

Abstract
Lazertinib, a novel third‐generation epidermal growth factor receptor tyrosine kinase inhibitor (EGFR‐TKI), demonstrates marked efficacy in EGFR‐mutant lung cancer. However, resistance commonly develops, prompting consideration of therapeutic strategies to overcome initial drug resistance mechanisms. This study aimed to elucidate the adaptive resistance to lazertinib and advocate novel combination treatments that demonstrate efficacy in preventing resistance as a first‐line treatment for EGFR mutation‐positive NSCLC. We found that AXL knockdown significantly inhibited lung cancer cell viability in the presence of lazertinib, indicating that AXL activation contributes to lazertinib resistance. However, long‐term culture with a combination of lazertinib and AXL inhibitors led to residual cell proliferation and increased the MCL‐1 expression level, which was mediated by the nuclear translocation of the transcription factor YAP. Triple therapy with an MCL‐1 or YAP inhibitor in combination with lazertinib and an AXL inhibitor significantly reduced cell viability and increased the apoptosis rate. These results demonstrate that AXL and YAP/MCL‐1 signals contribute to adaptive lazertinib resistance in EGFR‐mutant lung cancer cells, suggesting that the initial dual inhibition of AXL and YAP/MCL‐1 might be a highly effective strategy in eliminating lazertinib‐resistant cells.
Keywords: AXL, EGFR, EGFR‐TKI, MCL‐1, YAP
This report demonstrates that AXL and YAP/MCL‐1 signals contribute to adaptive lazertinib resistance in EGFR‐mutant lung cancer cells, suggesting that the initial dual inhibition of AXL and YAP/MCL‐1 might be a highly effective strategy for eliminating lazertinib‐resistant cells.
1. INTRODUCTION
Lung cancer is the leading cause of cancer‐related deaths worldwide, with non–small‐cell lung cancer (NSCLC) accounting for approximately 85% of all lung cancer cases. 1 , 2 The treatment approach for patients with NSCLC has recently shifted from a method based purely on tumor histology to a molecular‐targeted approach because of the discovery of targetable driver oncogenes and significant advancements in molecular‐targeted therapies. 3
Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (EGFR‐TKIs) have demonstrated efficacy in treating patients with NSCLC harboring EGFR mutations such as exon 19 deletion or L858R. In particular, the third‐generation EGFR‐TKI osimertinib significantly improved progression‐free survival (PFS) compared with that conferred under treatment with first‐generation EGFR‐TKIs in patients with untreated EGFR‐positive NSCLC. 4 Therefore, osimertinib has been approved for treating advanced untreated EGFR‐mutant NSCLC in various countries, including the United States and Japan. 4 , 5 , 6
Lazertinib, a relatively new third‐generation EGFR‐TKI and mutation‐selective EGFR inhibitor, demonstrates high selectivity for single (Ex19del, L858R, T790M) and double mutations (Ex19del/T790M, L858R/T790M). 7 In a phase III randomized, double‐blinded study, lazertinib significantly improved PFS compared with gefitinib (20.6 vs. 9.7 months; hazard ratio [HR], 0.45; 95% CI, 0.34–0.58; p < 0.001). 8 In addition, a recent phase III trial demonstrated that amivantamab, an EGFR‐MET proto‐oncogene bispecific antibody, in combination with lazertinib, showed significantly improved PFS compared with osimertinib (23.7 vs. 16.6 months; HR, 0.70; 95% CI, 0.58–0.85; p < 0.001). 9 Therefore, lazertinib is expected to undergo future clinical development for patients with EGFR‐mutant NSCLC. However, almost all patients inevitably acquire resistance to lazertinib after varying treatment periods. In addition, there is a lack of reports investigating the resistance mechanisms of lazertinib. Therefore, an understanding of the molecular mechanisms underlying intrinsic resistance and early refractoriness to lazertinib in EGFR‐mutant NSCLC is required.
This study aimed to elucidate the adaptive resistance to lazertinib and advocate novel combination treatments that demonstrate efficacy in preventing resistance as a first‐line treatment for EGFR mutation‐positive NSCLC.
2. MATERIALS AND METHODS
2.1. Cell culture and reagents
Seven human NSCLC cell lines harboring EGFR mutations were used. HCC4011 cells with the EGFR‐L858R mutation were generously provided by Dr. David P. Carbone (Ohio State University Comprehensive Cancer Center, Columbus, OH, USA) and Dr. John D. Minna (University of Texas Southwestern Medical Center, Dallas, TX). The H1975 human lung adenocarcinoma cell line with the EGFR‐L858R/T790M double mutation was kindly provided by Dr. Yoshitaka Sekido (Aichi Cancer Center Research Institute, Nagoya, Japan) and Dr. John D. Minna. The human cell lines HCC827 and HCC4006 (both with EGFR exon 19 deletion mutations) were purchased from the ATCC (Manassas, VA, USA), and the PC‐9 cell line with an EGFR exon 19 deletion was obtained from the RIKEN Cell Bank (Tsukuba, Japan). PC‐9GXR cells, with EGFR exon 19 deletion and T790M mutations, were established at Kanazawa University (Kanazawa, Japan) from PC‐9 cell xenograft tumors in nude mice that had acquired resistance to gefitinib. 10 We established the patient‐derived cell line KPP‐03 with an EGFR‐L858R mutation from tumor cells in pleural effusion, as described previously. 11
All cell lines were maintained in RPMI 1640 medium (Gibco, Grand Island, NY, USA) supplemented with 10% FBS, penicillin (100 U/mL), and streptomycin (50 g/mL) in a humidified CO2 incubator at 37°C. The cells were screened regularly for mycoplasma using a MycoAlert Mycoplasma Detection Kit (Lonza Bioscience, Basel, Switzerland). Lazertinib, ONO7475, S63845, and verteporfin were purchased from Selleck Chemicals (Houston, TX, USA).
2.2. Western blotting
Proteins (25‐μg aliquots) were resolved using SDS PAGE (Bio‐Rad Laboratories, Hercules, CA, USA). The electrophoresed protein samples were transferred to polyvinylidene difluoride membranes (Bio‐Rad Laboratories), washed three times with TBS, and incubated with a blotting‐grade blocker (Bio‐Rad Laboratories) for 1 h at 25°C before incubating overnight at 4°C with primary antibodies. The antibodies used in this study are listed in Table S1. The membranes were then washed three times with TBS and incubated with HRP‐conjugated species‐specific secondary antibodies for 1 h at 25°C, and the immunoreactive bands were visualized using SuperSignal West Dura Extended Duration substrate (Pierce Biotechnology, Waltham, MA, USA). Each experiment was performed independently at least thrice. All blots were obtained from the same experiment and processed in parallel.
2.3. Real‐time PCR analysis
Total RNA was extracted using NucleoSpin® RNA Plus (Takara Bio Inc., Shiga, Japan), and cDNA was synthesized using PrimeScript™ RT master mix (Perfect Real‐Time; TaKaRa Bio Inc.) according to the manufacturer's instructions. Real‐time PCR was performed using a TaKaRa PCR Thermal Cycler Dice® (Takara Bio Inc.) and SYBR Fast qPCR kit (Kapa Biosystems, Cape Town, South Africa) under the following amplification protocol: initial incubation at 95°C for 10 min, 40 cycles at 95°C for 15 s, and 60°C for 1 min, followed by melting curve analysis. Gene expression was calculated from relative standard curves, normalized to GAPDH levels, and analyzed using the method. 12 The primer sequences used in this study are listed in Table S2.
Additional methods are included in Supplementary Methods (Appendix S1).
3. RESULTS
3.1. AXL plays a pivotal role in the viability of EGFR‐mutant NSCLC cells treated with lazertinib
We examined the efficacy of lazertinib in seven EGFR‐mutant NSCLC cell lines (Figure 1A). Among these cell lines, HCC827 and HCC4006 were overly sensitive and showed low IC50 values for lazertinib. Although long‐term exposure to lazertinib resulted in higher sensitivity than that detected with exposure to gefitinib, treatment with lazertinib did not completely suppress the growth of PC‐9 cells, even at high concentrations (Figure 1B). To elucidate the survival mechanisms of the residual cells, a small interfering RNA (siRNA) library was used to screen for factors related to adaptive lazertinib resistance. Among the 56 siRNAs targeting receptor‐type tyrosine kinases, we focused on AXL knockdown because of its remarkable synergistic effect in reducing PC‐9 cell viability (Figure 1C). The proteins that were knocked down are listed in Table S3. We further confirmed the decrease in cell viability upon AXL knockdown in multiple EGFR‐mutant NSCLC cell lines. Therefore, AXL knockdown enhanced the inhibitory effects of lazertinib on the growth of multiple EGFR‐mutant NSCLC cells (Figure 1D, Figure S1).
FIGURE 1.
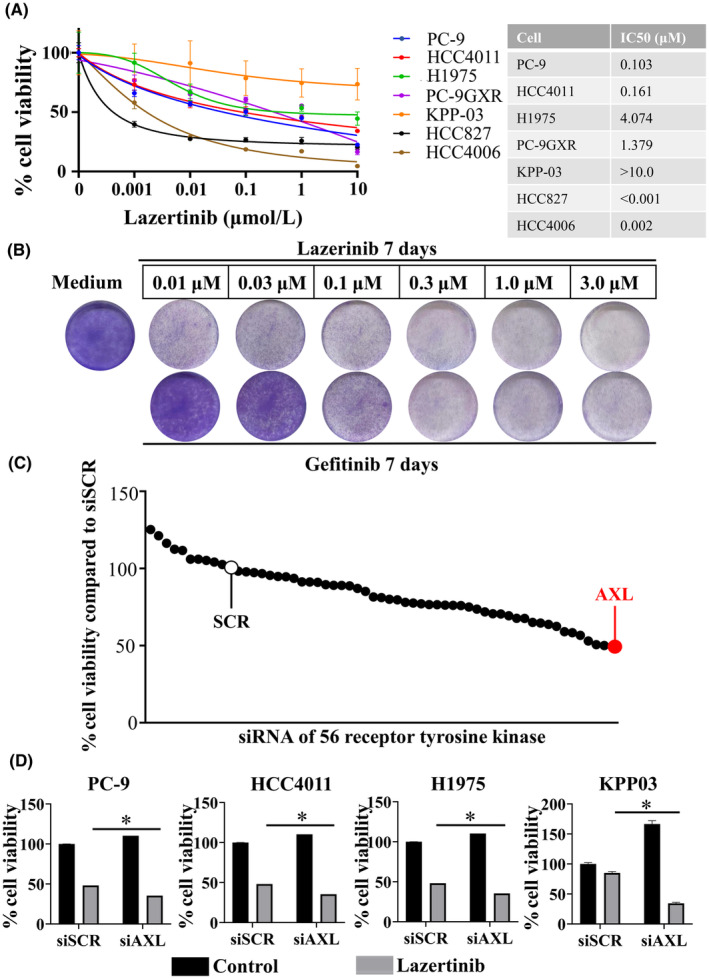
AXL plays a pivotal role in the viability of EGFR‐mutant NSCLC cells treated with lazertinib. (A) EGFR‐mutant NSCLC cell lines PC‐9, HCC4011, H1975, PC‐9GXR, KPP‐03, HCC827, and HCC4006 were incubated with indicated concentrations of lazertinib for 72 h. Cell growth was determined using MTT assays, and the IC50 values of lazertinib for each cell line were determined. *p < 0.05 (B) PC‐9 cell viability after 7 days of treatment with indicated concentrations of lazertinib or gefitinib, replenished every 72 h. (C) MTT assays evaluating the effect of a combination of EGFR inhibitor lazertinib (100 nmol/L) and knockdown of 56 receptor tyrosine kinases from Silencer® Select human kinase siRNA library V4 on PC‐9 cell viability. The 56 RTKs are rank ordered from highest to lowest according to their inhibitory effect on the viability of PC‐9 cells relative to nonspecific control siRNA. (D) PC‐9, HCC4011, H1975, and KPP‐03 cells were treated with nonspecific control siRNA or AXL‐specific siRNAs and were incubated with or without lazertinib (100 nmol/L) for 72 h. Cell viability was detected using MTT assays. *p < 0.05.
3.2. AXL inhibitor sensitizes AXL‐overexpressing EGFR‐mutant NSCLC cells to EGFR‐TKIs
In the assessment of AXL expression in EGFR‐mutant NSCLC cells, high AXL expression was observed in PC‐9, HCC4011, H1975, KPP‐03, and PC‐9GXR cells but not in HCC827 and HCC4006 cells (Figure 2A). To assess the role of AXL inhibition on cell viability, we examined the synergistic effect of the AXL inhibitor ONO7475 in combination with lazertinib in EGFR‐mutant NSCLC cells. This combination showed superior efficacy compared with that of lazertinib alone in the PC‐9, HCC4011, H1975, and KPP‐03 cell lines with high AXL expression (Figure 2B). A synergistic effect of this combination was also observed with other AXL inhibitors, BMS‐777607 or NPS‐1034, in cell lines exhibiting high AXL expression (Figure S2). In addition, ONO7475 enhanced the efficacy of lazertinib on the viability of the cell lines PC‐9, HCC4011, H1975, KPP‐03, and PC‐9GXR, all of which exhibited high AXL expression, but only had a marginal effect on the viability of the cell lines HCC827 and HCC4006, with low AXL expression (Figure 2C). These findings strongly emphasize the significance of AXL and its role as a biomarker for lazertinib resistance in EGFR‐mutant NSCLC cells. Western blot analysis was performed to evaluate the response to treatment with lazertinib alone and the combination of lazertinib with ONO7475. At 4 h of treatment, lazertinib effectively suppressed the phosphorylation of EGFR, as well as the activation of its downstream signaling pathways, including ERK and AKT (Figure 2D). However, the administration of lazertinib monotherapy resulted in the reactivation of ERK and AKT phosphorylation after 72 h (Figure 2E). In contrast, when lazertinib was combined with ONO7475, a more pronounced suppression of ERK and AKT phosphorylation was observed at 4 h compared with that detected in response to monotherapy (Figure 2D). Importantly, this enhanced inhibitory effect persisted even after 72 h, suggesting a potential synergistic action of the combination treatment. Continuous co‐treatment of PC‐9, HCC4011, and H1975 cells with lazertinib and ONO7475 for 9 days inhibited cell growth compared with lazertinib monotherapy, although complete inhibition was not achieved (Figure 2F). Furthermore, extended culture and cytotoxicity evaluation using the MTT assay demonstrated that combination therapy with lazertinib and ONO7475 temporarily suppressed the viability of AXL‐overexpressing cells. Nevertheless, eventual repopulation was observed after a specified period of lazertinib and ONO7475 combination therapy (Figure 2G).
FIGURE 2.
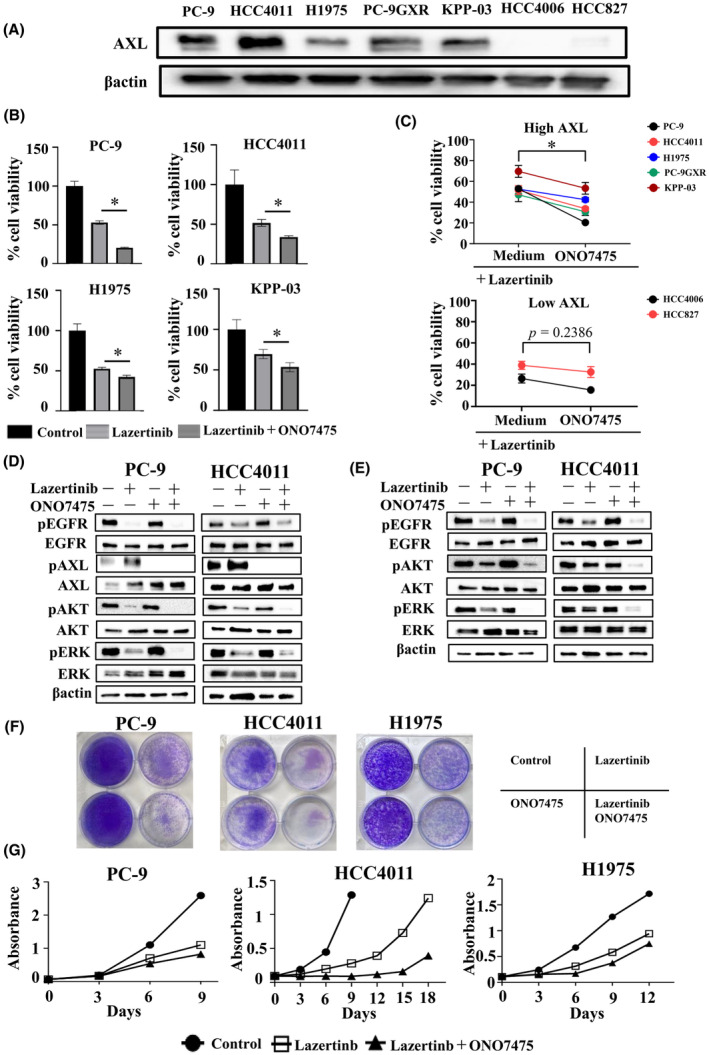
The AXL inhibitor sensitized AXL‐overexpressing EGFR‐mutant NSCLC cells to lazertinib. (A) EGFR‐mutant NSCLC cell lines PC‐9, HCC4011, H1975, PC‐9GXR, KPP‐03, HCC827, and HCC4006 were lysed, and the indicated proteins were detected using western blotting. (B) EGFR‐mutant NSCLC cell lines PC‐9, HCC4011, H1975, and KPP‐03 were incubated with lazertinib (100 nmol/L) or a combination of lazertinib (100 nmol/L) and AXL inhibitor ONO7475 (100 nmol/L) for 72 h. Cell growth was determined using MTT assays. *p < 0.05. (C) Quantitative determination of the inhibition of cell viability of high‐AXL‐expressing and low‐AXL‐expressing EGFR‐mutant cells treated with the EGFR‐TKI lazertinib (100 nmol/L) in the presence or absence of ONO7475 (100 nmol/L) for 72 h. Paired Student's t‐tests were used for comparisons. (D) PC‐9 and HCC4011 cells were treated with lazertinib (100 nmol/L), ONO7475 (100 nmol/L), or a combination of lazertinib and ONO7475 for 4 h. (E) PC‐9 and HCC4011 cells were treated with lazertinib (100 nmol/L), ONO7475 (100 nmol/L), or a combination of lazertinib and ONO7475 for 72 h. (F) PC‐9, HCC4011, and H1975 cells were visualized using crystal violet staining following 9 days of treatment with lazertinib (100 nmol/L), ONO7475 (100 nmol/L), or a combination of lazertinib and ONO7475 where the drugs were replenished every 72 h. (G) PC‐9, HCC4011, and H1975 cells were treated with lazertinib (100 nmol/L), ONO7475 (100 nmol/L), or a combination of lazertinib and ONO7475 in which the drugs were replenished every 72 h. Absorbance was measured every 3 days. *p < 0.05.
3.3. MCL‐1 knockdown sensitizes EGFR‐mutant NSCLC cells to combined lazertinib and ONO7475
Because the combination treatment with lazertinib and ONO7475 failed to completely control cancer cells, understanding the additional resistance mechanisms linked to the combination treatment is crucial. To achieve tumor eradication in combination therapy with lazertinib and ONO7475, we focused on inducing cell apoptosis and the expression of apoptotic and antiapoptotic proteins. Western blot analysis revealed increased MCL‐1 expression following lazertinib treatment as a common alternation in the three EGFR‐mutant NSCLC cell lines (Figure 3A). To further confirm the role of MCL‐1 in apoptosis resistance, we knocked down MCL‐1 expression using a specific siRNA, which led to enhanced cell apoptosis during combination therapy with lazertinib and ONO7475 (Figure 3B). Moreover, additional knockdown of MCL‐1 in combination with lazertinib and ONO7475 significantly enhanced the inhibition of cell viability (Figure 3C). Additionally, flow cytometry analysis showed that the addition of siMCL‐1 led to a significant increase in apoptotic cells compared to the combined treatment with lazertinib and ONO‐7475 (Figure 3D). These findings demonstrate the significance of the MCL‐1 protein in apoptosis resistance during combination therapy with lazertinib and AXL inhibitors in EGFR‐mutant NSCLC cells.
FIGURE 3.
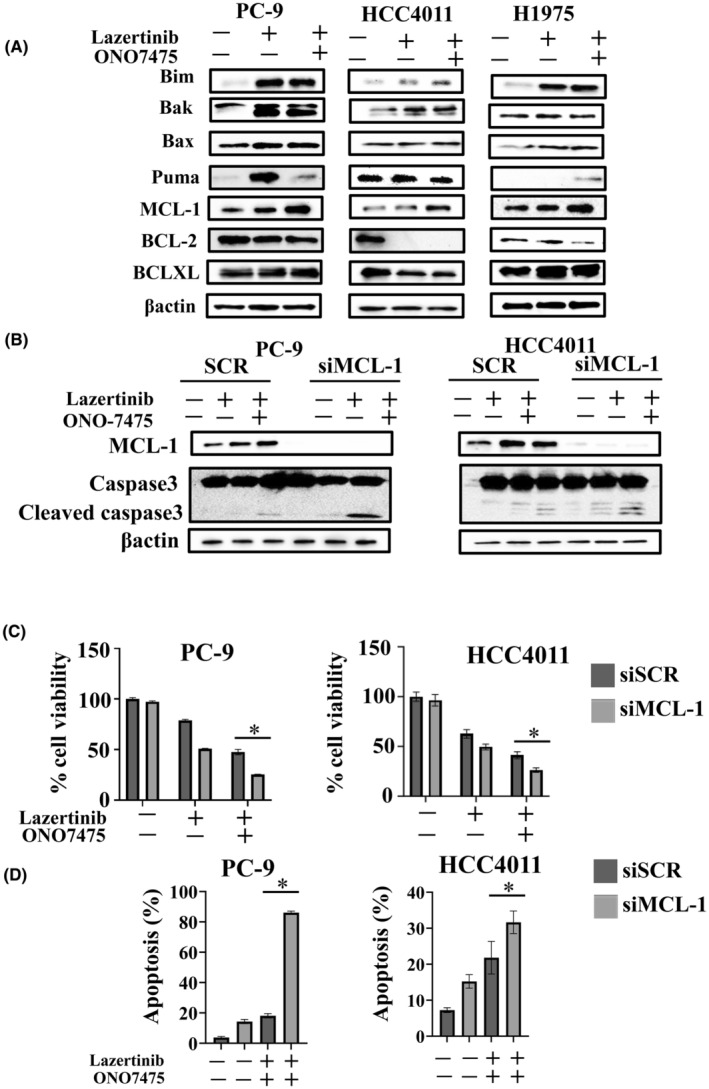
MCL‐1 knockdown sensitized EGFR‐mutant NSCLC cells to a combination of lazertinib and ONO7475. (A) PC‐9, HCC4011, and H1975 cells were treated with lazertinib (100 nmol/L), ONO7475 (100 nmol/L), or a combination of lazertinib and ONO7475 for 48 h. The cells were lysed, and the indicated proteins were detected using western blotting. (B) PC‐9 and HCC4011 cells were treated with nonspecific control siRNA or MCL‐1‐specific siRNA and incubated with lazertinib (100 nmol/L) or a combination of lazertinib (100 nmol/L) and ONO7475 (100 nmol/L) for 48 h. The cells were lysed, and the indicated proteins were detected using western blotting. (C) PC‐9 and HCC4011 cells treated with nonspecific control siRNA or MCL‐1‐specific siRNA and incubated with lazertinib (100 nmol/L) or a combination of lazertinib and ONO7475 (100 nmol/L) for 72 h. Cell growth was determined using MTT assays. *p < 0.05. (D) Apoptotic cell percentages in PC‐9 and HCC4011 cells double stained with annexin V and propidium iodide were determined by flow cytometry following treatment with lazertinib (100 nmol/L) and ONO7475 (100 nmol/L) with or without siMCL‐1 for 48 h. *p < 0.05.
3.4. Co‐treatment with MCL‐1 and AXL inhibitors and lazertinib promotes apoptosis through dissociation of MCL‐1 and Bak in EGFR‐mutant NSCLC cells
Given the involvement of MCL‐1 in apoptosis resistance, we performed a cell viability assay using the MCL‐1 inhibitor, S63845, in addition to lazertinib and ONO7475. S63845 significantly enhanced the therapeutic effects of the combination of lazertinib and ONO7475 with respect to reducing the viability of PC‐9, HCC4011, and H1975 cells (Figure 4A). Continuous triple therapy with lazertinib, ONO7475, and S63845 prevented the emergence of drug‐tolerant cells (Figure 4B). Furthermore, S63845 exerted enhanced inhibitory effects on cell proliferation, in combination with other EGFR‐TKIs such as osimertinib and afatinib, and not just with lazertinib (Figure S3).
FIGURE 4.
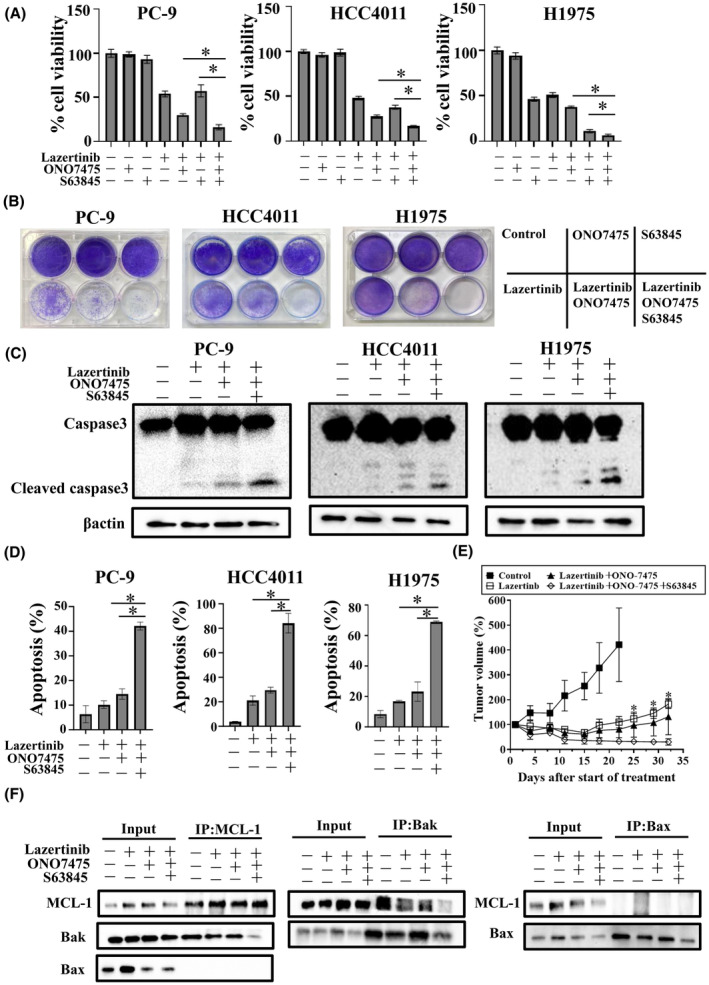
Co‐treatment with MCL‐1 inhibitor, AXL inhibitor, and lazertinib promoted apoptosis in EGFR‐mutant NSCLC cells. (A) PC‐9, HCC4011, and H1975 cells were treated with lazertinib (100 nmol/L), ONO7475 (100 nmol/L), MCL‐1 inhibitor S63845 (1 μmol/L), or combinations for 72 h. Cell growth was determined using MTT assays. *p < 0.05. (B) PC‐9, HCC4011, and H1975 cells were visualized using crystal violet staining following 9 days of treatment with lazertinib (100 nmol/L), ONO7475 (100 nmol/L), S63845 (1 μmol/L), or combinations where the drugs were replenished every 72 h. (C) PC‐9, HCC4011, and H1975 cells were treated with lazertinib (100 nmol/L) with or without ONO7475 (100 nmol/L), S63845 (1 μmol/L) for 48 h. The cells were lysed, and the indicated proteins were detected using western blotting. (D) Apoptotic cell percentages of PC‐9, HCC4011, and H1975 cells, which were double stained with annexin V and propidium iodide were detected using flow cytometry following treatment with lazertinib (100 nmol/L) with or without ONO7475 (100 nmol/L) and S63845 (1 μmol/L) for 48 h. *p < 0.05. (E) PC‐9 CDX tumors were treated with the vehicle control, 3 mg/kg lazertinib daily, 3 mg/kg lazertinib daily plus 10 mg/kg ONO‐7475 daily, and 3 mg/kg lazertinib daily plus 10 mg/kg ONO‐7475 daily plus 25 mg/kg S63845 twice a week (n = 6 per group). *Comparison of Lazertinib + ONO‐7475 with Lazertinib plus ONO‐7475 plus S63845 gave p < 0.05. (F) PC‐9 cells were treated with lazertinib (100 nmol/L) with or without ONO7475 (100 nmol/L) and S63845(1 μmol/L) for 48 h. Proteins were immunoprecipitated from the cell lysates and subjected to immunoblotting using anti‐MCL‐1, anti‐Bax, and anti‐Bak mAbs.
Western blot analysis revealed that S63845 in combination with lazertinib and ONO7475 enhanced the induction of the apoptotic factor cleaved caspase‐3 compared with the combination of lazertinib and ONO7475 alone, indicating the promotion of cell apoptosis (Figure 4C). Furthermore, flow cytometry analysis showed that the number of apoptotic cells increased with the combination of lazertinib and ONO7475 and did so more strongly under triple therapy with S63845 (Figure 4D). In addition, we evaluated the effect of the triple therapy in a PC‐9 cell CDX model. Mice were continuously administered lazertinib alone, lazertinib plus ONO‐7475, or lazertinib plus ONO‐7475 plus S64845 for 31 days. The lazertinib monotherapy and the lazertinib plus ONO‐7475 combination therapy exerted antitumor effects on subcutaneous tumors cell growth, and tumor regrowth was observed with both treatments within 3 and 4 weeks, respectively. However, tumor regrowth was not observed with the triple combination therapy throughout the observation period (Figure 4E). No apparent adverse events, including weight loss, were observed with any of these treatments (Figure S4). S63845 exerts its apoptotic effects by specifically inhibiting the interaction between the antiapoptotic protein, MCL‐1, and the pro‐apoptotic proteins, BAK and BAX. 13 To validate these pharmacological effects, an immunoprecipitation analysis was carried out to assess the impact of S63845 on the binding of MCL‐1 to BAK and BAX. MCL‐1 knockdown was found not to affect BAK expression (Figure S5). These findings demonstrated that incorporating S63845 resulted in MCL‐1 and BAK dissociation, but not in BAX dissociation (Figure 4F).
3.5. YAP–MCL‐1 axis induces tolerant cells with lazertinib and AXL inhibitor combination in EGFR‐mutant NSCLC cells
To further elucidate the underlying drug tolerance mechanisms, we focused on the transcription factors of MCL‐1 proteins. Several previous reports have shown the involvement of YAP, STAT3, and c‐JUN in regulating MCL‐1 as transcriptional co‐activators. 14 , 15 , 16 Western blot analysis showed the upregulation of YAP, STAT3, and c‐JUN expression upon combination treatment with lazertinib and ONO7475 in EGFR‐mutant NSCLC cells (Figure 5A). YAP knockdown using a specific siRNA in EGFR‐mutant NSCLC cells suppressed MCL‐1 expression when combined with lazertinib and ONO7475. In contrast, the knockdown of STAT3 or c‐JUN in EGFR‐mutant NSCLC cells did not significantly alter MCL‐1 expression (Figure 5B, Figure S6). YAP knockdown enhanced the efficacy of THE combination treatment with lazertinib and ONO7475 in inhibiting the viability of PC‐9 and HCC4011 cells, suggesting that YAP contributes to the resistance of lung cancer cells to lazertinib plus ONO7475 (Figure 5C). YAP translocates to the nucleus, where it binds to transcription factors such as TEAD, facilitating the transcription of various proteins. 17 Combined treatment with lazertinib and ONO7475 increased the translocation of YAP to the nucleus (Figure 5D). Furthermore, co‐localization analysis showed that the nuclear localization of YAP was induced by the combination of lazertinib and ONO7475 relative to lazertinib monotherapy (Figure 5E). YAP expression was observed throughout the cells in the control group, whereas in the group treated with a combination of lazertinib and ONO7475, YAP was localized within the nucleus. YAP knockdown significantly reduced MCL‐1 mRNA levels in PC‐9 and HCC4011 cells relative to those in the control (Figure 5F). These results demonstrated that resistance to the combination of lazertinib and an AXL inhibitor was enhanced by concurrent activation of the YAP–MCL‐1 axis in EGFR‐mutant NSCLC cells.
FIGURE 5.
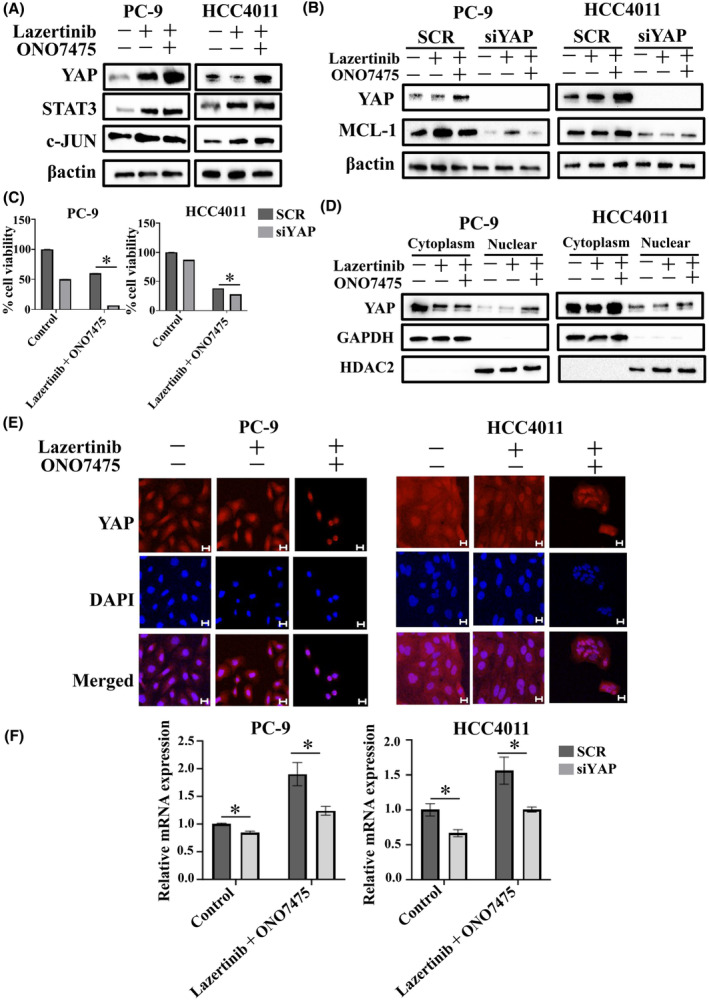
YAP–MCL‐1 axis‐induced tolerant cells with a combination of lazertinib and AXL inhibitor in EGFR‐mutant NSCLC cells. (A) PC‐9 and HCC4011 cells were treated with lazertinib (100 nmol/L), ONO7475 (100 nmol/L), or a combination of lazertinib and ONO7475 for 48 h. The cells were lysed, and the indicated proteins were detected using western blotting. (B) PC‐9 and HCC4011 cells were treated with nonspecific control siRNA or YAP‐specific siRNA and incubated with lazertinib (100 nmol/L) or a combination of lazertinib and ONO7475 (100 nmol/L) for 48 h. The cells were lysed, and the indicated proteins were detected using western blotting. (C) PC‐9 and HCC4011 cells were treated with nonspecific control siRNA or YAP‐specific siRNAs and incubated with lazertinib (100 nmol/L) or a combination of lazertinib and ONO7475 (100 nmol/L) for 72 h. Cell growth was determined using MTT assays. *p < 0.05. (D) PC‐9 and HCC4011 cells were treated with lazertinib (100 nmol/L) or a combination of lazertinib and ONO7475 (100 nmol/L) for 48 h. Proteins were fractured into the nucleus and cytoplasm using NE‐PER™ Nuclear and Cytoplasmic Extraction Reagents. (E) Immunofluorescence showing nuclear localization of YAP in EGFR‐mutant NSCLC cells exposed to lazertinib (100 nmol/L) or a combination of lazertinib and ONO7475 (100 nmol/L) for 48 h. Scale bars, 20 μm. (F) qPCR of MCL‐1 in PC‐9 and HCC4011 cells treated with nonspecific control siRNA or YAP‐specific siRNAs and incubated lazertinib (100 nmol/L) or a combination of lazertinib and ONO7475 (100 nmol/L) for 48 h. *p < 0.05.
3.6. Inhibition of YAP via MCL‐1 downregulation sensitizes EGFR‐mutant NSCLC cells to lazertinib and AXL inhibitor
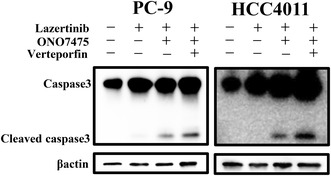
Next, we investigated the potential of verteporfin, a YAP inhibitor, to prevent drug tolerance. Western blot analysis showed that the triple therapy of PC‐9 and HCC4011 cells with verteporfin, lazertinib, and ONO7475 for 48 h inhibited MCL‐1 expression to a greater extent than detected under lazertinib monotherapy or in combination with ONO7475 (Figure 6A). Evaluation of MCL‐1 expression using real‐time PCR showed that treatment with verteporfin, lazertinib, and ONO7475 suppressed the mRNA levels of MCL‐1 compared with the combination of lazertinib and ONO7475 (Figure 6B). Triple therapy with verteporfin, lazertinib, and ONO7475 also reduced the viability of PC‐9 and HCC4011 cells (Figure 6C). Continuous triple therapy with lazertinib, ONO‐7475, and verteporfin induced the eradication of PC‐9 and HCC4011 cells (Figure 6D). Moreover, an increase in this cell viability‐suppressing effect was observed when verteporfin was combined with other EGFR‐TKIs, such as osimertinib, gefitinib, and erlotinib (Figure S7). Western blot analysis revealed enhanced expression of cleaved caspase‐3, indicating the induction of apoptosis through the concurrent administration of verteporfin (Figure 6E). These observations suggest that MCL‐1 might be involved in apoptotic resistance to combination treatment with lazertinib and ONO7475 via the accumulation of YAP in the nucleus (Figure 6F).
FIGURE 6.
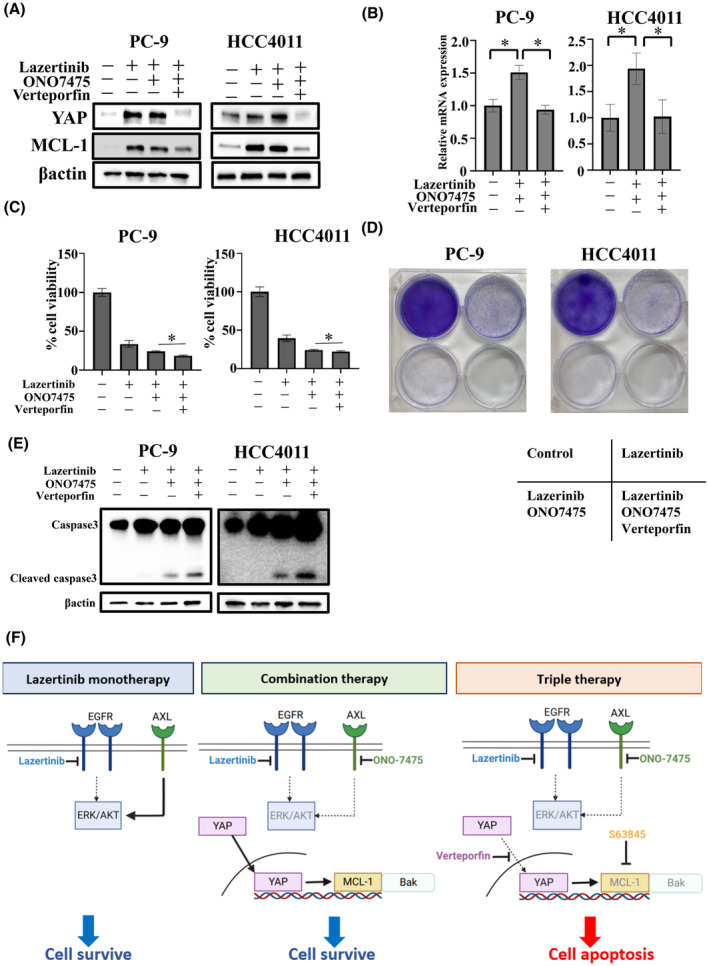
Inhibition of YAP via downregulation of MCL‐1‐sensitized EGFR‐mutant NSCLC cells to lazertinib and AXL inhibitor. (A) PC‐9 and HCC4011 cells were treated with lazertinib (100 nmol/L) with or without ONO7475 (100 nmol/L) and YAP inhibitor verteporfin (1 μmol/L) for 48 h. The cells were lysed, and the indicated proteins were detected using western blotting. (B) qPCR of MCL‐1 in PC‐9 and HCC4011 cells treated with lazertinib (100 nmol/L) and ONO7475 (100 nmol/L) with or without verteporfin (1 μmol/L) for 48 h. *p < 0.05. (C) PC‐9 and HCC4011 cells were treated with lazertinib (100 nmol/L) with or without ONO7475 (100 nmol/L) and verteporfin (1 μmol/L) for 72 h. Cell growth was determined using MTT assays. *p < 0.05. (D) PC‐9 and HCC4011 cells were visualized using crystal violet staining following 9 days of treatment with lazertinib (100 nmol/L), ONO7475 (100 nmol/L), verteporfin (1 μmol/L), or combinations, with the drugs replenished every 72 h. (E) PC‐9 and HCC4011 cells were treated with lazertinib (100 nmol/L) with or without ONO7475 (100 nmol/L) and verteporfin (1 μmol/L) for 48 h. The cells were lysed, and the indicated proteins were detected using western blotting. (F) Schematic presentation of the mechanism by which a combination of lazertinib and ONO7475 promote the YAP–MCL‐1 axis and facilitate the synthesis of antiapoptotic proteins in EGFR‐mutant NSCLC cells.
4. DISCUSSION
Although lung cancer cells harboring EGFR‐activating mutations display good initial responses to various classes of EGFR‐TKIs, a small percentage of cells nevertheless survive and expand under treatment, leading to acquired drug resistance and tumor heterogeneity, ultimately enhancing tumor recurrence. 18 , 19 Numerous studies have investigated the resistance mechanisms for EGFR‐TKIs, including osimertinib, a third‐generation EGFR‐TKI. The reported resistance mechanisms of osimertinib include “on‐target” effects, such as EGFR resistance mutations (EGFR‐C797X, −L792X, −G796X, and ‐S768I mutations), and “off‐target” effects, such as the activation of bypass signals (amplification of MET and HER2, and mutations in KRAS, BRAF, and PIK3CA), epithelial–mesenchymal transition (EMT), and transformation to small‐cell lung cancer and squamous cell carcinoma. 20 Some of the findings from preclinical experiments are currently being translated for clinical applications.
Lazertinib, a novel third‐generation EGFR‐TKI, is a promising treatment that is expected to be clinically developed for patients with EGFR‐mutant NSCLC; however, to date, no studies on its resistance mechanisms have been reported to date and elucidation of this mechanism is crucial for an effective therapeutic response. In this study, we revealed the role of AXL activation as a contributing factor in the development of adaptive resistance to lazertinib in AXL‐high‐expressing EGFR‐mutant NSCLC cells. AXL, a member of the Tyro3‐Axl‐Mer (TAM) receptor tyrosine kinase subfamily, has been identified as an important factor in numerous studies investigating therapeutic resistance in several cancers. 10 , 21 , 22 , 23 AXL also enhances EGFR‐induced signaling by interacting with EGFR and other members of the HER family, such as MET and PDGFR, leading to the development of resistance to EGFR‐TKIs. 10 Approximately 30% of cases exhibit AXL overexpression, which is associated with insensitivity to EGFR‐TKIs including osimertinib. 24 , 25 Our previous studies have shown that treatment of EGFR‐mutant NSCLC cells with EGFR‐TKIs leads to the inhibition of downstream ERK and AKT signaling pathways, thereby inactivating SPRY4, a suppressor of AXL, and facilitating AXL activation and resistance development. 10 In addition, combination with the AXL inhibitor ONO7475 sustained antitumor efficacy in AXL‐high expressing EGFR‐mutant NSCLC cells. 19 Clinical trials are currently underway to validate the safety profile of combination therapy with EGFR‐TKIs, which may be implemented in the future. In the present study, AXL was also identified as a crucial factor contributing to lazertinib resistance, and co‐administration of an AXL inhibitor enhanced the therapeutic efficacy of lazertinib. However, the initial combination of lazertinib and an AXL inhibitor only partially controlled cell proliferation, as the evasion of apoptosis regulated by transcription factors and their associated proteins hindered complete control. Therefore, the induction of apoptosis by direct or indirect suppression of antiapoptotic factors may completely suppress tumor cell survival, leading to tumor eradication.
Cell death signaling is closely related to the regulation of mitochondrial membrane permeability. This regulation involves the BCL‐2 family, which consists of BAX and BAK as pro‐apoptotic factors and BCL‐2, BCL‐XL, and MCL‐1 as antiapoptotic factors. 26 BAX and BAK inhibit mitochondrial outer membrane permeability as pro‐apoptotic factors, whereas MCL‐1 counteracts this effect by binding to BAK and BAX, thereby preventing apoptosis. To develop tumor eradication therapy, we investigated the antiapoptotic mechanisms to prevent complete eradication in AXL‐high‐expressing EGFR‐mutant NSCLC cells. As for MCL‐1, previous studies have shown that MCL‐1 expression is involved in drug resistance in various cancer types. 27 , 28 It has been reported that the interaction between MCL‐1 and BAK is related to the initial resistance to EGFR‐TKIs in EGFR‐positive lung cancer cells. 29 In the current study, we found that, when treated with a combination of lazertinib and an AXL inhibitor, MCL‐1 inhibition resulted in the dissociation of MCL‐1 and BAK, suggesting that the binding between MCL‐1 and BAK plays a crucial role in the apoptosis of high AXL expression EGFR‐mutant NSCLC cells. These observations indicate that the interaction between MCL‐1 and BAK plays an important role in apoptotic resistance in EGFR‐positive lung cancer in EGFR‐TKI monotherapy, as well as combination therapy with AXL inhibitors. In recent studies on lung cancer with driver oncogenes, a therapeutic strategy aimed at eradicating cancer cells proposed several initial combinations of multiple kinase inhibitors for specific tumor populations. 30 , 31 Importantly, we revealed that triple therapy with the MCL‐1 inhibitor lazertinib and an AXL inhibitor enhanced cell viability inhibition, and no repopulation was observed in long‐term cultures of AXL‐high‐expressing EGFR‐mutant NSCLC cells. In addition, the combination of the anaplastic lymphoma kinase (ALK)‐TKI alectinib and an MCL‐1 inhibitor reportedly enhanced tumor apoptosis in ALK‐positive lung cancer cells. 32 Based on these results, MCL‐1 may be a pivotal molecule for overcoming apoptosis resistance in molecularly targeted lung cancer therapeutics. Actually, triple therapy demonstrated significant antitumor effects in the CDX model. While the potential for toxicity exists with triple therapy, no evident adverse events were observed in this preclinical study. In addition, while the safety of lazertinib monotherapy and combination therapy with other agents has been evaluated in clinical trials, 9 there is a paucity of evidence on the safety of ONO‐7475 and S63845 in clinical settings. Additional assessment through clinical trials is imperative to further enhance our understanding.
YAP is a transcriptional co‐activator that shuttles between the nucleus and cytoplasm and reacts to various inputs in the Hippo‐dependent and non‐Hippo‐dependent pathways. To the best of our knowledge, this is the first report of the mechanism by which YAP mediates the upregulation of MCL‐1 expression in EGFR‐mutant NSCLC cells. Additionally, YAP inhibition induces cell apoptosis, similar to MCL‐1 inhibition. A previous study reported the mechanism by which YAP is translocated into the nucleus and the upregulation of its expression through the transcription of antiapoptotic proteins in ALK‐positive lung cancer cell lines. 33 Therefore, besides MCL‐1, YAP inhibition may be a promising targeted therapy for overcoming apoptotic resistance to the combination of lazertinib and AXL inhibitors. Further investigation of its clinical applications is required.
In conclusion, we revealed that AXL activation is involved in adaptive resistance to the novel EGFR‐TKI lazertinib in AXL‐expressing EGFR‐mutant NSCLC cells. The antiapoptotic protein MCL‐1, acting through YAP, is involved in apoptotic resistance to the combination of lazertinib and an AXL inhibitor. A novel triple therapy with an MCL‐1 inhibitor suppressed the viability and increased apoptosis of EGFR‐mutant NSCLC cells. Further development of this therapy is anticipated for clinical application.
AUTHOR CONTRIBUTIONS
Yohei Matsui: Conceptualization; data curation; investigation; software; supervision; visualization; writing – original draft. Tadaaki Yamada: Conceptualization; funding acquisition; methodology; project administration; resources; validation; visualization; writing – review and editing. Yuki Katayama: Conceptualization; methodology; software; validation. Soichi Hirai: Investigation; methodology; software; validation. Ryo Sawada: Investigation; methodology; validation; visualization. Yusuke Tachibana: Conceptualization; investigation; visualization. Masaki Ishida: Investigation; methodology; software; visualization. Hayato Kawachi: Data curation; formal analysis; methodology; software; visualization. Ryota Nakamura: Formal analysis; investigation; software; supervision. Naoya Nishioka: Data curation; investigation; methodology; supervision. Kenji Morimoto: Conceptualization; data curation; formal analysis; investigation; software; supervision; validation; visualization. Masahiro Iwasaku: Formal analysis; software; visualization. Mano Horinaka: Conceptualization; formal analysis; methodology; software; supervision; visualization. Toshiyuki Sakai: Data curation; investigation; methodology; validation; visualization. Shinsaku Tokuda: Data curation; investigation; methodology; software; validation; visualization. Koichi Takayama: Conceptualization; methodology; project administration; resources; supervision.
FUNDING INFORMATION
This work was supported by grants from Janssen Pharmaceutical K.K (to T. Yamada).
CONFLICT OF INTEREST STATEMENT
T. Yamada received speaking honoraria from Eli Lilly. T. Yamada received commercial research grants from Pfizer, Ono Pharmaceutical, Janssen Pharmaceutical K.K., AstraZeneca, and Takeda Pharmaceutical Company Limited. K. Takayama received research grants from Chugai‐Roche Co. and Ono Pharmaceutical Co. and personal fees from AstraZeneca Co., Chugai‐Roche Co., MSD‐Merck Co., Eli Lilly Co., Boehringer‐Ingelheim Co., and Daiichi‐Sankyo Co. The other authors have no conflict of interest.
ETHICS STATEMENTS
Approval of the research protocol by an Institutional Reviewer Board: N/A.
Informed Consent: N/A.
Registry and the Registration No. of the study/trial: N/A.
Animal Studies: Approval for the mouse experiments was obtained from the institutional review board of the University Hospital, Kyoto Prefectural University of Medicine (approval no. M2023‐124) and carried out according to the Animal Experimentation Regulation.
Supporting information
Appendix S1.
Figure S1.
Figure S2.
Figure S3.
Figure S4.
Figure S5.
Figure S6.
Figure S7.
Table S1.
Table S2.
Table S3.
ACKNOWLEDGMENTS
This work was supported by grants from Janssen Pharmaceutical K.K (to T. Yamada). We thank Editage (www.editage.com) for the English language editing. The animations in Figure 6E were created using BioRender.com.
Matsui Y, Yamada T, Katayama Y, et al. Initial AXL and MCL‐1 inhibition contributes to abolishing lazertinib tolerance in EGFR‐mutant lung cancer cells. Cancer Sci. 2024;115:3333‐3345. doi: 10.1111/cas.16292
DATA AVAILABILITY STATEMENT
All raw data are available from the corresponding author upon request.
REFERENCES
- 1. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73:17‐48. [DOI] [PubMed] [Google Scholar]
- 2. Govindan R, Page N, Morgensztern D, et al. Changing epidemiology of small‐cell lung cancer in the United States over the last 30 years: analysis of the surveillance, epidemiologic, and end results database. J Clin Oncol. 2006;24:4539‐4544. [DOI] [PubMed] [Google Scholar]
- 3. Recondo G, Facchinetti F, Olaussen KA, Besse B, Friboulet L. Making the first move in EGFR‐driven or ALK‐driven NSCLC: first‐generation or nextgeneration TKI? Nat Rev Clin Oncol. 2018;15:694‐708. [DOI] [PubMed] [Google Scholar]
- 4. Papadimitrakopoulou VA, Mok TS, Han JY, et al. Osimertinib versus platinum‐pemetrexed for patients with EGFR T790M advanced NSCLC and progression on a prior EGFR‐tyrosine kinase inhibitor: AURA3 overall survival analysis. Ann Oncol. 2020;31:1536‐1544. [DOI] [PubMed] [Google Scholar]
- 5. Ramalingam SS, Vansteenkiste J, Planchard D, et al. Overall survival with osimertinib in untreated, EGFR‐mutated advanced NSCLC. N Engl J Med. 2020;382:41‐50. [DOI] [PubMed] [Google Scholar]
- 6. Soria JC, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR‐mutated advanced non‐small‐cell lung cancer. N Engl J Med. 2018;378:113‐125. [DOI] [PubMed] [Google Scholar]
- 7. Yun J, Hong MH, Kim SY, et al. YH25448, an irreversible EGFR‐TKI with potent intracranial activity in EGFR mutant non‐small cell lung cancer. Clin Cancer Res. 2019;25:2575‐2587. [DOI] [PubMed] [Google Scholar]
- 8. Cho BC, Ahn MJ, Kang JH, et al. Lazertinib versus gefitinib as first‐line treatment in patients with EGFR‐mutated advanced non‐small‐cell lung cancer: results from LASER301. J Clin Oncol. 2023;41:4208‐4217. [DOI] [PubMed] [Google Scholar]
- 9. Cho BC. Amivantamab plus lazertinib vs osimertinib as first‐line treatment in patients with EGFR‐mutated, advanced non‐small cell lung cancer (NSCLC): primary results from MARIPOSA, a phase III, global, randomized, controlled trial. Presented at: ESMO Congress. 2023. Madrid, Spain, 34, S1306.
- 10. Taniguchi H, Yamada T, Wang R, et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat Commun. 2019;10:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nakamura R, Fujii H, Yamada T, et al. Analysis of tumor heterogeneity through AXL activation in primary resistance to EGFR tyrosine kinase inhibitors. JTO Clin Res Rep. 2023;4:100525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) method. Methods. 2001;25:402‐408. [DOI] [PubMed] [Google Scholar]
- 13. Kotschy A, Szlavik Z, Murray J, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538:477‐482. [DOI] [PubMed] [Google Scholar]
- 14. Banerjee K, Resat H. Constitutive activation of STAT3 in breast cancer cells: a review. Int J Cancer. 2016;138:2570‐2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kodama Y, Taura K, Miura K, Schnabl B, Osawa Y, Brenner DA. Antiapoptotic effect of c‐Jun N‐terminal Kinase‐1 through mcl‐1 stabilization in TNF‐induced hepatocyte apoptosis. Gastroenterology. 2009;136:1423‐1434. [DOI] [PubMed] [Google Scholar]
- 16. Fan F, Tonon G, Bashari MH, et al. Targeting mcl‐1 for multiple myeloma (MM) therapy: drug‐induced generation of mcl‐1 fragment mcl‐1(128‐350) triggers MM cell death via c‐Jun upregulation. Cancer Lett. 2014;343:286‐294. [DOI] [PubMed] [Google Scholar]
- 17. Dupont S, Morsut L, Aragona M, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179‐183. [DOI] [PubMed] [Google Scholar]
- 18. Takeuchi S, Yano S. Clinical significance of epidermal growth factor receptor tyrosine kinase inhibitors: sensitivity and resistance. Respir Investig. 2014;52:348‐356. [DOI] [PubMed] [Google Scholar]
- 19. Okura N, Nishioka N, Yamada T, et al. ONO‐7475, a novel AXL inhibitor, suppresses the adaptive resistance to initial EGFR‐TKI treatment in EGFR‐mutated non‐small cell lung cancer. Clin Cancer Res. 2020;26:2244‐2256. [DOI] [PubMed] [Google Scholar]
- 20. Blaquier JB, Ortiz‐Cuaran S, Ricciuti B, et al. Tackling osimertinib resistance in EGFR mutant non‐small cell lung cancer. Clin Cancer Res. 2023;29:3579‐3591. [DOI] [PubMed] [Google Scholar]
- 21. Yan D, Earp HS, DeRyckere D, Graham DK. Targeting MERTK and AXL in EGFR mutant non‐small cell lung cancer. Cancers (Basel). 2021;13:5639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Terry S, Dalban C, Rioux‐Leclercq N, et al. Association of AXL and PD‐L1 expression with clinical outcomes in patients with advanced renal cell carcinoma treated with PD‐1 blockade. Clin Cancer Res. 2021;27:6749‐6760. [DOI] [PubMed] [Google Scholar]
- 23. Abdel‐Rahman WM, Al‐Khayyal NA, Nair VA, Aravind SR, Saber‐Ayad M. Role of AXL in invasion and drug resistance of colon and breast cancer cells and its association with p53 alterations. World J Gastroenterol. 2017;23:3440‐3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Iida S, Miki Y, Suzuki T, et al. Activation of AXL and antitumor effects of a monoclonal antibody to AXL in lung adenocarcinoma. Anticancer Res. 2014;34:1821‐1827. [PubMed] [Google Scholar]
- 25. Cao B, Liu M, Wang L, et al. Remodelling of tumour microenvironment by microwave ablation potentiates immunotherapy of AXL‐specific CAR T cells against non‐small cell lung cancer. Nat Commun. 2022;13:6203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tsujimoto Y, Shimizu S. Bcl‐2 family: life‐or‐death switch. FEBS Lett. 2000;466:6‐10. [DOI] [PubMed] [Google Scholar]
- 27. Song L, Coppola D, Livingston S, Cress D, Haura EB. Mcl‐1 regulates survival and sensitivity to diverse apoptotic stimuli in human non‐small cell lung cancer cells. Cancer Biol Ther. 2005;4:267‐276. [DOI] [PubMed] [Google Scholar]
- 28. Williams MM, Cook RS. Bcl‐2 family proteins in breast development and cancer: could mcl‐1 targeting overcome therapeutic resistance? Oncotarget. 2015;6:3519‐3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Song KA, Hosono Y, Turner C, et al. Increased synthesis of MCL‐1 protein underlies initial survival of EGFR‐mutant lung cancer to EGFR inhibitors and provides a novel drug target. Clin Cancer Res. 2018;24:5658‐5672. [DOI] [PubMed] [Google Scholar]
- 30. Kurppa KJ, Liu Y, To C, et al. Treatment‐induced tumor dormancy through YAP‐mediated transcriptional reprogramming of the apoptotic pathway. Cancer Cell. 2020;37:104‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fernandes Neto JM, Nadal E, Bosdriesz E, et al. Multiple low dose therapy as an effective strategy to treat EGFR inhibitor‐resistant NSCLC tumours. Nat Commun. 2020;11:3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tanimoto A, Matsumoto S, Takeuchi S, et al. Proteasome inhibition overcomes ALK‐TKI resistance in ALK‐rearranged/TP53‐mutant NSCLC via Noxa expression. Clin Cancer Res. 2021;27:1410‐1420. [DOI] [PubMed] [Google Scholar]
- 33. Tsuji T, Ozasa H, Aoki W, et al. YAP1 mediates survival of ALK‐rearranged lung cancer cells treated with alectinib via pro‐apoptotic protein regulation. Nat Commun. 2020;11:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1.
Figure S1.
Figure S2.
Figure S3.
Figure S4.
Figure S5.
Figure S6.
Figure S7.
Table S1.
Table S2.
Table S3.
Data Availability Statement
All raw data are available from the corresponding author upon request.