

Abstract
Identification of bacterial protein–protein interactions and predicting the structures of these complexes could aid in the understanding of pathogenicity mechanisms and developing treatments for infectious diseases. Here we developed RoseTTAFold2-Lite, a rapid deep learning model that leverages residue–residue coevolution and protein structure prediction to systematically identify and structurally characterize protein–protein interactions at the proteome-wide scale. Using this pipeline, we searched through 78 million pairs of proteins across 19 human bacterial pathogens and identified 1,923 confidently predicted complexes involving essential genes and 256 involving virulence factors. Many of these complexes were not previously known; we experimentally tested 12 such predictions, and half of them were validated. The predicted interactions span core metabolic and virulence pathways ranging from post-transcriptional modification to acid neutralization to outer-membrane machinery and should contribute to our understanding of the biology of these important pathogens and the design of drugs to combat them.
Subject terms: Protein structure predictions, Machine learning, Bacterial structural biology
RoseTTAFold2-Lite uses residue–residue coevolution and protein structure prediction to identify and structurally characterize protein–protein interactions in bacterial pathogens.
Main
Understanding the biology of pathogenic bacteria is important for human health and therapeutics. Protein–protein interactions (PPIs) are central to biological processes, but many interactions remain unknown, especially for non-model organisms. High-throughput experiments such as the two-hybrid screen and affinity purification coupled with mass spectrometry have been used to identify PPIs in a variety of organisms1–3. However, such methods can fail to reveal transient interactions and be plagued by non-specific interactions in non-physiological conditions, which result in discrepancies between experiments along with high false-positive and false-negative rates4,5. Interacting proteins often co-evolve, and hence amino acid coevolution can be exploited to assess the likelihood that two proteins interact with each other. Coevolutionary information between proteins extracted from paired multiple sequence alignments (pMSAs) of orthologous proteins6–8 has been used to systematically identify PPIs in prokaryotes at an accuracy that rivals experimental screens7. Supplementing coevolution with deep-learning-based structure prediction methods has further increased the accuracy of PPI prediction, enabling large-scale prediction of PPIs in yeast9 and humans10,11.
We set out to systematically identify and structurally characterize PPIs in pathogenic bacteria. We selected 19 bacterial pathogens (Supplementary Table 1) that span 6 phyla and are the leading causes of pathogen-associated deaths in humans12. These organisms are associated with infections in skin (Staphylococcus aureus), gastrointestinal tract (Clostridioides difficile, Helicobacter pylori, Listeria monocytogenes and Salmonella typhimurium), respiratory system (Legionella pneumophila, Mycobacterium tuberculosis, Pseudomonas aeruginosa and Streptococcus pneumoniae) and urinary and genital tracts (Chlamydia trachomatis and Mycoplasma genitalium) and the plague (Yersinia pestis). For most of these organisms, large-scale experimental screens have identified essential genes and virulence factors; these results are summarized in the Database of Essential Genes12 and Virulence Factor DataBase (VFDB)13. We focused on essential genes and virulence factors because the former provides targets for drug development to inhibit essential cellular functions and treat infectious diseases, while the latter may explain molecular mechanisms of pathogenicity. Comparative analysis showed substantial overlap in the sets of essential genes between different pathogens, but each pathogen still harbours ~100 unique essential genes (Supplementary Table 2). In contrast, virulence factors differ considerably between species, suggesting a diversity of virulence mechanisms which we attempt to capture with our set of phylogenetically diverse species (Supplementary Table 2).
Results
Computational pipeline for proteome-wide PPI identification
To screen through hundreds of millions of protein pairs for PPIs, we first sought to increase the computational efficiency of PPI identification without compromising accuracy. We previously developed a two-track RoseTTAFold (RF 2-track) network that is a simplified version of RoseTTAFold14, which predicts 3D protein structure from amino acid sequence. Although RF 2-track was not trained to model protein complexes or distinguish interacting from non-interacting proteins, residue–residue distograms produced by this network enable the detection of PPIs on a proteome-wide scale at an accuracy that far exceeds statistical analysis of coevolution between proteins9. Similarly, we and others have used AlphaFold (AF)15 to evaluate interactions identified in lower-accuracy large-scale screens9–11,16,17; the computational cost of AF prohibits its application on a proteome-wide scale. AF-multimer (AFmm)18 was trained to model three-dimensional (3D) structures of known protein complexes, and consequently, it tends to predict PPIs between non-interacting pairs, showing a worse performance than AF in distinguishing true PPIs from random pairs (Fig. 1b, top).
Fig. 1. PPI identification by coevolution and deep learning methods.

a, Overview of the RF2-Lite network architecture. FAPE, frame-aligned point error. b, Benchmark performance of PPI prediction methods. Top: precision and recall curves of DCA (grey), RF 2-track (black), RF2-Lite (blue), AF (green) and AF-multimer (purple) in distinguishing true PPIs from random protein pairs. For different methods, we used the pMSAs generated by our bioinformatic pipeline (Supplementary Methods). We applied each method on a benchmark set of 1,000 randomly selected positive control pairs and 10,000 negative control pairs (Supplementary Methods). The precision and recall curve for this benchmark is in Supplementary Fig. 6a. Real signal-to-noise ratio for the PPI screen is on the order of 1:1,0001; to reflect the impact of a much larger set of non-interacting pairs, we upsampled the negative control set to 1,000,000 by randomly sampling 100 ‘pseudo’ interacting probabilities from the Gaussian distribution around each real interacting probability we obtained for the negative controls with a standard deviation of 0.1. Bottom: runtime comparison of different PPI identification methods. c, Schematic overview of our PPI screen pipeline. d, Precision and recall curves at different stages in the pipeline. Top: DCA on PPI prediction; solid black vertical line represents the recall cut-off in this stage. Middle: RF2-Lite screen procedure on the ‘pilot set’; solid black vertical line indicates the recall cut-off at this stage. Bottom: AF screen procedure on the ‘pilot set’; dashed horizontal line shows the precision cut-off, that is, 0.95. e, Summary of predicted PPIs for the ‘pilot set’ that focuses on essential genes and virulence factors. Left: interactions between interacting essential genes in the ‘pilot set’ based on different evidence: blue, green and orange circles represent our predicted pairs, functional interactions according to STRING (total score ≥900 and experimental score ≥400) and interacting pairs according to PDB (BLAST hit to complex in PDB e ≤ 0.00001, sequence identity ≥50% and coverage ≥50%), respectively. Right: PPIs involving virulence factors in the ‘pilot set’ supported by difference evidence: red, purple and yellow circles represent our predictions, pairs according to STRING and pairs according to PDB.
We hypothesized that a dedicated lighter-weight network trained on both interacting and non-interacting protein pairs that balances accuracy with speed could assist proteome-wide PPI screens. We revised the original RoseTTAFold network by introducing architectural improvements to increase accuracy while reducing the number of layers to enable the rapid computation necessary for large-scale screens (Fig. 1a and Supplementary Methods). We trained this network using a combination of (1) monomeric protein structures from Protein Data Bank (PDB), (2) AF models of UniRef50 sequences, (3) pairwise protein complex structures extracted from PDB and (4) random non-interacting protein pairs. The four types of training data were mixed at a ratio of 1:3:2:2 (Supplementary Table 3). The model was trained using the masked language model loss, distogram prediction loss, frame-aligned point error loss, accuracy estimation loss, bond geometry loss and van der Waals energy loss. For the negative interaction examples, we ignored the inter-chain region for frame-aligned point error calculation and required the network to predict the distogram to be in the ‘non-interacting bin’ for the inter-chain region. We designate the resulting network as RoseTTAFold2-Lite (RF2-Lite) as it resembles the RoseTTAFold2 architecture but has many fewer parameters, because we reduced the number of parameters by chunking the number of blocks19. RF2-Lite has improved performance in distinguishing true PPIs over the previous RF 2-track at the same precision, the recall for true PPIs by RF2-Lite is in between RF 2-track and AF (Fig. 1b, top, and Supplementary Figs. 6 and 7). Despite this increase in accuracy, RF2-Lite’s speed is still comparable to RF 2-track, and it requires about 20-fold less compute time than AF (Fig. 1b, bottom).
We combined direct coupling analysis (DCA)20, RF2-Lite and AF (Fig. 1c and Supplementary Methods) to identify and model interacting proteins and applied this pipeline to the 19 human pathogens listed in Supplementary Table 2. To monitor the performance of our pipeline, we assembled a set of positive controls and an ~700-fold larger negative set based on information from the STRING protein-protein interaction database (Supplementary Methods).
We constructed a database of 44,871 representative bacterial proteomes/genomes (one per species) obtained from the National Center for Biotechnology Information (NCBI) and used the reciprocal best hit criteria21 to identify an orthologue for every protein in each proteome (Supplementary Fig. 1). We aligned these orthologous sequences22,23, and for each protein pair in each of the 19 pathogens (Supplementary Fig. 2), we concatenated their multiple sequence alignments (MSAs) by connecting sequences of the same species to generate pMSAs (Supplementary Fig. 3). We removed proteins whose monomeric structure could not be confidently modelled by AF (average predicted local distance difference (pLDDT) test <50 in AFDB) and filtered the pMSAs based on their depth and quality (Supplementary Figs. 4 and 5): of the total 140.2 million protein pairs, we selected 77.9 million (56%) with higher monomer structure and MSA quality.
We assessed the residue–residue coevolution for the selected pairs using DCA and found that the 7.7 million (10%) high-scoring protein pairs by DCA contained 79% of the positive controls (Fig. 1d, top). Among these 7.7 million pairs, we initially focused on a ‘pilot set’ of 0.14 million pairs involving at least one virulence factor (according to VFDB) and 0.83 million pairs of essential genes (according to the Database of Essential Genes). We removed redundancy in this set by clustering proteins from the 19 species into orthologous groups using OrthoMCL v.6.10 (ref. 24). If the orthologues of a protein pair were present in multiple species, we selected only one pair with the highest DCA score, resulting in a total of 457,310 representative PPI candidates.
We used RF2-Lite to identify confident PPIs from the ‘pilot set’ and observed that we could achieve a recall of 28% at a precision of 95% when an RF2-Lite contact probability cut-off of 0.74 was used (Fig. 1d, middle). We investigated whether using a loose RF2-Lite cut-off (contact probability 0.05) to select candidate PPIs (46,609, around 10% selected) for AF could improve recall. The RF2-Lite → AF pipeline only improved the recall to 29% at 95% precision (Fig. 1d, bottom) at the cost of using 3 times more computer resources than simply relying on RF2-Lite to detect PPIs (Supplementary Table 4). Thus, the contribution of AF in distinguishing true PPIs from random pairs is limited, but it remains essential for obtaining high-quality 3D structures for the predicted protein complexes.
The successive use of DCA (selecting top 10%), RF2-Lite (cut-off, 0.05) and AF (cut-off, 0.9) collectively reduced the total number of random pairs by nearly 10,000-fold, resulting in 562 highly confident predictions from the ‘pilot set’. The identified binary protein complexes include 461 protein complexes involving essential genes (Fig. 1e, left) and 115 involving virulence factors (Fig. 1e, right). Further investigation of these interactions may be useful for understanding the mechanisms of pathogenicity and developing disease prevention and treatment strategies. The vast majority (19%) of predicted protein complexes from the ‘pilot set’ did not have experimental 3D structures in PDB (BLAST e ≤ 0.00001, identity ≥50% and coverage ≥50% for both proteins), and half do not have confident experimental support according to STRING25 (Supplementary Table 5).
To gain more structural and functional insights into these pathogens, we applied the RF2-Lite to AF pipeline to an additional 3.82 million pairs involving essential proteins and biological processes of therapeutic interest, such as the outer-membrane machinery (Supplementary Table 6). This search resulted in an additional 3,051 predicted PPIs. To facilitate downstream studies, we deposited all confident models to ModelArchive (see the Data availability statement) and provided additional metadata in the Supplementary Data 1. Inspection of the predicted PPIs revealed a small number of proteins (in particular ferredoxin and rubredoxin) with predicted interactions between many random proteins, likely constituting small false-positive hubs. We removed 405 PPIs involving such potential false-positive hubs before deposition to ModelArchive.
It is difficult to cover even a small fraction of the biological insights that can be revealed from these 3D structures of protein complexes in one paper. In the following sections, we first describe experimental validation for a subset of predictions and then highlight examples that illustrate some of the biological insights revealed by the identification of putative PPIs and computational modelling of protein complexes.
Experimental validation
To corroborate our benchmarking analyses, which suggest that our predicted interactions should be quite accurate, we selected two sets of predicted interactions for experimental characterization. We biased these selections towards PPIs with no previous experimental evidence or strong functional associations because validating such interactions could provide new biological insights. The first set (Supplementary Table 7) was selected based on statistical methods (GREMLIN) for PPI detection, before the development and application of the deep learning methods. This set was used to probe the accuracy of statistical (DCA and GREMLIN26) versus deep learning methods for PPI detection. The second set (Supplementary Table 8) was selected from our final set of predicted 3,613 PPIs, with a goal of evaluating the accuracy of our current entire pipeline.
We selected the first dataset using the following criteria: (1) at least 20 kb apart (with a minimum of 20 intervening genes), (2) not having homologous complexes in the PDB, (3) not predicted to have the same molecular function, (4) not annotated as part of the same biological pathway and (5) not strongly supported by STRING (combined score of <800). All 11 pairs show strong coevolution according to DCA and GREMLIN, but five pairs were not predicted to interact by RF2-Lite or AF (Supplementary Fig. 11). A bacterial two-hybrid (B2H) system27 coupled with a quantitative β-galactosidase assay28 was used to measure interactions for these 11 pairs (Supplementary Fig. 12).
Despite the strong support by DCA and GREMLIN, the five pairs not predicted to interact by RF2-Lite or AF did not show evidence of interaction using the B2H assay (Supplementary Fig. 11). Among the six pairs supported by RF2-Lite or AF, reporter activation indicative of interaction was detected for two: one is between iron-sulfur cluster binding protein lpg2881 (Uniprot: Q5ZRK0) and uncharacterized protein lpg0371 (Uniprot: Q5ZYK1) from L. pneumophila; another is between ribosomal silencing factor RsfS (PA4005; Uniprot: Q9HX22) and PhoH-like protein domain-containing protein YbeZ (PA3981; Uniprot: Q9HX38) from P. aeruginosa (Fig. 2a). For one additional pair, nucleoid-associated protein lmo2703 (Uniprot: Q8Y3X6) and signal recognition particle protein Ffh (Uniprot: Q8Y695) from L. monocytogenes, we were unable to assess the interaction experimentally due to false-positive reporter activation when only one protein was expressed (Supplementary Fig. 12). The remaining three pairs failed to generate a positive reporter signal; however, false-negative results from B2H assays do not necessarily rule out the existence of a genuine interaction due to possible failures in protein expression and folding of the fusion proteins, and lack of sensitivity of the screen to weak and transient interactions.
Fig. 2. Experimental validation of selected PPIs.

a, Interactions assessed by B2H that measures β-galactosidase activity resulting from activation of the lacZ reporter gene due to the interaction between two tested proteins that are fused to two domains of a transcription activator. E. coli expressing T25-zip and T18-zip fusion proteins was used as a positive control (+ control), and E. coli harbouring empty T25 and T18 plasmids was used as a negative control (− control). m/m, mix-and-match control. RU, relative unit (luminescence per optical density at 600 nm per h). Error bars indicate ±s.d. (n = 2 biological replicates each with 2 technical replicates). Computed models of experimentally validated PPIs (‘lpg2881 + lpg0371’ and ‘RsfS + YbeZ’) are shown on the right—top: iron-sulfur cluster binding protein lpg2881 (Q5ZRK0) and uncharacterized protein lpg0371 (Q5ZYK1) from L. pneumophila; bottom: ribosomal silencing factor RsfS (Q9HX22) and PhoH-like protein domain-containing protein YbeZ (Q9HX38) from P. aeruginosa. b–e, Interactions validated by Co-IP/pull-down. Predicted interacting partners in each PPI pair are heterologously expressed and tagged (–H, hexahistidine; –V, VSV-G epitope). A random bait protein was included as a negative control for each experiment. Control lanes correspond to samples with prey proteins and beads added without any bait proteins. Each positive interaction is supported by two independent Co-IP/pull-down experiments. b, Ubiquinone biosynthesis C-methyltransferase UbiE (P0A887) and protein of unknown function YcaR (P0AAZ7) from E. coli. c, Uncharacterized protein PA4106 (Q9HWS2) and a putative transcriptional factor PA4105 (Q9HWS3) from P. aeruginosa. d, lpg2881 and lpg0371 from L. pneumophila, a pair that is tested positive by B2H as well. e, Putative imidazole glycerol phosphate synthase subunit hisF2 (P72139) and lipopolysaccharide biosynthesis protein WbpG (Q9HZ78) from P. aeruginosa. In all the panels, connecting green bars are between representative residue–residue contacts at the interfaces predicted from the summed AF probability for distance bins below 12 Å. Ni-NTA, nickel-nitrilotriacetic acid; VSV-G, vesicular stomatitis virus glycoprotein epitope.
For both PPIs validated by our B2H assays, there are no published data directly supporting functional or physical interactions between the two proteins. However, in both cases, existing evidence indirectly suggests that the interactions could be biologically relevant. The pair of proteins from L. pneumophila (lpg2881–lpg0371; Q5ZRK0–Q5ZYK1) are homologous to proteins of the Rnf electron transport complex (RnfB with 53% sequence identity and RnfH with 36% sequence identity, respectively). The function of these proteins in L. pneumophila is unclear because this species appears to lack the other components of the complex, and one of the proteins, lpg0371, also shares homology with the antitoxin component of the RatAB toxin–antitoxin module. However, in species that encode the complete Rnf complex, RnfB and RnfH directly interact29. The interacting pair from P. aeruginosa consists of the ribosomal silencing factor RsfS and the PhoH-like protein domain-containing protein YbeZ. Under nutrient depletion or during stationary phase growth, RsfS binds to ribosomal protein L14, ultimately preventing the association of the 30S and 50S ribosomal subunits and repressing translation30. This facilitates adaptation to low-nutrient conditions and promotes survival during the stationary phase. The function of YbeZ is less well characterized, but it interacts with the RNase YbeY, and both proteins are required for processing and maturation of the 16S ribosomal RNA31. Our finding that YbeZ and RsfS interact suggests that the regulation of ribosome assembly and ribosome subunit processing may be linked in P. aeruginosa.
The second validation set, selected using the deep learning methods, consists of six protein pairs (Supplementary Table 8) lacking homologous protein complexes in the PDB, with little support in STRING (only one pair STRING > 600) and distant in the genome (separated by >100 genes) in half the cases. We focused on proteins consisting primarily of globular domains (percentage of residues from non-globular domains, <20%), as such proteins are more amenable to heterologous expression-based assays. Using co-immunoprecipitation (Co-IP) assays, we detected an interaction between four of the six pairs (Fig. 2b–e). These include a pair we had previously validated by B2H, Q5ZRK0–Q5ZYK1, a distally encoded pair from Escherichia coli, and two proximally encoded pairs from P. aeruginosa. E. coli UbiE catalyses a carbon-methyl transfer reaction in the biosynthesis of ubiquinone (coenzyme Q) and menaquinone (vitamin K2)32, while YcaR is a small protein detected as differentially expressed in multiple proteomics studies but to which no function has been assigned33,34. P. aeruginosa PA4105–PA4106 (Q9HWS3–Q9HWS2) are uncharacterized proteins with no clear homologues of known functions based on primary sequence comparisons, but a FoldSeek v.8 search35 revealed structural similarity between these proteins and TglI and TglH from Pseudomonas syringae pv. maculicola (P. syringae) which form a complex that catalyses the removal of cysteine β-methylene (β-CH2) from TglA–Cys, a step in the biosynthesis of the natural product 3-thiaglutamate (3-thiaGlu)36,37. P. aeruginosa Q9HZ78–P72139 are an amidotransferase essential in B-band lipopolysaccharide biosynthesis (WbpG, Q9HZ78) and a predicted imidazole glycerol phosphate synthase subunit (HisF2, P72139). It was previously proposed that HisF2, together with HisH2, delivers ammonia to WbpG38, a hypothesis our interaction finding supports. The PtsH–PtsN (Q9HVV2-Q9HVV4) pair with the highest support by STRING (score = 959) failed to generate a positive Co-IP signal (Supplementary Fig. 13); PtsH is a histidine-phosphorylatable phosphocarrier protein encoded adjacent to PtsN, a nitrogen regulatory protein with a phosphotransferase component, and the interaction between these proteins may be transient and thus difficult to detect by Co-IP.
These experimental data support the in silico benchmark in suggesting that the deep learning methods have greater accuracy than statistical methods in PPI discovery, identifying additional components for well-known biological pathways and accelerating the characterization of proteins of unknown function. In the following sections, we provide an overview of the much larger set of interactions predicted by the deep learning methods but not yet experimentally validated; to illustrate the insights that can be gained from these data, we provide biological context for selected interaction pairs and higher-order assemblies.
Binary interactions
From the total set of 3,613 predicted binary PPIs, 1,686 (47%) have homologous complexes in PDB (BLAST e ≤ 0.00001 for both proteins), 1,862 (52%) are supported by strong functional association according to STRING (total score ≥900), and 1,284 (36%) are supported by both PDB and STRING; the remaining 1,349 (37%, 3,613 − (1,686 + 1,862 − 1,284)), to our knowledge, are unknown PPIs. Although such previously unsupported PPIs might contain a higher fraction of false predictions, the high precision on our benchmark sets suggests the majority of the new predictions are likely correct. We identify 166 putative interactions that involve uncharacterized proteins (all Pfam domains are uncharacterized; Supplementary Methods), the majority of these pairs (149) include an interaction partner of known functional domains, and 131 (117 with known partners) not well described previously (STRING combined score <900 and BLAST e value to PDB chains >0.00001).
Of the predicted PPIs, 1,923 include one or more essential genes. Examples of predicted interactions among essential genes without homologous complexes in the PDB are highlighted in Fig. 3a–j and Supplementary Table 9. In some cases, the predicted PPIs support previous findings from the literature. For example, we predict an interaction between glucose-6-phosphate 1-dehydrogenase 2 (G6PD2) and OxPP (oxidative pentose pathway) cycle protein OpcA (Fig. 3a). G6PD2 is an isozyme of G6PD, a member of the pentose phosphate pathway, catalysing the oxidation of G6P to 6-phosphogluconolactone while converting NADP+ to NADPH and protecting cells from oxidative stress39. OpcA has been implicated as an allosteric activator of G6PD40, but, to our knowledge, the binding site remains unknown. Our predicted interface places OpcA away from the active site of G6PD, consistent with allosteric modulation of activity (Supplementary Fig. 19). We predict an interaction between 30S ribosomal protein S11 (rpsK), a surface-exposed ribosomal protein that forms part of the messenger RNA binding cleft which recognizes the Shine–Dalgarno sequence41,42, and YbeY, a highly conserved endoribonuclease which has been linked to numerous processes such as 16S rRNA maturation, 70S control, and regulation of mRNA43 (Fig. 3f). In some bacteria, YbeY plays a key role in virulence and cell stress44. Our predicted structure of S11–YbeY with an interface mediated by S11 β-strands agrees with previous work that identified S11–YbeY interaction by bacteria 2-hybrid, Co-IP and mutational analyses45. The 3D model of the S11–YbeY complex may lend further insights into how YbeY coordinates cleavage of the rRNA precursor during 16S maturation.
Fig. 3. Computed models of binary protein complexes.

a–j, Interactions involving essential genes. a, Interaction with an enzyme where the enzymatic site is highlighted in light green with an NAD moiety. b–d, Additional interactions involving essential genes. e,j Interactions involving transport pathways. f–i, Transcription and translation. k–t, Interactions involving virulence factors. u–y, Interactions with uncharacterized proteins. In all models, the first protein is in blue, and the second is in gold. Green bars are between representative residue–residue contacts at the interfaces predicted from the summed AF probability for distance bins below 12 Å. Additional information (organisms and UniProt annotations) is in Supplementary Table 9.
Of the predicted PPIs, 256 contain virulence factors (according to VFDB and Uniprot Keywords) that participate in pathogen colonization, nutrient acquisition and evasion of host immunity46. Secreted virulence factors rarely interact with endogenous proteins of a pathogen; consistent with this, we did not detect many PPIs involving virulence factors, and those we did identify mostly involve structural components of flagella (considered virulence factors in many bacteria40) and bacterial secretion systems (Fig. 3k–t). We also identified other interactions related to flagella function, for example, between the anti-sigma factor FlgM, a negative regulator of flagellin synthesis, and flagellar secretion chaperone (FliS) (Fig. 3p), an interaction supported by a previous experimental study47 but without 3D structure information. Our 3D models, in agreement with previous observations47, revealed that FlgM can compete with flagellin (FliC, major structural component of the flagella) for the same interface on FliS; FlgM uses its C-terminal helices to interact with FliS, which could prevent its interaction with the flagellar sigma factor FliA. The FliS–FlgM interaction might provide a negative feedback mechanism to control the expression of flagellin: when intracellular flagellin is abundant, it outcompetes FlgM in binding the anti-sigma factor FlgM, and the release of FlgM antagonizes the activity of sigma factor FliA, turning off the expression of late-stage flagellar genes, including flagellin (FliC).
We identify 149 putative interactions (Fig. 3u–y) between uncharacterized proteins (according to Pfam domains) and functionally annotated binding partners such as ketol-acid reductoisomerase IlvC, thiosulfate sulfurtransferase GlpE, ubiquinol-cytochrome c reductase, cell division protein FtsZ and bifunctional guanosine pentaphosphate [(p)ppGpp] synthase/hydrolase RelA. These predicted interaction partners provide contextual hypotheses about the function of these uncharacterized proteins, 72 of which are essential to pathogen survival, to guide further experimental studies aimed at elucidating their functions.
Multicomponent protein complexes
In many cases, the predicted binary interactions form larger sets, suggesting the formation of higher-order assemblies. For example, in our set of 3,613 predicted interactions, we found 206 trimeric protein complexes where each component is predicted to directly interact with the other two. Of the predicted binary interactions, 1,545 (40%) involve proteins that have multiple interacting partners, which allows us to build higher-order protein complexes by concatenating the MSAs of multiple proteins and modelling them together through AF.
Transfer RNA modification and sulfur transfers in the 2-thio modification complex of E. coli
Transfer RNAs (tRNA) play critical roles in protein synthesis and are often decorated with post-transcriptional modifications that contribute to efficient protein synthesis48. Wobble positions are hotspots of such modifications. In glutamate, glutamine and lysine tRNAs, the wobble uridine is modified to 5-methylaminomethyl-2-thiouridine (mnm5s2U) by tRNA 2-thiouridine synthesizing proteins (Tus)49; which include TusA, TusB, TusC, TusD, TusE and tRNA-specific 2-thiouridylase (MnmA). Cysteine desulfurase (IscS) is essential for 2-thio modification in E. coli49. IscS transfers sulfur from cysteine to TusA, which is transferred to TusD of the TusBCD complex via TusE and subsequently to MnmA, which incorporates the sulfur into the tRNA49,50. The structure of the IscS–TusA dimer and sulfur transfer mediating heterohexameric complex, TusBCD, has been co-crystallized50,51, but structural details for other components of this system are poorly understood. We predicted the structures of the TusE–MnmA and TusE–TusC complexes (Supplementary Fig. 20) and assembled a model of the full TusBCDE heterotetramer which contextualizes the interaction of TusE with TusBCD (Fig. 4a and Supplementary Fig. 21). Our model places TusE close to TusC and TusD, with a confidently predicted TusC–TusE interface (Supplementary Fig. 21e), and is consistent with the hypothesis that Cys108 of TusE accepts sulfur from Cys78 of TusD49,50 but also suggests that TusC serves as a scaffold to bring TusD and TusE to close proximity. We also predict the structure of the TusE–MnmA interaction and find that TusE cannot interact with TusBCD and MnmA simultaneously due to overlap in the interfaces with MnmA and TusD (Supplementary Fig. 20a–f).
Fig. 4. Computed models for multi-component protein complexes.
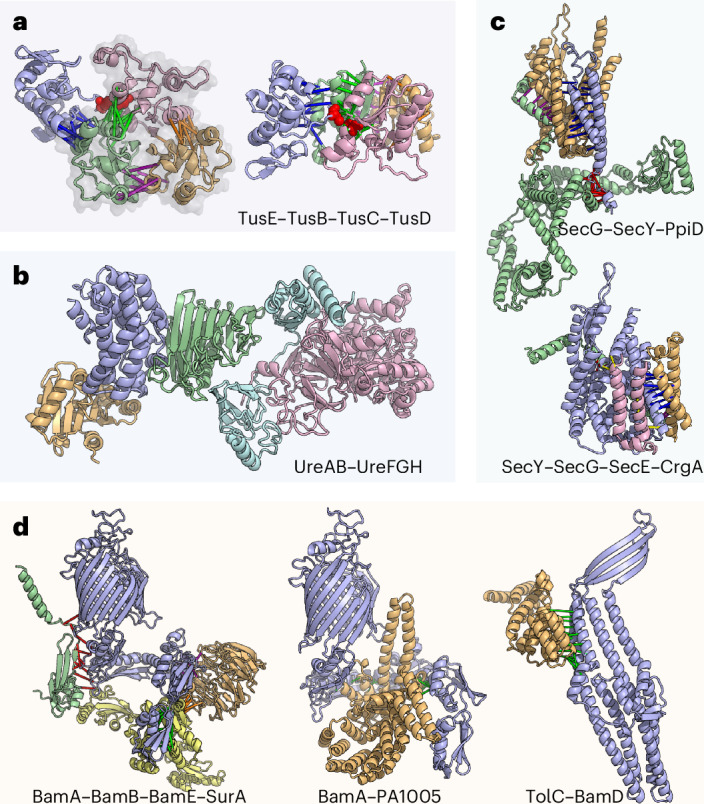
a, H. pylori tRNA 2-thiouridine synthesizing protein complex. Left: a model of the TusE(blue)–TusB(gold)–TusC(green)–TusD(pink) complex overlaid with the TusBCD PDB structure (2D1P, shown in semi-transparent grey). Right: an alternative view of this complex. b, The UreAB–UreFGH complex (coloured in cyan, pink, blue, gold and green, respectively) in H. pylori assembled through multiple subcomplexes: UreFGH, UreAB and UreAH. c, Accessory components of the Sec translocon. Top: P. aeruginosa SecG(blue)–SecY(gold)–PpiD(green) complex. Bottom: M. tuberculosis SecY(blue)–SecG(gold)–SecE(green)–CrgA(pink) complex. d, Accessory components of the P. aeruginosa and S. typhimurium outer-membrane β-barrel assembly machinery. Left: interaction between SurA (yellow) and Bam proteins (BamA, blue; BamB, gold; BamE, green). Middle: BamA (blue) and PA1005 (gold), a putative BepA orthologue. Right: interaction between TolC (blue) and BamD (gold). In all schematics, green, red, yellow and magenta bars connect representative residue–residue contacts at the interfaces predicted from the summed AF probability for distance bins below 12 Å.
A two-step nickel transfer in H. pylori urease complex
Urease hydrolyses urea into ammonia and is broadly conserved in bacteria and eukaryotes. In H. pylori, urease neutralizes gastric acid and facilitates gut colonization52, and thus proteins in the urease complex are considered virulence factors. While most bacterial ureases have three chains (UreA, UreB and UreC), H. Pylori urease has two due to the fusion of UreA and UreB orthologues53. The UreAB(C) system has four accessory proteins: UreE, UreF, UreG and UreH54. We predict a UreA–UreH interaction and use it to assemble a model of a UreAB–UreFGH pentamer (Fig. 4b and Supplementary Fig. 22e). The UreAB(C) and UreFGH substructures have been determined experimentally53,55, and our predictions are consistent with these (Supplementary Fig. 22a–d). During urease maturation, UreFGH receives nickel from UreE, but how this occurs remains poorly understood. Two hypotheses are (a) that UreE transfers nickel to UreFGH complex56 and (b) that upon binding guanosine-5′-triphosphate (GTP), UreG dissociates from UreFGH, receives nickel from UreE and subsequently interacts with the inactive UreFH to activate the complex55. Superimposing our UreE–UreG model onto the UreFGH complex shows that UreE clashes with UreF, indicating that UreE cannot directly interact with the UreFGH complex. Therefore, our observation supports the latter hypothesis wherein UreG likely receives nickel separately from UreFH (Supplementary Fig. 23)55.
Sec translocon interactors
The Sec translocon machinery transports proteins across the plasma membrane. The Sec translocon channel is a heterotrimeric complex composed of SecYEG, which operates in tandem with SecA, a RecA-like ATPase that moves peptides through the SecY channel in a process similar to Sec61 translocon in eukaryotes57. We predict interactions between the Sec translocon and peptidyl-prolyl cis/trans isomerase D (ppiD) (Fig. 4c, top, and Supplementary Fig. 24), which has been identified as the most prominent interactor of SecYEG by affinity purification coupled with mass spectrometry58 and Co-IP59. In our model of the SecYG-ppiD trimer, ppiD primarily interacts with SecY through the transmembrane helices while coming close to SecG via a small loop. We also predict interactions between Sec and CrgA, a transmembrane protein and a component of the divisome (Fig. 4c, bottom). We find that the CrgA–SecY interface occurs near the lateral gate of SecY60 (Supplementary Fig. 25a), potentially occluding Sec translocation. We hypothesize that during bacterial division, CrgA binds Sec to regulate and recruit translocation machinery near the cell division site; this latter hypothesis is further supported by the predicted interaction between CrgA and SecE (Supplementary Fig. 25) and a less confident prediction of CrgA–SecG interaction that fell slightly below our cut-off.
Outer-membrane β-barrel assembly machinery of P. aeruginosa and Vibrio cholerae
In Gram-negative bacteria, the β-barrel assembly machinery (BAM) is essential for the folding and insertion of outer-membrane β-barrel proteins61,62. BAM consists of an outer-membrane-spanning β-barrel, BamA, that interacts with four periplasmic lipoproteins, BamB, BamC, BamD and BamE, to form a five-component complex (computed interactions and structures agree with known experimental data (Supplementary Fig. 26))61–65. This complex has recently garnered increased attention as a potential therapeutic target, especially since the discovery of darobactin, a novel antimicrobial compound that binds along the lateral gate of BamA to inhibit outer-membrane protein (OMP) biogenesis66,67.
The function of BAM is assisted by several other proteins, including the chaperone survival factor A (SurA) and periplasmic chaperone 17 kDa protein (Skp). SurA plays an important role in facilitating the recruitment of unfolded OMPs from the periplasm to the BAM complex68. Both our BAM–SurA model and a recently published study using an orthogonal approach to ours69 place SurA in the same position to simultaneously interact with BamA, BamB and BamE (Fig. 4d, left). In addition, we predict an interaction between Skp and SurA (Supplementary Fig. 27), which, in addition to their roles in maintaining the solubility of unfolded OMP proteins, may act in tandem to disassemble oligomeric OMPs that have aggregated70.
We also predict an interaction between BamA and PA1005 (Uniprot: Q9I4W8) (Fig. 4d, middle), a possible orthologue of β-barrel assembly-enhancing protease (BepA) (Supplementary Fig. 28). E. coli BepA is a periplasmic zinc-metallopeptidase with an important role in outer-membrane homeostasis and is involved in the degradation of BamA in the absence of SurA71. BepA has been shown to interact with BAM72, and further cross-linking experiments suggest that BepA C-terminal tetratricopeptide repeat (TPR) domain is inserted into the periplasmic region of BamA, below the β-barrel71. Our computed model agrees with the proposed broad interface between BamA and BepA, provides structural details into the BamA-BepA interaction and also suggests that when BepA is in complex with BamA, BAM is unable to assemble into its active form due to steric clashes between BepA and periplasmic Bam lipoproteins.
TolC is an OMP that homo-trimerizes to form a large outer-membrane export tunnel that interacts with inner-membrane translocases73,74. The catalytic β-barrel domain of BamA binds substrates along the β-barrel seam during OMP folding, and in this process, the N-terminal of the β-barrel likely swings outward75,76. The interaction between BamA and TolC has been recognized as an essential step in the assembly of TolC which occurs in a SurA-independent manner77,78. We predict an interaction between BamD and TolC (Fig. 4d, right), which, when superimposed onto the BAM complex (Supplementary Fig. 29), depicts how the β-sheets of TolC interact with the N-terminal strand of the BamA β-barrel seam. Our computed model shows how TolC could be folded by the BAM complex and suggests that BamD may potentially replace SurA to stabilize or recruit TolC to BAM.
Discussion
RF2-Lite is a new deep learning network for PPI prediction that is optimized to balance the accuracy and speed necessary for large-scale applications. We integrated RF2-Lite into a pipeline for proteome-wide PPI detection and modelling. We applied this pipeline to an array of human bacterial pathogens, resulting in several thousand predicted PPIs and their 3D structure models. Over 1,000 of our predictions were previously unknown, and both our benchmark and experimental validation suggest that a large fraction of these new PPIs are likely correct and should provide novel biological insights. The 3D structure models of protein complexes generated in our study provide mechanistic details for numerous essential cellular pathways and virulence factors.
Our results show the potential of computational methods in elucidating the 3D interactome and gaining functional insights for any organism. However, there is still considerable room for improvement in reducing the false-positive and false-negative rates. As a consequence of the false negatives, our predictions are not comprehensive: the absence of interactions should not be overinterpreted. Although we sought to be conservative and predict only highly confident PPIs, false positives unavoidably exist in our datasets. If each protein on average interacts with only 1 partner, 80% of the predictions in our final dataset are expected to be correct based on our benchmark. Some predicted PPIs, if true, appear to be transient based on the function of proteins and hence could be difficult to detect with experimental methods such as Co-IP (without cross-linking). Based on our limited experimental validation, one should expect that two thirds of our predicted interactions would give a positive signal in Co-IP experiments. By directly training not only on the PDB but also on larger sets of protein pairs where direct interactions are confidently known to occur (and not occur), as was done by ref. 79 for peptide–MHC complexes, it should be possible to increase prediction accuracy across a broad spectrum of interaction modalities.
Methods
We have built upon our previously developed multi-step bioinformatics and deep learning pipelines for identifying pairs of interacting proteins within the proteome of an organism7,9 to improve the scalability and accuracy of predictions. The architecture of the new RF2-Lite, which was trained on both monomeric proteins and protein complexes, is outlined in Fig. 1a, and the major steps of the bioinformatics pipeline are listed in Fig. 1c. Based on our positive and negative controls, PPIs identified by our pipeline have a predicted precision of 95% based on the assumption that each protein directly interacts with five other proteins. However, if the signal-to-noise ratio is much lower, for example, the average number of direct interacting partners for each protein is 1, the estimated precision falls to 80%. A detailed description of our methodology is provided in Supplementary Methods.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Supplementary Figs. 1–29, Tables 1–9, Methods and Discussion.
Predictions, metadata and benchmark set.
Acknowledgements
We thank N. V. Grishin, E. Horvitz and H. Park for helpful discussions; L. Goldschmidt and A. Guillory for computing resource management; and L. Stewart and L. Stuart for logistical support. In addition, we are grateful to T. G. Bernhardt, Y. O. Elshenawi, X. Liu, G. V. Mukamolova, K. M. Ottemann, M. L. Reniere and N. R. Salama for their correspondence and biological expertise. We acknowledge funding from Bill and Melinda Gates Foundation #OPP1156262 (to I.R.H.) and Washington Research Foundation and Translational Research Fund (to M.B.). This work was supported by the National Research Foundation of Korea grant funded by the Korea government (Ministry of Science and ICT) (number RS-2023-00210147 to M.B.), and J.Z. was supported by Cancer Prevention and Research Institute of Texas (CPRIT) training grant RP210041. The Defense Threat Reduction Agency grant HDTRA1-21-1-0007 (to A.K. and I.A.), Audacious Project at the Institute for Protein Design (to A.K.), Spark Therapeutics (to I.A.) and National Institute of Allergy and Infectious Diseases Federal Contracts HHSN272201700059C and 75N93022C00036 (to I.A.) are also acknowledged. We likewise thank National Institute of Health R01AI145954 (to J.D.M.), Defense Advanced Research Projects Agency Biological Technologies Office Program: Harnessing Enzymatic Activity for Lifesaving Remedies (HEALR) under cooperative agreement number HR0011-21-2-0012 (to J.D.M.) and I-2095-20220331 (to Q.C.) from the Welch Foundation. J.D.M. and D.B. are Howard Hughes Medical Institute investigators, and Q.C. is a Southwestern Medical Foundation endowed scholar.
Author contributions
Q.C. and D.B. conceived the research and contributed equally; I.R.H. and J.Z. prepared the sequence alignments used in the screen; M.B. designed and trained RoseTTAFold2-Light; I.R.H., J.Z., J.P., I.A. and Q.C. designed the PPI screening procedure; I.R.H. and J.Z. carried out the screen; I.R.H., J.Z., M.B., A.K. and Q.C. analysed and presented computational results; Y.W., C.A.T. and B.A.J. conducted laboratory experiments; Y.W. analysed and presented experimental results; I.R.H., Y.W., T.W., D.T.H., S.B.P. and J.D.M. provided biological insights on specific examples; I.R.H., Y.W., S.B.P., J.D.M., Q.C. and D.B. drafted the manuscript; all authors discussed the results and commented on the manuscript.
Peer review
Peer review information
Nature Microbiology thanks Ylva Ivarsson and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Data availability
Structures of highly confident pairs with accompanying metadata are available via ModelArchive at https://modelarchive.org/doi/10.5452/ma-bak-evip. Other high-order protein complexes are shared at https://conglab.swmed.edu/pathogens/. RF2-Lite is available at https://github.com/SNU-CSSB/RF2-Lite. AlphaFold was obtained from https://github.com/deepmind/alphafold on 16 July 2021 (v2.0.0).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Ian R. Humphreys, Jing Zhang, Minkyung Baek, Yaxi Wang.
Contributor Information
Minkyung Baek, Email: minkbaek@snu.ac.kr.
Qian Cong, Email: qian.cong@utsouthwestern.edu.
David Baker, Email: dabaker@uw.edu.
Supplementary information
The online version contains supplementary material available at 10.1038/s41564-024-01791-x.
References
- 1.Rajagopala, S. V. et al. The binary protein–protein interaction landscape of Escherichia coli. Nat. Biotechnol.32, 285–290 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Uetz, P. et al. A comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae. Nature403, 623–627 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Butland, G. et al. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature433, 531–537 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Edwards, A. M. et al. Bridging structural biology and genomics: assessing protein interaction data with known complexes. Trends Genet.18, 529–536 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Mackay, J. P., Sunde, M., Lowry, J. A., Crossley, M. & Matthews, J. M. Protein interactions: is seeing believing? Trends Biochem. Sci.32, 530–531 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Ovchinnikov, S., Kamisetty, H. & Baker, D. Robust and accurate prediction of residue–residue interactions across protein interfaces using evolutionary information. Elife3, e02030 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cong, Q., Anishchenko, I., Ovchinnikov, S. & Baker, D. Protein interaction networks revealed by proteome coevolution. Science365, 185–189 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Green, A. G. et al. Large-scale discovery of protein interactions at residue resolution using co-evolution calculated from genomic sequences. Nat. Commun.12, 1396 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Humphreys, I. R. et al. Computed structures of core eukaryotic protein complexes. Science374, eabm4805 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pei, J., Zhang, J. & Cong, Q. Human mitochondrial protein complexes revealed by large-scale coevolution analysis and deep learning-based structure modeling. Bioinformatics38, 4301–4311 (2022). [DOI] [PubMed] [Google Scholar]
- 11.Zhang, J., Pei, J., Durham, J., Bos, T. & Cong, Q. Computed cancer interactome explains the effects of somatic mutations in cancers. Protein Sci.31, e4479 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang, R., Ou, H.-Y. & Zhang, C.-T. DEG: a database of essential genes. Nucleic Acids Res.32, D271–D272 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen, L. et al. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res.33, D325–D328 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science373, 871–876 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao, M., Nakajima An, D., Parks, J. M. & Skolnick, J. AF2Complex predicts direct physical interactions in multimeric proteins with deep learning. Nat. Commun.13, 1744 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burke, D. F. et al. Towards a structurally resolved human protein interaction network. Nat. Struct. Mol. Biol.10.1038/s41594-022-00910-8 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evans, R. et al. Protein complex prediction with AlphaFold-Multimer. Preprint at bioRxiv10.1101/2021.10.04.463034 (2022).
- 19.Baek, M. et al. Efficient and accurate prediction of protein structure using RoseTTAFold2. Preprint at bioRxiv10.1101/2023.05.24.542179 (2023).
- 20.Weigt, M., White, R. A., Szurmant, H., Hoch, J. A. & Hwa, T. Identification of direct residue contacts in protein–protein interaction by message passing. Proc. Natl Acad. Sci. USA106, 67–72 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wall, D. P., Fraser, H. B. & Hirsh, A. E. Detecting putative orthologs. Bioinformatics19, 1710–1711 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Eddy, S. R. Accelerated profile HMM searches. PLoS Comput. Biol.7, e1002195 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol.7, 539 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen, F., Mackey, A. J., Stoeckert, C. J. Jr & Roos, D. S. OrthoMCL-DB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res.34, D363–D368 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szklarczyk, D. et al. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res.43, D447–D452 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kamisetty, H., Ovchinnikov, S. & Baker, D. Assessing the utility of coevolution-based residue–residue contact predictions in a sequence- and structure-rich era. Proc. Natl Acad. Sci. USA110, 15674–15679 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karimova, G., Pidoux, J., Ullmann, A. & Ladant, D. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc. Natl Acad. Sci. USA95, 5752–5756 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karimova, G., Gauliard, E., Davi, M., Ouellette, S. P. & Ladant, D. Protein–protein interaction: bacterial two-hybrid. Methods Mol. Biol.1615, 159–176 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Zhang, L. & Einsle, O. Architecture of the RNF1 complex that drives biological nitrogen fixation. Nat. Chem. Biol.20, 1078–1085 (2024). [DOI] [PubMed] [Google Scholar]
- 30.Häuser, R. et al. RsfA (YbeB) proteins are conserved ribosomal silencing factors. PLoS Genet.8, e1002815 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xia, Y. et al. Endoribonuclease YbeY is essential for RNA processing and virulence in Pseudomonas aeruginosa. MBio11 10.1128/mBio.00659-20 (2020). [DOI] [PMC free article] [PubMed]
- 32.Lee, P. T., Hsu, A. Y., Ha, H. T. & Clarke, C. F. A C-methyltransferase involved in both ubiquinone and menaquinone biosynthesis: isolation and identification of the Escherichia coli ubiE gene. J. Bacteriol.179, 1748–1754 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Božik, M. et al. Stress response of Escherichia coli to essential oil components—insights on low-molecular-weight proteins from MALDI-TOF. Sci. Rep.8, 13042 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sultonova, M. et al. Integrated changes in thermal stability and proteome abundance during altered nutrient states in Escherichia coli and human cells. Proteomics22, e2100254 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Kempen, M. Fast and accurate protein structure search with Foldseek. Nat. Biotechnol.10.1038/s41587-023-01773-0 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ting, C. P. et al. Use of a scaffold peptide in the biosynthesis of amino acid-derived natural products. Science365, 280–284 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng, Y. et al. Structures of the holoenzyme TglHI required for 3-thiaglutamate biosynthesis. Structure31, 1220–1232.e5 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feng, L. et al. Structural and genetic characterization of enterohemorrhagic Escherichia coli O145 O antigen and development of an O145 serogroup-specific PCR assay. J. Bacteriol.187, 758–764 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sandoval, J. M., Arenas, F. A. & Vásquez, C. C. Glucose-6-phosphate dehydrogenase protects Escherichia coli from tellurite-mediated oxidative stress. PLoS ONE6, e25573 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hagen, K. D. & Meeks, J. C. The unique cyanobacterial protein OpcA is an allosteric effector of glucose-6-phosphate dehydrogenase in Nostoc punctiforme ATCC 29133. J. Biol. Chem.276, 11477–11486 (2001). [DOI] [PubMed] [Google Scholar]
- 41.Kaminishi, T. et al. A snapshot of the 30S ribosomal subunit capturing mRNA via the Shine–Dalgarno interaction. Structure15, 289–297 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Yusupova, G. Z., Yusupov, M. M., Cate, J. H. & Noller, H. F. The path of messenger RNA through the ribosome. Cell106, 233–241 (2001). [DOI] [PubMed] [Google Scholar]
- 43.Jacob, A. I., Köhrer, C., Davies, B. W., RajBhandary, U. L. & Walker, G. C. Conserved bacterial RNase YbeY plays key roles in 70S ribosome quality control and 16S rRNA maturation. Mol. Cell49, 427–438 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vercruysse, M. et al. The highly conserved bacterial RNase YbeY is essential in Vibrio cholerae, playing a critical role in virulence, stress regulation, and RNA processing. PLoS Pathog.10, e1004175 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vercruysse, M. et al. Identification of YbeY-protein interactions involved in 16S rRNA maturation and stress regulation in Escherichia coli. mBio7 10.1128/mBio.01785-16 (2016). [DOI] [PMC free article] [PubMed]
- 46.Finlay, B. B. & Falkow, S. Common themes in microbial pathogenicity revisited. Microbiol. Mol. Biol. Rev.61, 136–169 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barembruch, C. & Hengge, R. Cellular levels and activity of the flagellar sigma factor FliA of Escherichia coli are controlled by FlgM-modulated proteolysis. Mol. Microbiol.65, 76–89 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Suzuki, T. The expanding world of tRNA modifications and their disease relevance. Nat. Rev. Mol. Cell Biol.22, 375–392 (2021). [DOI] [PubMed] [Google Scholar]
- 49.Ikeuchi, Y., Shigi, N., Kato, J.-I., Nishimura, A. & Suzuki, T. Mechanistic insights into sulfur relay by multiple sulfur mediators involved in thiouridine biosynthesis at tRNA wobble positions. Mol. Cell21, 97–108 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Numata, T., Fukai, S., Ikeuchi, Y., Suzuki, T. & Nureki, O. Structural basis for sulfur relay to RNA mediated by heterohexameric TusBCD complex. Structure14, 357–366 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Shi, R. et al. Structural basis for Fe-S cluster assembly and tRNA thiolation mediated by IscS protein–protein interactions. PLoS Biol.8, e1000354 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eaton, K. A., Brooks, C. L., Morgan, D. R. & Krakowka, S. Essential role of urease in pathogenesis of gastritis induced by Helicobacter pylori in gnotobiotic piglets. Infect. Immun.59, 2470–2475 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ha, N. C. et al. Supramolecular assembly and acid resistance of Helicobacter pylori urease. Nat. Struct. Biol.8, 505–509 (2001). [DOI] [PubMed] [Google Scholar]
- 54.Carter, E. L., Flugga, N., Boer, J. L., Mulrooney, S. B. & Hausinger, R. P. Interplay of metal ions and urease. Metallomics1, 207–221 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fong, Y. H. et al. Structure of UreG/UreF/UreH complex reveals how urease accessory proteins facilitate maturation of Helicobacter pylori urease. PLoS Biol.11, e1001678 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Farrugia, M. A., Macomber, L. & Hausinger, R. P. Biosynthesis of the urease metallocenter. J. Biol. Chem.288, 13178–13185 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rapoport, T. A. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature450, 663–669 (2007). [DOI] [PubMed] [Google Scholar]
- 58.Jauss, B. et al. Noncompetitive binding of PpiD and YidC to the SecYEG translocon expands the global view on the SecYEG interactome in. J. Biol. Chem.294, 19167–19183 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Götzke, H. et al. YfgM is an ancillary subunit of the SecYEG translocon in Escherichia coli. J. Biol. Chem.289, 19089–19097 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Van den Berg, B. et al. X-ray structure of a protein-conducting channel. Nature427, 36–44 (2004). [DOI] [PubMed] [Google Scholar]
- 61.Voulhoux, R., Bos, M. P., Geurtsen, J., Mols, M. & Tommassen, J. Role of a highly conserved bacterial protein in outer membrane protein assembly. Science299, 262–265 (2003). [DOI] [PubMed] [Google Scholar]
- 62.Wu, T. et al. Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell121, 235–245 (2005). [DOI] [PubMed] [Google Scholar]
- 63.Noinaj, N. et al. Structural insight into the biogenesis of β-barrel membrane proteins. Nature501, 385–390 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Han, L. et al. Structure of the BAM complex and its implications for biogenesis of outer-membrane proteins. Nat. Struct. Mol. Biol.23, 192–196 (2016). [DOI] [PubMed] [Google Scholar]
- 65.Gu, Y. et al. Structural basis of outer membrane protein insertion by the BAM complex. Nature531, 64–69 (2016). [DOI] [PubMed] [Google Scholar]
- 66.Imai, Y. et al. A new antibiotic selectively kills Gram-negative pathogens. Nature576, 459–464 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kaur, H. et al. The antibiotic darobactin mimics a β-strand to inhibit outer membrane insertase. Nature593, 125–129 (2021). [DOI] [PubMed] [Google Scholar]
- 68.Sklar, J. G., Wu, T., Kahne, D. & Silhavy, T. J. Defining the roles of the periplasmic chaperones SurA, Skp, and DegP in Escherichia coli. Genes Dev.21, 2473–2484 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schiffrin, B. et al. Dynamic interplay between the periplasmic chaperone SurA and the BAM complex in outer membrane protein folding. Commun. Biol.5, 1–15 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chamachi, N. et al. Chaperones Skp and SurA dynamically expand unfolded OmpX and synergistically disassemble oligomeric aggregates. Proc. Natl Acad. Sci. USA 119 10.1073/pnas.2118919119 (2022). [DOI] [PMC free article] [PubMed]
- 71.Daimon, Y. et al. The TPR domain of BepA is required for productive interaction with substrate proteins and the β-barrel assembly machinery complex. Mol. Microbiol.106, 760–776 (2017). [DOI] [PubMed] [Google Scholar]
- 72.Narita, S.-I., Masui, C., Suzuki, T., Dohmae, N. & Akiyama, Y. Protease homolog BepA (YfgC) promotes assembly and degradation of β-barrel membrane proteins in Escherichia coli. Proc. Natl Acad. Sci. USA110, E3612–E3621 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thanabalu, T., Koronakis, E., Hughes, C. & Koronakis, V. Substrate-induced assembly of a contiguous channel for protein export from E. coli: reversible bridging of an inner-membrane translocase to an outer membrane exit pore. EMBO J.17, 6487–6496 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Koronakis, V., Sharff, A., Koronakis, E., Luisi, B. & Hughes, C. Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature405, 914–919 (2000). [DOI] [PubMed] [Google Scholar]
- 75.Tomasek, D. et al. Structure of a nascent membrane protein as it folds on the BAM complex. Nature583, 473–478 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Doyle, M. T. et al. Cryo-EM structures reveal multiple stages of bacterial outer membrane protein folding. Cell185, 1143–1156.e13 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Werner, J. & Misra, R. YaeT (Omp85) affects the assembly of lipid-dependent and lipid-independent outer membrane proteins of Escherichia coli. Mol. Microbiol.57, 1450–1459 (2005). [DOI] [PubMed] [Google Scholar]
- 78.Bennion, D., Charlson, E. S., Coon, E. & Misra, R. Dissection of β-barrel outer membrane protein assembly pathways through characterizing BamA POTRA 1 mutants of Escherichia coli. Mol. Microbiol.77, 1153–1171 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Motmaen, A. et al. Peptide-binding specificity prediction using fine-tuned protein structure prediction networks. Proc. Natl Acad. Sci. USA120, e2216697120 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figs. 1–29, Tables 1–9, Methods and Discussion.
Predictions, metadata and benchmark set.
Data Availability Statement
Structures of highly confident pairs with accompanying metadata are available via ModelArchive at https://modelarchive.org/doi/10.5452/ma-bak-evip. Other high-order protein complexes are shared at https://conglab.swmed.edu/pathogens/. RF2-Lite is available at https://github.com/SNU-CSSB/RF2-Lite. AlphaFold was obtained from https://github.com/deepmind/alphafold on 16 July 2021 (v2.0.0).