms within the coding region.
Conclusion
As most sequence variants were also found in controls we conclude that mutations in PITX2 are not a common cause of dTGA.
Background
With a frequency of up to 1%, congenital heart disease represents one of the most common major congenital anomalies [1-3]. Transposition of the great arteries (TGA) accounts for 5% of all congenital heart defects [4]. TGA manifests during the early fifth week of development affecting the septation of the common outflow tract into aorta and pulmonary arteries, and has been suggested to represent a laterality defect of the heart [5]. The more common dTGA (dextro-looped TGA) represents a complete inversion of the great vessels (atrioventricular concordance and ventriculoarterial discordance). In the less common lTGA (laevo-looped TGA), both atrioventricular and ventriculoarterial discordance is present. Despite the high prevalence and clinical importance of TGA, we are just beginning to unravel the etiology of this heterogeneous disease. Up to now, three genes have been suggested to be involved in the etiology of dTGA in humans: PROSIT240, a novel TRAP240-like gene, has been recently isolated and several mutations are suggested to be responsible for a subset of TGA patients [6]. Isolated mutations in ZIC3 [7] and CFC1 (human CRYPTIC gene) [8,9] have also been detected in patients with TGA. ZIC3 and CFC1 have been shown before to be involved in laterality defects in humans [8,10]. However the total number of mutations detected so far within these three genes is not sufficient to explain the high incidence of dTGA and point towards strong heterogeneity.
As cardiac neural crest cells contribute to the formation of the outflow septum that divides the common outflow tract, an association between neural crest disturbance and TGA has been suggested. Extirpation experiments in chick could show that neural crest cells contribute to normal aorticopulmonary septation. Deletion of those cells causes malformation of the aorticopulmonary septum resulting in common arterial outflow channels or transposition of the great arteries [11,12]. Pitx2, a bicoid-related homeodomain transcription factor involved in eye, heart and craniofacial development and establishment of left-right asymmetry, is expressed in several tissues of the developing mouse embryo including neural crest derived organs [13]. In humans, PITX2 haploinsufficiency causes Axenfeld-Rieger Syndrome (ARS), an autosomal dominant disorder involving ocular, dental and umbilical defects [14] and, in some patients with unknown mutations, also cardiac defects [15,16]. Most interestingly, Pitx2 loss of function experiments in mice cause severe cardiovascular defects including transposition of the great arteries [17-20]. Kioussi et al. reported that Pitx2-/- mice, that survive up to E15, invariantly exhibit major cardiac outflow tract abnormalities, amongst which 30% show incomplete septation of the great arteries, that may develop with double outlet right ventricle (DORV) or transposition of the great arteries [20]. Deletion of the Dvl2 gene [21], which is regulated by the same pathway as Pitx2, leads to the same severe outflow tract malformations, indicating a strong implication of this pathway in the outflow tract phenotype. These lines of evidence prompted us to investigate whether PITX2 mutations in humans can also contribute to the etiology of TGA.
Methods
Human subjects and genomic DNA
Peripheral-blood samples were taken from healthy individuals and patients with simple dTGA after informed consent had been obtained, after approval by the institutional review board of ethics of the Medical Department of the University of Heidelberg and the Newcastle and North Tyneside Health Authority Joint Ethics Committee. Genomic DNA was prepared using the Puregene DNA Isolation Kit (Gentra, Inc., USA).
PCR and mutation screening
Amplifications were performed using the High Fidelity System (Roche) according to the manufacturer's protocol. Primers were designed according to the PITX2 sequence gene bank accession number AF238048 and respective sequences are given in table 1. Mutation screening was performed using denaturing high performance liquid chromatography (DHPLC). A WAVE DNA-Fragment Analysis System (Transgenomic Inc., Cheshire) was used.
Table 1.
Primer pairs used for mutation analysis, covering the coding region of PITX2.
| exon | primer name | sequence 5'> 3' | TA°C | reference |
| 2 | PITX2-exon2for: PITX2-exon2rev: |
tag tct cat ctg agc cct gc gcg att tgg ttc tga ttt cct |
60 | Ref: [25] this paper |
| 3 | PITX2-exon3bfor: PITX2-exon3brev: |
ttg ctc ttt gtc cct ctt tct cct cgg agt gtc taa gtt caa gca gca |
60 | this paper |
| 4a | PITX2-exon4afor: PITX2-exon4arev: |
ccg cct ctg gtt tta aga tg gca aag acc ccc ttc ttc tc |
60 | this paper |
| 4b | PITX2-exon4bfor: PITX2-exon4brev: |
ctt gac act tct ctg tca gg aag cgg gaa tgt ctg cag g |
60/56/52* | Ref: [25] |
| 5 | PITX2-exon5for: PITX2-exon5rev: |
cag ctc ttc cac ggc ttc t ttc tct cct ggt cta ctt gg |
60 | Ref: [25] |
| 6 | PITX2-exon6for: PITX2-exon6rev: |
gta atc tgc act gtg gca tc agt ctt tca agg gcg gag tt |
65 | Ref: [25] |
TA: Annealing temperature
* step down PCR was performed with three temperatures for 10/10/15 cycles.
Sequencing
Sequencing was performed on a MegaBACE sequencer (Amersham Bioscience, Piscataway) using the DYEnamic™ ET terminator Cycle Sequencing Kit following the manufacturer's protocol. Sequencing reactions were performed on both DNA strands. Sequences were analyzed using the Clustal program (German Cancer Research Center, Biocomputing Facility HUSAR, Heidelberg).
Results and Discussion
96 patients with dTGA were analyzed for mutations in PITX2 by DHPLC and direct sequencing. All coding exons of PITX2 (exon 2 to 6, including both alternatively spliced exons 4a and b) were amplified by intron-specific exon-flanking primers to screen exon-intron junctions (table 1, figure 1). Non-coding regions (exon 1 and the 3'part of exon 6), intronic regions beyond the intronic sequences covered by the amplification, and promoter elements were not examined. We identified seven intronic and UTR variants, two silent mutations and two polymorphisms within the coding region. Most of these variants were found also in similar frequencies in 100 control individuals and are therefore unlikely to be of functional relevance. The missense mutation detected in exon 4b (204C>A, P65T) most likely represents a polymorphic variant compared to the sequence in the database, as the heterozygous form was invariantly detectable in all tested patients and controls. This finding also excludes large deletions in the patients affecting the whole gene locus. Three intronic (IVS2+7A>G, IVS3+11G>T, IVS4a-62C>A) and one silent mutations (30G>C Ser10Ser) were not detectable in 100 controls. One variant in the 5'UTR (2–40T>C) and one missense mutation (30C>T S27F) were only found once in control individuals (table 2).
Figure 1.
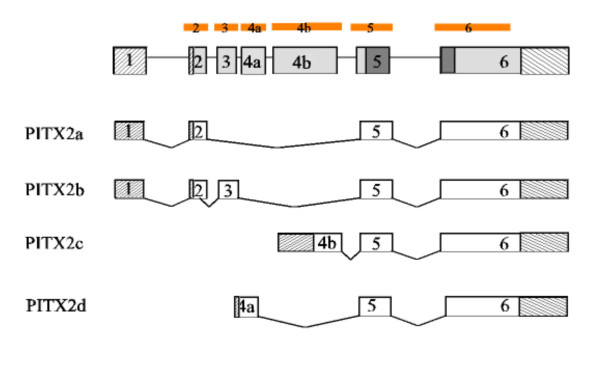
Schematic diagram of PITX2 isoforms a, b, c and d. The genomic organization of the PITX2 gene is given on top, exons are numbered, 5' and 3' UTRs of the different possible transcripts are indicated by striped boxes and the homeodomain is shaded in dark grey. Modified from Cox et al, 2002 [24]. The regions of the PITX2 gene included in the mutational screening are indicated as orange bars at the very top of the scheme.
Table 2.
Summary of PITX2 sequence variations in the dTGA study cohort
| patients (n = 96) | controls | |||
| type of variation: | specific variation | variant frequency (%) | number of controls | frequency (%) |
| intronic/UTR variations: | 2–40T>C (5'UTR exon 2) 2–18T>C (5'UTR exon 2) IVS2+7A>G (intron 2) IVS2-106C>A (intron 2) IVS3+11G>T (intron 3) IVS4a+11G (intron 4a) IVS4a-62C>A (intron 4a) |
10 (10.4%) 0 (0%) 1 (1.04%) 17 (17.7%) 1 (1.04%) 30 (31,25%) 1 (1.04%) |
100 100 100 100 100 100 100 |
12 (12%) 1 (1%) 0 (0%) 20 (20%) 0 (0%) 39 (39%) 0 (0%) |
| silent mutations: | 30G>C (S10S) (exon 2) 63C>T (A21A) (exon 4b) |
1 (1.04%) 1 (1.04%) |
100 100 |
0 (0%) 2 (2%) |
| polymorphism within coding region: | 30C>T (S27F) (exon 3) 204C>A (P65T) (exon 4b) |
0 (0%) 96 (100%) |
100 100 |
1 (1%) 100 (100%) |
UTR: untranslated region
We report on the mutation screening of PITX2, as we considered it to be an interesting candidate gene for TGA due to its role in regulating asymmetric cardiac morphogenesis [22] and interesting data from mouse studies. Impaired Pitx2 function in mice leads to severe cardiac malformations [17-20]. It has been suggested that altered PITX2 expression in the outflow tract could underlie either TGA or DORV [22].
PITX2 comprises three major isoforms, formed by differential splicing or alternative promotor usage: PITX2a, b, c, as well as one minor isoform PITX2d (Fig 1). We have included all coding exons in our screening as all forms exhibit a differential expression pattern [18,19]. Pitx2c is of special interest, as only this isoform is asymmetrically expressed within the lateral plate mesoderm and the heart and governs asymmetric organ morphogenesis in a dose-dependent manner [23,19]. Furthermore, the newly identified minor isoform, PITX2d, that in fact does not bind to DNA, was included in the study since it may influence expression levels of the other splice variants and also regulate the transcriptional activity of the major isoforms on protein level [24]. As only low amounts of PITX2 are required for normal cardiac development and as the different isoforms can possibly compensate for each other in some cell populations, it might require a combination of different sequence variants within different isoforms of the gene to dramatically reduce PITX2 function and therefore manifest a cardiac phenotype.
Conclusion
To address whether altered PITX2 function could also contribute to the formation of dTGA in humans, we screened the coding regions as well as exon-intron boundaries of the PITX2 gene for mutations in 96 patients with dTGA. The majority of detected variants, however, were also found in controls with comparable frequency. Three intronic and one silent mutation could not be detected in 100 controls. As they were only found once in the cohort of 96 patients and as none of the variants was found within the evolutionary conserved homeodomain, we consider them to be rare polymorphisms rather than functional mutations, although we cannot totally exclude the latter possibility. Further investigations will have to evaluate whether these sequence variants might change splicing processes. Due to the study design we can also not exclude mutations in the very 5'and 3' UTRs and within introns as well as the promoter regions of the gene. Nevertheless, we conclude that the detected mutations in PITX2 are not a common cause of dTGA.
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
NM designed the study, BN participated in the mutation analysis and drafted the manuscript, RR and KS performed PCRs and sequencing reactions. HJR, JG and EG provided patient care and collected blood samples. GR was involved in study design and supervision and finalized the manuscript. All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Acknowledgments
Acknowledgements
We would like to thank D. Driscoll, Children's Hospital of Philadelphia, for support in collecting DNA samples. N.M. was supported by the Landesgraduiertenförderung, Baden-Württemberg, Germany.
Contributor Information
Nadja Muncke, Email: nadja_muncke@med.uni-heidelberg.de.
Beate Niesler, Email: beate_niesler@med.uni-heidelberg.de.
Ralph Roeth, Email: ralph_roeth@med.uni-heidelberg.de.
Karin Schön, Email: karin_schoen@med.uni-heidelberg.de.
Heinz-Juergen Rüdiger, Email: Heinz-Juergen_Ruediger@med.uni-heidelberg.de.
Elizabeth Goldmuntz, Email: goldmuntz@email.chop.edu.
Judith Goodship, Email: j.a.goodship@ncl.ac.uk.
Gudrun Rappold, Email: gudrun_rappold@med.uni-heidelberg.de.
References
- Hoffman JI. Incidence of congenital heart disease: I. Postnatal incidence. Pediatr Cardiol. 1995;16:103–113. doi: 10.1007/BF00801907. [DOI] [PubMed] [Google Scholar]
- Hoffman JI. Incidence of congenital heart disease: II. Prenatal incidence. Pediatr Cardiol. 1995;16:155–165. doi: 10.1007/BF00801907. [DOI] [PubMed] [Google Scholar]
- Samanek M. Congenital heart malformations: prevalence, severity, survival, and quality of life. Cardiol Young. 2000;10:179–185. doi: 10.1017/s1047951100009082. [DOI] [PubMed] [Google Scholar]
- Fyler DC, Buckley LP, Hellenbrand WE, et al. Report of the New England regional infant cardiac program. Pediatrics. 1980;65:375–461. [PubMed] [Google Scholar]
- Digilio MC, Casey B, Toscano A, Calabro R, Pacileo G, Marasini M, Banaudi E, Giannotti A, Dallapiccola B, Marino B. Complete transposition of the great arteries: patterns of congenital heart disease in familial precurrence. Circulation. 2001;104:2809–2814. doi: 10.1161/hc4701.099786. [DOI] [PubMed] [Google Scholar]
- Muncke N, Jung C, Rudiger H, Ulmer H, Roeth R, Hubert A, Goldmuntz E, Driscoll D, Goodship J, Schon K, Rappold G. Missense mutations and gene interruption in PROSIT240, a novel TRAP240-like gene, in patients with congenital heart defect (transposition of the great arteries) Circulation. 2003;108:2843–2850. doi: 10.1161/01.CIR.0000103684.77636.CD. [DOI] [PubMed] [Google Scholar]
- Megarbane A, Salem N, Stephan E, Ashoush R, Lenoir D, Delague V, Kassab R, Loiselet J, Bouvagnet P. X-linked transposition of the great arteries and incomplete penetrance among males with a nonsense mutation in ZIC3. Eur J Hum Genet. 2000;8:704–708. doi: 10.1038/sj.ejhg.5200526. [DOI] [PubMed] [Google Scholar]
- Bamford RN, Roessler E, Burdine RD, Saplakoglu U, dela Cruz J, Splitt M, Towbin J, Bowers P, Ferrero GB, Marino B, Schier AF, Shen MM, Muenke M, Casey B. Loss-of-function mutations in the EGF-CFC gene CFC1 are associated with human left-right laterality defects. Nat Genet. 2000;26:365–369. doi: 10.1038/81695. [DOI] [PubMed] [Google Scholar]
- Goldmuntz E, Bamford R, Karkera JD, dela Cruz J, Roessler E, Muenke M. CFC1 mutations in patients with transposition of the great arteries and double-outlet right ventricle. Am J Hum Genet. 2002;70:776–780. doi: 10.1086/339079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebbia M, Ferrero GB, Pilia G, Bassi MT, Aylsworth A, Penman-Splitt M, Bird LM, Bamforth JS, Burn J, Schlessinger D, Nelson DL, Casey B. X-linked situs abnormalities result from mutations in ZIC3. Nat Genet. 1997;17:305–308. doi: 10.1038/ng1197-305. [DOI] [PubMed] [Google Scholar]
- Kirby ML, Gale TF, Stewart DE. Neural crest cells contribute to normal aorticopulmonary septation. Science. 1983;220:1059–1061. doi: 10.1126/science.6844926. [DOI] [PubMed] [Google Scholar]
- Creazzo TL, Godt RE, Leatherbury L, Conway SJ, Kirby ML. Role of cardiac neural crest cells in cardiovascular development. Annu Rev Physiol. 1998;60:267–286. doi: 10.1146/annurev.physiol.60.1.267. [DOI] [PubMed] [Google Scholar]
- Hjalt TA, Semina EV, Amendt BA, Murray JC. The Pitx2 protein in mouse development. Dev Dyn. 2000;218:195–200. doi: 10.1002/(SICI)1097-0177(200005)218:1<195::AID-DVDY17>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Semina EV, Reiter R, Leysens NJ, Alward WL, Small KW, Datson NA, Siegel-Bartelt J, Bierke-Nelson D, Bitoun P, Zabel BU, Carey JC, Murray JC. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat Genet. 1996;14:392–399. doi: 10.1038/ng1296-392. [DOI] [PubMed] [Google Scholar]
- Sadeghi-Nejad A, Senior B. Autosomal dominant transmission of isolated growth hormone deficiency in iris-dental dysplasia (Rieger's syndrome) J Pediatr. 1974;85:644–648. doi: 10.1016/s0022-3476(74)80507-x. [DOI] [PubMed] [Google Scholar]
- Brooks JK, Coccaro PJ, Jr, Zarbin MA. The Rieger anomaly concomitant with multiple dental, craniofacial, and somatic midline anomalies and short stature. Oral Surg Oral Med Oral Pathol. 1989;68:717–724. doi: 10.1016/0030-4220(89)90161-8. [DOI] [PubMed] [Google Scholar]
- Gage PJ, Suh H, Camper SA. Dosage requirement of Pitx2 for development of multiple organs. Development. 1999;126:4643–4651. doi: 10.1242/dev.126.20.4643. [DOI] [PubMed] [Google Scholar]
- Kitamura K, Miura H, Miyagawa-Tomita S, Yanazawa M, Katoh-Fukui Y, Suzuki R, Ohuchi H, Suehiro A, Motegi Y, Nakahara Y, Kondo S, Yokoyama M. Mouse Pitx2 deficiency leads to anomalies of the ventral body wall, heart, extra- and periocular mesoderm and right pulmonary isomerism. Development. 1999;126:5749–5758. doi: 10.1242/dev.126.24.5749. [DOI] [PubMed] [Google Scholar]
- Liu C, Liu W, Lu MF, Brown NA, Martin JF. Regulation of left-right asymmetry by thresholds of Pitx2c activity. Development. 2001;128:2039–2048. doi: 10.1242/dev.128.11.2039. [DOI] [PubMed] [Google Scholar]
- Kioussi C, Briata P, Baek SH, Rose DW, Hamblet NS, Herman T, Ohgi KA, Lin C, Gleiberman A, Wang J, Brault V, Ruiz-Lozano P, Nguyen HD, Kemler R, Glass CK, Wynshaw-Boris A, Rosenfeld MG. Identification of a Wnt/Dvl/beta-Catenin – > Pitx2 pathway mediating cell-type-specific proliferation during development. Cell. 2002;111:673–685. doi: 10.1016/S0092-8674(02)01084-X. [DOI] [PubMed] [Google Scholar]
- Hamblet NS, Lijam N, Ruiz-Lozano P, Wang J, Yang Y, Luo Z, Mei L, Chien KR, Sussman DJ, Wynshaw-Boris A. Dishevelled 2 is essential for cardiac outflow tract development, somite segmentation and neural tube closure. Development. 2002;129:5827–5838. doi: 10.1242/dev.00164. [DOI] [PubMed] [Google Scholar]
- Franco D, Campione M. The role of Pitx2 during cardiac development. Linking left-right signaling and congenital heart diseases. Trends Cardiovasc Med. 2003;13:157–163. doi: 10.1016/S1050-1738(03)00039-2. [DOI] [PubMed] [Google Scholar]
- Schweickert A, Campione M, Steinbeisser H, Blum M. Pitx2 isoforms: involvement of Pitx2c but not Pitx2a or Pitx2b in vertebrate left-right asymmetry. Mech Dev. 2000;90:41–51. doi: 10.1016/S0925-4773(99)00227-0. [DOI] [PubMed] [Google Scholar]
- Cox CJ, Espinoza HM, McWilliams B, Chappell K, Morton L, Hjalt TA, Semina EV, Amendt BA. Differential regulation of gene expression by PITX2 isoforms. J Biol Chem. 2002;277:25001–25010. doi: 10.1074/jbc.M201737200. [DOI] [PubMed] [Google Scholar]
- Martin DM, Probst FJ, Fox SE, Schimmenti LA, Semina EV, Hefner MA, Belmont JW, Camper SA. Exclusion of PITX2 mutations as a major cause of CHARGE association. Am J Med Genet. 2002;111:27–30. doi: 10.1002/ajmg.10473. [DOI] [PubMed] [Google Scholar]