

Abstract
Background:
KBG syndrome is a monogenic disorder caused by heterozygous pathogenic variants in ANKRD11. A recent single-case study suggested that the clinical spectrum of KBG syndrome, classically defined by distinctive craniofacial traits and developmental delay, may include movement disorders.
Case report:
We report a 24-year-old patient harboring a pathogenic de novo ANKRD11 frameshift variant. The phenotype was dominated by a progressive tremor-dominant movement disorder, characterized by rest, intention and postural tremor of the hands, voice tremor, head and tongue tremor, increased muscle tone and signs of ataxia. Additionally, the patient had a history of mild developmental delay and epilepsy.
Discussion:
Adding to the recently described individual, our present patient highlights the relevance of movement disorders as a clinically relevant manifestation of KBG syndrome. ANKRD11 pathogenic variants should be considered in the differential diagnosis of combined tremor syndromes.
Keywords: KBG syndrome, ANKRD11, tremor, combined tremor syndrome
Introduction
KBG syndrome (MIM 148050, ORPHA 2332) is a genetic neurodevelopmental disorder characterized by developmental delay (DD), learning difficulties or intellectual disability (ID) and distinct craniofacial features [1,2]. Additional observed features are seizures, microcephaly, changes in brain anatomy, psychiatric and behavioral abnormalities, conductive hearing disorders, eye abnormalities and congenital heart defects [1,3]. KBG syndrome is caused by single nucleotide pathogenic variants and small indels in ANKRD11 or lager copy number variants (mostly deletions) at 16q24.3 involving ANKRD11. Most variants are loss-of-function variants, with haploinsufficiency of ANKRD11 considered to be the pathogenic mechanism [1]. ANKRD11 encodes a protein that plays an important role in chromatin remodeling via histone acetylation during neuronal development and thus regulates gene expression. Definitive genotype-phenotype correlations do not exist [1].
Recently, the phenotypic spectrum of KBG syndrome was proposed to be extended by the umbrella term “epileptic dyskinetic encephalopathy” due to the description of a patient with KBG syndrome characterized by an early-onset developmental disorder with epileptic encephalopathy and dyskinetic movement disorder [4]. This patient had persistent daily seizures despite treatment with multiple antiepileptic drugs, whereas in most patients with KBG-syndrome the epilepsy responds well to antiepileptic treatment [1,4]. In addition, this was the first report of an association of variants in ANKRD11 with hyperkinetic movements. The movement disorder was characterized as infantile-onset jittery movements of the limbs and head, progressing into a mixed hyperkinetic movement disorder including choreoathetosis. Until then, movement disorders were not considered part of the phenotypic spectrum of KBG syndrome and are largely unreported in this context [4]. We now present a second patient of an ANKRD11-associated movement disorder and the first patient of an ANKRD11-associated tremor-dominant syndrome. We describe a 24-year-old male patient with a de novo frameshift duplication in ANKRD11, who displayed progressive and therapy-resistant intention, postural and resting tremor.
After review of the literature, this unusual presentation expands the phenotypic spectrum of KBG syndrome and adds ANKRD11 variants to the list of genetic alterations associated with tremor-dominant movement disorders.
Patient description
The boy first came to medical attention at the age of six weeks with pronounced sleep myoclonus. In addition, fluttering of the tongue when drinking and trembling of the lower lip, especially when excited, were observed. Subsequently, the infant had tremor of the hands (left>right), microcephaly and mild developmental delay, particularly in the development of motor skills with gait instability. Free walking was achieved at 18 months. The intensity of the tremor gradually increased with age. Awake-sleep EEGs performed at the age of 6 weeks and 8 weeks, as well as at 2 years after a fever-associated seizure, showed normal results without epileptic activity. An extended examination at the age of 16 years revealed epileptic activity on the EEG in the form of low- to medium-amplitude generalized, frontally accentuated (poly) spike-wave paroxysms. Follow-up EEGs continued to show the above described pathologic pattern, as a result of which antiepileptic therapy with levetiracetam (2× 1500 mg/d) was supplemented with topiramate (2× 50 mg/d) and carbamazepine (400 mg/d). An MRI examination of the head showed unremarkable findings apart from a suspected small arachnoid cyst in the left sylvian fissure and single small medullary lesions. Thyroid dysfunction or Wilson’s disease were excluded as the cause of the tremor by biochemical screening.
Currently, at the age of 24 years, the patient presented with a rest tremor and a fine-beating postural tremor of the arms and legs, as well as an intention tremor of the hands (Figure 1 and Video 1). In addition, he exhibited a voice tremor, mild head tremor, mild tongue tremor and increased muscle tone. There were significant restrictions in daily life due to tremor-related fine motor impairment (use of cutlery, drinking, writing). A response of the tremor to alcohol was not reported. A therapeutic approach with propranolol (2 × 40 mg/d) was unsuccessful, while primidone was not tolerated due to nausea. In the course of further detailed diagnostics, finger-to-nose test and heel-to-shin test showed bilateral dysmetria and hyperreflexia and spasticity of the upper and lower extremities were observed. In addition to the tremor and epilepsy, the patient presented with a dysplastic right kidney, chronic obstipation (especially in childhood), astigmatism with strabismus and a myopia (–3 Dpt.). Due to proteinuria, therapy with candesartan was carried out, the kidney function was unimpaired. The patient showed distinctive craniofacial features (triangular face, microcephalus, thin lips, protruding ears), an abduction deficit of the third and fourth finger and clinodactyly of the fifth fingers (Video 1). The second incisors and premolars in all four quadrants were absent. Since childhood the patient experienced recurrent middle ear infections (currently 2–3 times a year), so tympanostomy tubes were inserted. Hearing was not impaired.
Figure 1.
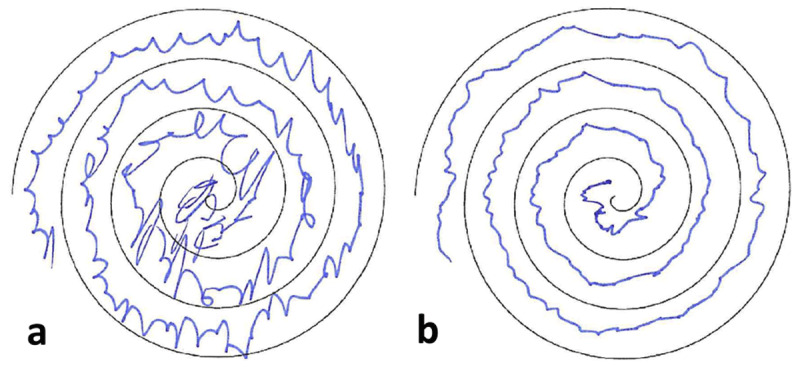
Visualization of the patient’s action tremor using Achimedean spiral. a left hand b right hand. The patient is right-handed.
Video 1.
Video documentation of the patient’s tremor-dominant movement disorder (including a jerky component) upon consultation at our institution. Segment A shows the patient’s resting tremor of the upper and lower extremities. Segment B documents the patient’s postural tremor of the hands. Segment C demonstrates the action tremor of the left hand. The patient showed pronounced action tremor of both hands (left>right) when tracing the Archimedean spiral.
Note characteristic facial features: microcephaly, triangular face, thin lips, protruding ears.
To further evaluate an underlying cause of the patient’s symptoms, trio whole-exome sequencing was performed at the age of 24 years. The analysis revealed a de novo heterozygous frameshift variant (NM_001256182.2: c.5469dup, p.(Pro1824SerfsTer126)) in ANKRD11 that was classified as pathogenic (ACMG criteria PVS1, PS2, PM2_supp and PP5). The variant predicts a frameshift and premature termination of protein translation, resulting in KBG syndrome in the patient.
Discussion
The patient’s symptoms were overall consistent with the previously described phenotype of KBG syndrome. However, the progressive tremor-dominant movement disorder, which significantly determined the phenotype of our patient, broadens the previously known phenotypic spectrum of the condition. To date, only one case of a movement disorder in association with an ANKRD11 variant has been described [4].
Tremor is one of the most common movement disorders in adults [5] and is defined as an “involuntary, rhythmic, oscillatory movement of a body part”. The consensus statement of the International Parkinson and Movement Disorder Society published in 2018 introduces a new biaxial tremor classification, whereby tremor is categorized according to clinical features (Axis 1) and etiology (Axis 2) [6]. Within Axis 1, a distinction is made between the presence of an “isolated” tremor (such as essential tremor, ET) or the occurrence of tremor as part of a more complex (neurological) disorder (“combined tremor”) [6,7].
Tremor is genetically heterogeneous despite high phenotypic similarity. Most monogenic tremors occur as part of an overarching syndrome and are part of a neurologic phenotypic spectrum (“combined” tremor syndromes) [6,7]. The Online Mendelian Inheritance in Man (OMIM) database lists 532 results (04/2024) when searching for the keyword “tremor”. In particular, numerous monogenic neurodegenerative, neuropathic and metabolic diseases manifest with tremor often accompanied by signs of other movement disorders, especially of a dystonic (e.g. ANO3, SGCE), ataxic (e.g. PPPP2R2B, ATX3, TBP) or parkinsonian (e.g. LRRK2, FMR1, ATX3) type. In some of these conditions, including autosomal dominant cranio-cervical dystonia (DYT-ANO3) and different subtypes of spinocerebellar ataxia (including SCA12 and SCA40), an initial presentation as an “isolated” tremor before an onset of further neurological symptoms in the form of dystonia or ataxia has been described, potentially leading to a misdiagnosis of ET (ET-like syndromes). This is further complicated by the fact that in many cases of ET other neurological findings occur in the course of time, which, however, in their severity do not allow for an alternative neurological diagnosis (so called soft signs). This concept of ET with other neurological signs of uncertain significance such as tandem gait impairment or mild memory impairment was recently introduced as “ET plus” [5,7].
On this basis, monogenic tremor syndromes are categorized into “isolated” tremor syndromes (ET-like syndromes), when the tremor is (yet) the only manifestation of the disease and “combined” tremor syndromes (either as “ET plus syndromes” or “tremor combined with other neurological and non-neurological features”) [7]. ET on the other hand, is defined as isolated action tremor of the upper extremity of a duration of at least three years in the absence of other neurological symptoms. Other parts of the body (lower limb, head and voice) may also be affected in the context of ET [8].
The fact that the here presented patient did not have an isolated action tremor but also a pronounced postural tremor, rest tremor and minor intention tremor in combination with accompanying spasticity, signs of ataxia and other systemic abnormalities clearly distinguishes the clinical picture from that of an ET. We therefore diagnose a monogenic combined tremor syndrome classified as “tremor combined with other neurological and non-neurological features” in our patient in the context of a KBG syndrome. Consequently, we propose ANKRD11-KBG syndrome to be included in the list of monogenic causes of a combined tremor syndrome. Retrospective and prospective analyses of cohorts with pathogenic ANKRD11 variants will be crucial to confirm the occurrence of movement disorders in this condition, which will be important for guiding clinical management and identifying potential therapeutic targets based on the gene’s known function. It should be noted that although trio exome sequencing revealed no other significant findings related to the patient’s symptoms, another monogenic cause for the tremulous movement disorder, while unlikely, cannot be completely ruled out due to technical limitations of exome sequencing.
Acknowledgements
The authors thank the patient and his family for consenting to participate in this report.
Funding Statement
JW and MZ receive research support from the German Research Foundation (DFG 458949627; WI-1820/14-1; ZE 1213/2-1). MZ acknowledges grant support by the European Joint Programme on Rare Diseases (EJP RD Joint Transnational Call 2022), and the German Federal Ministry of Education and Research (BMBF, Bonn, Germany), awarded to the project PreDYT (PREdictive biomarkers in DYsTonia, 01GM2302), by the Federal Ministry of Education and Research (BMBF) and the Free State of Bavaria under the Excellence Strategy of the Federal Government and the Länder, as well as by the Technical University of Munich – Institute for Advanced Study. M.Z. is a member of the Medical and Scientific Advisory Council of the Dystonia Medical Research Foundation and a member of the Governance Council of the International Cerebral Palsy Genomics Consortium. MZ’s research is supported by a “Schlüsselprojekt” grant from the Else Kröner-Fresenius-Stiftung (2022_EKSE.185).
Ethics and Consent
Ethical approval for genetic studies and publication of deidentified clinical and molecular data was obtained according to ethical guidelines.
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this work is consistent with those guidelines. The manuscript submitted for publication has been performed following ethical standards stated in the 1964 Declaration of Helsinki and its later amendments. Written informed consent was obtained from the patient for the publication of this case report and the publication of all iconographic and video materials.
Competing Interests
The authors have no competing interests to declare.
Author Contributions
AMS and TK contributed equally to this work.
FH and MZ contributed equally to this work.
References
- 1.Morel Swols D, Foster J, Tekin M. KBG syndrome. Orphanet J Rare Dis. Dec 19 2017; 12(1): 183. DOI: 10.1186/s13023-017-0736-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sirmaci A, Spiliopoulos M, Brancati F, et al. Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am J Hum Genet. Aug 12 2011. 89(2): 289–94. DOI: 10.1016/j.ajhg.2011.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Low K, Ashraf T, Canham N, et al. Clinical and genetic aspects of KBG syndrome. Am J Med Genet A. Nov 2016. 170(11): 2835–2846. DOI: 10.1002/ajmg.a.37842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Donnellan EP, Gorman KM, Shahwan A, Allen NM. Epileptic dyskinetic encephalopathy in KBG syndrome: Expansion of the phenotype. Epilepsy Behav Rep. 2024; 25: 100647. DOI: 10.1016/j.ebr.2024.100647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lenka A, Jankovic J. Tremor Syndromes: An Updated Review. Front Neurol. 2021; 12: 684835. DOI: 10.3389/fneur.2021.684835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhatia KP, Bain P, Bajaj N, et al. Consensus Statement on the classification of tremors. from the task force on tremor of the International Parkinson and Movement Disorder Society. Mov Disord. Jan 2018. 33(1): 75–87. DOI: 10.1002/mds.27121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Magrinelli F, Latorre A, Balint B, et al. Isolated and combined genetic tremor syndromes: a critical appraisal based on the 2018 MDS criteria. Parkinsonism Relat Disord. Aug 2020. 77: 121–140. DOI: 10.1016/j.parkreldis.2020.04.010 [DOI] [PubMed] [Google Scholar]
- 8.Lenka A, Pandey S. Essential Tremor: Five New Things. Neurol Clin Pract. Apr 2022; 12(2): 183–186. DOI: 10.1212/cpj.0000000000001145 [DOI] [PMC free article] [PubMed] [Google Scholar]