

Abstract

The phytochemical investigation of Euphorbia desmondii resulted in the isolation of 15 previously undescribed triterpenoids (desmondiins A, C-P) and 8 already described compounds. The structures of the isolated compounds were determined by extensive spectroscopic analyses. The compounds were identified as tirucallane and euphane triterpenes based on 7-keto-8-ene, 11-keto-8-ene, or 7,11-diketo-8-ene skeletons. Additionally, the selective trypanocidal activities of these compounds against Trypanosoma cruzi were evaluated. Desmondiins A, C, D, F, H, and M exhibited IC50 values in the range of 3–5 μM, and selectivity indices between 5–9, against T. cruzi epimastigotes over the host cell (RAW264.7 macrophages). Furthermore, desmondiin A efficiently inhibited amastigote replication in host cells (IC50 = 2.5 ± 0.3 μM), which was comparable to that of the positive control, benznidazole (3.6 ± 0.4 μM). Overall, the isolated euphane and tirucallane triterpenoids could act as antichagasic lead scaffolds.
Trypanosoma cruzi, a parasitic protozoan, is the causative agent of Chagas disease (American trypanosomiasis). It is believed that 6–7 million people worldwide, primarily in Latin America, suffer from this disease, which accounts for 12,000 fatalities annually. Further, 10% of patients with persistent infections experience neurological or digestive disorders or both, and up to one-third of these patients experience cardiac changes that may require special care.1,2 The dermatologic symptoms of acute infections include conjunctivitis, widespread morbilliform eruption, and localized swelling at the inoculation site (chagoma).3 Chagas disease can be transmitted by the triatomine bug (vector-borne) as well as orally (food-borne). Furthermore, it can spread during pregnancy or childbirth, through blood or blood products, during organ transplants, and through laboratory accidents.4
The proliferative forms of T. cruzi are known as epimastigotes. In the gut of kissing bugs (Triatoma spp.), epimastigotes transform into metacyclic trypomastigotes, which are capable of infecting mammals. The disease is primarily transmitted through bites by an infected T. cruzi bug. The infected T. cruzi bug, which feeds on the blood of mammals (including humans), contaminates human skin with metacyclic trypomastigotes, causing an infection. Thereafter, the parasites exploit a process that is facilitated by self-inflicted scratching and use the proteolytic enzymes present in the saliva of the infected bug to penetrate the bloodstream through the skin.5,6
Even though Chagas’ disease was discovered over a century ago, only two nitro derivatives (nifurtimox and benznidazole) have been identified for its treatments. However, both substances are significantly limited by their severe side effects, need a prolonged treatment duration, show selective drug sensitivities toward various T. cruzi strains, and are ineffective at the chronic stage of the disease.7 Therefore, it is crucial to discover new, less toxic, and more effective therapeutic options for treating the disease.8,9
Over 600 plant species belonging to different families have been evaluated for their trypanocidal activities. Screening of extracts and compounds from species of the families Asteraceae, Poaceae, Piperaceae, Euphorbiaceae, Polygalaceae, and Solanaceae have yielded promising results.10,11 Following the extant reports on the trypanocidal effects of chemically different triterpenoids,10Euphorbia desmondii Keay & Milne-Redh (Euphorbiaceae), which is native to West and Central African countries (Ghana, Benin Republic, Nigeria, Niger, Cameroun, and Chad), was investigated in this study.12,13 This plant is a shrub or tree that typically grows to a height of 5.2 m. It is called a “male gu” to distinguish it from E. kamerunica, which is called “female gu”. Economically, its products are used to produce gums and resins.14 Previous phytochemical investigations provided the identification of ingenol in E. desmondii.15,16 The present study focuses on the isolation of triterpenoids from E. desmondii Keay & Milne-Redh (Euphorbiaceae) and evaluation of the trypanocidal activities of the isolated compounds.
Results and Discussion
Isolation and Structure Determination of the Compounds
The CHCl3 fraction of the MeOH extract prepared from the aerial parts of E. desmondii was separated by multistep chromatography to afford 23 pure compounds (1–23).
Desmondiin A (1) was isolated as a
white, amorphous powder. Its molecular formula was determined as C30H48O4 by HR-ESI-MS analysis (m/z 473.3637 [M + H]+, calcd.
for C30H49O4+: 473.3625).
The 1H and 13C NMR JMOD spectra of 1 revealed the presence of one secondary and seven tertiary methyl
groups, eight methylenes, and six methines of which one was oxygenated
(Tables 1 and 2). The 13C NMR JMOD and HSQC spectra
indicated eight quaternary carbons (δC 198.2, 165.2,
139.1, 82.4, 47.9, 44.9, 39.5, and 38.8). Further, the presence of
two olefinic groups in the molecule was evident from the proton (δH 5.54 d and 5.69 m) and carbon chemical shifts (δC 130.6, 134.9, 139.1, and 165.2), and one keto group was revealed
by the carbon signal at δC 198.2. The structure of 1 was further analyzed by 1H–1H COSY spectrum, which enabled the elucidation of four structural
fragments: C-1–C-3, C-5–C-6, C-11–C-12, and C-15–C-16–C-17–C-20(C-21)–C-22–C-23–C-24
based on the sequences of the correlated protons.
Table 1. 1H NMR (500 MHz) Data for Compounds 1–7 in CDCl3.
|
δH,mult. (Jin Hz) |
|||||||
|---|---|---|---|---|---|---|---|
| No. | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| 1 | 1.44, m | 1.43, m | 1.43, m | 1.43, m | 1.54, m | 1.56, m | 1.78, m |
| 1.87, dt (13.2, 3.6) | 1.86, m | 1.86, dt (13.0, 3.4) | 1.86, dt (13.0, 3.4) | 2.32, m | 2.45, m | 2.42, m | |
| 2 | 1.75, m | 1.77, m | 1.76, m, | 1.75, m | 1.78, m | 1.77, m (2H) | 1.74, m |
| 1.68, m | 1.67, m | 1.68, m | 1.67, m | 1.72, m | |||
| 3 | 3.30, m | 3.29, dd (11.6, 4.4) | 3.30, dd (11.2, 4.3) | 3.30, dd (11.5, 4.3) | 3.31, dd (11.0, 4.7) | 3.32, dd (10.1, 5.8) | 3.32, m |
| 5 | 1.68, m | 1.68, dd (13.5, 4.7) | 1.67, dd (13.6, 4.7) | 1.67, dd (13.5, 4.6) | 1.65, dd (14.1, 5.2) | 1.65, dd (12.5, 5.8) | 1.65, dd (12.3, 6.0) |
| 6 | 2.47, m | 2.44, dd (18.6, 4.7) | 2.42, m | 2.40, dd (18.7, 4.6) | 2.40, m | 2.45, m (2H) | 2.45, m (2H) |
| 6 | 2.33, m | 2.36, dd (18.6, 13.5) | 2.32, m | 2.33, dd (18.7, 13.5) | 2.46, m | ||
| 11 | 2.41, dt (20.1, 9.0) | 2.41, m | 2.40, m | 2.37, m | 5.04, t (7.7) | 4.70, t (8.2) | 4.70, t (7.8) |
| 2.24, m | 2.24, dd (20.7, 7.2) | 2.22, ddd (19.4, 6.7, 1.5) | 2.22, dd (20.3, 6.6) | ||||
| 12 | 1.78, m (2H) | 1.82, m | 1.76, m | 1.77, m | 2.16, dd (13.4, 7.3) | 1.79, m | 1.81, m |
| 2.34, m | 2.39, m | 2.36, m | |||||
| 15 | 1.54, m | 1.51, m | 1.56, m | 1.52, m | 1.44, m | 1.44, m | 1.44, m |
| 2.15, m | 2.13, tt (10.8, 2.4) | 2.13, m | 2.12, ddd (12.2, 9.8, 1.7) | 2.08, m | 2.10, ddd (12.2, 9.6, 2.0) | 2.12, m | |
| 16 | 1.38, m | 1.37, m | 1.35, m | 1.35, m | 1.33, m | 1.32, m | 1.34, m |
| 1.95, m | 1.94, m | 1.95, m | 2.00, m | 1.92, m | 1.92, m | 1.95, m | |
| 17 | 1.55, m | 1.49, m | 1.46, m | 1.46, m | 1.67, m | 1.60, m | 1.51, m |
| 18 | 0.76, s | 0.75, s | 0.73, s | 0.72, s | 0.73, s | 0.74, s | 0.73, s |
| 19 | 1.06, s | 1.05, s | 1.06, s | 1.05, s | 1.17, s | 1.26, s | 1.26, s |
| 20 | 1.57, m | 1.50, m | 1.42, m | 1.42, m | 1.59, m | 1.55, s | 1.46, m |
| 21 | 0.86, d (6.2) | 0.86, d (6.4) | 0.94, d (6.3) | 0.93, d (6.1) | 0.91, d (6.4) | 0.86, d (6.4) | 0.92, d (6.2) |
| 22 | 1.88, m | 1.94, m | 1.05, m | 0.96, m | 2.06, m | 1.89, m | 1.20, m |
| 2.38, m | 2.14, m | 2.32, m | 1.42, m | 2.31, m | 2.39, m | 1.85, m | |
| 23 | 5.69, m | 2.43, m | 1.47, m | 1.62, m | 5.63, ddd (15.5, 8.3, 4.9) | 5.65, ddd (15.8, 7.7, 6.3) | 2.28 m |
| 1.66, m | 1.42, m | 2.54 m | |||||
| 24 | 5.54, d (15.6) | 2.39, m | 4.02, t (6.6) | 5.59, d (15.5) | 5.55, d (15.8) | 6.48, t (7.2) | |
| 25 | 2.61, sept (6.8) | ||||||
| 26 | 1.34, s | 1.10, d (6.8) | 2.13, s | 4.84, br s, 4.93, br s | 1.30, s | 1.33, s | 9.40, s |
| 27 | 1.34, s | 1.10, d (6.8) | – | 1.72, s | 1.38, s | 1.34, s | 1.76, s |
| 28 | 1.00, s | 0.99, s | 1.00, s | 0.96, s | 1.00, s | 1.00, s | 1.00, s |
| 29 | 0.89, s | 0.88, s | 0.89, s | 0.88, s | 0.92, s | 0.91, s | 0.92, s |
| 30 | 0.98, s | 0.98, s | 0.97, s | 0.99, s | 1.12, s | 1.14, s | 1.16, s |
Table 2. 13C NMR (125 MHz) Data for Compounds 1–7 in CDCl3.
|
δC,type |
|||||||
|---|---|---|---|---|---|---|---|
| No. | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| 1 | 34.8, CH2 | 34.8, CH2 | 34.8, CH2 | 34.8, CH2 | 34.1, CH2 | 33.8, CH2 | 33.8, CH2 |
| 2 | 27.6, CH2 | 27.7, CH2 | 27.6, CH2 | 27.6, CH2 | 27.5, CH2 | 27.6, CH2 | 27.5, CH2 |
| 3 | 78.3, CH | 78.3, CH | 78.3, CH | 78.2, CH | 78.4, CH | 78.4, CH | 78.4, CH |
| 4 | 38.8, C | 39.0, C | 39.0, C | 39.0, C | 39.2, C | 39.2, C | 39.2, C |
| 5 | 48.5, CH | 48.5, CH | 48.5, CH | 48.4, CH | 49.4, CH | 49.4, CH | 49.4, CH |
| 6 | 36.0, CH2 | 35.8, CH2 | 35.9, CH2 | 35.9, CH2 | 36.0, CH2 | 36.0, CH2 | 36.0, CH2 |
| 7 | 198.2, C | 198.3, C | 198.3, C | 198.4, C | 199.7, C | 200.2, C | 200.1, C |
| 8 | 139.1, C | 139.1, C | 139.1, C | 139.1, C | 143.5, C | 140.5, C | 140.5, C |
| 9 | 165.2, C | 165.4, C | 165.4, C | 165.5, C | 157.3, C | 161.3, C | 161.1, C |
| 10 | 39.5, C | 39.5, C | 39.5, C | 39.4, C | 38.4, C | 39.7, C | 39.7, C |
| 11 | 23.8, CH2 | 23.9, CH2 | 23.8, CH2 | 23.8, CH2 | 81.1, CH | 68.2, CH | 68.2, CH |
| 12 | 30.3, CH2 | 30.2, CH2 | 30.1, CH2 | 30.0, CH2 | 38.2, CH2 | 43.0, CH2 | 43.1, CH2 |
| 13 | 44.9, C | 44.9, C | 44.8, C | 44.8, C | 46.4, C | 46.4, C | 46.3, C |
| 14 | 47.9, C | 47.9, C | 47.8, C | 47.8, C | 48.0, C | 48.1, C | 48.2, C |
| 15 | 31.6, CH2 | 31.6, CH2 | 31.6, CH2 | 31.6, CH2 | 32.0, CH2 | 31.9, CH2 | 31.9, CH2 |
| 16 | 28.5, CH2 | 28.8, CH2 | 28.8, CH2 | 28.7, CH2 | 27.6, CH2 | 27.7, CH2 | 28.0, CH2 |
| 17 | 48.4, CH | 48.6, CH | 48.9, CH | 48.8, CH | 48.3, CH | 48.6, CH | 48.9, CH |
| 18 | 16.1, CH3 | 15.9, CH3 | 15.7, CH3 | 15.7, CH3 | 17.1, CH3 | 16.6, CH3 | 16.5, CH3 |
| 19 | 18.8, CH3 | 18.8, CH3 | 18.8, CH3 | 18.8, CH3 | 19.2, CH3 | 19.9, CH3 | 19.9, CH3 |
| 20 | 36.0, CH | 35.7, CH | 36.4, CH | 36.4, CH | 35.5, CH | 35.9, CH | 35.8, CH |
| 21 | 19.3, CH3 | 19.0, CH3 | 18.9, CH3 | 19.0, CH3 | 19.6, CH3 | 19.3, CH3 | 18.8, CH3 |
| 22 | 38.8, CH2 | 30.1, CH2 | 36.0, CH2 | 31.8, CH2 | 38.1, CH2 | 38.6, CH2 | 34.6, CH2 |
| 23 | 130.6, CH | 37.4, CH2 | 20.9, CH2 | 32.1, CH2 | 129.2, CH | 130.1, CH | 26.2, CH2 |
| 24 | 134.9, CH | 215.3, CH2 | 44.4, CH2 | 76.5, CH | 135.8, CH | 135.1, CH | 155.0, CH |
| 25 | 82.4, C | 41.1, CH | 209.2, C | 147.6, C | 82.5, C | 82.4, C | 139.9, C |
| 26 | 24.6, CH3 | 18.8, CH3 | 30.0, CH3 | 111.6, CH2 | 24.9, CH3 | 24.5, CH3 | 195.4, CH |
| 27 | 24.6, CH3 | 18.8, CH3 | – | 17.3, CH3 | 23.8, CH3 | 24.5, CH3 | 9.4, CH3 |
| 28 | 27.5, CH3 | 27.3, CH3 | 27.5, CH3 | 24.5, CH3 | 27.8, CH3 | 27.7, CH3 | 27.7, CH3 |
| 29 | 15.2, CH3 | 15.2, CH3 | 15.2, CH3 | 15.2, CH3 | 15.4, CH3 | 15.3, CH3 | 15.3, CH3 |
| 30 | 24.6, CH3 | 24.3, CH3 | 24.5, CH3 | 27.4, CH3 | 25.3, CH3 | 25.9, CH3 | 25.9, CH3 |
These structural units, along with the quaternary carbons, were connected by long-range heteronuclear correlations extracted from an HMBC spectrum. The most informative HMBC correlations were those of the methyl groups as follows: H3-18 to C-12, C-13, C-14, and C-17; H3-19 to C-1, C-5, C-10, and C-9; H3-21 to C-17, C-20, and C-22; H3-26 and H3-27 to C-24 and C-25; H3-28 and H3-29 to C-3, C-4, and C-5; and H3-30 to C-8, C-13, C-14, and C-15 (Figure 1). These correlations allowed the establishment of the planar structure as a tetracyclic euphane- or tirucallane-type triterpene. The position of the keto group at C-7 was confirmed by the HMBC cross peaks of H2-6 (δH 2.33, 2.47) with C-7 (δC 198.2). A tetrasubstituted olefin group was placed at position C-8–C-9, as indicated by the correlations of H3-19 with C-9 and H3-30 with C-8; the disubstituted olefin was positioned at C-23–C-24, as revealed by the correlations of H3-26 and H3-27 with C-24. The 26- and 27-methyl groups displayed correlations with the quaternary carbon at δC 82.4 (C-25); this high chemical shift value combined with the molecular formula confirmed the presence of a hydroperoxy group (OOH) at C-25.17
Figure 1.

Key 1H–1H COSY (−) and HMBC correlations (H → C) of compounds 1, 9, and 12.
The relative configuration of compound 1 was determined using the NOESY experiment. The Overhauser effect observed between H-3 and H-5, as well as H3-18 and H-11α indicated the α orientation of these groups and hydrogens (Figure 3). Conversely, the NOESY correlations between H-11β and H3-19 and H3-30 and H-17 revealed the stereochemistry of rings A–D bearing an α-oriented 18-methyl group and β-oriented 19- and 30-methyl groups. To determine the stereochemistry of C-20, two arguments were considered: the NOESY correlation of H3-21 with H-16α (δH 1.38 m) exhibiting an α-oriented 21-methyl group and the 1H NMR chemical shift of H3-21 at δH 0.86, which is characteristic to the euphane triterpenes (instead of H-21 at δH 0.94, which is characteristic to tirucallane triterpenes).18−21 Therefore, compound 1 was confirmed to be (23E)-25-hydroperoxyeupha-8,23-diene-3β-ol-7-one, which was named desmondiin A. Compound 1 is a stereoisomer of (23E)-25-hydroperoxytirucalla-8,23-dien-3β-ol-7-one (19) isolated from Euphorbia micractina.17
Figure 3.

Inhibition activity of 1 against amastigote replication in the host cells. Compound 1 efficiently inhibited amastigote replication, thereby limiting the transmission of the parasite from infected cells, as observed with benznidazole. (A) Representative brightfield microscopic image of CCL39 fibroblasts infected with Trypanosoma cruzi with amastigotes (arrow). (B) Infected cells exhibiting the GFPY fluorescence signal of the analyzed T. cruzi strain. (C) Inhibition curves of compound 1 and the positive control, benznidazole, of the GFPY parasite release from the infected RAW264.7 cells were measured by fluorescence-activated cell sorting (FACS) after 6 days. The data show the mean values ± SD of three independent experiments (each performed in triplicates).
Desmondiin C (2) was isolated as a white, amorphous powder. Its molecular formula was C30H48O3, as determined by HR-ESI-MS analysis (m/z 457.3688 [M + H]+, calcd. for C30H49O3+: 457.3676). The 1D- and 2D-NMR spectra of 2 revealed that it shares the same structural series as 1. Further, the 1H and 13C JMOD NMR spectra of 2 indicated two keto groups (δC = 198.3 and 215.3) (Tables 1 and 2). The C-23–C-24 olefin bond and hydroxyl group at C-25 were absent, and a 24-keto group and an isopropyl group [C-25(C-26)–C-27] were detected. The position of the keto group at C-24 was supported by the HMBC correlations of H-23 (δH 2.43 m) with C-24 (δC 215.3), and the isopropyl group connected to the keto group was shown by the HMBC cross peaks of H3-26 and H3-27 (both at δH 1.10 d) with C-24. Thus, it was confirmed that desmondiin C (2) was eupha-8-ene-3β-ol-7,24-dione.
Desmondiin D (3) was isolated as a white, amorphous powder. Its molecular formula was C29H46O3, as confirmed by HR-ESI-MS analysis (m/z 443.3527 [M + H]+, calcd. for C29H47O3+: 443.3520). The 1H and 13C JMOD NMR spectra of 3 indicated that it exhibited the same 3-hydroxy-7-keto-8-ene-substituted skeleton as 1 and 2, but in 3, a seven-carbon-containing moiety at C-17 was elucidated. This side chain comprised two methyls, three methylenes, one methine, and one keto group (Tables 1 and 2). The keto group was positioned at C-25 because of its HMBC correlation with the terminal 26-methyl group (δH = 2.13 s). 1H–1H COSY correlation between H2-24 and H2-23, and HMBC cross-peaks between H3-21 and C-22, and H3-26 and C-25 provided evidence to the structure of the C7 side chain. Further, diagnostic NOESY correlation between H-11α/H3-18, H-11β/H3-19, H3-19/H-1β, H-1α/H-3α, H-3α/H-5α, H3-30/H-17, H-17/H3-21 (Figure 2) indicated the stereostructure of desmondiin D (3).
Figure 2.
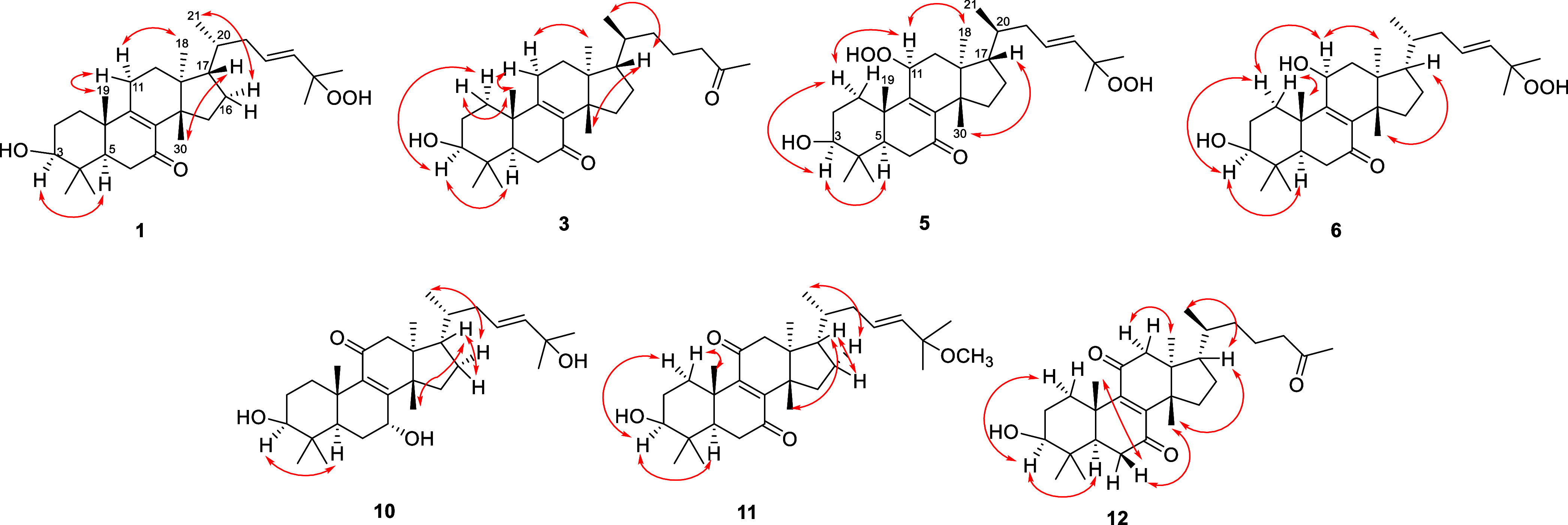
Key NOESY correlations of compounds 1, 3, 5, 6, 10–12.
Desmondiin E (4) was isolated as a white, amorphous powder. HR-ESI-MS data provided its molecular formula C30H48O3 (m/z 457.3685 [M + H]+, calcd. for C30H49O3+: 457.3676). The 1D and 2D NMR spectral data of 4 displayed the same tirucallan-8-en-7-one structure as that of 3 (Tables 1 and 2), the differences being in the C-24–C-27 part of the molecule. This part comprised a quaternary carbon (δC 147.6), a methine (δH 4.02 t; δC 76.5), a methylidene (δH 4.84 br s, 4.93 br s; δC 111.6), and one methyl group (δH 1.72 s; δC 17.3). HMBC correlations of this methyl group with C-25, C-26, and C-24), in addition to the correlations of the methine proton (δH 4.02) with C-25 and C-22 confirmed the presence of the 24-hydroxy-25-ene-substituted side chain and allowed the structure of desmondiin E, as depicted in the structural formula 4. This compounds was synthetised by reduction of (24R)-24-hydroperoxytirucalla-8,25-dien-3-ol-7-one by Xu et al.17 The stereoisomer of 4, the euphane sooneuphanone was isolated from Euphorbia soongarica.22
The molecular formula of desmondiin F (5) was C30H48O6, as determined by the HR-APCI-MS analysis (m/z 505.3519 [M + H]+, calcd. for C30H49O6+: 505.3524). The evaluation of the 1D- and 2D-NMR spectra of 5 revealed that it was similar to the known compound, (23E)-25-hydroperoxytirucalla-8,23-dien-3β-ol-7-one (19),17 also isolated in this study. However, the resonances attributed to the 11-methylene of 19 were replaced by that of a methine group (δH 5.04 t; δC 81.1) in 5 (Tables 1 and 2). The position of this oxygenated methine at C-11 was supported by the HMBC correlations between the proton signal at δH 5.04 (H-11) with the carbon signals at δC 157.3 (C-9), 143.5 (C-8), and 38.2 (C-12) in the HMBC spectrum. The deshielded 13C NMR signal of C-11 (δC 81.1) indicated the presence of a hydroxyperoxy group in this position,23 corresponding to the molecular formula. The NOESY experiment of 5 confirmed the tirucallane skeleton and α-position of H-11 by demonstrating the NOE correlation between H-11α and H3-18 (Figure 2). The β-oriented 11-hydroxyperoxy group was consistent with the coupling constant of H-11, i.e., 7.7 Hz.24 Therefore, the structure of desmondiin F (5) was elucidated as (23E)-11,25-dihydroperoxytirucalla-8,23-dien-3β-ol-7-one.
Desmondiin G (6) had the molecular formula C30H48O5 based on the HR-APCI-MS analysis (m/z 489.3568 [M + H]+, calcd. for C30H49O5+: 489.3575; m/z 455.3512 [M – OOH]+, calcd. for C30H47O3+: 455.3520). The 1D- and 2D-NMR spectra of 6 revealed that this compound was an euphane triterpene substituted with one keto and two hydroxy groups, and contained a tetrasubstituted olefin and the same side chain at C-17, as in compound 1 (Tables 1 and 2). The position of the keto group (δC 200.2) at C-7 was determined by its HMBC correlation with those of H-5 and H-6. The presence of the 11β-hydroxy group was confirmed by the HMBC cross peaks of H2-12 with C-11 and the NOESY correlation of H-11 with H-1α (δH 1.56) and H3-18 (Figure 2). The presence of a C-8–C-9 double bond was confirmed from the long-range correlations of H3-19 and H3-30 with carbon signals at δC 140.5 (C-8) and 161.3 (C-9), respectively. The 23-ene-25-hydroperoxy structure of 7 corresponded to the MS data as well as the 1H and 13C chemical shift assignments of 1. Thus, desmondiin G was confirmed as (23E)-25-hydroperoxyeupha-8,23-diene-3β,11β-diol-7-one (6).
Desmondiin H (7) exhibited a quasi-molecular ion at m/z 471.3465 [M + H]+ (calcd. for C30H47O4 471.3469) in the positive HR-APCI-MS spectrum, indicating that its molecular formula was C30H46O4. The NMR spectroscopic features of 7 were similar to those of 6, except for the aliphatic chain at C-17. In this part of compound 7, a trisubstituted olefin group (δH 6.48 t; δC 155.0, 139.9) and an aldehyde group (δH 9.40 s; δC 195.4) were detected (Tables 1 and 2). The methyl group at δH 1.76 s (H3-27) exhibited HMBC correlation with the olefinic and aldehyde carbons, indicating that it exhibited a 24-en-26-al structure. The key NOESY correlations were the same as for compound 5 indicating that desmondiin H (7) comprised a tirucallane skeleton.
Desmondiin I (8) had a molecular formula C30H48O3, as confirmed the HR-ESI-MS analysis (m/z 457.3687 [M + H]+, calcd. for C30H49O3+: 457.3676). The 1H and 13C NMR spectra displayed signals that were attributable to an euphane triterpene containing two hydroxylated methines (δH 3.31 dd, 4.27 dd; δC 78.8, 65.6), one trisubstituted (δH 5.08 t; 131.4) and one tetrasubstituted olefin group (δC 141.6, 157.3), and one keto group (δC 200.4) (Tables 3 and 4). The HMBC spectrum facilitated the determination of the positions of these functionalities (Figure 1): the hydroxy groups were positioned at C-3 and C-7 with respect to the correlations of H-3 with C-28 and C-29 and H-7 with C-5, C-8, and C-9. The position of the keto group at C-11 was elucidated based on its HMBC cross peaks with H2-12.
Table 3. 1H NMR (500 MHz) Data for Compounds 8–10 in CDCl3.
|
δH,mult. (Jin Hz) |
|||
|---|---|---|---|
| No. | 8 | 9 | 10 |
| 1 | 1.01, m | 1.01, m | 1.00, m |
| 2.56, dt (13.4, 3.4) | 2.57, dt (13.3, 3.2) | 2.58, m | |
| 2 | 1.70, m | 1.71, m | 1.70, m |
| 1.67, m | |||
| 3 | 3.31, dd (11.3, 5.2) | 3.32, dd (10.4, 3.4) | 3.32, dd (11.2, 5.0) |
| 5 | 1.42, m | 1.43, m | 1.40, m |
| 6 | 1.83, m | 1.83, d (14.1) | 1.82, d (14.0) |
| 1.53, m | 1.69, m | ||
| 7 | 4.27, dd (3.6, 1.7) | 4.27, br s | 4.27, br s |
| 12 | 2.60, d (18.9) | 2.62, m | 2.60, d (18.8) |
| 2.44, d (18.9) | 2.42, m | 2.42, d (18.8) | |
| 15 | 2.36, m | 2.36, m | 2.37, m |
| 1.39, m | 1.40, m | 1.41, m | |
| 16 | 1.42, m | 1.42, m | 1.43, m |
| 2.01, m | 2.00, m | 2.00, m | |
| 17 | 1.66, m | 1.68, m | 1.69, m |
| 18 | 0.92, s | 0.92, s | 0.95, s |
| 19 | 1.17, s | 1.17, s | 1.17, m |
| 20 | 1.46, m | 1.47, m | 1.53, m |
| 21 | 0.82, d (6.4) | 0.89, d (6.3) | 0.86, d (6.8) |
| 22 | 1.06, m | 1.07, m | 1.71, m |
| 1.49, m | 1.68, m | 2.25, dt (13.3, 3.9) | |
| 23 | 1.86, m | 1.43, m | 5.60, d (15.6) |
| 2.09, m | 1.62, m | ||
| 24 | 5.08, t (7.3) | 4.02, t (6.3) | 5.55, dd (16.1, 6.6) |
| 26 | 1.68, s | 4.85, s | 1.30, s |
| 4.94, s | |||
| 27 | 1.60, s | 1.72, s | 1.30, s |
| 28 | 1.04, s | 1.05, s | 1.05, s |
| 29 | 0.85, s | 0.85, s | 0.85, s |
| 30 | 1.01, s | 1.01, s | 1.02, s |
Table 4. 13C NMR (125 MHz) Data for Compounds 8–10 in CDCl3.
|
δC,type |
|||
|---|---|---|---|
| No. | 8 | 9 | 10 |
| 1 | 33.9, CH2 | 34.0, CH2 | 33.9, CH2 |
| 2 | 28.0, CH2 | 28.0, CH2 | 28.0, CH2 |
| 3 | 78.8, CH | 78.8, CH | 78.8, CH |
| 4 | 38.5, C | 38.6, C | 38.6, C |
| 5 | 45.5, CH | 45.6, CH | 45.6, CH |
| 6 | 28.6, CH2 | 28.6, CH2 | 28.6, CH2 |
| 7 | 65.6, CH | 65.6, CH | 65.6, CH |
| 8 | 157.3, C | 157.2, C | 157.2, C |
| 9 | 141.6, C | 141.6, C | 141.6, C |
| 10 | 38.6, C | 38.6, C | 38.6, C |
| 11 | 200.4, C | 200.3, C | 200.2, C |
| 12 | 51.8, CH2 | 51.7, CH2 | 51.6, CH2 |
| 13 | 44.3, C | 44.3, C | 44.4, C |
| 14 | 51.1, C | 51.1, C | 51.1, C |
| 15 | 29.7, CH2 | 29.7, CH2 | 29.7, CH2 |
| 16 | 27.8, CH2 | 27.8, CH2 | 27.6, CH2 |
| 17 | 50.8, CH | 50.8, CH | 50.4, CH |
| 18 | 18.3, CH3 | 18.5, CH3 | 18.5, CH3 |
| 19 | 18.1, CH3 | 18.4, CH3 | 18.1, CH3 |
| 20 | 36.0, CH | 36.1, CH | 36.3, CH |
| 21 | 18.6, CH3 | 18.8, CH3 | 18.9, CH3 |
| 22 | 35.3, CH2 | 30.9, CH2 | 38.0, CH2 |
| 23 | 25.3, CH2 | 32.1, CH2 | 125.1, CH |
| 24 | 124.8, CH | 76.1, C | 139.9, CH |
| 25 | 131.4, C | 147.9, C | 70.9, C |
| 26 | 25.8, CH3 | 111.3, CH2 | 30.1, CH3 |
| 27 | 17.8, CH3 | 17.7, CH3 | 30.1, CH3 |
| 28 | 28.3, CH3 | 28.3, CH3 | 28.3, CH3 |
| 29 | 16.2, CH3 | 16.3, CH3 | 16.2, CH3 |
| 30 | 26.0, CH3 | 26.1, CH3 | 26.0, CH3 |
The 8,9-olefin group was confirmed by the HMBC correlations of H3-30 and H2-6 with C-8 and H3-19 with C-9. The 24,25 double bond was proved by the correlations of H3-27, H3-26, and H-23 with C-24 and C-25. The NOESY experiment allowed the stereochemical assignment of 8 considering the H-3/H-7, H-7/H3-30, H-20/H-12β, H-16α/H3-21, H-16β/H-17, and H3-30/H-17 cross-peaks Thus, the structure of desmondiin I corresponded to eupha-8,24-dien-3β,7α-diol-11-one (8).
Desmondiin J (9) was isolated as a white amorphous powder with a molecular formula C30H48O4, as established from the HR-APCI-MS analysis (m/z 473.3620 [M + H]+, calcd. for C30H49O4+: 473.3625). This compound displayed structural patterns similar to those of 8, only differentiated by the side chain at C-17 which was similar to that of compound 4 (Tables 3 and 4). The key NOESY correlation of H3-30 with H-7, and H-3 with H-5 confirmed the presence of 3β- and 7α-hydroxy groups.
HR-APCI-MS spectrum of desmondiin K (10) displayed a protonated molecular ion peak at m/z 473.3620 [M + H]+ (calcd. for C30H49O4+: 473.3625), corresponding to a molecular formula C30H46O4. A comparison of the 1D- and 2D-NMR data of 10 with those of 8 indicated that these compounds only differed in positions C-23–C-27 (Tables 3 and 4). This structural part comprised a disubstituted olefin group [δH 5.55 dd (J = 15.8, 6.6 Hz), 5.60 d (J = 15.8 Hz), δC 125.1, 139.9.], one oxygen-substituted quaternary carbon (δC 70.9), and two methyl groups (δH 2 × 1.30 s, δC 2 × 30.1). HMBC correlations of H-23, H3-26, and H3-27 with C-25 and those of H-24 with C-26 and C-27 demonstrated the presence of a double bond at C-23–C-24 and a hydroxyl group at C-25. The stereochemical assignment of desmondiin K (10) was made based on the NOESY correlations as depicted on Figure 2.
The molecular formula of desmondiin L (11) was C31H48O4, as provided by the HR-APCI-MS analysis (m/z 485.3618 [M + H]+, calcd. for C31H49O4+: 485.3625; m/z 471.3462 [M + H – CH2]+, calcd. for C30H47O4+: 471.3470); m/z 453.3359 [M + H – CH3OH]+, calcd. for C30H45O3+: 453.3363). The 1H NMR and 13C NMR spectra revealed that 11 was a triterpene comprising two keto groups, two olefin bonds, one methoxy, and one hydroxy group, as indicated by the carbon at δC 78.1 (C-3) (Tables 5 and 6). The keto groups were positioned at C-7 and C-11 based on the HMBC connectivities of H-5 and H2-6 to C-7 and H2-12 to C-11 (Figure 1). The positions of the olefin groups at C-8–C-9 and C-23–C-24 were confirmed by the HMBC correlations H3-30/C-8, H3-19/C-9, and H2-22/C-23, H3-26, and H3-27/C-24, respectively. The 25-methoxy substitution was evident from the long-range correlation of the OCH3 protons (δH 3.15) with C-25 (δC 74.9). The NOESY correlations shown in Figure 2 led to the conclusion that desmondiin L is eupha-8,23-dien-3β-hydroxy-25-methoxy-8,11-dione (11).
Table 5. 1H NMR (500 MHz) Data for Compounds 11–15 in CDCl3.
|
δH,mult. (Jin Hz) |
||||
|---|---|---|---|---|
| No. | 11 | 12 | 13 | 14 + 15 |
| 1 | 1.10, m | 1.11, m | 1.05, m | 1.11, m |
| 2.55, m | 2.52, m | 2.45, m | 2.52, m | |
| 2 | 1.67, m | 1.73, m | 1.71, m | 1.74, m |
| 1.76, m | 1.74, m | |||
| 3 | 3.30, dd (10.3, 5.5) | 3.30, m | 3.28, m | 3.30, dd (10.4, 5.5) |
| 5 | 1.64, m | 1.64, dd (13.5, 4.9) | 1.65, m | |
| 6 | 2.44, m | 2.48, m | 2.49, m | 1.65, m |
| 2.55, m | 2.52, m | 2.54, m | 2.48, m | |
| 12 | 2.48, d (18.7) | 2.43, d (19.0) | 2.44, d (18.7) | 2.44, m |
| 2.67, d (18.7) | 2.65, d (19.0) | 2.68, d (18.7) | 2.64, m | |
| 15 | 1.66, m | 1.66, m | 1.65, m | 1.65, m |
| 2.15, t (11.2) | 2.13, m | 2.16, m | 2.14, m | |
| 16 | 1.41, m | 1.38, m | 1.40, m | 1.40, m |
| 2.02, m | 2.02, m | 2.04, m | 2.02, m | |
| 17 | 1.68, m | 1.65, m | 1.67, m | 1.65, m |
| 18 | 0.95, s | 0.92, s | 0.93, s | 0.92, s |
| 19 | 1.31, s | 1.31, s | 1.31, s | 1.30, s |
| 20 | 1.53, m | 1.46, m | 1.55, m | 1.46, m |
| 21 | 0.87, d, 6.5 | 0.92, d (6.8) | 0.94, d (6.4) | 0.87, d (6.4) |
| 22 | 1.73, m | 0.99, m | 1.24, m | 1.10, m |
| 2.27, dt, (13.7, 3.3) | 1.44, m | 1.65, m | 1.50, m | |
| 23 | 5.48, m | 1.46, m | 2.29, m | 1.37, m |
| 1.65, m | 2.42, m | 1.65, m | ||
| 24 | 5.40, d (15.8) | 2.40, t (7.0) | 6.45, t (7.3) | 4.27, t (6.5) |
| 25 | 1.25, s | |||
| 26 | 1.25, s | 2.13, s | 9.40, s | 5.02, br s |
| 5.04, m | ||||
| 27 | 1.75, s | 1.74, s | ||
| 28 | 1.03, s | 1.03, s | 1.03, s | 1.03, s |
| 29 | 0.90, s | 0.91, s | 0.90, s | 0.90, s |
| 30 | 1.09, s | 1.08, s | 1.09, s | 1.08, s |
| 25-OCH3 | 3.15, s | |||
Table 6. 13C NMR (125 MHz) Data for Compounds 11–15 in CDCl3.
|
δC,type |
||||
|---|---|---|---|---|
| No. | 11 | 12 | 13 | 14, 15 |
| 1 | 34.1, CH2 | 34.1, CH2 | 34.1, CH2 | 34.1, CH2 |
| 2 | 27.6, CH2 | 27.6, CH2 | 27.6, CH2 | 27.6, CH2 |
| 3 | 78.1, CH | 78.2, CH | 78.1, CH | 78.1, CH |
| 4 | 38.7, C | 38.8, C | 38.7, C | 38.7, C |
| 5 | 48.7, CH | 48.7, CH | 48.6, CH | 48.6, CH |
| 6 | 35.9, CH2 | 36.0, CH2 | 35.9, CH2 | 35.9, CH2 |
| 7 | 200.0, C | 199.9, C | 200.0, C | 200.0, C |
| 8 | 149.9, C | 150.0, C | 149.9, C | 149.9, C |
| 9 | 154.9, C | 154.9, C | 154.9, C | 155.0, C |
| 10 | 38.2, C | 38.3, C | 38.2, C | 38.2, C |
| 11 | 201.8, C | 202.0, C | 201.7, C | 202.0, C |
| 12 | 51.5, CH2 | 51.6, CH2 | 51.6, CH2 | 51.5/51.6, CH2 |
| 13 | 45.3, C | 45.3, C | 45.2, C | 45.3, C |
| 14 | 48.0, C | 48.1, C | 48.1, C | 48.1, C |
| 15 | 31.9, CH2 | 32.0, CH2 | 31.9, C | 31.9, CH2 |
| 16 | 28.1, CH2 | 28.3, CH2 | 28.3, CH2 | 27.9, CH2 |
| 17 | 49.2, CH | 49.5, CH | 48.6, CH | 48.6, CH |
| 18 | 18.8, CH3 | 18.7, CH3 | 18.8, CH3 | 18.6, CH3 |
| 19 | 17.9, CH3 | 17.9, CH3 | 17.9, CH3 | 17.9, CH3 |
| 20 | 36.2, CH | 36.1, CH | 35.9, CH | 35.8, CH |
| 21 | 18.9, CH3 | 18.5, CH3 | 18.5, CH3 | 18.8, CH3 |
| 22 | 38.2, CH2 | 34.6, CH2 | 34.1, CH2 | 30.7/31.0, CH2 |
| 23 | 128.2, CH | 21.0, CH2 | 26.4, CH2 | 27.6, CH2 |
| 24 | 137.3, CH | 44.0, CH2 | 154.4, CH | 89.5/90.1, CH |
| 25 | 74.9, C | 208.8, C | 139.6, C | 144.0/143.7, C |
| 26 | 26.0, CH3 | 30.0, CH3 | 195.3, CH | 114.3/114.8, CH2 |
| 27 | 26.0, CH3 | 9.4, CH3 | 17.2/17.4, CH3 | |
| 28 | 27.7, CH3 | 27.8, CH3 | 27.7, CH3 | 27.7, CH3 |
| 29 | 15.2, CH3 | 15.2, CH3 | 15.3, CH3 | 15.2, CH3 |
| 30 | 24.1, CH3 | 24.2, CH3 | 24.1, CH3 | 24.2, CH3 |
| 25–OCH3 | 50.4 | |||
HR-ESI-MS analysis of desmondiin M (12) displayed an [M + H]+ ion at m/z 457.3325, corresponding to a molecular formula C29H44O4 (calcd. for C29H44O4+: 457.3312), indicating that it was a nor-triterpene. The 1H and 13C JMOD NMR spectra of 12 were similar to those of 11, except for the side chain connected at C-17 (Tables 5 and 6). The C-23–C-24 double bond in 12 was saturated, and an acetyl group (δC 30.0, 208.8, δH 2.13 s) was detected at C-24. This was supported by the HMBC correlations of H2-24 (δH 2.40 t) and H3-26 (δH 2.13 s) with C-25 (δC 208.8). Further, HMBC correlations between H2-24 and C-22, C-23 and between H3-21 and C-22 justified the shortened (C7) side chain at C-17. NOESY correlations (Figure 2) proved the tirucallane skeleton by the cross-peaks of H-17 with H3-21 and H3-30, and H-20 with H3-18 thus, the structure 3β-hydroxy-27-nor-tirucalla-8-en-7,11,25-trione (12) was elucidated for desmondiin M (Figure 1).
Desmondiin N (13), a white amorphous powder, had a molecular formula C30H44O4 as confirmed by HR-ESI-MS analysis (m/z 469.3308 [M + H]+, calcd for C30H45O4+: 469.3312). The NMR data of 13 (Tables 5 and 6) indicated that it was very similar to those of 11 and 12, except for the side chain at C-17, which was elucidated as the same as that of desmondiin G (7). Accordingly, the structure of desmondiin N was determined as 13.
The isomeric compounds desmondiin O and P (14, 15) were isolated as a mixture. Their molecular formulas are C30H46O5 each, as calculated using the HR-ESI-MS peak at m/z 487.3430 [M + H]+ (calcd. for C30H47O5+: 487.3418). A comparison of the 1H and 13C JMOD NMR spectra of 14 and 15 (Tables 5 and 6) revealed that they are different in the C8 aliphatic chain, as indicated by the duplicated carbon signals assigned to this part (δC-22 30.7/31.0, δC-24 89.5/90.1, δC-25 143.7/144.0, δC-26 114.3/114.8, and δC-27 17.2/17.4). Compounds 14 and 15 are structurally similar to euphorol A isolated from Euphorbia resinifera latex, although the 1H (δH24 4.27 t) and 13C NMR signals (δC-24 89.5/90.1) of 24-methine group of 14 and 15 were downfield-shifted compared to those of euphorol A (δH-24 4.01 t; δC-24 76.5).23 Such deshielding of the NMR signals indicated the presence of a hydroperoxy group instead of a hydroxy group.17 Based on the above data, desmondiins O and P were identified as 24α (14) and 24β (15) stereoisomers of eupha-8,25-dien-3β,24-dihydroxy-8,11-dione.
Compounds 16 (named desmondiin B),2517,2118,17,1819,1720 (neritriterpenol J),2321 (neritriterpenol K),2322,17,26 and 23(24) were previously reported from E. sikkimensis, E. kansui, E. micractina, Monadenium lugardae, E. neriifolia, E. humifusa, and E. resinifera.
Determination of the Trypanocidal Activity
To biologically characterize the isolated triterpenoids for their trypanocidal activities, their cytotoxicities in RAW264.7 macrophages (mammalian host cells) and T. cruzi epimastigotes were analyzed. As presented in Table 7, eight of the compounds displayed relatively potent and selective trypanocidal activities, with good selectivity indices (>5).
Table 7. Compounds Showing Trypanocidal Effects on Trypanosoma cruzi with IC50 Values <5 μM and a Selectivity Index of 5.
| Compound | RAW 264.7 IC50(μM) | T. cruziepimastigote WT-Y IC50(μM) | Selectivity Index |
|---|---|---|---|
| 1 | 38.1 | 4.5 | 8.5 |
| 2 | 31.5 | 5 | 6.3 |
| 3 | 31 | 4 | 7.8 |
| 5 | 18.4 | 3.4 | 5.4 |
| 7 | 23.5 | 3.7 | 6.4 |
| 8 | 28.4 | 4.9 | 5.8 |
| 12 | 24.4 | 3.7 | 6.6 |
| 20 + 21 | 26.7 | 3.1 | 8.6 |
Interestingly, some compounds (5, 8, and 12) at the lower micromolar concentrations increased the proliferation of the epimastigotes by 15–30%; however, they became trypanocidal (inhibiting the epimastigotes) at higher concentrations (Supporting Information Figure S130). To determine the efficiencies of these compounds in a cellular infection system in which the amastigote stage was targeted (Figures 3A–B), desmondiin A (1) was analyzed for its effects against amastigotes, which displayed the best potency and selectivity toward the epimastigotes. As shown in Figure 3C, desmondiin A (1) efficiently and potently inhibited parasite replication in the host cells, as confirmed by the concentration-dependent reduction of parasite release after 6 days (see Experimental section), yielding an IC50 value of 2.4 ± 0.3 μM. Thus, desmondiin A (1) was as potent as the positive control, benznidazole, in the infection assay (IC50 = 3.2 ± 0.5 μM, with a robust 10-fold selective toxicity over the host cells). The structure–activity relationships of the desmondiins appeared complex and related to the absolute configuration of the side chain of the tetracyclic triterpenoid scaffold, combined with the functional group substitutions. Although studies have demonstrated the trypanocidal effects of triterpenoids on T. cruzi,27 their mode of action remains elusive. Desmondiin A (1) is currently among the most potent antichagasic triterpene when compared to previous reports on different triterpenoids,28,29 including ursolic acid and betulinic acid.30−32 Subsequent studies will focus on elucidating the action mechanism of these compounds, as well as the utilization of natural scaffolds to generate more potent antichagasic triterpenoids.
Experimental Section
General Experimental Procedures
The optical rotations were measured by using a Jasco P-2000 polarimeter (Jasco International Co. Ltd., Hachioji, Tokyo, Japan). NMR spectra were recorded in CDCl3 using a Bruker Avance DRX 500 spectrometer at 500 MHz (1H) and 125 MHz (13C). Signals of residue nondeuterated solvents were used as references. The HR-ESI-MS spectra were recorded on a Thermo Scientific Q-Exactive Plus Orbitrap mass spectrometer equipped with an ESI- or APCI-ion source in positive ionization mode. The MassLynx software was used for data acquisition and processing. To separate the extract and fractions, VLC was performed using silica gel (15 μm, Merck); LiChroprep RP-18 (40–63 μm, Merck) stationary phase was used for RP-VLC; OCC was performed with polyamide (MP Biomedicals). Further, flash chromatography (FC) was performed using a CombiFlash Rf+ Lumen instrument with integrated ultraviolet (UV), UV–visible (UV–Vis), and evaporative light scattering detection using an RP column (RediSep C18 Bulk 950, Teledyne Isco, Lincoln, NE, USA). PTLC was performed on silica gel 60 F254 plates (Merck). HPLC was performed using WUFENG, WATERS, and Agilent HPLC instruments equipped with LiChrospher Si 60 (4 mm × 250 mm, 5 μm) and Luna (R) Silica (2) 100 (250 mm × 21.2 mm, 5 μm), as well as RP Kinetex C18 100A (4.6 mm × 150 mm, 5 μm) and Agilent ZORBAX ODS C18 100A (9.4 mm × 250 mm, 5 μm) columns. TLC plates were detected under UV light at 254 nm and sprayed with concentrated sulfuric acid followed by heating for 5 min. Solvents employed for the extraction, OCC, VLC, FC, and PTLC were of analytical grade (VWR Ltd., Hungary).
Plant Material
The aerial parts of Euphorbia desmondii Keay and Milne-Redh (Euphorbiaceae) were collected in June 2020 from the outskirts of Zaria City (Kaduna State, Nigeria). They were identified by Umar Shehu Gallah (National Research Institute for Chemical Technology, NARICT, Zaria, Nigeria). A voucher specimen was deposited in NARICT (number: Narict/Biores/322) and in the Herbarium of the Department of Pharmacognosy, University of Szeged (Szeged, Hungary; No. 898).
Extraction and Isolation
The experiment aimed to isolate triterpenes, so we designed all separation steps adequate to triterpenes, and the progress of the compound isolation was monitored using TLC with conc. sulfuric acid detection. The air-dried powdered plant material (1950 g) was extracted with MeOH (63 L) via percolation at room temperature. Thereafter, the MeOH extract was concentrated in vacuo to yield a greenish brown oily material (207.4 g). This extract was dissolved in MeOH–H2O (1:1) and subjected to solvent–solvent partitioning using CHCl3 (4 × 1000 mL). The organic phase (109.8 g) was subjected to OCC on polyamide (320 g) using MeOH–H2O (1:4, 2:3, 3:2, 4:1, and 5:0) mixtures as eluents to yield 20% (5.5 g), 40% (11.4 g), 60% (22.4 g), 80% (14.2 g), and 100% (31.2 g) MeOH fractions, respectively. The 60% MeOH fraction displayed a unique TLC profile and was selected for further chromatographic separations. VLC was performed on the 60% MeOH fraction using silica gel and a gradient system comprising cyclohexane–EtOAc–EtOH (100:0:0, 90:10:0, 80:20:0, 70:30:0, 75:25:1, 75:25:2, 75:25:4, 75:25:6, 75:25:8, 75:25:10, 75:25:12, 75:25:15, and 75:25:20). The obtained fractions were analyzed by TLC, and those with similar profiles were combined, yielding combined fractions A–H. Fraction C (1.14 g) was separated by VLC on reversed-phase Si gel using MeOH–H2O mixtures (50:50, 60:40, 65:35, 70:30, 75:35, 80:20, 85:15, and 90:10) as eluents, thus affording fractions C/I–C/VII. Next, fraction C/VI was subjected to NP-VLC separation using a gradient system comprising petroleum ether–EtOAc as the mobile phase to afford subfractions C/IV/1–4. Furthermore, C/IV/3 was further separated by RP-HPLC using an isocratic system comprising MeCN–H2O (58:42) as the eluent to yield fractions C/IV/3/A–C. Additionally, the NP-HPLC purification of C/IV/3/C via isocratic elution with cyclohexane–EtOAc (65:35) resulted in the isolation of 12 (4.9 mg) and fraction C/IV/3/C/III; they were separated by PTLC on silica gel using CHCl3–acetone (9:1) as the developing system to isolate 2 (2.9 mg). Similarly, subfraction C/V was subjected to NP-VLC separation using a gradient system comprising petroleum ether–EtOAc as the eluent to yield fractions C/V/1–3. The RP-HPLC separation of C/V/3 using a gradient system comprising MeCN–H2O (from 85:15 to 100:0) yielded subfractions C/V/3/A–C. The two-step purification of C/V/3/B by NP-HPLC (gradient system: cyclohexane/EtOAc, from 70:30 to 65:35) and NP-PLC (isocratic eluent: CHCl3–acetone, 9:1) facilitated the isolation of 8 (2.0 mg). The NP-HPLC purification of fraction C/V/3/C using an isocratic system comprising cyclohexane/EtOAc (65:35) facilitated the isolation of 16 (4.3 mg), 17 (11.6 mg), and 18 (7.3 mg).
Fraction D (3.21 g) was separated by FC using MeOH–H2O mixtures (from 50:50 to 100:0) as eluents to afford subfractions D/I–XIV. The four-step purification of fraction D/VII by NP-VLC (gradient elution with petroleum ether–EtOAc mixtures), RP-HPLC [gradient system of MeCN–H2O (91:9–100:0)], NP-HPLC (gradient system: cyclohexane–EtOAc, from 55:45 to 52:48), and NP-PTLC (isocratic elution: CHCl3–acetone, 9:1) resulted in the isolation of 12 (4.5 mg). Conversely, the chromatographic separation of D/VIII by NP-VLC using a gradient system comprising petroleum ether–EtOAc as the eluent afforded fractions D/VIII/1–6. The RP-HPLC separation of D/VIII/2 using a gradient system comprising MeCN–H2O (96:4–100:0) afforded fractions D/VIII/2/A–D. The two-step purification of D/VIII/2/B by NP-HPLC (gradient system: cyclohexane–EtOAc, 60:40–55:45) and RP-HPLC (gradient system: MeOH–H2O, 89:11–91:9) resulted in the isolation of 22 (8.4 mg) and the mixture of 14 and 15 (2.3 mg). The two-step purification of D/VIII/2/C/III by NP-HPLC (gradient system: cyclohexane–EtOAc, 50:50–45:55) and NP-PLC (developing system: CHCl3–acetone, 87:13) resulted in the isolation of 3 (2.3 mg) and 4 (5.2 mg). The NP-HPLC separation of fraction D/VIII/2/C yielded D/VIII/2/C/I–III; the three fractions were purified by NP-PTLC using a mobile phase comprising CHCl3–acetone (87:13). Employing this approach, D/VIII/2/C/I, D/VIII/2/C/II, and D/VIII/2/C/III facilitated the isolation of 13 (2.3 mg), 19 (4.3 mg), and 1 (11.3 mg), respectively. Furthermore, fraction D/VIII/3 was subjected to RP-HPLC using an isocratic mobile system comprising MeCN–H2O (50:50), affording D/VIII/3/A–C. The NP-HPLC purification of D/VIII/3/A using a gradient system comprising cyclohexane–EtOAc (50:50–47:53) resulted in the isolation of 5 (2.5 mg). The further purification of D/VIII/3/C by NP-PTLC using a CHCl3–acetone (86:14) developing system resulted in the isolation of compound 23 (6.3 mg).
Additionally, fraction E (3.28 g) was subjected to FC using MeOH/H2O mixtures (from 55:45 to 100:0) as eluents. This separation afforded subfractions E/I–E/VI. The NP-VLC separation of E/IV using a gradient system comprising CHCl3–acetone (from 98:2 to 80:20) afforded E/IV/1–3. Thereafter, fraction E/IV/2 was subjected to RP-HPLC via gradient elution using MeCN–H2O mixtures (41:59–100:0) to yield E/IV/2/A–P. The two-step purification of E/IV/2/H by NP-HPLC (gradient system: cyclohexane–EtOAc, 50:50–45:55) and RP-HPLC (gradient system: MeOH–H2O, 88:12–92:8) resulted in the isolation of mixtures of 20 and 21 (2.2 mg). Similarly, the two-step purification of E/IV/2/P by NP-HPLC (gradient system: cyclohexane–EtOAc, 50:50–40:60) and RP-HPLC (gradient system: MeOH–H2O, 88:12–92:8) resulted in the isolation of 9 (2.3 mg) and 7 (1.9 mg). Further, the purification of fraction E/IV/3 by RP-HPLC (gradient system: MeCN–H2O, 71:29–78:22) and NP-HPLC (gradient system: cyclohexane–EtOAc, 50:50–30:70) resulted in the isolation of 6 (3.9 mg) and 10 (4.9 mg).
Desmondiin A (1)
White amorphous powder; [α]D26 – 7.9 (c 0.1, CHCl3); UV (MeOH) λmax (log ε) = 257 (3.85) nm; 1H and 13C NMR data, see Tables 1 and 2; HR-ESI-MS + m/z 473.3637 [M + H]+ (calcd. for C30H49O4+: 473.3625).
Desmondiin C (2)
White amorphous powder; [α]D27 + 11.6 (c 0.1, CHCl3); UV (MeOH) λmax (log ε) = 256 (3.80) nm; 1H and 13C NMR data, see Tables 1 and 2; HR-ESI-MS + m/z 457.3688 [M + H]+ (calcd. for C30H49O3+: 457.3676).
Desmondiin D (3)
White amorphous powder; [α]D25 – 6.3 (c 0.1, CHCl3); UV (MeOH) λmax (log ε) = 258 (3.81) nm; 1H and 13C NMR data, see Tables 1 and 2; HR-ESI-MS + m/z 443.3527 [M + H]+ (calcd. for C29H47O3+: 443.3520).
Desmondiin E (4)
White amorphous powder; [α]D30 – 10.7 (c 0.1, CHCl3); UV (MeOH) λmax (log ε) = 258 (3.80) nm; 1H and 13C NMR data, see Tables 1 and 2; HR-ESI-MS + m/z 457.3685 [M + H]+ (calcd. for C30H49O3+: 457.3676).
Desmondiin F (5)
White amorphous powder; [α]D26 + 9.1 (c 0.1, CHCl3); UV (MeOH) λmax (log ε) = 257 (3.79) nm; HR-APCI-MS + m/z 505.3519 [M + H]+ (calcd. for C30H49O6+: 505.3524).
Desmondiin G (6)
White amorphous powder; [α]D25 + 17.5 (c 0.1, CHCl3); UV (MeOH) λmax (log ε) = 258 (3.88) nm; 1H and 13C NMR data, see Tables 1 and 2; HR-APCI-MS + m/z 489.3568 [M + H]+ (calcd. for C30H49O5+: 489.3575) and 455.3512 [M – OOH]+ (calcd. for C30H47O3+: 455.3520).
Desmondiin H (7)
White amorphous powder; [α]D25 + 9.8 (c 0.05, CHCl3); UV (MeOH) λmax (log ε) = 256 (3.82) nm; 1H and 13C NMR data, see Tables 1 and 2; HR-APCI-MS + m/z 471.3465 [M + H]+ (calcd. for C30H47O4: 471.3469).
Desmondiin I (8)
White amorphous powder; [α]D25 – 11.5 (c 0.1, CHCl3); UV (MeOH) λmax (log ε) = 254 (3.80) nm; 1H and 13C NMR data, see Tables 3 and 4; HR-ESI-MS + m/z 457.3687 [M + H]+ (calcd. for C30H49O3+: 457.3676).
Desmondiin J (9)
White amorphous powder; [α]D25 – 8.4 (c 0.05, CHCl3); UV (MeOH) λmax (log ε) = 254 (3.78) nm; 1H and 13C NMR data, see Tables 3 and 4; HR-APCI-MS + m/z 473.3620 [M + H]+ (calcd. for C30H49O4+ 473.3625).
Desmondiin K (10)
White amorphous powder; [α]D25 + 6.3 (c 0.1, CHCl3); UV (MeOH) λmax (log ε) 253 (3.77) nm; 1H and 13C NMR data, see Tables 3 and 4; HR-APCI-MS + m/z 473.3620 [M + H]+ (calcd. for C30H49O4+: 473.3625) and 455.3513 [M + H – H2O]+ (calcd. for C30H47O3+: 455.3520).
Desmondiin L (11)
White amorphous powder; [α]D27 + 29.8 (c 0.1, CHCl3); UV (MeOH) λmax (log ε) = 270 (3.92) nm; 1H and 13C NMR data, see Tables 5 and 6; HR-APCI-MS + m/z 485.3618 [M + H]+ (calcd. for C31H49O4+: 485.3625), 471.3462 [M + H – CH2]+ (calcd. for C30H47O4+: 471.3470), and 453.3359 [M + H – CH3OH]+ (calcd. for C30H45O3+: 453.3363).
Desmondiin M (12)
White amorphous powder; [α]D26 + 26.3 (c 0.05, CHCl3); UV (MeOH) λmax (log ε) = 272 (3.95) nm; 1H and 13C NMR data, see Tables 5 and 6; HR-ESI-MS + m/z 457.3325 (calcd. for C29H44O4+: 457.3312).
Desmondiin N (13)
White amorphous powder; [α]D26 + 14.2 (c 0.1, CHCl3); UV (MeOH) λmax (log ε) = 219 (3.90), 269 (3.92) nm; 1H and 13C NMR data, see Tables 5 and 6; HR-ESI-MS m/z 469.3308 (calcd. for C30H45O4+: 469.3312).
Desmondiins O + P (14 + 15)
White amorphous solid; UV (MeOH) λmax (log ε) = 271 (3.85) nm; 1H and 13C NMR data, see Tables 5 and 6; HR-ESI-MS m/z 487.3430 [M + H]+ (calcd. for C30H47O5+: 487.3418).
Biological Assays
Epimastigotes of T. cruzi [Y strain (ATCC 50832)] were maintained in weekly passages in a liver infusion tryptose (LIT) medium supplemented with 10% heat-inactivated fetal bovine serum (hiFBS) at 28 °C, as described previously.33 The epimastigote cultures in the logarithmic-growth phase (1–5 × 107 parasites/mL) were used for the experiments unless stated otherwise. The green fluorescent protein (GFP)-overexpressing epimastigotes were generated by transfection using pTREX-n-eGFP, a gift from Rick Tarleton (Addgene plasmid # 62544; http://n2t.net/addgene:62544;RRID:Addgene_62544), as described previously. The GFP-expressing epimastigotes were cultured in LIT medium supplemented with 10% hiFBS and 500 μg/mL G418.
According to the Drugs for Neglected Diseases initiative (DNDi), an ideal lead compound must exhibit activity against trypanosomes at concentrations of less than or equal to 10 μM as well as exhibit a 10-fold-greater potency toward T. cruzi over mammalian cells (SI).33 To assess the selectivities of the isolated compounds, their trypanocidal activities against the epimastigotes were assessed by the Cell Proliferation Kit II (XTT) assay, as previously described.11 Briefly, 1.5 × 105 epimastigotes were seeded per well in 96-well plates. The compounds were added at a single concentration, 10 μM (for the initial screening experiments) or six concentrations ranging from 10–0.3 μM for 72 h at 28 °C. In all the experiments, benznidazole was used as the positive control. Following the incubation period, the plates were examined under a microscope for sterility and growth of controls. To the plates, 50 μL of XTT and PMS (Phenazine methosulfate, Sigma-Aldrich, MO, USA) solution (XTT and PMS at 0.5 and 0.025 mg/mL, respectively) was added, and the plates were then incubated at 28 °C for 2.5 h. The parasites were fixed by the addition of 50 μL of MeOH for 15 min before the absorbance was measured at 490 nm on a Tecan plate reader. Results were expressed as percentage cell viability relative to vehicle control or IC50 values calculated by GraphPad Prism version 8.0. All assessments were performed in triplicate in at least two independent experiments.
Fluorescence-activated cell sorting (FACS)-based quantitation of released parasites from infected RAW264.7 cells: The host cells were seeded in 24-well plates at densities of 40,000 and 10,000 cells/mL. The cells were allowed to adhere for 24 h, after which they were infected with trypomastigotes that were harvested by the swim-out procedure described above at a multiplicity of 10. On the next day, the noninternalized trypomastigotes were washed off, and fresh RPMI with 2% fetal bovine serum was added to the wells along with the test substance. In all of the experiments, the screening was performed in triplicate, including tests with DMSO and benznidazole controls. The released parasites were fixed using 4% paraformaldehyde in phosphate-buffered saline for 1 h, after which the samples were analyzed by FACS (BD Biosciences), as described previously.11
Microscopy
The host CCL39 cells were seeded in 24-well plates at a density of 20,000 cells/mL. The cells were allowed to adhere for 24 h, after which they were infected with trypomastigotes at a multiplicity of 10. On the next day, the noninternalized trypomastigotes were washed off, and fresh RPMI with 2% hiFBS was added to the wells along with the test substance. On the fourth day, following infection, the cells were fixed with 4% paraformaldehyde for 1 h and scanned using a Nikon Eclipse TS2 microscope. Thereafter, the images were analyzed on ImageJ.
Acknowledgments
This study was supported by National Research, Development and Innovation Office, Hungary (NKFIH; K-135845). The authors thank the financial support provided by the Ministry of Innovation and Technology of Hungary from NKFIH Fund, project no. TKP2021-EGA-32.
Data Availability Statement
The NMR data for 1–16 have been deposited in the Natural Products Magnetic Resonance Database (NP-MRD; www.np-mrd.org) and can be found at NP0333436 (1), NP0333438 (2), NP0333439 (3), NP0333440 (4), NP0333441 (5), NP0333442 (6), NP0333443 (7), NP0333444 (8), NP0333445 (9), NP0333446 (10), NP0333447 (11), NP0333448 (12), NP0333449 (13), NP0333450 (14), NP0333451 (15), and NP0333451 (16).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jnatprod.4c00730.
UV, HR-ESI-MS, and NMR spectra of the isolated compounds; data of antiproliferative assay on T. cruzi epimastigoge (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Coura J. R.; Viñas P. A.; Junqueira A. C. Mem. Inst. Oswaldo Cruz. 2014, 109, 856–62. 10.1590/0074-0276140236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://www.who.int/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis) retrived on 29 05 2024.
- Hemmige V.; Tanowitz H.; Sethi A. Int. J. Dermatol. 2012, 51, 501–508. 10.1111/j.1365-4632.2011.05380.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Albizu C.; Rivero R.; Ballering G.; Freilij H.; Santini M. S.; Bisio M. M. C. Front. Parasitol. 2023, 2, 138375 10.3389/fpara.2023.1138375. [DOI] [Google Scholar]
- Kemmerling U.; Bosco C.; Galanti N. Biol. Res. 2010, 43, 307–316. 10.4067/S0716-97602010000300007. [DOI] [PubMed] [Google Scholar]
- Cortes V.; Cruz A.; Onetti S.; Kinzel D.; Garcia J.; Ortiz S.; Lopez A.; Cattan P. E.; Botto-Mahan C.; Solari A. PLoS Negl. Trop. Dis. 2021, 15, e0009729 10.1371/journal.pntd.0009729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meymandi S.; Hernandez S.; Park S.; Sanchez D. R.; Forsyth C. Curr. Treat. Options Infect. Dis. 2018, 10, 373–388. 10.1007/s40506-018-0170-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarin-Bidóia D.; Garcia F. P.; Ueda-Nakamura T.; Silva S. O.; Nakamura C. V. Mem. Inst. Oswaldo Cruz. 2022, 117, e220396 10.1590/0074-02760220396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Huertas P.; Cardona-Castro N. Biomed. Pharmacother. 2021, 142, 112020 10.1016/j.biopha.2021.112020. [DOI] [PubMed] [Google Scholar]
- Izumi E.; Ueda-Nakamura T.; Dias Filho B. P.; Veiga Junior V. F.; Nakamura C. V. Nat. Prod. Rep. 2011, 28, 809–823. 10.1039/c0np00069h. [DOI] [PubMed] [Google Scholar]
- Salm A.; Krishnan S. R.; Collu M.; Danton O.; Hamburger M.; Leonti M.; Almanza G.; Gertsch J. iScience 2021, 24, 102310 10.1016/j.isci.2021.102310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govaerts R.; Frodin D. G.; Radcliffe-Smith A.. World Checklist and Bibliography of Euphorbiaceae (and Pandaceae) Vol. 4; Royal Botanic Gardens: Kew, 2000, pp 1–1622. [Google Scholar]
- Flora of West Tropical Africa. In Kew Bulletin, Vol 139, 1955. Retrieved from https://powo.science.kew.org/taxon/urn:lsid:ipni.org:names:346272-1#distributions on 4th January 2024.
- Burkill H. M.The useful plants of West Tropical Africa, Vol 2, 1985. Retrieved from https://plants.jstor.org/search?Date_fld=1968 on 4th January, 2024.
- Abo K. A. Afr. J. Med. Med. Sci. 1994, 23, 161–163. [PubMed] [Google Scholar]
- Hammadi R.; Kúsz N.; Mwangi P. W.; Kulmány Á.; Zupkó I.; Orvos P.; Tálosi L.; Hohmann J.; Vasas A. Nat. Prod. Commun. 2019, 14, 1–5. 10.1177/1934578X19863509. [DOI] [Google Scholar]
- Xu W.; Zhu C.; Cheng W.; Fan X.; Chen X.; Yang S.; Guo Y.; Ye F.; Shi J. J. Nat. Prod. 2009, 72, 1620–1626. 10.1021/np900305j. [DOI] [PubMed] [Google Scholar]
- Pettit G. R.; Ye Q.; Herald D. L.; Knight J. C.; Hogan F.; Melody N.; Mukku V. J.; Doubek D. L.; Chapuis J. C. Isolation and structure of cancer cell growth inhibitory tetracyclic triterpenoids from the Zimbabwean Monadenium lugardae. J. Nat. Prod. 2016, 79, 1598–1603. 10.1021/acs.jnatprod.6b00107. [DOI] [PubMed] [Google Scholar]
- Jiang Z. H.; Tanaka T.; Hirata H.; Fukuoka R.; Kouno I. Tetrahedron 1997, 53, 16999–17008. 10.1016/S0040-4020(97)10150-8. [DOI] [Google Scholar]
- Xu J.; Xiao D.; Lin Q. H.; He J. F.; Liu W. Y.; Xie N.; Feng F.; Qu W. J. Nat. Prod. 2016, 79, 1899–1910. 10.1021/acs.jnatprod.5b01137. [DOI] [PubMed] [Google Scholar]
- Wang L. Y.; Wang N. L.; Yao X. S.; Miyata S.; Kitanaka S. J. Nat. Prod. 2003, 66, 630–633. 10.1021/np0205396. [DOI] [PubMed] [Google Scholar]
- Gao J.; Aisa H. A. J. Nat. Prod. 2017, 80, 1767–1775. 10.1021/acs.jnatprod.6b01099. [DOI] [PubMed] [Google Scholar]
- Chang S. S.; Huang H.-T.; Wei W.-C.; Lo I.-W.; Lin Y.-C.; Chao C.-H.; Liao G.-Y.; Shen Y.-C.; Chen J.-J.; Li T.-L.; Lin L.-T.; Tai C.-J.; Kuo Y.-H.; Liaw C.-C. Front. Chem. 2023, 11, 223335 10.3389/fchem.2023.1223335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Liang H.; Zhao Y.; Wang G.; Yao H.; Kasimu R.; Wu Z.; Li Y.; Huang J.; Wang J. Fitoterapia 2016, 108, 33–40. 10.1016/j.fitote.2015.11.009. [DOI] [PubMed] [Google Scholar]
- Fang C.-H.; Li Y.-P.; Li Y.; Yue J.-M.; Bao J.; Yu J.-H. Phytochemistry 2023, 211, 113684 10.1016/j.phytochem.2023.113684. [DOI] [PubMed] [Google Scholar]
- Lu Z. Q.; Chen G. T.; Zhang J. Q.; Huang H. L.; Guan S. H.; Guo D. A. Helv. Chim. Acta 2007, 90, 2245–2250. 10.1002/hlca.200790233. [DOI] [Google Scholar]
- Durão R.; Ramalhete C.; Madureira A. M.; Mendes E.; Duarte N. Pharmaceuticals 2022, 15, 340. 10.3390/ph15030340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Araujo J. I. F.; Aires N. L.; Almeida-Neto F. W. Q.; Marinho M. M.; Marinho E. M.; Paula Magalhaes E.; de Menezes R. R. P. P. B.; Sampaio T. L.; Maria Costa Martins A.; Teixeira E. H.; et al. J. Biomol. Struct. Dyn. 2022, 40, 12302–12315. 10.1080/07391102.2021.1970025. [DOI] [PubMed] [Google Scholar]
- Meira C. S.; Barbosa-Filho J. M.; Lanfredi-Rangel A.; Guimarães E. T.; Moreira D. R.; Soares M. B. Exp. Parasitol. 2016, 166, 108–115. 10.1016/j.exppara.2016.04.007. [DOI] [PubMed] [Google Scholar]
- Vanrell M. C.; Martinez S. J.; Muñoz L. I.; Salassa B. N.; Gambarte Tudela J.; Romano P. S. Front. Cell. Infect. Microbiol. 2022, 12, 919096 10.3389/fcimb.2022.919096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leite A. C.; Ambrozin A. R.; Fernandes J. B.; Vieira P. C.; da Silva M. F.; de Albuquerque S. Planta Med. 2008, 74, 1795–1799. 10.1055/s-0028-1088323. [DOI] [PubMed] [Google Scholar]
- Mazoir N.; Benharref A.; Bailén M.; Reina M.; González-Coloma A.; Martínez-Díaz R. A. Z. Naturforsch. C J. Biosci. 2011, 66, 360–366. 10.1515/znc-2011-7-807. [DOI] [PubMed] [Google Scholar]
- Ioset J. R.; Brun R.; Wenzler T.; Kaiser M.; Yardley V.. Drug Screening for Kinetoplastid Diseases: A Training Manual for Screening in Neglected Diseases; DNDi Pan-Asian Screening Network, April 2009.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The NMR data for 1–16 have been deposited in the Natural Products Magnetic Resonance Database (NP-MRD; www.np-mrd.org) and can be found at NP0333436 (1), NP0333438 (2), NP0333439 (3), NP0333440 (4), NP0333441 (5), NP0333442 (6), NP0333443 (7), NP0333444 (8), NP0333445 (9), NP0333446 (10), NP0333447 (11), NP0333448 (12), NP0333449 (13), NP0333450 (14), NP0333451 (15), and NP0333451 (16).