

Abstract
The unfolded protein response (UPR) detects and mitigates the harmful effects of dysregulated endoplasmic reticulum (ER) function. The UPR has been best characterized as a protein quality control response, and the sole UPR sensor in yeast, Ire1, is known to detect misfolded ER proteins. However, recent work suggests the UPR can also sense diverse defects within the ER membrane, including increased fatty acid saturation and altered phospholipid abundance. These and other lipid-related stimuli have been referred to as lipid bilayer stress and may be sensed independently through Ire1’s transmembrane domain. Here, we show that the loss of Isc1, a phospholipase that catabolizes complex ceramides, causes UPR induction, even in the absence of exogenous stress. A series of chemical and genetic approaches identified a requirement for very long-chain fatty acid (VLCFA)-containing phytoceramides for UPR induction. In parallel, comprehensive lipidomics analyses identified large increases in the abundance of specific VLCFA-containing phytoceramides in the isc1Δ mutant. We failed to identify evidence of an accompanying defect in protein quality control or ER-associated protein degradation. These results extend our understanding of lipid bilayer stress in the UPR and provide a foundation for mechanistic investigation of this fascinating intersection between ceramide metabolism, membrane homeostasis, and the UPR.
The unfolded protein response (UPR) has been traditionally viewed as a protein quality control response, but recent evidence suggests that it also responds to defects in the endoplasmic reticulum membrane.
The authors show that loss of Isc1, a phospholipase for complex ceramides, induces the UPR and this induction is mediated by very long chain fatty acid-containing phytoceramides.
These results support the notion of this new mode of UPR signaling and provide a mechanistic foundation for further study of the intersection between ceramides, membrane homeostasis, and the UPR.
INTRODUCTION
The unfolded protein response (UPR) is a broadly acting stress response that monitors for defects in the function of the endoplasmic reticulum (ER) and orchestrates an adaptive cellular program to mitigate the toxic effects of ER stress (Shamu and Walter, 1996; Cox et al., 1997; Kimata et al., 2003; Adams et al., 2019). The UPR has been best characterized in yeast, although it is broadly conserved from yeast to humans. In yeast, the central sensor of ER stress is Ire1, an ER resident single-pass transmembrane protein with dual kinase and endoribonuclease activities present within its large C-terminal cytoplasmic domain. Activation of Ire1 triggers dimerization and autophosphorylation of Ire1, which then activates its endoribonuclease activity to perform the unconventional cytoplasmic splicing of mRNA for the transcription factor HAC1 (XBP1 in mammals). HAC1’s 3′-intron normally represses its own expression by folding back onto and obscuring its 5′-untranslated region (UTR), thereby preventing translation. Once activated and translated, Hac1 translocates to the nucleus, where it drives a complex transcriptional program involving hundreds of genes (Cox and Walter, 1996; Travers et al., 2000). Many of its targets include regulators of ER protein quality control, such as ER chaperones and components of the ER-associated protein degradation (ERAD) machinery. Interestingly, the UPR also induces the expression of various genes involved in lipid metabolism.
Traditionally, ER stress has come to be viewed as almost synonymous with ER proteotoxic stress. And, indeed, Ire1 possesses a luminal domain that is thought to monitor for misfolded or unfolded proteins within the ER (Promlek et al., 2011). Furthermore, many of the canonical chemical inducers of the UPR are thought to affect protein folding, including tunicamycin (which interferes with glycosylation within the ER) and dithiothreitol (DTT; which reduces disulfide bonds). An unexpected and exciting development in recent years has been the recognition that the UPR may also respond to defects in the ER membrane. By now, many different lipid-associated UPR triggers have been identified, including increased fatty acid saturation (Pineau et al., 2009; Ariyama et al., 2010; Volmer et al., 2013; To et al., 2017; Micoogullari et al., 2020), alterations in the relative abundance of different phospholipid species (Thibault et al., 2012; Shyu et al., 2019), inositol depletion (Promlek et al., 2011), dysregulation of sterol metabolism (Pineau et al., 2009; Thibault et al., 2012; Cohen et al., 2017), and dysregulation of sphingolipid metabolism (Lépine et al., 2011; Tam et al., 2018). Collectively, these and other stress inputs have been referred to as lipid bilayer stress. Recent work suggests that these stressors may be sensed independently from Ire1’s luminal sensing domain. Indeed, these membrane defects may be sensed through Ire1’s own transmembrane domain (Halbleib et al., 2017; Cho et al., 2019). While this new aspect of the UPR is still emerging, and many of its mechanistic details remain uncertain, its recognition has suggested a broader purview of the UPR as a general sensor of ER homeostasis rather than a narrower proteotoxic stress response.
Among the many distinct classes of lipids, ceramides are a diverse and poorly understood group (Figure 1A). Ceramides consist of a C18 sphingosine backbone linked through an amide bond to a fatty acid group, which can be of variable length and saturation (Figure 1B). Phytoceramides are the predominant ceramide species in yeast and are characterized by an additional hydroxyl group within the sphingosine backbone (Figure 1B). More complex ceramides can be generated through modification by polar head groups, yielding species such as inositol-phosphorylceramide (IPC), mannosyl-inositol-phosphorylceramide (MIPC), and mannosyl-diinositol-phosphorylceramide (M(IP)2C; Figure 1C). Ceramides have been implicated in a variety of cellular processes, including protein trafficking, plasma-membrane function, heat-shock response, autophagy, cell-cycle regulation, and ER-associated degradation (Obeid et al., 1993; Jenkins et al., 1997; Spassieva et al., 2009; Epstein et al., 2012; Rego et al., 2012; Uemura et al., 2014; Rodriguez-Gallardo et al., 2020; Hwang et al., 2023).
FIGURE 1:

Schematic diagram of sphingolipid biosynthesis in S. cerevisiae. (A) Detailed enzymatic pathway of sphingolipid synthesis. Steps inhibited by myriocin, Aureobasidin A, and Fumonisin B are indicated. CoA, coenzyme A; -OH, hydroxyl. Figure adapted from (Dickson, 2010). (B) Generic structure of a phytoceramide. The length and saturation of the fatty acyl moiety are variable. Dihydroceramides lack the hydroxyl group shown in purple font. The hydroxyl group (OH) indicated in red font represents the side of attachment of polar head groups in complex ceramides (see panel C). (C) Structures of the indicated complex ceramides in yeast. Figure adapted from (Yamagata et al., 2013).
Isc1 is the inositol phosphosphingolipid phospholipase C that hydrolyzes IPC, MIPC, and M(IP)2C to yield free ceramides and free polar head groups in yeast. Although these three specific complex ceramides are not present in humans, Isc1 itself is conserved, with its putative human orthologue being sphingomyelin phosphodiesterase 2 (SMPD2; Sawai et al., 2000; Yi et al., 2023). SMPD2 uses a similar phospholipase C-type enzymatic mechanism but acts on sphingomyelins to yield free ceramides and free phospholipid head groups (e.g., choline, ethanolamine). While Isc1’s specific chemical reaction is well understood, its detailed roles in cellular homeostasis remain poorly understood. Loss of Isc1 has been found to result in a number of different phenotypes, including defects in protein trafficking, mitochondrial function, genotoxic stress signaling, chromosome segregation, and decreased chronological lifespan (Almeida et al., 2008; Kitagaki et al., 2009; Teixeira et al., 2016; Rego et al., 2018; Matmati et al., 2020; Balazova et al., 2022), consistent with the diverse cellular distribution and functions of ceramides themselves.
Here, we show that loss of Isc1 induces the UPR. Using a combination of chemical, genetic, and lipidomic approaches, we identify the precise class of lipid mediator responsible for UPR induction in the isc1Δ mutant. UPR stimulation appears to be primarily caused by dysregulated lipid metabolism without evidence of obvious proteotoxicity or a protein quality control defect. These results strongly support the emerging notion of a broader UPR that senses membrane defects as well as misfolded proteins and identifies both an unexpected trigger of ER stress and an unexpected lipid inducer of Ire1.
RESULTS
Loss of Isc1 causes induction of the UPR
To monitor for induction of the UPR, we employed a previously generated reporter construct that consists of four copies of the UPR promoter element (UPRE) fused to the open reading frame of GFP (Figure 2A) (Jonikas et al., 2009). This reporter is induced by canonical small molecule inducers of ER stress, such as tunicamycin and DTT, and induction is entirely dependent on Ire1. We examined UPR induction in the isc1Δ mutant under steady-state conditions (i.e., in the absence of any exogenous stressor). There was strong induction of the UPR which could be observed by flow cytometry (Figure 2B) and by fluorescence microscopy (Figure 2C). To ensure that UPR induction derived specifically from the loss of Isc1 (as opposed to any other unanticipated mutation within the strain), we expressed ISC1 from a low-copy (centromeric) expression vector under the control of its own promoter. Restoration of physiologic ISC1 expression in the isc1Δ strain fully suppressed UPR induction back to wild-type levels (Figure 2D). Similarly, deletion of IRE1 completely abrogated isc1Δ-dependent UPR induction, which could be fully rescued by a low-copy centromeric plasmid expressing IRE1 from its endogenous promoter (Figure 2E). Isc1Δ-dependent UPR induction was also fully abrogated in a hac1Δ mutant (Figure 2F). Thus, UPR induction in isc1Δ appears to represent bona fide Ire1/Hac1-dependent UPR signaling.
FIGURE 2:

Loss of ISC1 constitutively activates the UPR. (A). Schematic of the UPR reporter system. Four copies of the Hac1 recognition sequence (UPRE) are linked to the open reading frame for GFP. The reporter is integrated genomically. (B) Constitutive induction of UPR in the isc1∆ mutant, as determined by flow cytometry. (C) Live cell fluorescence microscopy of WT and isc1∆ cells expressing the UPR reporter. (D) UPR induction in wild-type harboring empty vector (EV), isc1∆ harboring EV, and isc1∆ harboring a centromeric ISC1 plasmid, as determined by flow cytometry. (E) UPR induction in wild-type harboring empty vector (EV), isc1∆ harboring EV, isc1∆ire1∆ harboring EV, and isc1∆ire1∆ harboring centromeric IRE1 plasmid, as determined by flow cytometry. (F) UPR induction in the WT, isc1∆, hac1∆ and isc1∆hac1∆, as determined by flow cytometry. (G) UPR induction in various mutants of sphingolipid metabolism, as determined by flow cytometry. Error bars represent standard deviations from technical triplicates. In panels B, F, and G, asterisks indicate p ≤ 0.001 by two-tailed Student’s t test for comparisons relative to isc1∆; for panels D and E, asterisks indicate p ≤ 0.001 by two-tailed Student’s t test for comparisons relative to isc1∆ + EV. Similar results were obtained in >20 (panel B), three (panel C), eight (panel D), five (panel E), three (panel F), and two (panel G) independent experiments, respectively.
To assess the physiologic relevance of UPR induction in the isc1Δ mutant, we compared the extent of UPR induction to that seen upon loss of the ER chaperone calnexin (cne1Δ), a known inducer of the UPR (Jonikas et al., 2009). The extent of UPR induction was comparable for both isc1Δ and cne1Δ (Supplemental Figure S1A). Next we examined UPR induction by the canonical chemical inducer DTT. Again, the extent of induction in isc1Δ was comparable to that seen with DTT doses up to 1 mM (Supplemental Figure S1B). Finally, we examined the specificity of UPR induction for the isc1Δ mutant, which was highly specific because UPR induction was not observed in six other mutants of sphingolipid metabolism (Figure 2G).
Identification of the UPR inducer in isc1Δ
Isc1’s primary substrates are the complex sphingolipids IPC, MIPC, and M(IP)2C (Figure 1A). To determine whether the accumulation of these species was important for UPR induction, we treated isc1Δ cells with Aureobasidin A, a small molecule inhibitor of Aur1 (Hashida-Okado et al., 1996). Because Aur1 is responsible for the synthesis of IPC, Aureobasidin A treatment is expected to reduce the levels of these complex sphingolipids. We confirmed that Aureobasidin A was able to enter cells as treatment reduced growth rates (Supplemental Figure S2). However, UPR induction persisted in the isc1Δ mutant (Figure 3A), suggesting an alternate basis for UPR stimulation.
FIGURE 3:

Identification of the lipid species mediating UPR induction in the isc1∆ mutant. (A and B) UPR induction in wild-type and isc1∆ cells treated with the indicated concentrations of Aureobasidin A (Aba; panel A) or myriocin (Myr; panel B), as determined by flow cytometry. Error bars represent standard deviations from triplicates. ***, p ≤ 0.001, two tailed Student’s t test for comparison of treated cells relative to control. Drug treatments were for 4 h. (C–E) UPR induction in the indicated single and double mutants, as determined by flow cytometry. Error bars represent standard deviations from technical triplicates. ***, p ≤ 0.001, two tailed Student’s t test for comparison relative to isc1Δ. Similar results were obtained in two (panel A), four (panel B), five (panel C), three (panel D), and five (panel E) independent experiments, respectively.
Next, we examined the effect of myriocin, a small molecule that acts at the beginning of the sphingolipid synthesis pathway by inhibiting serine palmitoyltransferase, which is rate-determining for the pathway (Figure 1A; Miyake et al., 1995). Myriocin treatment resulted in essentially complete abrogation of UPR induction in the isc1Δ mutant (Figure 3B). Together, these results suggest that a lipid mediator downstream of serine palmitoyltransferase, but upstream of Aur1 (Figure 1A), mediates UPR induction in the isc1Δ mutant.
To further narrow the search, we switched to a genetic approach. We knocked out SUR2, which is responsible for the generation of phytosphingosine (Figure 1A), in the isc1Δ background. This resulted in a near-total abrogation of UPR induction (Figure 3C), suggesting that the UPR inducer was downstream of Sur2. We then knocked out the sphingolipid hydroxylase, SCS7, which functions downstream of SUR2. Here, UPR induction was maintained in the isc1Δscs7Δ double mutant (Figure 3D), suggesting that the active species was a phytosphingosine, dihydroceramide, or phytoceramide, and not a hydroxylated ceramide species.
Two ceramide classes can be synthesized from dihydrosphingosine or phytosphingosine, respectively, by the addition of a free fatty acid, which can be of variable length (Figure 1A). Very long chain fatty acids (VLCFA; C>20) are unabundant and poorly understood species (Erdbrügger and Fröhlich, 2021) but are components of some important ceramide species. These VLCFAs are synthesized from shorter-chain fatty acids through an enzymatic cycle that includes Elo3 (Figure 1A; Oh et al., 1997; Tehlivets et al., 2007; Kihara, 2012). We, therefore, knocked out ELO3 in the isc1Δ background, which abrogated UPR induction (Figure 3E) and, therefore, suggested that the critical mediator contained a VLCFA. Interestingly, the elo3Δ mutant alone showed UPR induction (Figure 3E), consistent with earlier reports (Jonikas et al., 2009), although we did not study this further.
Dihydroceramides can be converted to dihydrosphingosines through the action of the dihydroceramidase Ydc1 (Figure 1). Similarly, the ceramidase Ypc1 can return phytoceramides to phytosphingosine (Figure 1). We, therefore, overexpressed either Ydc1 or Ypc1 in the isc1Δ background and monitored UPR induction. Strikingly, Ypc1 overexpression strongly abrogated UPR induction (Figure 4A), while Ydc1 had no effect (Figure 4B), specifically implicating phytoceramides in isc1Δ-dependent UPR induction. To further substantiate this result, we supplemented cells with the phytoceramide precursor, phytosphingosine (Figure 1). This resulted in a modest but reproducible increase in UPR signaling in the isc1Δ mutant (Figure 4C). Conversely, when we chemically inhibited the ceramide synthase complex using Fumonisin B (Figure 1A), we saw a reduction in UPR signaling (Figure 4D). Altogether, these results strongly suggest that the critical UPR inducer upon loss of Isc1 is a VLCFA-containing phytoceramide species.
FIGURE 4:

Identification of phytoceramides as critical UPR inducers in the isc1∆ mutant. (A and B) Induction of the UPR in strains lacking or containing overexpression plasmids for YPC1 (panel A) or YDC1 (panel B), as determined by flow cytometry. Error bars represent standard deviations from technical triplicates. ***, p ≤ 0.001, two tailed Student’s t test for comparison relative to isc1∆ harboring an empty vector (EV) plasmid. (C and D) UPR induction of WT and isc1∆ cells treated with 50 µM phytosphingosine (PHS; panel C) or 100 µM Fumonisin B (Fum B; panel D), as determined by flow cytometry. Error bars represent standard deviations from technical triplicates. ***, p ≤ 0.001, two tailed Student’s t test for comparison of treated cells relative to control. Similar results were obtained in three (panels A–C) and five (panel D) independent experiments, respectively.
Lipidomic analysis of the isc1Δ mutant
To directly measure the abundance of various lipid species, we purified total lipids from wild-type and isc1Δ cells and compared their lipidomic profiles using a comprehensive mass spectrometry-based approach (Aluri et al., 2021; Bonney and Prentice, 2021; Hossain et al., 2024). Only a handful of lipid species showed statistically significant differences between the two strains (Supplemental Table S1). Remarkably, three of the species showing increased relative abundance in isc1Δ cells corresponded to VLCFA-containing ceramide species (C42:0;3, C44:0;3, and C46:0;3; Figure 5A). These species are compatible with VLCFA-containing phytoceramides. In principle, these species could also represent hydroxylated dihydroceramides, although the functional data in Figures 3 and 4 argue against that assignment. These species were strongly increased (5–10-fold) relative to wild-type (Figure 5B). In contrast, the corresponding dihydroceramide species showed no change relative to wild-type (Figure 5B), again consistent with the genetic and chemical analyses. Thus, UPR induction in the isc1Δ mutant is likely attributable to the accumulation of one or more VLCFA-containing phytoceramides. We sought to determine whether such a species was sufficient to induce the UPR in wild-type cells but could not achieve cell permeability under any conditions due to the extreme hydrophobicity of this lipid class.
FIGURE 5:

Increased abundance of specific phytoceramide species in the isc1∆ mutant. (A) Volcano plot analysis of comprehensive lipidomic profiles from wild-type and isc1∆ cells. The mean intensities of the m/z values ± 0.005 from triplicates are shown normalize to the wild-type values. Species showing increased abundance in the isc1∆ mutant are present on the right side of the graph, with species showing decreased abundance present to the left. Statistical significance, as determined by p < 0.005 is shown on the vertical access. Other lipid species identified above the cutoff are DG44:2, DG44:1, DG 44:0, DG 42:2. Lipid species with m/z values above the cutoff (p < 0.005) were annotated based on the LIPID MAPS Lipidomics Gateway (Wellcome Trust) Database. (B) Relative abundance of the indicated phytoceramide and dihydroceramide species are shown normalized to their wild-type abundance. Error bars represent standard deviations from technical triplicates. ***, p ≤ 0.001 by two-tailed student’s t test comparison relative to wild-type cells. Similar results were obtained in two independent experiments.
Several of the remaining upregulated species in isc1Δ (Figure 5A) corresponded to neutral lipids, particularly diacylglycerol species. We did not study this finding further, although it is worth noting that a previous study identified increased phosphatidylglycerol phospholipase (Pgc1) activity in the isc1∆ mutant (Balazova et al., 2022), which could account for this result.
Lack of evidence of proteotoxic stress in the isc1Δ mutant
It remains unclear whether the proteotoxic stress and lipid bilayer stress arms of the UPR are truly independent or whether there might be functional overlap between these stressors. For example, defects in the membrane could compromise the ERAD pathway, which destroys misfolded ER proteins and which itself requires the function of many membrane-bound proteins including Hrd1, the putative retrotranslocon for misfolded ER proteins (Huyer et al., 2004; Schoebel et al., 2017; Vasic et al., 2020). Furthermore, VLCFA-containing ceramides have been specifically implicated in ERAD function (Hwang et al., 2023). Thus, we sought to determine whether isc1Δ showed evidence of an overwhelmed or otherwise defective ERAD pathway. To do this, we expressed CPY* in wild-type and isc1Δ cells. CPY* is a widely studied misfolded protein that is recognized by the ER quality control machinery and rapidly destroyed by ERAD (Wolf and Fink, 1975; Stolz and Wolf, 2012). There was no evidence of a degradation defect in isc1Δ (Figure 6A).
FIGURE 6:

Lack of evidence for a protein quality control defect in isc1Δ mutant. (A) Cycloheximide chase analysis of CPY* turnover in wild-type and isc1Δ as determined by SDS–PAGE followed by immunoblotting with anti-HA antibody (upper panel) or anti-Hog1 antibody (lower panel; loading control). (B) Analysis of Gas1 maturation in wild-type and isc1Δ as determined by SDS–PAGE, subjected to immunoblotting with anti-HA antibody (upper panel) or anti-GAPDH antibody (lower panel; loading control). Eri1 is involved in Gas1 protein maturation process, and eri1Δ mutant serves as a control. The mature form of Gas1 is ∼125kDA and the precursor form of Gas1 is ∼105kDA. (C) Viability of the indicated strains upon expression of a toxin ricin protein that requires normal ERAD function for its toxicity. Cells were spotted in threefold serial dilutions and grown at 30°C for 2–6 d. Expression is stimulated in galactose-containing media and repressed in glucose-containing media. Hrd1 is required for ERAD and serves as an internal control. Similar results were obtained in three (panel A) and two (panel B and C) independent experiments, respectively.
As a second test of this model, we employed a modified Ricin A protein (Li et al., 2010). This protein is normally taken up by endocytosis followed by retrograde transport through the secretory pathway to the ER where it then retrotranslocates via the ERAD machinery into the cytoplasm and can then exert its toxic cellular effects. To directly target this protein to the ER during protein synthesis, an N-terminal signal sequence has been added. Under conditions of adequate ERAD function, the expression of this protein is lethal to cells. Various mutants with defects in the ERAD pathway, including the hrd1Δ mutant, prevent the toxic protein from reaching the cytoplasm and, therefore, result in increased cell viability (Li et al., 2010). We expressed the toxic ricin protein in isc1Δ cells and observed no increase in cell survival (Figure 6B).
Defects in the biogenesis of glycosylphosphatidylinositol (GPI)-anchored proteins, such as Gas1, can lead to proteotoxic stress (Castillon et al., 2011). We, therefore, monitored biogenesis of Gas1 by immunoblotting. In wild-type cells, Gas1 was present primarily as the mature (∼125 kDa) protein, while in the eri1Δ mutant, which is known to be defective in Gas1 biogenesis, the abundance of immature Gas1 protein (105 kDa) was increased, as previously described (Sobering et al., 2004). The isc1Δ mutant showed no defect in Gas1 biogenesis (Figure 6C). While these results cannot formally exclude an ERAD defect or a component of proteotoxic stress in the isc1Δ mutant, they support the notion that UPR induction reflects dysregulated lipid metabolism.
Rescue of the isc1Δ mutant by human sphingomyelinase SMPD2
The human sphingomyelinase SMPD2 has been proposed to be the human orthologue of Isc1 (Sawai et al., 2000; Yi et al., 2023). We, therefore, sought to determine whether SMPD2 expression in yeast could revert UPR induction in the isc1Δ mutant. Indeed, plasmid-based expression of SMPD2 almost completely abrogated UPR induction in the isc1Δ background (Figure 7), suggesting that the two enzymes are closely related.
FIGURE 7:
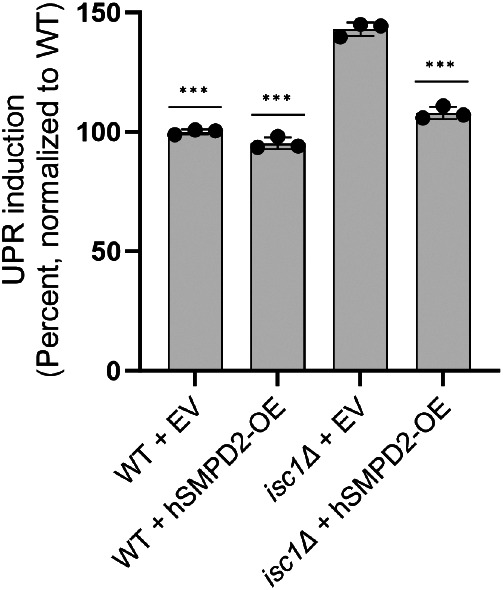
Rescue of isc1Δ by the human sphingomyelinase SMPD2. UPR induction in strains lacking or containing overexpression plasmids for SMPD2, as determined by flow cytometry. Error bars represent standard deviations from technical triplicates. ***, p ≤ 0.001, two tailed Student’s t test for comparison relative to isc1∆ + empty vector (EV) strain. Similar results were obtained in three independent experiments.
DISCUSSION
Despite more traditional conceptions of the UPR as a proteotoxic stress response, a growing body of literature suggests that the UPR may also respond to a wide array of defects in lipid metabolism, including increased fatty acid saturation, changes in the relative abundance of phospholipids, defective cholesterol synthesis, and others (Pineau et al., 2009; Promlek et al., 2011; Volmer et al., 2013; Shyu et al., 2019; Micoogullari et al., 2020). Whether this so-called lipid bilayer stress is truly independent of proteotoxic stress remains an interesting open question. Some evidence suggests that Ire1 can sense lipid-related defects independently of its luminal protein-sensing domain (Promlek et al., 2011; Halbleib et al., 2017, Ho et al., 2020), which tends to support this model. It is also possible that lipid-related defects may themselves cause proteotoxic stress, leading to conceptions of “anticipatory” ER stress signaling (Rutkowski and Hegde, 2010). In most cases of lipid bilayer stress, rigorous assessment of accompanying proteotoxic stress has not been performed, which has contributed to some of the uncertainty in this area and is inevitably complicated by the difficulty in interpretation of negative results.
Here, we show that the loss of a complex ceramide phospholipase C, Isc1, induces the UPR. Several lines of investigation failed to identify an accompanying ER protein quality control defect, although we cannot exclude that possibility based on these data. Our data are distinguished by the precision with which we have identified the specific lipid mediator of UPR induction and by the impressive convergence of the chemical/genetic functional data that established a requirement for VLCFA-containing phytoceramide synthesis in UPR signaling and the lipidomic data that showed large increases in abundance of three specific VLCFA-containing phytoceramides in the isc1Δ mutant. Consistent with our results, a prior report also identified an increased abundance of VLCFA-containing phytoceramides in the isc1Δ mutant (Kitagaki et al., 2007).
These data raise several interesting questions regarding this fascinating aspect of UPR signaling. The first concerns the mechanism whereby phytoceramide species accumulate in the isc1Δ mutant. Their apparent central role in UPR induction was surprising because loss of Isc1 would be predicted to deplete some phytoceramide species rather than increase their abundance, as free ceramides are products of Isc1’s enzymatic activity (Figure 1A). One possibility, still speculative, is that dysregulation of ceramide metabolism in the isc1Δ mutant leads to compensatory upregulation of de novo ceramide synthesis, thus leading to an indirect accumulation of specific phytoceramides. Membranes are also highly dynamic structures and can respond to alterations in their biophysical properties by changing their compositions (Ernst et al., 2018). A classic example is so-called homeoviscuous adaptation, whereby, cells respond to heat stress by altering the degree of fatty acid saturation to maintain the appropriate level of membrane fluidity (Sinensky, 1974; Ernst et al., 2016). Thus, a second possibility is that membrane defects caused by loss of Isc1 are compensated for by increased abundance of certain phytoceramide species.
Interestingly, some evidence suggests that UPR induction itself can stimulate ceramide synthesis (Epstein et al., 2012), which might further exacerbate the situation in isc1Δ cells. However, the abrogation of UPR induction in the isc1Δ mutant by various chemical or genetic inhibitors of ceramide synthesis strongly supports the notion that increased phytoceramide abundance causes UPR induction in this mutant rather than being a simple outcome of UPR induction.
A second important question concerns the mechanism whereby phytoceramides can induce the UPR. Recent evidence suggests that Ire1 can directly sense defects in the membrane through its own transmembrane domain (Halbleib et al., 2017; Cho et al., 2019). In principle, Ire1 might somehow sense the increased abundance of phytoceramides within the membrane, either directly through the binding of ceramides to Ire1 or indirectly through other changes in the membrane, including alterations in its overall hydrophobicity, fluidity, or stiffness. Because the implicated phytoceramide species contain very long chain fatty acids, they might also alter the length or curvature of the membrane. To date, our efforts to demonstrate the sufficiency of individual purified phytoceramides for UPR induction have been thwarted by one of the key biophysical properties of phytoceramides, their extreme hydrophobicity, which prevents their cellular uptake in yeast. Similarly, our attempts to identify a putative phytoceramide binding site within Ire1’s transmembrane domain have been hampered by the importance of the transmembrane domain for overall Ire1 function, and to date, we have not been able to develop a mutant that shows specific abrogation of UPR induction in the isc1Δ background without also destroying more general proteotoxic stress signaling. Thus, these issues remain important open questions for future work. Higher organisms contain two other UPR sensors in addition to Ire1, and it is worth noting that one of these sensors, ATF6, has been proposed to activate the UPR through direct binding to ceramides (Tam et al., 2018).
The relatively recent recognition that membrane homeostasis falls squarely within the purview of the UPR’s stress sensing program represents an exciting development suggesting a broader basis for the UPR in general ER homeostasis. Our results provide a foundation for a detailed mechanistic investigation into the fascinating intersection between ceramide metabolism, membrane homeostasis, and the UPR.
MATERIALS AND METHODS
Strains and plasmids
Yeast strains and plasmids are listed in Tables 1 and 2. Standard PCR-based techniques were used for strain constructions. YPD medium consisted of 1% yeast extract, 2% Bacto peptone, and 2% dextrose. Synthetic medium consisted of 0.7% Difco yeast nitrogen base supplemented with adenine, uridine, amino acids, and 2% dextrose. For plasmid selection, the relevant amino acids or nucleic acids were omitted. For galactose-inducible expression, 2% galactose was used instead of dextrose.
TABLE 1:
Yeast strains.
| Name | Genotype | Source |
|---|---|---|
| BY4741 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | RGC |
| sTR055 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 isc1::HYG | This study |
| YMY284 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 4XUPRE2-ndegY-GFP::LEU2 | This study |
| YMY315 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 4XUPRE2-ndegY-GFP::LEU2 isc1Δ::HYG | This study |
| sTR022 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 leu2Δ0 4XUPRE2-ndegY-GFP::LEU2 ire1::KAN | This study |
| sTR023 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 4XUPRE2-ndegY-GFP::LEU2 isc1::HYG ire1::KAN | This study |
| sTR177 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 4XUPRE2-ndegY-GFP::LEU2 hac1::KAN | This study |
| sTR178 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 4XUPRE2-ndegY-GFP::LEU2 isc1::KAN hac1::KAN | This study |
| sTR083 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 4XUPRE2-ndegY-GFP::LEU2 sur2::KAN | This study |
| sTR098 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 4XUPRE2-ndegY-GFP::LEU2 sur2::KAN isc1::HYG | This study |
| sTR117 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 4XUPRE2-ndegY-GFP::LEU2 scs7::KAN | This study |
| sTR118 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 4XUPRE2-ndegY-GFP::LEU2 scs7::NAT isc1:: HYG | This study |
| sTR081 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 4XUPRE2-ndegY-GFP::LEU2 elo3::KAN | This study |
| sTR082 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 4XUPRE2-ndegY-GFP::LEU2 elo3::KAN isc1:HYG | This study |
| sTR138 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 4XUPRE2-ndegY-GFP::LEU2 ipt1::KAN | This study |
| sTR139 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 4XUPRE2-ndegY- GFP:: LEU2 ypc1::KAN | This study |
| sTR140 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 4XUPRE2-ndegY-GFP::LEU2 ydc1::KAN | This study |
| sTR141 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 4XUPRE2-ndegY-GFP::LEU2 csg1::KAN | This study |
| sTR142 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 4XUPRE2-ndegY-GFP::LEU2 csh1::KAN | This study |
| sTR143 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 4XUPRE2-ndegY-GFP::LEU2 csg2::KAN | This study |
| sTR171 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 4XUPRE2-ndegY-GFP::LEU2 cne1::NAT | This study |
RGC, Research Genetics Collection (available from Thermo Fisher Scientific).
TABLE 2:
Yeast Plasmids.
| Name | Description | Source |
|---|---|---|
| YCplac33 | Single copy cloning vector (CEN/URA3) | Gietz and Sugino, 1988 |
| pKT10 | 2 μ cloning vector under GAPDH promoter (URA3) | Hwang et al., 2006 |
| pTR001 | Isc1 (in YCplac33) | This study |
| pJH233 | Ire1 (in YCplac33) | Guerra-Moreno et al., 2019 |
| pTR004 | Ypc1 (in pKT10) | This study |
| pTR005 | Ydc1 (in pKT10) | This study |
| pSM1763 | CPY*-HA (CEN/URA3) | Huyer et al., 2004 |
| pCM-HA-Gas1 | N-terminal HA tagging under endogenous promoter | McLellan et al., 2012 |
| pJH209 | PGAL1-ss-RTA-E177A | Li et al., 2010 |
| pTR013 | SMPD2 (in pKT10) | This study |
Drug treatment
Myriocin (#M1177; Sigma) was reconstituted to a 2.5 mg/ml stock concentration in DMSO and used at the indicated concentration. Aureobasidin A (#TA9H97F31429; Sigma,) and Fumonisin B (#62580; Cayman Chemicals) were dissolved in ethanol at 0.5 mg/ml and 11µM, respectively, and used at the indicated concentrations. Phytosphingosine (#860499; Avanti Polar Lipids) was stored at a stock concentration of 5 mM in ethanol and used at 50 µM. To facilitate cellular uptake, Fumonisin B and PHS-treated cells were grown in the presence of 0.5% tergitol (#NP40S; Sigma). Cells were standardized by optical density, grown for 2 h in synthetic media, and then treated with drugs for 4 h.
Flow cytometry
Exponentially growing cultures were prepared for flow cytometry analysis as previously described (Micoogullari et al., 2020). Overnight cultures were diluted in synthetic media to an optical density (OD600) of 0.08 and grown for 6 h, yielding a final optical density of ∼0.2–0.4. Fluorescence was measured using BD LSRFortessa (Becton Dickinson) across 10,000 total events. Data analysis was performed using the Bioconductor FlowCore and ggcyto (Hahne et al., 2009) packages in R. Dead cells were excluded from analysis using propidium iodide (#P4170; Sigma) staining. Median signal intensities were calculated, normalized to wild-type (which was set to 100%), averaged across technical triplicates, and plotted with standard deviations. Statistical analysis was performed using two-tailed student’s t test with the indicated p values.
Fluorescence microscopy
Confocal microscopy was performed on live exponentially growing cells using glass slides and a Zeiss LSM710 confocal microscope equipped with 63X oil-immersion objective. GFP imaging was performed at 483 nM (excitation) and 583 nM (emission). Images were processed using ImageJ (National Institutes of Health).
Lipid extraction
Total lipids were extracted as previously described (Knittelfelder and Kohlwein, 2017). In brief, cultures were grown overnight in YPD, diluted the next day in fresh YPD to a starting of OD600 = 0.2, and grown to log phase (OD600∼0.8). Cells were washed with sterile water and resuspended in CHCl3 (2:1) solution before disruption with glass beads at 3200 rpm at 4°C for 30 min. MgCl2 (0.034%) was added to the suspension and mixed for 10 min at 4°C. Samples were centrifuged, and the aqueous phase was discarded. MeOH/H2O/CHCl3 (48:47:3) was added to the extracts, which were then vortexed and centrifuged. The lower organic phase was mixed with CHCl3/MeOH (2:1) and MeOH/H2O/CHCl3 (48:47:3). Samples were centrifuged, and the aqueous phase was again discarded. The organic lower phase-containing lipid fraction was transferred to a fresh tube. Extracts were desiccated in a vacuum chamber and stored at −80°C. Samples were dissolved in CHCl3/ MeOH (2:1) solution for lipidomic analysis.
Lipid analysis using TIMS-ToF Mass Spectrometer (MS)
The samples were prepared as previously described (Bonney and Prentice, 2021). Briefly, lyophilized samples were resuspended in 0.5 ml methanol/chloroform (2:1, vol/vol), vortexed, and sonicated for 10 min. Samples were centrifuged at 12,000 rpm for 10 min and the supernatant transferred using a glass syringe to HPLC tubes analysis.
Supernatants were analyzed via direct infusion to a timsTOF flex (Bruker Daltonics, Billerica, MA) MS as previously described (Aluri et al., 2021; Hossain et al., 2024). Samples were acquired for ∼1 min with an infusion rate of 5 μl/min. timsControl software v4.1.13 and DataAnalysis software (Bruker Daltonics, Billerica, MA) were used to control the instrument and to perform data analysis, respectively. The instrument was calibrated before data collection and samples were analyzed in triplicate. The instrument was operated in negative ion mode with a capillary voltage of 4500 V, collision energy of 15 eV, and collision RF 1500 Vpp at 200°C. Peak intensities with less than 500 units were excluded from the analysis. Mean intensities of the m/z values ± 0.005 from triplicates were normalize to wild-type values. Lipid species were annotated based on the LIPID MAPS Lipidomics Gateway (Wellcome Trust) Database for m/z values that were above the p value cutoff (p < 0.005).
Cycloheximide chase assay
Cycloheximide chase assay was performed as previously described by (Weisshaar et al., 2017; Jochem et al., 2019). Cells were grown to log phase at 30°C and transferred to 37°C. After 1 h, cycloheximide (C7698; Sigma) was added to a final concentration of 100 μg/ml to inhibit protein synthesis. Cells were normalized by optical density, 1.5 OD600 equivalents were collected, and cells were treated with 2M lithium acetate on ice for 5 min followed by 0.4M NaOH on ice for 5 min. Cell pellets were resuspended in 1X Laemmli loading buffer and boiled for 5 min. Analysis was by standard SDS–PAGE and immunoblotting with anti-Pgk1 (#459250; Invitrogen; 1:1000) and anti-HA peroxidase (#12013819001; Roche; 1:2000) antibodies. Antimouse secondary antibodies (NA931; GE). Detection was by enhanced chemiluminescence.
Phenotypic analysis
Exponentially growing cultures were standardized by optical density, spotted in threefold serial dilutions (starting at OD600 = 0.4) onto glucose- or galactose-containing plates, and incubated at 30°C. Expression of the toxin ricin protein is under the control of the pGAL1 promoter, which is suppressed in the presence of glucose.
Gas1 biogenesis assay
Gas1 maturation assay was performed as previously described (McLellan et al., 2012). Briefly, cells were grown to log phase at 30°C in selective media to maintain the plasmid, pCM-HA-Gas1 (Addgene #38313). Cells were washed with sterile water and lysed with 1M phenylmethylsulfonyl fluoride in ethanol. Cells were disrupted using zirconia beads and incubated at –80°C for 1 h. Samples were dried using a vacuum concentrator before adding 200 µl of 2% sodium dodecyl sulfate in 20 mM Tris-HCl pH 6.8. Samples were boiled for 5 min. Five micrograms of protein were analyzed by 4–12% Bis-Tris Gel electrophoresis (Invitrogen #NP0322BOX), followed by immunoblotting with anti-HA peroxidase (Roche #12013819001; 1:10,000) and anti-GAPDH (Sigma #G9545; 1:4000) antibodies. Antirabbit secondary antibodies (GE #NA934; 1:10,000) were used. Detection was by enhanced chemiluminescence.
Liquid culture growth assay
Cultures were grown overnight in minimal media and diluted the next day in fresh minimal media to a starting of OD600 = 0.1 in the absence or presence of 100 ng/mL Aureobasidin A. Cells were grown at 30°C while shaking continuously at 215 rpm. The optical density of the cultures was measured every 2 h for 16 h.
Supplementary Material
Acknowledgments
We thank Susan Michaelis, Lynne Roberts, and Susan Lindquist for the CPY*, Ricin, and Gas1 plasmids, respectively. Nathalie Y. R. Agar is key opinion leader for Bruker Daltonics, and receives support from Thermo Finnegan and EMD Serono. This work was supported by National Institutes of Health grant RO1-GM135337 (to J.H. and N.A.) and a Capital Award from the Massachusetts Life Sciences Center (to N.A.).
Abbreviations used:
- DTT
dithiothreitol
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- EV
empty vector
- GFP
green fluorescent protein
- GPI
glycosylphosphatidylinositol
- HA
hemagglutinin
- IPC
inositol-phosphorylceramide
- M(IP)2C
mannosyl-diinositol-phosphorylceramide
- MIPC
mannosyl-inositol-phosphorylceramide
- OE
overexpressor
- PHS
phytosphingosine
- UPR
unfolded protein response
- UPRE
UPR promoter element
- UTR
untranslated region
- VLCFA
very long chain fatty acid
- WT
wild-type
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E24-03-0121) on July 18, 2024.
REFERENCES
- Adams CJ, Kopp MC, Larburu N, Nowak PR, Ali MMU (2019). Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front Mol Biosci 6, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida T, Marques M, Mojzita D, Amorim MA, Silva RD, Almeida B, Rodrigues P, Ludovico P, Hohmann S, Moradas-Ferreira P, et al. (2008). Isc1p Plays a Key Role in Hydrogen Peroxide Resistance and Chronological Lifespan through Modulation of Iron Levels and Apoptosis. Mol Biol Cell 19, 865–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aluri KC, Hossain MA, Kanetkar N, Miller BC, Dowgiallo MG, Sivasankar D, Sullivan MR, Manetsch R, Konry T, Ekenseair A, et al. (2021). Cyclic Thiosulfinates as a Novel Class of Disulfide Cleavable Cross-Linkers for Rapid Hydrogel Synthesis. Bioconjug Chem 32, 584–594. [DOI] [PubMed] [Google Scholar]
- Ariyama H, Kono N, Matsuda S, Inoue T, Arai H (2010). Decrease in Membrane Phospholipid Unsaturation Induces Unfolded Protein Response*. J Biol Chem 285, 22027–22035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balazova M, Vesela P, Babelova L, Durisova I, Kanovicova P, Zahumensky J, Malinsky J (2022). Two Different Phospholipases C, Isc1 and Pgc1, Cooperate To Regulate Mitochondrial. Microbiol Spectr 10, e02489–e02522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonney JR, Prentice BM (2021). Perspective on Emerging Mass Spectrometry Technologies for Comprehensive Lipid Structural Elucidation. Anal Chem 93, 6311–6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillon GA, Aguilera-Romero A, Manzano-Lopez J, Epstein S, Kajiwara K, Funato K, Watanabe R, Riezman H, Muñiz M (2011). The yeast p24 complex regulates GPI-anchored protein transport and quality control by monitoring anchor remodeling. Mol Biol Cell 22, 2924–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Stanzione F, Oak A, Kim GH, Yerneni S, Qi L, Sum AK, Chan C (2019). Intrinsic Structural Features of the Human IRE1α Transmembrane Domain Sense Membrane Lipid Saturation. Cell Rep 27, 307–320.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen N, Breker M, Bakunts A, Pesek K, Chas A, Argemí J, Orsi A, Gal L, Chuartzman S, Wigelman Y, et al. (2017). Iron affects Ire1 clustering propensity and the amplitude of endoplasmic reticulum stress signaling. J Cell Sci 130, 3222–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox JS, Chapman RE, Walter P (1997). The unfolded protein response coordinates the production of endoplasmic reticulum protein and endoplasmic reticulum membrane. Mol Biol Cell 8, 1805–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox JS, Walter P (1996). A Novel Mechanism for Regulating Activity of a Transcription Factor That Controls the Unfolded Protein Response. Cell 87, 391–404. [DOI] [PubMed] [Google Scholar]
- Dickson RC (2010). Roles for Sphingolipids in Saccharomyces cerevisiae. Adv Exp Med Biol 688, 217–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein S, Castillon GA, Qin Y, Riezman H (2012). An essential function of sphingolipids in yeast cell division. Mol Microbiol 84, 1018–1032. [DOI] [PubMed] [Google Scholar]
- Erdbrügger P, Fröhlich F (2021). The role of very long chain fatty acids in yeast physiology and human diseases. Biol Chem 402, 25–38. [DOI] [PubMed] [Google Scholar]
- Ernst R, Ballweg S, Levental I (2018). Cellular mechanisms of physicochemical membrane homeostasis. Curr Opin Cell Biol 53, 44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst R, Ejsing CS, Antonny B (2016). Homeoviscous Adaptation and the Regulation of Membrane Lipids. J Mol Biol 428, 4776–4791. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Sugino A (1988). New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74, 527–534. [DOI] [PubMed] [Google Scholar]
- Guerra-Moreno A, Ang J, Welsch H, Jochem M, Hanna J (2019). Regulation of the Unfolded Protein Response in Yeast by Oxidative Stress. FEBS Lett 593, 1080–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahne F, LeMeur N, Brinkman RR, Ellis B, Haaland P, Sarkar D, Spidlen J, Strain E, Gentleman R (2009). flowCore: a Bioconductor package for high throughput flow cytometry. BMC Bioinformatics 10, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbleib K, Pesek K, Covino R, Hofbauer HF, Wunnicke D, Hänelt I, Hummer G, Ernst R (2017). Activation of the Unfolded Protein Response by Lipid Bilayer Stress. Mol Cell 67, 673–684.e8. [DOI] [PubMed] [Google Scholar]
- Hashida-Okado T, Ogawa A, Endo M, Yasumoto R, Takesako K, Kato I (1996). AUR1, a novel gene conferring aureobasidin resistance on Saccharomyces cerevisiae: a study of defective morphologies in Aur1p-depleted cells. Mol Gen Genet 251, 236–244. [DOI] [PubMed] [Google Scholar]
- Ho N, Yap WS, Xu J, Wu H, Koh JH, Goh WWB, George B, Chong SC, Taubert S, Thibault G (2020). Stress sensor Ire1 deploys a divergent transcriptional program in response to lipid bilayer stress. J Cell Biol 219, e201909165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain MA, Sarin R, Donnelly DP, Miller BC, Weiss A, McAlary L, Antonyuk SV, Salisbury JP, Amin J, Conway JB, et al. (2024). Evaluating protein cross-linking as a therapeutic strategy to stabilize SOD1 variants in a mouse model of familial ALS. PLoS Biol 22, e3002462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huyer G, Piluek WF, Fansler Z, Kreft SG, Hochstrasser M, Brodsky JL, Michaelis S (2004). Distinct machinery is required in Saccharomyces cerevisiae for the endoplasmic reticulum-associated degradation of a multispanning membrane protein and a soluble luminal protein. J Biol Chem 279, 38369–38378. [DOI] [PubMed] [Google Scholar]
- Hwang G-W, Ishida Y, Naganuma A (2006). Identification of F-box proteins that are involved in resistance to methylmercury in Saccharomyces cerevisiae. FEBS Lett 580, 6813–6818. [DOI] [PubMed] [Google Scholar]
- Hwang J, Peterson BG, Knupp J, Baldridge RD (2023). The ERAD system is restricted by elevated ceramides. Sci Adv 9, eadd8579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins GM, Richards A, Wahl T, Mao C, Obeid L, Hannun Y (1997). Involvement of Yeast Sphingolipids in the Heat Stress Response of Saccharomyces cerevisiae *. J Biol Chem 272, 32566–32572. [DOI] [PubMed] [Google Scholar]
- Jochem M, Ende L, Isasa M, Ang J, Schnell H, Guerra-Moreno A, Micoogullari Y, Bhanu M, Gygi SP, Hanna J (2019). Targeted Degradation of Glucose Transporters Protects against Arsenic Toxicity. Mol Cell Biol 39, e00559–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonikas MC, Collins SR, Denic V, Oh E, Quan EM, Schmid V, Weibezahn J, Schwappach B, Walter P, Weissman JS, et al. (2009). Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science 323, 1693–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara A (2012). Very long-chain fatty acids: elongation, physiology and related disorders. J Biochem 152, 387–395. [DOI] [PubMed] [Google Scholar]
- Kimata Y, Kimata YI, Shimizu Y, Abe H, Farcasanu IC, Takeuchi M, Rose MD, Kohno K (2003). Genetic Evidence for a Role of BiP/Kar2 That Regulates Ire1 in Response to Accumulation of Unfolded Proteins. Mol Biol Cell 14, 2559–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagaki H, Cowart LA, Matmati N, Montefusco D, Gandy J, de Avalos SV, Novgorodov SA, Zheng J, Obeid LM, Hannun YA (2009). ISC1-dependent Metabolic Adaptation Reveals an Indispensable Role for Mitochondria in Induction of Nuclear Genes during the Diauxic Shift in Saccharomyces cerevisiae*. J Biol Chem 284, 10818–10830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagaki H, Cowart LA, Matmati N, Vaena de Avalos S, Novgorodov SA, Zeidan YH, Bielawski J, Obeid LM, Hannun YA (2007). Isc1 regulates sphingolipid metabolism in yeast mitochondria. BBA - Biomembranes 1768, 2849–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knittelfelder OL, Kohlwein SD (2017). Thin-Layer Chromatography to Separate Phospholipids and Neutral Lipids from Yeast. Cold Spring Harb Protoc 2017, pdb.prot085456. [DOI] [PubMed] [Google Scholar]
- Lépine S, Allegood JC, Park M, Dent P, Milstien S, Spiegel S (2011). Sphingosine-1-phosphate phosphohydrolase-1 regulates ER stress-induced autophagy. Cell Death Differ 18, 350–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Spooner RA, Allen SCH, Guise CP, Ladds G, Schnöder T, Schmitt MJ, Lord JM, Roberts LM (2010). Folding-competent and Folding-defective Forms of Ricin A Chain Have Different Fates after Retrotranslocation from the Endoplasmic Reticulum. Mol Biol Cell 21, 2543–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matmati N, Hassan BH, Ren J, Shamssedine AA, Jeong E, Shariff B, Snider J, Rødkær SV, Chen G, Mohanty BK, et al. (2020). Yeast Sphingolipid Phospholipase Gene ISC1 Regulates the Spindle Checkpoint by a CDC55-Dependent Mechanism. Mol Cell Biol 40, e00340–e00419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLellan CA, Whitesell L, King OD, Lancaster AK, Mazitschek R, Lindquist S (2012). Inhibiting GPI Anchor Biosynthesis in Fungi Stresses the Endoplasmic Reticulum and Enhances Immunogenicity. ACS Chem Biol 7, 1520–1528. [DOI] [PubMed] [Google Scholar]
- Micoogullari Y, Basu SS, Ang J, Weisshaar N, Schmitt ND, Abdelmoula WM, Lopez B, Agar JN, Agar N, Hanna J (2020). Dysregulation of very-long-chain fatty acid metabolism causes membrane saturation and induction of the unfolded protein response. Mol Biol Cell 31, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake Y, Kozutsumi Y, Nakamura S, Fujita T, Kawasaki T (1995). Serine palmitoyltransferase is the primary target of a sphingosine-like immunosuppressant, ISP-1/myriocin. Biochem Biophys Res Commun 211, 396–403. [DOI] [PubMed] [Google Scholar]
- Obeid LM, Linardic CM, Karolak LA, Hannun YA (1993). Programmed Cell Death Induced by Ceramide. Science 259, 1769–1771. [DOI] [PubMed] [Google Scholar]
- Oh C-S, Toke DA, Mandala S, Martin CE (1997). ELO2 and ELO3, Homologues of theSaccharomyces cerevisiae ELO1 Gene, Function in Fatty Acid Elongation and Are Required for Sphingolipid Formation*. J Biol Chem 272, 17376–17384. [DOI] [PubMed] [Google Scholar]
- Pineau L, Colas J, Dupont S, Beney L, Fleurat-Lessard P, Berjeaud J-M, Bergès T, Ferreira T (2009). Lipid-Induced ER Stress: Synergistic Effects of Sterols and Saturated Fatty Acids. Traffic 10, 673–690. [DOI] [PubMed] [Google Scholar]
- Promlek T, Ishiwata-Kimata Y, Shido M, Sakuramoto M, Kohno K, Kimata Y (2011). Membrane aberrancy and unfolded proteins activate the endoplasmic reticulum stress sensor Ire1 in different ways. Mol Biol Cell 22, 3520–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rego A, Cooper KF, Snider J, Hannun YA, Costa V, Côrte-Real M, Chaves SR (2018). Acetic acid induces Sch9p-dependent translocation of Isc1p from the endoplasmic reticulum into mitochondria. Biochimica et biophysica acta. Molecular and cell biology of lipids 1863, 576–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rego A, Costa M, Chaves SR, Matmati N, Pereira H, Sousa MJ, Moradas-Ferreira P, Hannun YA, Costa V, Côrte-Real M (2012). Modulation of Mitochondrial Outer Membrane Permeabilization and Apoptosis by Ceramide Metabolism. PLOS ONE 7, e48571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Gallardo S, Kurokawa K, Sabido-Bozo S, Cortes-Gomez A, Ikeda A, Zoni V, Aguilera-Romero A, Perez-Linero AM, Lopez S, Waga M, et al. (2020). Ceramide chain length–dependent protein sorting into selective endoplasmic reticulum exit sites. Sci Adv 6, eaba8237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski DT, Hegde RS (2010). Regulation of basal cellular physiology by the homeostatic unfolded protein response. J Cell Biol 189, 783–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawai H, Okamoto Y, Luberto C, Mao C, Bielawska A, Domae N, Hannun YA (2000). Identification of ISC1 (YER019w) as Inositol Phosphosphingolipid Phospholipase C inSaccharomyces cerevisiae *. J Biol Chem 275, 39793–39798. [DOI] [PubMed] [Google Scholar]
- Schoebel S, Mi W, Stein A, Ovchinnikov S, Pavlovicz R, DiMaio F, Baker D, Chambers MG, Su H, Li D, et al. (2017). Cryo-EM structure of the protein-conducting ERAD channel Hrd1 in complex with Hrd3. Nature 548, 352–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamu CE, Walter P (1996). Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J 15, 3028–3039. [PMC free article] [PubMed] [Google Scholar]
- Shyu P, Ng BSH, Ho N, Chaw R, Seah YL, Marvalim C, Thibault G (2019). Membrane phospholipid alteration causes chronic ER stress through early degradation of homeostatic ER-resident proteins. Sci Rep 9, 8637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinensky M (1974). Homeoviscous adaptation–a homeostatic process that regulates the viscosity of membrane lipids in Escherichia coli. Proc Natl Acad Sci USA 71, 522–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobering AK, Watanabe R, Romeo MJ, Yan BC, Specht CA, Orlean P, Riezman H, Levin DE (2004). Yeast Ras Regulates the Complex that Catalyzes the First Step in GPI-Anchor Biosynthesis at the ER. Cell 117, 637–648. [DOI] [PubMed] [Google Scholar]
- Spassieva SD, Mullen TD, Townsend DM, Obeid LM (2009). Disruption of ceramide synthesis by CerS2 down-regulation leads to autophagy and the unfolded protein response. Biochem J 424, 273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz A, Wolf DH (2012). Use of CPY* and Its Derivatives to Study Protein Quality Control in Various Cell Compartments. In: Ubiquitin Family Modifiers and the Proteasome: Reviews and Protocols, ed. Dohmen RJ, and Scheffner M, Totowa, NJ: Humana Press, 489–504. [DOI] [PubMed] [Google Scholar]
- Tam AB, Roberts LS, Chandra V, Rivera IG, Nomura DK, Forbes DJ, Niwa M (2018). The UPR Activator ATF6 Responds to Proteotoxic and Lipotoxic Stress by Distinct Mechanisms. Dev Cell 46, 327–343.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tehlivets O, Scheuringer K, Kohlwein SD (2007). Fatty acid synthesis and elongation in yeast. Biochim Biophys Acta 1771, 255–270. [DOI] [PubMed] [Google Scholar]
- Teixeira V, Medeiros TC, Vilaça R, Ferreira J, Moradas-Ferreira P, Costa V (2016). Ceramide signaling targets the PP2A-like protein phosphatase Sit4p to impair vacuolar function, vesicular trafficking and autophagy in Isc1p deficient cells. BBA - Molecular and Cell Biology of Lipids 1861, 21–33. [DOI] [PubMed] [Google Scholar]
- Thibault G, Shui G, Kim W, McAlister GC, Ismail N, Gygi SP, Wenk MR, Ng DTW (2012). The Membrane Stress Response Buffers Lethal Effects of Lipid Disequilibrium by Reprogramming the Protein Homeostasis Network. Mol Cell 48, 16–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To M, Peterson CWH, Roberts MA, Counihan JL, Wu TT, Forster MS, Nomura DK, Olzmann JA (2017). Lipid disequilibrium disrupts ER proteostasis by impairing ERAD substrate glycan trimming and dislocation. Mol Biol Cell 28, 270–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P (2000). Functional and Genomic Analyses Reveal an Essential Coordination between the Unfolded Protein Response and ER-Associated Degradation. Cell 101, 249–258. [DOI] [PubMed] [Google Scholar]
- Uemura S, Shishido F, Tani M, Mochizuki T, Abe F, Inokuchi J (2014). Loss of hydroxyl groups from the ceramide moiety can modify the lateral diffusion of membrane proteins in S. cerevisiae. J Lipid Res 55, 1343–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasic V, Denkert N, Schmidt CC, Riedel D, Stein A, Meinecke M (2020). Hrd1 forms the retrotranslocation pore regulated by auto-ubiquitination and binding of misfolded proteins. Nat Cell Biol 22, 274–281. [DOI] [PubMed] [Google Scholar]
- Volmer R, van der Ploeg K, Ron D (2013). Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc Natl Acad Sci USA 110, 4628–4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisshaar N, Welsch H, Guerra-Moreno A, Hanna J (2017). Phospholipase Lpl1 links lipid droplet function with quality control protein degradation. Mol Biol Cell 28, 716–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf DH, Fink GR (1975). Proteinase C (carboxypeptidase Y) mutant of yeast. J Bacteriol 123, 1150–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata M, Obara K, Kihara A (2013). Unperverted synthesis of complex sphingolipids is essential for cell survival under nitrogen starvation. Genes Cells 18, 650–659. [DOI] [PubMed] [Google Scholar]
- Yi J, Qi B, Yin J, Li R, Chen X, Hu J, Li G, Zhang S, Zhang Y, Yang M (2023). Molecular basis for the catalytic mechanism of human neutral sphingomyelinases 1 (hSMPD2). Nat Commun 14, 7755. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.