

Abstract
Interpreting the clinical significance of putative splice-altering variants outside canonical splice sites remains difficult without time-intensive experimental studies. To address this, we introduce Parallel Splice Effect Sequencing (ParSE-seq), a multiplexed assay to quantify variant effects on RNA splicing. We first apply this technique to study hundreds of variants in the arrhythmia-associated gene SCN5A. Variants are studied in ‘minigene’ plasmids with molecular barcodes to allow pooled variant effect quantification. We perform experiments in two cell types, including disease-relevant induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs). The assay strongly separates known control variants from ClinVar, enabling quantitative calibration of the ParSE-seq assay. Using these evidence strengths and experimental data, we reclassify 29 of 34 variants with conflicting interpretations and 11 of 42 variants of uncertain significance. In addition to intronic variants, we show that many synonymous and missense variants disrupted RNA splicing. Two splice-altering variants in the assay also disrupt splicing and sodium current when introduced into iPSC-CMs by CRISPR-Cas9 editing. ParSE-seq provides high-throughput experimental data for RNA-splicing to support precision medicine efforts and can be readily adopted to study other loss-of-function genotype-phenotype relationships.
Subject terms: Molecular medicine, Genetics research, RNA splicing
Interpreting the significance of putative splice-altering variants outside canonical splice sites remains challenging. Here, the authors describe ParSE-seq, a high-throughput assay to annotate the effect of germline variants on RNA-splicing. They calibrate the assay and deploy it to study hundreds of variants in the arrhythmia-associated gene SCN5A.
Introduction
Understanding the clinical consequences of genetic variation is one of the greatest challenges in contemporary human genetics1. While most research has focused on understanding exonic variants at the protein level, there is an increasing need to explore variant effects on splicing2–4. Sequencing studies have estimated that ~10% of pathogenic variants may act through disruptions of splicing outside of the canonical splice acceptor (AG) or donor (GT) splice sites, known as cryptic splicing (Fig. 1A)5,6. Despite these estimates, both intronic and exonic splice-altering variants have been less well-studied compared to protein-coding missense variants, largely due to the difficulty of identifying and functionally investigating this class of variants. Several assays exist for assessing variant-associated splicing, including direct measurements from patient tissue7–9, minigene assays10–12, and genome editing at the endogenous locus13–15. However, these assays are often not calibrated with large numbers of control benign and pathogenic variants and are too low-throughput to decipher the thousands of potential splice-altering variants in Mendelian disease genes. An alternative approach is high-throughput multiplexed assays that use deep sequencing to read out pools of splicing outcomes. Several successful implementations of this approach have enabled splicing investigation through saturation mutagenesis of specific genes16,17 and medium to large variant libraries by pooled oligonucleotide synthesis6,18–21. Building upon these seminal contributions, we sought to develop a versatile, accurate, multiplexed assay of putative splice-altering variants that could facilitate clinical classification of variants2.
Fig. 1. Splice-altering variants and ParSE-seq assay schematic.

A Splicing regulatory sequences disrupted or introduced by cis-genetic variants. A acceptor site. D donor site. ESE exonic splicing enhancer. ISE intronic splicing enhancer. Y pyrimidine. B Quantification of percent spliced in (PSI) from transcripts associated with WT- or variant-containing transcripts. Canonical reads are divided by the total amount of reads for a given exon triplet cassette. C Lollipop diagram of ClinVar reported SCN5A splice-altering variant locations23. Green track shows SCN5A exons (rectangles) and introns (narrow line). Most pathogenic (red) and likely pathogenic (orange) variants are located near the canonical splice sites, and are distributed throughout the gene product. There is only one variant of uncertain significance (yellow) and four conflicting interpretation variants (CI; gray) that are annotated as “splice-altering”. OMIM indicates Online Mendelian Inheritance of Man. Annotated genetrack obtained from https://www.ncbi.nlm.nih.gov/clinvar/. D Most SCN5A splice variants in ClinVar are associated with canonical splice sites, with only one ClinVar example of a non-canonical splice variant. Data in Source Data. E Schematic of ParSE-seq assay. A clonal gene library is cloned into a minigene vector (modified pET01), pooled, and barcoded. MCS indicates “multiple cloning site” for construct insertion (red triangle). Barcode are inserted into the downstream exon (purple); yellow circle represents restriction site for barcode in downstream exon. Barcodes (NNNN) are then assigned to the WT or variant (Var; red asterisk) inserts by long-read sequencing through assembly, and the splicing outcomes are determined with short-read sequencing after transfection into two cell types—human embryonic kidney cells (HEK) and induced pluripotent stem cell-cardiomyocytes (iPSC-CM). F The ParSE-seq SCN5A variant library was designed to evaluate three categories of variants: assay calibration, classification of clinically relevant variants, and testing of exonic variants with high SpliceAI scores. VUS variant of uncertain significance. CI conflicting interpretation. SNV single nucleotide variants. Complete descriptions of all variants studied are available in Supplementary Data 2.
In the 2015 American College of Medical Genetics and Genomics (ACMG) classification guidelines, well-validated functional data could be implemented at the strong level for abnormal assay results (PS3) or normal assay results (BS3)22. In 2020, the ClinGen Sequence Variant Interpretation Working Group developed a quantitative approach to calibrate such functional assays. The working group recommended that functional assays be deployed on many control benign/likely benign (B/LB) and pathogenic/likely pathogenic (P/LP) variants. These results are used to calculate an odds of pathogenicity (OddsPath), the likelihood ratio of variant pathogenicity given normal or abnormal assay results23,24. The OddsPath value can be converted into a quantitative evidence strength in the ACMG classification scheme, ranging from supporting to very strong evidence, depending on the number of tested control variants and the concordance of the assay results. Several high-throughput assays of protein-coding variants have been calibrated using this scheme25–27, but to our knowledge, functional assays of splice-altering variants have thus far not. A clinically calibrated, high-throughput splicing assay would facilitate the reclassification of variants of uncertain significance (VUS) and conflicting interpretation (CI) variants in disease-associated genes.
SCN5A encodes the main voltage-gated sodium channel in the heart, NaV1.5. Although Brugada syndrome (BrS) has a strong polygenic influence28, loss-of-function variants in SCN5A are the leading monogenic cause of the arrhythmia disorder BrS (MIM #601144)29, and are also associated with other cardiac disorders30. The majority of BrS-associated SCN5A variants reported to date are missense, frameshift, indels, or canonical splice site variants (variants in 2-bp exon-adjacent splice sites)31. We and others have previously deployed low-throughput functional assays to demonstrate splicing defects in several SCN5A exonic and intronic variants outside the 2-bp splice sequences14,32,33. Enabling larger scale investigations into splice-altering variants in SCN5A would decrease clinical uncertainty when managing patients at risk for this potentially fatal arrhythmia syndrome.
In this work, we present a multiplexed splicing assay, Parallel Splice Effect-sequencing (ParSE-seq), to determine the splice-altering consequences of hundreds of intronic and exonic variants in SCN5A. We implement ParSE-seq for 244 SCN5A variants in human embryonic kidney (HEK293) cells and 224 variants in induced pluripotent-derived cardiomyocyte cells (iPSC-CM). We calibrate the assay with nearly 50 ClinVar-annotated benign and pathogenic variants and compare our experimental outcomes to the in silico tool SpliceAI. Using our calibrated strength of evidence, we propose reclassifications for 9 VUS, and contribute functional data to help adjudicate 29 CI variants in ClinVar. Furthermore, we demonstrate that some missense variants may be incorrectly described as having normal function by conventional cDNA-based patch clamping assays that cannot assess splicing outcomes.
Results
Methodology overview and feasibility
We envisioned a high-throughput minigene-based assay that would build upon labor-intensive single-variant minigene studies and extend previous high-throughput functional assays by establishing a platform to investigate variants for higher clinical relevance. Variants disrupt RNA splicing through disruption or creation of splice sites or splicing regulatory elements3 (Fig. 1A; Supplementary Figs. 1 and 2). Our assay used a minigene plasmid containing a test construct bearing an exon and surrounding native intronic sequence, flanked by rat insulin exons 1 and 2. When feasible, we included large (250 bp) native intronic flanking regions on either side of the exon (lengths in Supplementary Table 1). We barcoded and pooled these minigene plasmids and used high-throughput RNA sequencing to quantify percent spliced in (PSI) of the wildtype (WT) or variant test exons (Fig. 1B). We designed a library to test the splicing effects of variants in SCN5A. Most ClinVar-annotated splice-altering variants in SCN5A are at the canonical splice sites, motivating us to explore other variants that may affect splicing in this gene (Fig. 1C, D). Toward this goal, we introduced two restriction sites into the 3′ exon of the minigene vector pET01 to allow for a barcode to be included in the spliced RNA-sequencing read14 (Fig. 1E; Supplementary Data 1; Supplementary Table 2 for primers; see “Methods” section for full details). This design enabled the linkage of reads to variants even if the variant was not included in the spliced transcript.
Assay implementation
We studied variants from two groups: (1) exonic and intronic variants clinically observed in individuals; and (2) exonic variants predicted by SpliceAI to disrupt splicing (Fig. 1F). Clinically observed variants came from gnomAD, ClinVar, and literature reports (some of which were not present in ClinVar). B/LB and P/LP variants from ClinVar were used to calibrate the assay (see “Methods” section; N = 69), after which we applied calibrated functional evidence criteria to facilitate classification of VUS and CI variants (N = 119). These included 258 single nucleotide variants and 12 indels. There were 58 exonic single nucleotide variants with high aggregate SpliceAI scores (>0.80), of which 3 had been observed in ClinVar (2 VUS, 1 CI). This library design allows us to ask three primary questions: (1) How well does the assay perform on ClinVar P/LP and B/LB control variants? (2) Can these results help reclassify VUS or resolve variants with CI? (3) What is the prospective accuracy of SpliceAI for exonic synonymous and missense variants? Accordingly, we adopted a previously published minigene to incorporate a site for barcoding to enable multiplexed experiments (Fig. 2A). We performed two major tasks to analyze our sequencing data: first, assembly (long-read sequencing linking the barcode to the construct/mutation) and the assay (inserting the library into cells and, second, short-read targeted RNA-seq to determine the PSI for each barcode; Fig. 1B; Supplementary Fig. 3). We first linked barcodes to WT or variant-containing plasmids, and then use barcode sequencing to quantify PSI for each plasmid (PSI; Fig. 2B). After introducing an 18 bp poly-N barcode, we used long-read sequencing of the plasmid pool to link each barcode to the insert (assembly). We identified 3238 unique barcodes linked to 284 unique inserts (290 possible; median 11 barcodes/insert; Fig. 2C). Comprehensive library characteristics are presented in Supplementary Data 2. Barcode frequencies were highly correlated when quantified by PacBio or Illumina sequencing (R = 0.82, p value = 4.0 × 10−125; Supplementary Fig. 4).
Fig. 2. ParSE-seq assay in HEK cells and iPSC-CMs.

A Detailed schematic of the ParSE-seq barcodable minigene plasmid. A previously used minigene vector was mutagenized to introduce a restriction site into the 3′ rat insulin exon 2. After digestion of a pool of minigene plasmids with SCN5A WT and variant inserts (middle), an 18-mer barcode was subcloned into the downstream exon (see Supplementary Fig. 2 for complete barcoding steps). Nts nucleotides. B Overview schematic of assembly and assay steps, and subsequent integration. Dashed lines represent amplicons for long- and short-read next generation sequencing (NGS). Following barcode insertion into the digested plasmid pool, a long-read PCR amplicon was used to link the barcode to the SCN5A insert (assembly). The pool was transfected into HEK cells and iPSC-CMs, after which short-read RNA-seq was used to link the barcode to splicing outcomes (assay; PSI percent spliced in). Assembly and assay data were merged by barcode for subsequent analysis steps (Supplementary Fig. 3). C Barcode counts for the assembly, and recovered barcodes present across three replicates in HEK and iPSC-CM assays. More barcodes were detected from the more easily transfected HEK cells than iPSC-CMs. Raw data available in Source Data and Supplementary Data 3. Black (library, pretransfection), red (iPSC-CMs), green (HEK). D Unique WT or variant inserts covered by barcodes in (C). Despite lower total barcode recovery in (C), most inserts are still recovered with the high stoichiometry of barcodes: inserts. Raw data available in Source Data and Supplementary Data 3. E PSI for all WT exons in iPSC-CMs and HEK cells. Data are averaged across three replicates and error bars represent the standard error of the mean. Red indicates iPSC-CMs, green indicates HEK cells. Raw data available in Source Data.
We transfected HEK cells and iPSC-CM cells each in three biological replicates with the barcoded library, isolated RNA, performed RT-PCR, and used targeted RNA-seq to quantify the impact of variants on minigene splicing. After quality control (see “Methods” section), we measured splicing efficiency in HEK cells for 2218 barcodes associated with 263 unique inserts (244 variants and 19 WT exons, Fig. 2C, D; raw data in Supplementary Data 3). Although the transfection efficiency in iPSC-CMs was low (~2% of cells were GFP+), we nonetheless measured splicing efficiency for a total of 927 unique barcodes associated with 243 unique inserts (224 variants and 19 WT exons; Fig. 2C, D; all PSI raw data in Supplementary Data 3). PSI values between replicates were highly correlated (Supplementary Figs. 5 and 6). We calculated a PSI for each WT exon in HEK and iPSC-CMs, and observed slightly higher PSIs in iPSC-CMs than in HEK cells (Fig. 2E). The WT exons for three exons that undergo alternative splicing in the heart (exons 6B, 18, and 24) had low PSI in the ParSE-seq assay (Fig. 2E; Genotype Tissue Expression Database transcript composition in Supplementary Fig. 7)34.
For each variant we calculated ΔPSI_norm, the normalized change in PSI of the variant compared to the PSI of the corresponding WT exon. Figure 3A–C shows the location and ΔPSI_norm of all variants quantified in the HEK and iPSC-CM assays. In Fig. 3D, we show the continuous distribution of variant ΔPSI_norm among all studied variants in iPSC-CMs, which ranges from severe abrogation of splicing, to WT-like splicing, to enhanced PSI for a few variants (Fig. 3C). Transfection of the barcoded library revealed high correlation of ΔPSI_norm scores between HEK cells and iPSC-CMs (R2 = 0.84, p = 6.4 × 10−95; Fig. 3E). We considered variants with a false discovery rate (FDR) < 0.1 and a ΔPSI_norm < −50% to be “abnormal”, and variants to be “normal” with an FDR > 0.1 and change in ΔPSI_norm > −20%; all other variants were considered indeterminate in our assay (Fig. 3F; see “Methods” section and Supplementary Fig. 8 for control variant performance and sensitivity analysis in Supplementary Table 3)26. 105/94/45 variants had normal/abnormal/indeterminate splicing in the HEK293 assay, and 104/78/42 variants had normal/abnormal/indeterminate splicing in the iPSC-CM assay. Of the 244 and 224 variants studied in HEK and iPSC-CMs, 207 were present in both cell types. We observed no discordance in variant splicing results between the two cell types (variants with a normal splicing result in one cell type and an abnormal splicing result in the other cell type; Supplementary Fig. 9). Due to the high concordance between assay results in the two cell types, we focused on the more physiologically relevant iPSC-CM results for the rest of the analyses. We observed variable effects on splicing by variant class in Fig. 3G. Among non-indeterminant variants, we observed splicing disruptions among 24/25 2-bp canonical splice sites variants, 20/48 missense variants, 18/57 synonymous variants, 5/6 stop-gain, and 11/45 non-canonical intronic variants.
Fig. 3. ParSE-seq results for a library of SCN5A variants.

A Example lollipop diagram showing variants superimposed along construct. iPSC-CM results for Exon 23 are shown. The y-axis represents mean ΔPSI_norm across three biological replicates, and the x-axis represents genomic position along the exonic (box) and intronic (line) segments of the synthetic insert. Purple indicates exonic variants, blue intronic variants outside the 2-bp canonical splice sites, and red canonical splice site variants. PSI percent spliced in. B Lollipop diagram showing distribution of ParSE-seq investigated variants in HEK cells. Axes are defined as in (A). An average of three experimental replicates is shown. C Lollipop diagram showing distribution of ParSE-seq investigated variants in iPSC-CMs. Axes are defined as in (A). An average of three experimental replicates is shown. D Waterfall plot of mean ΔPSI_norm by variant (N = 243). Red dashed line corresponds to −50% normalized ΔPSI, and blue to −20% normalized ΔPSI. E Spearman correlation (two-sided test) of mean ΔPSI_norm between HEK and iPSC-CMs (N = 207). Error bars refer to 95% confidence interval. F Volcano plot of normalized ΔPSI and −log10(FDR). Each dot represents a variant studied in iPSC-CMs (N = 243). Most variants fall within normal (blue) or abnormal (red) quadrants, but some remain indeterminant due to statistical or biological ambiguity (gray). FDR indicates false discovery rate. G Barplot of ParSE-seq variant outcomes by variant mutation type in iPSC-CMs, 2-bp indicates the conserved 2-base pair canonical splice sites AG-GT. Raw data for plots available in Source Data.
To analyze the molecular impacts of splice-altering variants, we examined the composition of non-canonical splicing reads (Supplementary Fig. 10). We observed exon truncation events, intron retention events, and exon skipping events, consistent with our prior low-throughput study14. For some variants, multiple aberrant splicing events were observed at appreciable levels. Many of these altered transcripts resulted in changes to the reading frame. In addition, some small and large in-frame insertions and deletions were also observed.
Most clinically ascertained variants tested in the ParSE-seq assay are rare among patients. In a recently published international BrS patient cohort35, there were 614 SCN5A-BrS patients harboring a variety of SCN5A variants, i.e., missense, splice-site, frameshift, nonsense, intronic. Of these, there were a total of 27 splice-altering variants affecting 43 patients (functionally abnormal non-consensus splice variants or consensus splice site variants). In the current study, we proactively investigated 18 unique variants harbored by 36 patients, which complemented our previous low-throughput investigations.
Functional studies and in silico splice variant effect predictors
We next examined the concordance between our experimental splicing scores and the in silico predictor SpliceAI5. In Fig. 4A, we show the distributions of the aggregate SpliceAI scores by ClinVar classification. Variants with higher SpliceAI scores were more likely be classified as P/LP compared to B/LB variants, with an intermediate distribution for VUS and CI variants. Across quintiles of SpliceAI scores, we observed different fractions of normal/abnormal/indeterminant variants, with the highest fraction of indeterminant variants in the intermediate three quintiles (14/51 indeterminant variants; Fig. 4B). To further explore the robustness of SpliceAI, we evaluated its ability to predict splice-altering exonic variants. Overall, 41 variants with SpliceAI scores >0.80 had normal or abnormal splicing in the iPSC-CM assay, including two ClinVar VUS’s (c.1338G > A/p.Glu446Glu, ClinVar: 451631 and c.4297G > T/p.Gly1433Trp, ClinVar: 519412). In our ParSE-seq assay, 37 of these 41 prospectively ascertained variants disrupted splicing (Fig. 4C), suggesting a high positive predictive value of SpliceAI-prioritized variants. In addition to SpliceAI, we also examined correlation of experimental data with in silico predictions from ABSplice36 and Pangolin37 (scores in Supplementary Data 2; three-way in silico correlations presented in Supplementary Fig. 11). We examined the correlation of each predictor with ΔPSI_norm for all non-canonical splice site variants in Fig. 4D–F. SpliceAI provided the highest correlation with experimental data (Spearman ρ = −0.81), followed by Pangolin (Spearman ρ = −0.67) and AbSplice (Spearman ρ = −0.61). Correlations between effect predictors and ParSE-seq outcomes for all variants, including canonical splice sites, are presented in Supplementary Fig. 12.
Fig. 4. Comparison of experimental data and in silico splicing predictors.

A Aggregate SpliceAI scores for each ClinVar variant class (N = 269). Raw scores available in Supplementary Data 2. B/LB benign and likely benign, CI conflicting interpretation, P/LP pathogenic likely pathogenic, VUS variant of uncertain significance. B Categorical distribution of variant effect in ParSE-seq stratified by SpliceAI score quintiles. Raw scores available in Supplementary Data 3. Red indicates abnormal assay result, gray indeterminate result, and blue normal result (for B and C). C Results of prospectively identified exonic variants by SpliceAI score >0.8 stratified by mutation type and ParSE-seq outcome. Raw data available in Supplementary Data 3. Correlation of normalized ΔPSI for non-canonical splice site variants against aggregate SpliceAI scores (N = 140; D), Pangolin scores (N = 141; E), and ABSplice scores (N = 137; F). Confidence interval fit using LOESS (see “Methods” section). P values were determined using a Pearson correlation. Raw data available in Source Data.
In addition to aggregate SpliceAI scores and ParSE-seq ΔPSI_norm comparisons across the library, we also compared specific SpliceAI molecular predictions to observed ParSE-seq splice outcomes (Supplementary Fig. 13). SpliceAI predictions typically matched experimental data for activation of cryptic splice sites (c.3358A > T, c.1891-5C > G, c.2024-11T > A), although in the ParSE-seq assay a different cryptic site may become activated (c.4220C > G) or exon skipping may result (c.4298G > T and c.3564G > T). Some variants led to multiple splice aberrations in the ParSE-seq experiment (e.g., exon skipping and exon truncation/intron retention), despite only having a single SpliceAI predicted aberrant event (c.3358A > T and c.2024-11T > A).
ACMG assay calibration
We tested ClinVar B/LB and P/LP control variants to provide a calibrated strength of evidence for the assay. We followed the ClinGen Sequence Variant Interpretation Working Group scheme for converting control assay results into likelihood ratios of pathogenicity, termed OddsPath24. A total of 47 ClinVar B/LB and P/LP control variants were recovered from our splicing experiment in iPSC-CMs with normal or abnormal splicing results. Most known P/LP variants for calibration came from canonical splice sites, due to a lack of previously annotated SCN5A P/LP splice-altering variants. We observed a near-perfect concordance of ClinVar classifications and assay outcomes in iPSC-CMs: 24 of 25 P/LP variants had abnormal splicing and 21 of 22 B/LB variants had normal splicing (Fig. 5A; Supplementary Table 4; derivation in “Methods” section). Applying these values, the OddsPathPathogenic (strength of evidence of abnormal assay results) was 21.1 and the OddsPathBenign (strength of evidence of normal assay results) was 0.042. These values correspond to strong application of abnormal functional evidence toward pathogenicity (PS3 strong; OddsPath > 18.7) and strong application of normal functional evidence toward a benign classification (BS3 strong; OddsPath < 0.053). The HEK cell assay performed similarly well with 30/31 concordant P/LP variants and 27/27 concordant B/LB variants, yielding a OddsPathPathogenic of 26.13 and an OddsPathBenign of 0.032 (PS3 strong and BS3 strong; Fig. 5B). The discrepant B/LB control in iPSC-CMs was an indeterminant variant in the HEK cell assay, with a ΔPSI_norm of 0.36 and FDR of 0.001 in HEK293 cells.
Fig. 5. Assay calibration and evidence-based variant classification.
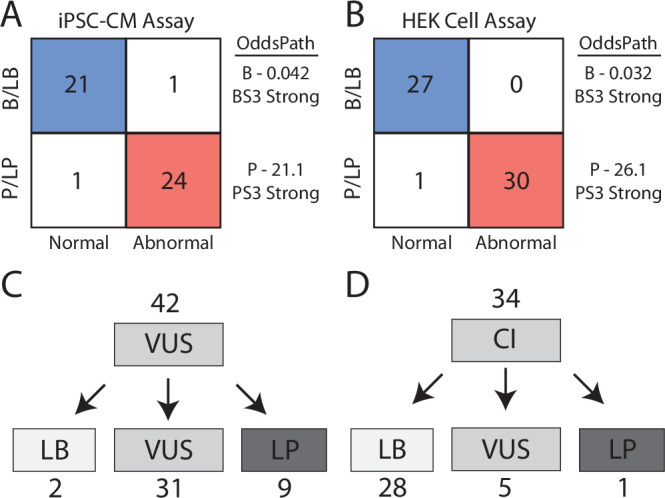
ParSE-seq results for B/LB and P/LP controls in iPSC-CMs (A) and HEK cells (B). We calculated two OddsPath values (B benign, P pathogenic) to implement BS3 and PS3 (ACMG benign and pathogenic functional evidence codes), both at a strong level of evidence. More barcodes were recovered for HEK cells, enabling additional control sample size (N = 47) vs (N = 58). Blue indicates concordant benign annotation and assay result, and red concordant pathogenic annotation and assay result. C Variants of uncertain significance (VUS) were reclassified using functional data from ParSE-seq at the strong level of evidence. BS3 evidence was applied exclusively to synonymous and intronic VUS. PS3 evidence is applied to all VUS. Evidence weights for classification provided in Supplementary Table 5. LB likely benign and LP likely pathogenic. D Classifications of conflicting interpretation (CI) variants using functional evidence. BS3/PS3 applied as for VUS in (C). Calibration control outcomes and ClinVar classifications in Source Data.
Variant reclassification
After identifying a calibrated strength of evidence to implement functional data (PS3 strong and BS3 strong), we sought to provide updated clinical interpretations for 42 VUS and 34 CI variants for which we obtained normal or abnormal splicing results in the iPSC-CM assay. We identified 8 VUS that were abnormal in our splicing assay (5 exonic, 3 intronic). Applying these functional data and in silico predictions from SpliceAI, we reclassified 9 abnormally splicing variants to LP, and 2 normally splicing variant to LB (Fig. 5C; all ACMG criteria in Supplementary Table 5). Since a normal splicing result in the ParSE-seq assay does not address possible dysfunction of the translated protein product for missense variants, we limited application of benign functional data to synonymous and intronic variants. Variants with annotations of CIs are a growing fraction of variants in ClinVar38,39. We anticipated that functional studies could provide additional data to adjudicate variants with CIs, especially for synonymous and intronic variants. Applying the ParSE-seq results from iPSC-CMs, we classified 28 of 34 CI variants as LB (Fig. 5D). We classified 1 CI variant as LP due to its splice-altering effect.
Cryptic splicing effects of missense variants
Missense variants that disrupt gene function and lead to disease are usually presumed to disrupt protein function. Functional assays of missense variants are often performed using cDNA, which may obscure missense variant effects on splicing (Fig. 6A)40. We hypothesized that some SCN5A missense variants may cause Mendelian disorders such as BrS through an aberrant splicing mechanism rather than isolated disruption of protein function. In iPSC-CMs, we recovered determinate data for 48 missense variants; 28 were listed as VUS in ClinVar, and 20 were identified prospectively with high SpliceAI scores (>0.8). Of these, we identified 18 splice-altering missense variants, created by 20 unique splice-altering single nucleotide variants. These variants were distributed throughout the protein, but often clustered in hotspots near exon boundaries. To explore differences in missense variant splicing and protein function in cDNA and endogenous assays, we studied the clinically relevant missense variant c.4220C > G/p.Ala1407Gly. This variant was reported in a patient undergoing genetic evaluation for SCN5A-related disease in ClinVar and disrupted splicing in the ParSE-seq assay by causing a large out-of-frame exon truncation event (Fig. 6B, C). We then performed cDNA-based automated patch clamping experiments on this variant using HEK293 cells stably expressing SCN5A cDNA (Fig. 6C, D). The variant had near-normal electrophysiologic function in this assay, with a normalized peak current density of 121.9 ± 9.1% (n = 46) of WT (Fig. 6C, D). In addition to peak current, we also studied additional electrophysiological functions of voltages of half-activation, voltages of half-inactivation, recovery from inactivation, time constant τ of inactivation, and late current (Supplementary Fig. 14 and Supplementary Table 6). We did not observe large differences in these parameters, except for a left-shift of activation for p.Ala1407Gly (P = 0.09, two-tailed T test) and a right-shift of inactivation for p.Ala1407Gly (P = 0.069, two-tailed T test), which are both predicted to cause gain-of-function (Supplementary Fig. 14; Supplementary Table 6). We also characterized a second missense variant, c.3392C > T/p.Thr1131Ile, which disrupted splicing in the ParSE-seq assay but had near-normal electrophysiological functions in the cDNA assay (Supplementary Fig. 14; Supplementary Table 6).
Fig. 6. A missense variant disrupts NaV1.5 function through a splice-altering mechanism.

A Schematic showing that assays using complementary DNA (cDNA) do not account for splice-altering variant effects. Left: Schematic of genomic locus with large intronic sequences; right: cDNA-based sequence without introns used in many SCN5A functional assays. Adjacent exons are annotated in green and blue in an alternating pattern for clarity. B Molecular analysis of the ParSE-seq assay showed activation of an upstream cryptic splice donor site, resulting in a 31-bp exon truncation. Raw data in Supplementary Data 3. Red portion of exon indicates truncated exon section. C Quantification of mean canonical PSI among reads for the WT exon construct and variant construct. Error bar corresponds to standard error of the mean across three biological replicates. P values were calculated from a two-tailed t-test. Raw data in Supplementary Data 3. D Quantification of mean sodium channel current densities for WT NaV1.5 and variant NaV1.5 using the SyncroPatch automated patch clamping system (cDNA assay), in stably expressing HEK293 cells. Error bar corresponds to standard error of the mean across biological replicate cells. N = 90 cells (WT) and N = 46 cells (c.4220C > G/p.Ala1407Gly). P values were calculated from a two-tailed t-test. Data are available in Source Data and Supplementary Table 6. E Representative single-cell sodium current traces for a WT and variant HEK cell. cDNA assessment of this missense variant did not show an effect on protein function when assessed by automated patch clamping, a system that cannot assess splicing impact. Currents are measured in nano amperes (nA). Raw data are available in Source Data. F CRISPR editing of a population control induced pluripotent stem cell (iPSC) line was performed to make a heterozygous edit of the line. G The WT and heterozygote variant iPSCs were chemically differentiated into cardiomyocytes (iPSC-CMs). H Differentiated iPSC-CMs were treated with dimethylsulfoxide (DMSO) or the nonsense-mediated decay (NMD) inhibitor cycloheximide (CHX), followed by RNA-isolation and RNA-seq (N = 3 for each condition). We observed aberrant splicing consistent with the ParSE-seq molecular event (exon truncation) in the variant, but not WT lines. Notably, treatment with cycloheximide increases the ratio of WT splicing to exon truncation, consistent with NMD degradation of the aberrant transcript. Raw counts are available from NIH BioProject accession #1106089. I Manual patch clamp of the WT and variant iPSC-CMs was performed to test the effect of aberrant splicing on protein function. Sodium currents were abrogated in the presence of the variant compared to WT, consistent with haploinsufficiency from loss-of-splicing in (F). Error bars represent standard error of the mean across biological replicate cells (N = 10, WT and N = 12, variant). Raw data is presented in Supplementary Table 7.
Given the discrepancy in in vitro assays, we further studied the effect on splicing and protein function by introducing the c.4220C > G/p.Ala1407Gly variant at the endogenous locus in iPSC-CMs41. Using CRISPR-Cas9, we generated a heterozygous edit of c.4220C > G in a population control iPSC line (Fig. 6E). The variant and WT population control lines were differentiated into iPSC-CMs using a chemical differentiation method and then studied at the RNA level (RNA-seq) and protein level (patch clamp; Fig. 6F). To avoid confounding by nonsense-mediated decay (NMD) in the endogenous locus of heterozygous iPSC-CMs42,43, we treated both isogenic lines with the NMD inhibitor cycloheximide (CHX) alongside a vehicle control, dimethylsulfoxide (DMSO). We performed RNA-seq and observed reads corresponding to the ParSE-seq exon truncation event in the variant line treated with vehicle control (Fig. 6H). The aberrant reads were increased after treatment with CHX (consistent with NMD degradation of out-of-frame transcript; Fig. 6H). This truncation event was not observed in isogenic control lines treated with either DMSO or CHX (Fig. 6H). To test whether variant-induced aberrant splicing affected protein-level function, isogenic pairs of iPSC-CMs were studied by patch clamping to measure sodium channel current (Fig. 6I). In addition to peak current density, we also provide membrane resistance for the current CRISPR-edited iPSC-CM lines and reference membrane potential data for our population control line (Supplementary Table 7)44. We observed a decrease in peak sodium current across a range of voltages in the c.4220C > G line compared to the isogenic control, confirming the hallmark loss-of-function phenotype for SCN5A-linked BrS45,46 (Fig. 6I). We also determined voltages of half-activation for each iPSC-CM line, and observed a statistically significant decrease in the mean voltage of half-activation for c.4220C > G, consistent with loss-of-function (−35.6 vs −42.1 mV, p < 0.01, N = 10–12; Supplementary Fig. 15).
Thus, ParSE-seq can help identify a class of missense, splice-altering variants for which cDNA-based assays of protein function yield incorrect conclusions about variant pathogenicity. This result highlights that for missense variants, ParSE-seq can be used to complement traditional cDNA-based assays of protein function.
To further validate the ParSE-seq assay at the endogenous locus, we studied an intronic variant, c.1891-5C>G, by CRISPR editing of a population control iPSC line (Supplementary Figs. 15 and 16). This variant was selected from the set of splice-disrupting variants that appeared in heterozygotes undergoing evaluation for SCN5A-related disease in ClinVar. We found concordance between the ParSE-seq predictions of aberrant splicing and iPSC-CM RNA splicing, with introduction of a frame-shifting 4-base pair intron retention event. Moreover, this aberrant splicing event severely abrogated peak current density by patch clamp of the iPSC-CMs, consistent with the primary mechanism of SCN5A-BrS (Supplementary Fig. 16).
Discussion
In this study, we developed a high-throughput method to assess the splicing consequences of hundreds of variants (ParSE-seq). We implemented barcoding of a pool of minigene plasmids to enable multiplexed splicing readouts using high-throughput sequencing and applied the method to study variants in the cardiac sodium channel gene SCN5A. In iPSC-CMs, we quantified variant effects on splicing for 224 variants, and detected 78 variants with abnormal splicing. We observed concordance of splicing results for 45/47 B/LB and P/LP variants, and we determined that our assay could be applied at the strong level in the ACMG classification scheme (BS3 and PS3) when assuming ClinVar as ground truth. Leveraging these calibrated strengths of evidence, we classified SCN5A VUS and CI variants. We determined that the in silico tool SpliceAI has high, but not perfect, concordance with experimentally measured splicing effects. We also demonstrated examples of missense variants that had normal electrophysiology using conventional heterologous expression cDNA-based approaches but disrupt splicing. Lastly, we showed that our ParSE-seq results predict aberrant splicing in a disease-relevant iPSC-CM model, with consequences at both the RNA and protein levels. We envision that ParSE-seq will be applicable to many disease genes and will be accessible using openly available computational pipelines and democratized gene synthesis available to the community.
A recent survey of clinical patient variant data highlighted the need for accurate splicing assays to improve variant classification and support the implementation of personalized medicine2. Our assay identified 43 exonic variants and 11 intronic variants outside canonical splice sites which disrupt splicing in iPSC-CMs. This data is of immediate clinical relevance to individuals who are heterozygous for these variants, and they should be screened for BrS and cardiac conduction defects seen with SCN5A loss-of-function variants. In addition, we identified 30 VUS or CI variants that could be classified as LB after incorporating our splicing data, which reduces clinical uncertainty for these variant carriers2. There is only limited clinical data currently available for most of the variants in this study, and some of the variants have not yet been detected in any individuals to our knowledge (high SpliceAI prediction variants). However, with the rapid rise of genetic sequencing and data sharing (especially regarding patient phenotypes), many future individuals will likely be discovered with variants in this study. These phenotypes of these individuals could be examined to further examine the validity of our variant classifications. Recently, standardization of techniques and harmonization of classifications have improved the clinical integration of splicing results from primary or peripheral tissue9. We anticipate that lab-based methods like ParSE-seq and clinical studies of peripheral tissue will be complementary. In vitro functional data will be especially useful when tissue is not available, when there are no other heterozygotes to segregate a phenotype, or expression of the relevant transcript is not sufficiently high in the clinically accessible tissue. Clinical potential will be further improved by the integration of functional assays and AI-based in silico tools for splicing predictions5,37.
ParSE-seq builds on pioneering work in the field of high-throughput splicing assays. For example, multiplexed splicing readouts were recently applied to LMNA, MYBPC3, and TTN variants using minigene-based large oligo constructs and barcoding20,21. Although some previous studies have examined splice outcomes for thousands of multiplexed variants, these experiments are typically limited to only small exons with minimal adjacent intronic sequence due to the size limits of chip-based oligonucleotide synthesis6,18,19,47. This approach often necessitates the inclusion of only small adjacent intronic regions and precludes studying large exons. ParSE-seq builds on these approaches by considering a more complete set of aberrant splicing patterns through the triplet exon cassette design, as well as enabling the testing of much larger clinically relevant exons feasible with commercially available clonal gene synthesis technology. For most tested exons we included 250 bp of intronic sequence on each side of the target exon. Inclusion of larger segments of flanking introns has been shown to increase assay validity due to more complete capture of cis-regulatory elements18. A major advantage of this framework is the standardization of library preparation, wherein a library can quickly be assembled using genomic coordinates of variants of interest. In addition, previous high-throughput splicing methods have not been calibrated using known B/LB and P/LP standards for clinical variant classification in the ACMG scheme. Further, we perform the assay in a disease-relevant cell type (iPSC-CMs). Most minigene-based methods are limited by which constructs they can study. Mutually exclusive exon splicing and multiple exon skipping events are increasingly recognized as causes of disease48,49. Although our current study focuses on a single study exon and surrounding intronic sequence, the ParSE-seq method should be amenable to inclusion of multiple study exons, constrained by cost of synthesis and transfection efficiency of large plasmids. Although CRISPR-edited iPSC-CMs are rising in frequency as a cellular model to study variant effects, these experiments are still relatively low-throughput and resource-intensive. Heterologous expression in HEK293 cells is the most commonly used cell model to study SCN5A variant function at the protein level (440 of 524 experiments in a large literature review we performed in 2018)46.
Loss-of-function variants in ion channel genes have been classified into four categories: Class I (RNA degradation or reduced expression), Class II (diminished trafficking), Class III (changes in gating), or Class IV (changes in ion permeability)50. Traditionally, many missense variants in SCN5A have acted through a Class III or Class IV mechanism46. However, our data show that the missense variant c.4220C > G/p.Ala1407Gly appears to mainly act through a Class I mechanism. ParSE-seq may also complement other functional assays such as automated patch clamp and multiplexed assays of variant effect (MAVEs), proactive high-throughput assays of variant function51. For example, MAVEs subject libraries of many variants at each position of a protein to a fitness assay, which allows prospective functional evaluation of a variant even before it is clinically observed. This approach holds great promise for reducing the VUS problem52. Functional assays of missense variants are often performed using cDNA. As cDNA contains only exons without intervening intronic sequence, any exonic variant effect on splicing will not be annotated (exon skipping and/or cryptic splice site activation causing exon truncation). Therefore, current cDNA assays such as patch clamp electrophysiology can mis-annotate the molecular impact of a splice-altering variant. Here we show the limits of these approaches for certain missense variants, which could produce disparate outcomes depending on the assay. In some previous cDNA-based deep mutational scans, investigators have excluded their functional results for variants with SpliceAI scores above a certain threshold53, or assumed deleterious splicing based on prediction-alone when applying results to clinical cohorts27. We also note that for missense variants, we did not use a normal splicing result in the ParSE-assay to reclassify variants, because ParSE-seq cannot rule out effects of missense variants on protein function. We envision that future high-throughput studies of missense variants will use complementary cDNA-based functional assays and ParSE-seq to comprehensively annotate variant effects. In silico tools could also be used to prioritize which candidate splice-disrupting variants should be investigated. Alternatively, we demonstrate, albeit in low-throughput for only two variants, that candidate splice variants can be introduced at the endogenous locus by CRISPR into relevant differentiated cell types. These cells can be studied to measure both splice-altering and protein-altering effects (Fig. 6). We additionally show the feasibility of obtaining prospective splicing results of many exonic variants prioritized by an in silico predictor. Our current implementation of splice prediction tools aggregates predictions of acceptor and donor loss/gain to a single value to enable comparisons across tools. However, manual interrogation of such predictors may provide insight into exact molecular vs in silico predictions of spliced RNA (Supplementary Fig. 13). A recently published tool allows for the visualization of any SpliceAI prediction to facilitate molecular interpretation54. Ultimately, comprehensive splicing data for predicted splice-altering variants in Mendelian loss-of-function genes could complement cDNA-based MAVEs in progress for these genes55,56.
ParSE-seq can rapidly assess hundreds of candidate splice-altering variants. Given the plethora of VUS and CI variants that may disrupt splicing2, this method may help classify large sets of variants in Mendelian disease-associated genes that act through a loss-of-function mechanism. Although we validate two ParSE-seq splice-altering variants by CRISPR editing of the iPSC-CMs, most variants were tested only in multiplexed minigene assays. While we anticipate most splice-altering variants to result in loss-of-function (NaV1.5 peak current abrogation for SCN5A variants), there may be alternative mechanisms revealed by functional assessment of the CRISPR-edited iPSC-CM model. For example, while ParSE-seq quantifies broad molecular impacts such as exon skipping, exon truncation, and intron retention, it is possible that some splicing abnormalities may not have a detrimental effect, or otherwise altered effect, on downstream protein function (protein tolerant in-frame insertion/deletions). In the current version of gnomAD v4, there are no observed indel variants >3 amino acids that are reported as B/LB. The frequency of affected variant heterozygotes is difficult to ascertain based off ClinVar data alone, as detailed patient phenotypes and case counts are not routinely reported from submitting centers. There may be examples where the ParSE-seq minigene assay does not fully capture all nuances of biology at the endogenous locus. For example, splicing regulatory motifs in the native context may have a long-distance effect not captured in the minigene-based assay. This incomplete ascertainment may lead to discordant results with in silico predictors for a subset of variants. The first and last exons of a gene, exons using non-canonical 2-bp splice sites, and exons that were difficult to synthesize due to high GC content or restriction enzyme incompatibility were not included in the library. Exons that undergo extensive alternative splicing in the endogenous tissue (e.g., SCN5A exons 6B, 18, and 24) may have low intrinsic PSI in the minigene assay, which may limit the use of the assay for these exons. Despite these limitations, we anticipate that ParSE-seq will be a useful method for to rapidly assessing variant splicing effects. Given the plethora of variants that may act through disrupting splicing, our method can be used to efficiently characterize the splicing effects of variants in disease-associated genes.
Methods
Ethical statement
We performed variant-level analyses in iPSC-CMs from an established cell line44. The participant (male age 30–40) from which these cells were derived provided informed consent. As we were interested in variant-level effects, we did not consider sex/gender or genetic ancestry in the selection of this cell line. The Vanderbilt University Medical Center IRB (#9047) approved the use of the induced pluripotent stem cells used in this study.
Selection of variants
Variants were selected from three categories (Fig. 1F)—(1) B/LB and P/LP ClinVar variants to calibrate the assay; (2) VUS and CI variants in SCN5A that could be reclassified with splicing functional data; (3) exonic missense and synonymous variants with a strong prediction of splice-disruption (aggregate SpliceAI5 score > 0.8). These included 258 single nucleotide variants and 12 indels. Control B/LB and P/LP variants were chosen from ClinVar, accessed on October 2, 2022. We excluded B/LB variants with gnomAD allele count of 4 or less (approximate minor allele frequency < 2.5 × 10−5), due to the possibility that such very rare variants may nevertheless be associated with BrS57,58. We used gnomAD v3.1.2 accessed on October 6th 2022, with Ensembl ID ENST00000333535.9. All selected variants were within 170 base pairs of an exon/intron junction. We selected P/LP variants that were annotated in ClinVar to act through aberrant splicing rather than by altering protein function. We included BrS patient case counts when available from a previously published international cohort study35. For ClinVar variants, we conservatively assumed at least one affected patient per reported variant.
SCN5A wild-type exons and library coverage
The ParSE-seq assay requires an acceptor and donor splice site on each end of the insert plasmid’s exon. The SCN5A transcript ENST00000333535.9 contains 27 coding exons (exons 2–28). We interrogated splicing effects of variants in 19 of these exons (see Fig. 3B, C). The minigene-based assay requires an acceptor and donor splice site on each end of the test exon, and is therefore incompatible with the first or last coding exons (2 and 28). In addition, SCN5A uses two instances of non-canonical AC/AT splice sites between exons 3 and 4, and exons 25 and 26. Therefore, we did not study variants in these four exons or in adjacent intronic locations. Furthermore, we were unable to include plasmids with exon 15 due to synthesis incompatibility (high GC content) and exon 17 due to overlap of restriction enzymes used for barcoding.
Construction of plasmid library
The exon trapping vector pET01 (MoBiTec) was mutagenized to introduce two restriction sites (AscI and MfeI) in the 3′ rat insulin exon to produce pAG424 (Supplementary Fig. 1)59. We used an inverse PCR mutagenesis method with primers ag491 and ag492 to introduce the restriction sites which were then verified by Sanger sequencing (all primers are presented in Supplementary Table 2)59. In total, 290 clonal genes containing an exon surrounded by 100–250 bp of intronic sequence (mean = 230) were directly synthesized, cloned into pAG424, and sequence verified by Twist Biosciences (South San Francisco, CA). This rapid Twist clonal gene synthesis method was a modification of our low-throughput, restriction-enzyme-based minigene protocol14. Plasmids containing exons 6, 10, and 12 were shortened from 250 bp due to closely adjacent exons (exon 6) or high GC content at distant intronic sequence which was incompatible with Twist synthesis (exons 10 and 12). We studied the adult form of SCN5A exon 6 (exon 6B). All construct sizes (exon and flanking intronic lengths) are presented in Supplementary Table 1, and full sequences in Supplementary Data 2. We resuspended each plasmid in water to a concentration of 25 ng/μl. We pooled 50 ng of each plasmid to form a library pool. We also added two additional plasmids (V291 and V304—Supplementary Data 2) that failed Twist synthesis using PCR amplification of the WT insert from genomic DNA and QuikChange mutagenesis14. In this case, primers with AscI/MfeI sequences flanking the region of interest were used in a Q5 PCR reaction (NEB) following manufacturer’s protocol. The amplicon was PCR purified (Qiagen) per manufacturer’s protocol. pAG424 and the amplicon were then each digested with AscI and MfeI (NEB) at 37 °C for 1 h, followed by separation on a 1% agarose gel. The bands were removed, and then purified following instructions from a Gel Extraction Kit (QIAGEN). Each component was then ligated with supplies from a T4 ligation kit (NEB) for 1 h, followed by heat inactivation at 65 °C for 10 min. A 1 μL aliquot was then used to transform 50 μl competent cells (NEB), followed by incubation at 37 °C overnight, and DNA extraction using a Spin Miniprep Kit (QiIAGEN), per manufacturer’s protocol. The plasmids were sequence verified by Genewiz before use in the ParSE-seq assay.
Adding a barcode to the plasmid library
We digested the plasmid pool pMO515 with AscI (NEB) and MfeI (NEB) followed by incubation with Calf Intestinal Phosphatase (NEB). An insert containing random 18-mer barcodes was produced (Supplementary Fig. 2)56. Briefly, ag1371 and ag1372 were annealed, followed by extension to make fully double stranded DNA using Klenow polymerase (NEB)60. Due to its small size, the double stranded DNA was then phenol/chloroform extracted and digested using AscI and MfeI (NEB), and was again purified by phenol/chloroform extraction. The pool of minigene plasmids was also digested with AscI and MfeI and cleaned by gel extraction (QIAGEN). The digested vector pool and barcode insert were ligated using T4 ligase (NEB). The ligation product was PCR purified (QIAGEN) and electroporated into ElectroMax DH10B cells (ThermoFisher) using a Gene Pulser Electroporator (BioRad; 2.0 kV, 25 μF, 200 Ω). The resulting bacterial culture was then grown overnight, and DNA was isolated by a maxiprep (QIAGEN) to yield the barcoded plasmid library. Barcode diversity was estimated by plating dilutions of the library on LB-ampicillin plates and counting colonies.
Assembly steps
We established the relationship between barcodes and inserts (WT or variant exon and introns) using PacBio long-read SMRT sequencing61. We used the pooled barcoded library as template for a PCR with primers mo37 and ag489 using Q5 polymerase (NEB) per the manufacturer’s protocol. Amplification was split among eight individual reactions and cycle number was minimized to reduce PCR-mediated recombination. The PCR protocol had one denaturation step of 98 °C for 30 s, followed by 20 cycles of 98 °C for 10 s, 55 °C for 15 s, and 72 °C for 1 min, followed by a final 72 °C hold for 5 min62. The reactions were pooled, and used to generate a SMRT Bell 3.0 library (PacBio) according to the manufacturer’s instructions. The library was sequenced with PacBio Sequel II 8M SMRT Cell by Maryland Genomics.
We recorded 30 h of PacBio SMRT cell sequencing. To mitigate sequencing errors in the raw PacBio data, we only analyzed Circular Consensus Sequence (CCS) reads. A total of 4,136,990 CCS reads were obtained as fastq files, with an average size of 1312 base pairs. The median Q score was 48 across CCS reads.
Computational pipeline—assembly
The full computational pipeline is diagrammed in Supplementary Fig. 3. FASTQ files from the PacBio sequencing were filtered for reads containing barcodes with the predicted 8-nt prefix and 6-nt suffix sequences immediately adjacent to the 18-bp barcode using Unix and Python. Unique barcodes were identified, and then reads were parsed by barcode into new FASTQ files. For each barcode, the Unix grep command was used to count the number of reads containing each designed insert. To be included in the assembly, a barcode was required to be present in at least 50 reads in the assembly sequencing (N = 3303). To ensure a high-quality assembly, we only analyzed filtered reads with a perfect match to one of the candidate WT or mutant constructs across the entire sequence insert using “grep” in a Unix bash script (mean 636 nt; Supplementary Fig. 3). The barcode identity was assigned as the most frequently aligned insert if that insert represented more than 50% of the read counts. After implementation of these quality control cutoffs, 284 of the 290 targeted plasmids were successfully detected in the plasmid pool.
Splicing assay and preparation of Illumina library
HEK cells were grown in HEK media: Dulbecco’s Eagle’s medium supplemented with 10% fetal bovine serum, 1% non-essential amino acids, and 1% penicillin/streptomycin. For the HEK cell assay, three wells of a 6-well plate with HEK cells at 30–40% confluency were independently transfected with the barcoded plasmid library using FuGENE 6 (Promega) per manufacturer’s instructions. For iPSC-CM transfections, we used a “C2” healthy population control line63. iPSCs were differentiated into a monolayer of iPSC-CMs using a chemical differentiation method64. Following 30 days of differentiation, iPSC-CMs were dissociated with TrypLE™ Select (ThermoFisher) and replated as single cells. Cells were cultured for an additional 2 days, after which the pooled library was transfected using Viafect (Promega)65. For both HEK293 and iPSC-CM experiments, RNA was isolated 24 h post-transfection using the RNeasy Plus Mini Kit (QIAGEN) and reverse transcribed using the primer mo38 (which targets a constant region of the transcribed minigene) with SuperScript III (Invitrogen). Libraries were prepared for Illumina sequencing by touchdown PCR using Q5 polymerase (NEB) with primers binding reference minigene exons, sequence containing Illumina i7 and i5 indexing sequences and Illumina NovaSeq dual indexing motifs (Supplementary Table 2). The PCR protocol included a single denaturation step of 98 °C for 30 s, touchdown with 10 cycles of 98 °C for 10 s, 10 cycles of 65–55 °C (decreasing by 1 °C/cycle for 15 s, followed by extension at 72 °C for 1 min), followed by an additional 20 cycles of 98 °C 10 s, 55 °C for 15 s, and 72 °C 1 min, followed by a final extension of 72 °C for 5 min. PCR amplicons were purified with a PCR purification kit (QIAGEN). Libraries were then sequenced using Illumina NovaSeq paired-end 150 base sequencing to ~50M reads/sample.
Computational pipeline—assay
A diagram of the computational pipeline is presented in Supplementary Fig. 3. Reads were filtered for correct barcode prefix and suffix sequences and were divided into separate files by barcode as described above. In all splicing assay studies, each barcode was required to be present in at least 25 reads in each replicate to be included. The PSI metric was calculated using grep searches for splice junctions corresponding to the WT exon splicing to the reference exons in the R1 and R2 reads:
| 1 |
computationally implemented as:
| 2 |
For variants that would alter the coding sequence of the WT exon, a bespoke R1 or R2 junction was created and used for those specific variants. PSIs were then averaged across barcodes for each variant, using the barcode-variant lookup table from the assembly step described above. We used the assigned PSI as the average PSI for each variant across 3 independent transfections into HEK or iPSC-CMs. For each variant, a ΔPSI value was calculated:
For each variant, a normalized ΔPSI value was calculated:
| 3 |
This ΔPSI_norm value represents the change in splicing of the variant compared to the level of splicing of the corresponding WT exon. The value is normalized to the level of WT splicing to determine the percent change of splicing regardless of the baseline level of WT PSI. For example, if a WT exon had a PSI of 80% and the variant exon had a PSI of 40%, the ΔPSI_norm would equal −50%. A ΔPSI_norm value of −100% indicates a complete loss of normal splicing, whereas a value of 0 indicates identical splicing to WT.
Statistical analysis
Statistical analyses were performed in R. Comparison of WT and variant PSI (using mean of three replicate samples) used a two-sided t-test implemented in R. For the set of studied variants, FDR were calculated using the R command p.adjust.
Thresholds for normal, abnormal, and indeterminant function were derived from assay performance on B/LB controls as recommended by the ClinGen Sequence Variant Interpretation Working Group24. We used the mean (−2.1%) and standard deviation (17%) of ΔPSI_norm among B/LB variants and created corresponding z-score thresholds (Supplementary Fig. 8). To assign functional outcomes, we considered variants with a z-score < −3 (−53%) to be abnormal, and those with a z-score > −1 (−19%) to be normal. These corresponded roughly to −50% and −20%, which we used as our primary cutoffs for generalizability across different genes. We considered the range < −20% and > −50% as indeterminant without additional studies to avoid dichotomization of a continuous score. The benign and pathogenic calibrated strengths of evidence (OddsPath) were robust and consistent across a range of thresholds (Supplementary Tables 3 and 4).
To also account for statistical significance, variants with FDR < 0.1 and ΔPSI_norm < −50% were considered splice-altering. Variants with FDR > 0.1 and ΔPSI_norm ≥ −20% were considered non-splice altering. All other variants were labeled indeterminate in the assay. We excluded variants from our analysis if the standard error of the PSI among the three replicates was >0.15. Violin plots, best fit curves, and barplots were plotted in R using ggplot2 (see GitHub for code).
We used locally estimated scatterplot smoothing (LOESS) as a non-parametric regression model for comparing in silico splicing predictors with experimental ParSE-seq data. LOESS was selected as a smoothing method due to the largely bimodal distribution of our experimental data. A 95% confidence interval is displayed alongside the line of fit. LOESS was implemented in ggplot2 using default settings with ΔPSI_normvariant plotted as a function of aggregate SpliceAI predictions. Full code describing this analysis is available on GitHub.
ACMG OddsPath Calculation
We performed assay calibration according to the framework developed by Brnich et al.24 and extended by Fayer et al.25. For the high-throughput splicing assay, we calculate the likelihood ratio of pathogenicity (termed OddsPath) for both splice-altering (pathogenic) and non-splice-altering (benign) assay results. First, we removed all variants with indeterminate scores. Then, we calculated benign and pathogenic OddsPath values using the equations:
| 4 |
| 5 |
Where, the Prior P1 was defined as:
| 6 |
The benign and pathogenic posterior P2 was then calculated:
| 7 |
| 8 |
Following the approach recommended by Brnich et al.24, if P2Pathogenic = 1 it was conservatively estimated to have one additional discordant variant (a functionally abnormal benign variant) by the following equation:
| 9 |
Similarly, if P2Benign = 0, an additional discordant variant was included (a functionally normal pathogenic variant):
| 10 |
Each posterior was combined with the prior to derive an OddsPath and assign evidence for PS3 and BS3 criteria, respectively.
In silico predictors
For intronic variants, SpliceAI was implemented on the Unix command line using an input VCF file consisting of Human Genome Variation Society GRCh38 coordinates for all variant positions in the splicing library5. In addition, pre-computed SpliceAI scores for all possible SCN5A exonic variants were downloaded from the SpliceAI dataset hosted at Illumina BaseSpace. We computed an aggregate SpliceAI score to incorporate contributions from each of the four predicted categories using the formula45:
| 11 |
where AG = probability of acceptor gain, AL = probability of acceptor loss, DG = probability of donor gain, and DL = probability of donor loss. These aggregate scores were used in the analysis of SpliceAI correlations with experimental ΔPSI scores, as well as to select exonic variants above a threshold of 0.8 (high prediction of altered splicing). Receiver Operator Characteristic curves were plotted in R using the library pROC, comparing aggregate SpliceAI scores for the binary outcome of a normal or abnormal splice assay result, removing indeterminate assay results66.
We accessed pre-computed computed ABSplice data from https://zenodo.org/records/787180936. We analyzed ENSG00000183873 and restricted the analysis to splicing results for the specific tissue “Heart—Left Ventricle”. Pangolin scores were obtained using the command line Pangolin tool with default settings and a .csv file of chromosomal locations as input37.
ACMG variant classification
We determined clinical classifications of variants using the ACMG criteria using an online tool hosted by the University of Maryland (https://www.medschool.umaryland.edu/Genetic_Variant_Interpretation_Tool1.html/)67. We implemented functional evidence at the calibrated strength of evidence based on the results of the calibration described above (PS3 strong and BS3 strong). We applied PP3 for an aggregate SpliceAI >0.5, and BP3 for SpliceAI <0.2. We applied PM2 for variants with an allele frequency of <2.5 × 10−5 per recommendations for BrS57.
High-throughput electrophysiology
We used an established workflow for generating automated patch clamp data for variants in HEK293T landing pad cells (gift of Kenneth Matreyek)68–70. Briefly, we cloned SCN5A variants into cDNA-containing plasmids using Quikchange mutagenesis (Agilent), integrated these plasmids into landing pad HEK293 cells68,69, used negative and positive selection with iCasp and blasticidin to generate stable lines, and studied the stable lines on the SyncroPatch 384PE automated patch clamping platform71 (Nanion). The cloning, patch clamping, and data analysis process has been extensively described by our group in previous publications70–72. In the SyncroPatch HEK293 and manual iPSC-CM patch experiments, the series resistance (Rs) was monitored using the Seal Resistance QC variable to achieve a range of 5–10 MΩ. Multiple protocols were applied sequentially to each cell, including current–voltage (IV) curves to measure INa activation, inactivation, recovery from inactivation, and late current protocols. A full description of this method has been published70. Two independent transfections were performed for SyncroPatch and manual experiments for each mutant, and data were averaged across all cells passing QC criteria (capacitance and seal resistance)70. In the manual iPSC-CM patch clamp experiments, the series resistance (Rs) was monitored using Seal Test (Clampex 10.9 software) to achieve a range of 5–10 MΩ. Current–voltage curves were generated by repeated voltage changes to the same cells. Two trials were performed for each cell line and data were then averaged across all measured cells
iPSC-CM maintenance and differentiation
We performed variant-level analyses in iPSC-derived CMs from an established cell line44. The participant (male age 30–40) from which these cells were derived provided informed consent. As we were interested in variant-level effects, we did not consider sex/gender or genetic ancestry in the selection of this cell line. The Vanderbilt University Medical Center IRB (#9047) approved the use of the induced pluripotent stem cells used in this study. iPSCs were cultured at 37 °C in a humidified 95% air/5% CO2 incubator on Matrigel coated plates (BD BioSciences). Cells were maintained in mTeSR plus media (STEMCELL) and passaged every 3–4 days. Stem cell experiments were carried out in the “C2” line previously characterized by our laboratory63. At 60–80% confluency, stem cells began differentiation to cardiomyocytes using a chemical differentiation method44,64. RNA analyses and patch clamp studies were performed at days 35–40 of differentiation. NMD inhibition assays were carried out by exposing cells to 100 μM CHX (Sigma-Aldrich) or 1% DMSO (Sigma-Aldrich) as a vehicle control for 6 h, followed by harvesting of RNA as described below73.
CRISPR-Cas9 gene editing
We designed CRISPR-Cas9 guide plasmids based on the 20-bp NGG SpCas9 editor using the online tool CRISPOR74. PAM sites were altered in the repair template to a synonymous amino acid for exonic variants and an intronic SNV when intronic; in either case, the disrupting variant was chosen to have a predicted minimal impact on splicing (aggregate SpliceAI <0.05). CRISPR guides were cloned into (pX458)20 plasmid (Addgene #48138, a gift of Feng Zhang)14. Two complementary oligonucleotides were phosphorylated and annealed using T4 PNK (NEB) per protocol, followed by simultaneous digestion and ligation of pX458 with BbsI-HF (NEB) and T4 ligase (NEB) per manufacturer’s protocol. The sample was transformed into competent E. coli cells, followed by incubation, plating on an ampicillin agar plate, and then colony expansion and miniprep (Qiagen) as described above. The cloned guide plasmid and a 151-nucleotide repair template bearing the desired change and PAM site variant were co-electroporated into dissociated iPSCs using the Neon Transfection System (ThermoFisher MPK5000). After 72 h, cells were dissociated, singularized, and sorted for GFP+ cells using a BD Fortessa 5-laser instrument. Single colonies were picked, DNA extracted using QuickExtract (Lucigen), PCR amplified using primers mo198 and mo199, and Sanger sequenced to identify a colony with a heterozygous edit.
RNA-seq and visualization of splice junctions
RNA was isolated from iPSC-CMs between day 30 and 35 of differentiation using the RNeasy Minikit (QAIGEN). Integrity of RNA was determined on a 2100 Bioanalyzer (Agilent). The Vanderbilt Technologies for Advanced Genomics core prepared a polyA-selection library and sequenced the library to a depth of 50 million PE100 reads on an Illumina Novaseq sequencer. FASTQ files were analyzed with fastp75, reads were aligned with hisat276, bam files indexed with SAMtools77, and quantified in Integrative Genomics Viewer78.
Whole-cell voltage-clamp recording of sodium current
During manual patch clamp experiments on iPSC-CMs, we selected iPSC-CMs at age of Days 30–40. During the experiments, membrane resistance (Rm) was monitored throughout using the Membrane Test (Clampex 10.9). We first optimized the electrode capacitance compensation on the amplifier, performed following giga-seal formation and before achievement of the whole-cell configuration. In this way, the capacitive transients were completely and well-compensated by ~80% when whole-cell capacitance compensation was enabled. Next, we used Seal Test (Clampex 10.9) as an oscilloscope window for monitoring the current signal to achieve a reading close to 10 MΩ. Optimization of the capacitance compensation was an extremely important step for accurate Cm and Rm measurements in Membrane Test. To achieve high quality giga-seal formation before cell membrane break-in, we chose cells with giga-seal of 1–2 GΩ for the experiments.
Whole-cell voltage-clamp experiments in iPSC-CMs were conducted at room temperature (22–23 °C). Glass microelectrodes were heat polished to tip resistances of 0.5–1 MΩ. Data acquisition was carried out using MultiClamp 700B patch clamp amplifier and pCLAMP 10 software suite (Molecular Device Corp., San Jose, CA, USA). To record sodium currents in iPSC-CMs, currents were filtered at 5 kHz (−3 dB, four-pole Bessel filter) and digitized at frequency of 2–20 kHz by using an analog-to-digital interface (DigiData 1550B, Molecular Device Corp., San Jose, CA, USA). To minimize capacitive transients, capacitance and series resistance were corrected ~80%. Voltage-clamp protocols used are shown on the figures. Electrophysiological data were analyzed using Clampfit 10 software and the figures were prepared by using Graphing & Analysis software OriginPro 8.5.1 (OriginLab Corp., Northampton, MA, USA). To provide better voltage control of sodium current, the external solution was K+-free and Ca2+-free with a lower sodium concentration (50 mmol/L), containing (in mM) NaCl 50, NMDG 85, glucose 10, HEPES 10, adjusted to pH 7.4 by NaOH. The pipette (intracellular) solution had (in mM) NaF 5, CsF 110, CsCl 20, EGTA 10, and HEPES 10, adjusted to pH 7.3 by CsOH. To eliminate the overlapped L-/T-type inward calcium currents and outward potassium currents, different blockers (1 µM nisoldipine, 200 µM NiCl2, and 200 µM 4-aminopyride) were added into the cell bath solution. To record sodium current, cells were held at −100 mV and current was elicited with a 50-ms pulse from −100 to +40 mV in 10 mV increments. Current densities were expressed in the unit of pA/pF after normalization to cell size (pF), generated from the cell capacitance calculated by the function of Membrane Test (OUT 0) in pCLAMP 10 software.
The average capacitances of the iPSC-CMs were 56.9 ± 4.8 pF (WT), 50.6 ± 2.5 pF (c.1891-5G > C), and 45.9 ± 0.8 pF (c.4220G > C). The average membrane resistances of the iPSC-CMs were 1.54 ± 0.09 GΩ (WT), 1.53 ± 0.08 GΩ (c.1891-5G > C variant), and 1.55 ± 0.1 GΩ (c.4220G > C variant). These parameters were not statistically significantly different from each other (p > 0.05). We did not measure membrane potentials during the sodium current (INa) measurements under modified experimental conditions (see below). However differentiated iPSC-CMs from the same population control iPSC line studied with physiological intra- and extracellular solutions had potentials ranging from −75 to −90 mV, with an average potential of −82.2 ± 1.2 mV.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Source data
Acknowledgements
This research was funded by American Heart Association fellowship 907581 (M.J.O.), and by the National Institutes of Health: 1F30HL163923-01 (M.J.O.), T32GM007347 (M.J.O.), R00 HG010904 (A.M.G.), R35 GM150465 (A.M.G.), R01 HL149826 (D.M.R.), and R01 HL164675 (D.M.R.). Flow cytometry experiments were performed in the Vanderbilt Flow Cytometry Shared Resource. The Vanderbilt Flow Cytometry Shared Resource is supported by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404). The Nanion SyncroPatch 384PE is housed and managed within the Vanderbilt High-Throughput Screening Core Facility, an institutionally supported core, and was funded by NIH Shared Instrumentation Grant 1S10OD025281. The HTS Core receives support from the Vanderbilt Institute of Chemical Biology and the Vanderbilt Ingram Cancer Center (P30 CA68485). We thank Maryland Genomics at the Institute for Genome Sciences, University of Maryland School of Medicine for performing PacBio SMRT sequencing. We thank Kenneth Matreyek and Doug Fowler for the HEK293 landing pad cells. We thank Yuko Wada for assistance with genome editing and Jeremy Smith for computational support. We thank Servier Medical Art, under CC by 4.0, for supply images for Fig. 6.
Author contributions
D.M.R. and A.M.G. oversaw the project. M.J.O. developed the ParSE-seq experimental methodology and completed all computational analysis. T.Y. provided experimental support for manual patch clamp of iPSC-CMs. J.L. and M.L.H. assisted in iPSC-CM CRISPR editing and cell culture. J.S. and M.C. assisted in APC experiments. M.J.O., D.M.R., and A.M.G. wrote the manuscript. All authors reviewed and edited the manuscript. D.M.R. and A.M.G. provided funding support for the study.
Peer review
Peer review information
Nature Communications thanks Kaoru Ito, Olga Kopach and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Data availability
All DNA and RNA sequencing data are available at the NCBI Sequence Read Archive (NIH BioProject accession number 1106089). Processed data are available on GitHub (https://github.com/GlazerLab/ParSE-seq; permanently copied at Zenodo79). All processed data are also available in the accompanying Supplementary Information/Source Data file. We have deposited variant classifications on ClinVar (SUB14636616 [www.ncbi.nlm.nih.gov/clinvar/?term=SUB14636616]). Source data are provided with this paper.
Code availability
Code used to analyze raw data and generate figures and tables is available in GitHub: https://github.com/GlazerLab/ParSE-seq79.
Competing interests
A.M.G. is a consultant for BioMarin, Inc. All remaining authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors jointly supervised this work: Dan M. Roden, Andrew M. Glazer.
Contributor Information
Dan M. Roden, Email: dan.roden@vumc.org
Andrew M. Glazer, Email: andrew.m.glazer@vumc.org
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-52474-4.
References
- 1.Green, E. D. et al. Strategic vision for improving human health at The Forefront of Genomics. Nature586, 683–692 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Truty, R. et al. Spectrum of splicing variants in disease genes and the ability of RNA analysis to reduce uncertainty in clinical interpretation. Am. J. Hum. Genet.108, 696–708 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sibley, C. R., Blazquez, L. & Ule, J. Lessons from non-canonical splicing. Nat. Rev. Genet.17, 407–421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vaz-Drago, R., Custódio, N. & Carmo-Fonseca, M. Deep intronic mutations and human disease. Hum. Genet.136, 1093–1111 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Jaganathan, K. et al. Predicting splicing from primary sequence with deep learning. Cell176, 535–548.e524 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Soemedi, R. et al. Pathogenic variants that alter protein code often disrupt splicing. Nat. Genet.49, 848–855 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cummings, B. B. et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci. Transl. Med.910.1126/scitranslmed.aal5209 (2017). [DOI] [PMC free article] [PubMed]
- 8.Kremer, L. S. et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat. Commun.8, 15824 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bournazos, A. M. et al. Standardized practices for RNA diagnostics using clinically accessible specimens reclassifies 75% of putative splicing variants. Genet. Med.24, 130–145 (2022). [DOI] [PubMed] [Google Scholar]
- 10.Gaildrat, P. et al. Use of splicing reporter minigene assay to evaluate the effect on splicing of unclassified genetic variants. Methods Mol. Biol.653, 249–257 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Valenzuela-Palomo, A. et al. Splicing predictions, minigene analyses, and ACMG-AMP clinical classification of 42 germline PALB2 splice-site variants. J. Pathol.10.1002/path.5839 (2021). [DOI] [PMC free article] [PubMed]
- 12.Fraile-Bethencourt, E. et al. Functional classification of DNA variants by hybrid minigenes: identification of 30 spliceogenic variants of BRCA2 exons 17 and 18. PLoS Genet.13, e1006691 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanses, U. et al. Intronic CRISPR repair in a preclinical model of Noonan syndrome-associated cardiomyopathy. Circulation142, 1059–1076 (2020). [DOI] [PubMed] [Google Scholar]
- 14.O’Neill, M. J. et al. Functional assays reclassify suspected splice-altering variants of uncertain significance in Mendelian channelopathies. Circ. Genom. Precis. Med.10.1161/circgen.122.003782 (2022). [DOI] [PMC free article] [PubMed]
- 15.Tobert, K. E. et al. Genome sequencing in a genetically elusive multi-generational long QT syndrome pedigree identifies a novel LQT2-causative deeply intronic KCNH2 variant. Heart Rhythm10.1016/j.hrthm.2022.02.004 (2022). [DOI] [PubMed]
- 16.Georgics, P. et al. High-throughput splicing assays identify missense and silent splice-disruptive POU1F1 variants underlying pituitary hormone deficiency. Am J Hum Genet.108, 1526–1539 (2021). [DOI] [PMC free article] [PubMed]
- 17.Cortés-López, M. et al. High-throughput mutagenesis identifies mutations and RNA-binding proteins controlling CD19 splicing and CART-19 therapy resistance. Nat. Commun.13, 5570 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheung, R. et al. A multiplexed assay for exon recognition reveals that an unappreciated fraction of rare genetic variants cause large-effect splicing disruptions. Mol. Cell73, 183–194.e188 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adamson, S. I., Zhan, L. & Graveley, B. R. Vex-seq: high-throughput identification of the impact of genetic variation on pre-mRNA splicing efficiency. Genome Biol.19, 71 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patel, P. N. et al. Contribution of noncanonical splice variants to TTN truncating variant cardiomyopathy. Circ. Genom. Precis. Med.14, e003389 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ito, K. et al. Identification of pathogenic gene mutations in LMNA and MYBPC3 that alter RNA splicing. Proc. Natl. Acad. Sci. USA114, 7689–7694 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med.17, 405–424 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Landrum, M. J. et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res.42, D980–D985 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brnich, S. E. et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med.12, 3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fayer, S. et al. Closing the gap: systematic integration of multiplexed functional data resolves variants of uncertain significance in BRCA1, TP53, and PTEN. Am. J. Hum. Genet.108, 2248–2258 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang, C. et al. A calibrated functional patch-clamp assay to enhance clinical variant interpretation in KCNH2-related long QT syndrome. Am. J. Hum. Genet.109, 1199–1207 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scott, A. et al. Saturation-scale functional evidence supports clinical variant interpretation in Lynch syndrome. Genome Biol.23, 266 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barc, J. et al. Genome-wide association analyses identify new Brugada syndrome risk loci and highlight a new mechanism of sodium channel regulation in disease susceptibility. Nat. Genet.54, 232–239 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hosseini, S. M. et al. Reappraisal of reported genes for sudden arrhythmic death: evidence-based evaluation of gene validity for Brugada syndrome. Circulation138, 1195–1205 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peters, S. et al. Arrhythmic phenotypes are a defining feature of dilated cardiomyopathy-associated SCN5A variants: a systematic review. Circ. Genom. Precis. Med.15, e003432 (2022). [DOI] [PubMed] [Google Scholar]
- 31.Kapplinger, J. D. et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm7, 33–46 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bardai, A. et al. Sudden cardiac arrest associated with use of a non-cardiac drug that reduces cardiac excitability: evidence from bench, bedside, and community. Eur. Heart J.34, 1506–1516 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Hong, K. et al. Cryptic 5′ splice site activation in SCN5A associated with Brugada syndrome. J. Mol. Cell. Cardiol.38, 555–560 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Battle, A., Brown, C. D., Engelhardt, B. E. & Montgomery, S. B. Genetic effects on gene expression across human tissues. Nature550, 204–213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walsh, R. et al. Enhancing rare variant interpretation in inherited arrhythmias through quantitative analysis of consortium disease cohorts and population controls. Genet. Med.23, 47–58 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wagner, N. et al. Aberrant splicing prediction across human tissues. Nat. Genet.55, 861–870 (2023). [DOI] [PubMed] [Google Scholar]
- 37.Zeng, T. & Li, Y. I. Predicting RNA splicing from DNA sequence using Pangolin. Genome Biol.23, 103 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glazer, A. M. Genetics of congenital arrhythmia syndromes: the challenge of variant interpretation. Curr. Opin. Genet. Dev.77, 102004 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosamilia, M. B., Lu, I. M. & Landstrom, A. P. Pathogenicity assignment of variants in genes associated with cardiac channelopathies evolve toward diagnostic uncertainty. Circ. Genom. Precis. Med.15, e003491 (2022). [DOI] [PubMed] [Google Scholar]
- 40.Cartegni, L., Chew, S. L. & Krainer, A. R. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat. Rev. Genet.3, 285–298 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Sayed, N., Liu, C. & Wu, J. C. Translation of human-induced pluripotent stem cells: from clinical trial in a dish to precision medicine. J. Am. Coll. Cardiol.67, 2161–2176 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kurosaki, T., Popp, M. W. & Maquat, L. E. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat. Rev. Mol. Cell Biol.20, 406–420 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lykke-Andersen, S. & Jensen, T. H. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol.16, 665–677 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Bersell, K. R. et al. Transcriptional dysregulation underlies both monogenic arrhythmia syndrome and common modifiers of cardiac repolarization. Circulation10.1161/circulationaha.122.062193 (2022). [DOI] [PMC free article] [PubMed]
- 45.Ishikawa, T. et al. Functionally validated SCN5A variants allow interpretation of pathogenicity and prediction of lethal events in Brugada syndrome. Eur. Heart J.42, 2854–2863 (2021). [DOI] [PubMed] [Google Scholar]
- 46.Kroncke, B. M., Glazer, A. M., Smith, D. K., Blume, J. D. & Roden, D. M. SCN5A (NaV1.5) variant functional perturbation and clinical presentation: variants of a certain significance. Circ. Genom. Precis. Med.11, e002095 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chiang, H. L. et al. Mechanism and modeling of human disease-associated near-exon intronic variants that perturb RNA splicing. Nat. Struct. Mol. Biol.29, 1043–1055 (2022). [DOI] [PubMed] [Google Scholar]
- 48.Dawes, R. et al. SpliceVault predicts the precise nature of variant-associated mis-splicing. Nat. Genet.55, 324–332 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen, X. et al. Antisense oligonucleotide therapeutic approach for Timothy syndrome. Nature628, 818–825 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Delisle, B. P., Anson, B. D., Rajamani, S. & January, C. T. Biology of cardiac arrhythmias: ion channel protein trafficking. Circ. Res.94, 1418–1428 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Fowler, D. M. & Fields, S. Deep mutational scanning: a new style of protein science. Nat. Methods11, 801–807 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Starita, L. M. et al. Variant interpretation: functional assays to the rescue. Am. J. Hum. Genet.101, 315–325 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jia, X. et al. Massively parallel functional testing of MSH2 missense variants conferring Lynch syndrome risk. Am. J. Hum. Genet.108, 163–175 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Sainte Agathe, J. M. et al. SpliceAI-visual: a free online tool to improve SpliceAI splicing variant interpretation. Hum. Genomics17, 7 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kozek, K. A. et al. High-throughput discovery of trafficking-deficient variants in the cardiac potassium channel KV11.1. Heart Rhythm17, 2180–2189 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Glazer, A. M. et al. Deep mutational scan of an SCN5A voltage sensor. Circ. Genom. Precis. Med.13, e002786 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Whiffin, N. et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet. Med.19, 1151–1158 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jain, P. C. & Varadarajan, R. A rapid, efficient, and economical inverse polymerase chain reaction-based method for generating a site saturation mutant library. Anal. Biochem.449, 90–98 (2014). [DOI] [PubMed] [Google Scholar]
- 60.Fowler, D. M., Stephany, J. J. & Fields, S. Measuring the activity of protein variants on a large scale using deep mutational scanning. Nat. Protoc.9, 2267–2284 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eid, J. et al. Real-time DNA sequencing from single polymerase molecules. Science323, 133–138 (2009). [DOI] [PubMed] [Google Scholar]
- 62.Potapov, V. & Ong, J. L. Examining sources of error in PCR by single-molecule sequencing. PLoS ONE12, e0169774 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wada, Y. et al. Common ancestry-specific ion channel variants predispose to drug-induced arrhythmias. Circulation10.1161/circulationaha.121.054883 (2022). [DOI] [PMC free article] [PubMed]
- 64.Burridge, P. W. et al. Chemically defined generation of human cardiomyocytes. Nat. Methods11, 855–860 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bodbin, S. E., Denning, C. & Mosqueira, D. Transfection of hPSC-cardiomyocytes using Viafect™ transfection reagent. Methods Protoc.310.3390/mps3030057 (2020). [DOI] [PMC free article] [PubMed]
- 66.Robin, X. et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform.12, 77 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kleinberger, J., Maloney, K. A., Pollin, T. I. & Jeng, L. J. An openly available online tool for implementing the ACMG/AMP standards and guidelines for the interpretation of sequence variants. Genet. Med.18, 1165 (2016). [DOI] [PMC free article] [PubMed]
- 68.Matreyek, K. A., Stephany, J. J., Chiasson, M. A., Hasle, N. & Fowler, D. M. An improved platform for functional assessment of large protein libraries in mammalian cells. Nucleic Acids Res.48, e1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Matreyek, K. A., Stephany, J. J. & Fowler, D. M. A platform for functional assessment of large variant libraries in mammalian cells. Nucleic Acids Res.45, e102 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Glazer, A. M. et al. High-throughput reclassification of SCN5A variants. Am. J. Hum. Genet.107, 111–123 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.O’Neill, M. J. et al. Dominant negative effects of SCN5A missense variants. Genet. Med.24, 1238–1248 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Glazer, A. M. et al. Arrhythmia variant associations and reclassifications in the eMERGE-III sequencing study. Circulation10.1161/circulationaha.121.055562 (2021). [DOI] [PMC free article] [PubMed]
- 73.O’Neill, M. J. et al. Multicenter clinical and functional evidence reclassifies a recurrent noncanonical filamin C splice-altering variant. Heart Rhythm10.1016/j.hrthm.2023.05.006 (2023). [DOI] [PMC free article] [PubMed]
- 74.Concordet, J. P. & Haeussler, M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res.46, W242–w245 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics34, i884–i890 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol.37, 907–915 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol.29, 24–26 (2011). [DOI] [PMC free article] [PubMed]
- 79.Matthew J. O’Neill, D. M. R., & Andrew M. Glazer. 10.5281/zenodo.13170911 (Zenodo, 2024).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
All DNA and RNA sequencing data are available at the NCBI Sequence Read Archive (NIH BioProject accession number 1106089). Processed data are available on GitHub (https://github.com/GlazerLab/ParSE-seq; permanently copied at Zenodo79). All processed data are also available in the accompanying Supplementary Information/Source Data file. We have deposited variant classifications on ClinVar (SUB14636616 [www.ncbi.nlm.nih.gov/clinvar/?term=SUB14636616]). Source data are provided with this paper.
Code used to analyze raw data and generate figures and tables is available in GitHub: https://github.com/GlazerLab/ParSE-seq79.