
Fig. 4.
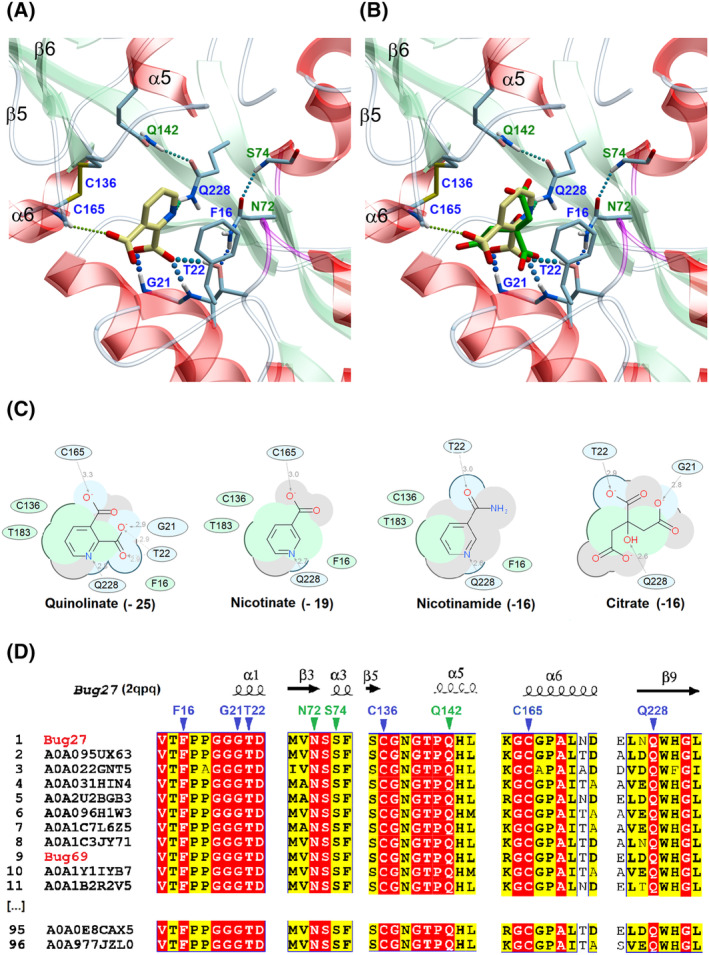
In silico binding analysis of Bug27. (A) Close‐up view of Bordetella pertussis Bug27 (PDB: 2qpq) binding site in ribbon representation with docked Qa (in yellow sticks). Secondary structure elements are color‐coded as follows: light green for β‐sheets, red for α‐helices, and white for loops or turns. Binding residues in direct contact with the ligand (with H‐bonds or hydrophobic interactions) are displayed in sticks with blue labels. Additional residues that indirectly contribute to the ligand stabilization (e.g., by forming an H‐bonding network) are displayed with green labels. H‐bonds are shown as colored dotted lines, while π‐π interactions as black dotted lines. (B) Docked Qa (yellow) overlaid with co‐crystallized citrate molecule (green) from PDB 2qpq. (C) 2D interaction diagram between tested ligands and receptor. Green and blue shading represents hydrophobic and H‐bonding contacts, respectively. Dashed arrows represent hydrogen bonds. The docking score is in brackets. The ICM software docking Score is a Generalized Born Surface Area (GBSA)/Molecular Mechanics (MM)‐type scoring function augmented with a directional hydrogen bonding term [46]. (D) Partial views of the structure‐based sequence alignment of the NAD‐related Bug27/69 TctC subfamily (Blue cluster in Fig. 2C). The alignment was performed using 96 representative sequences out of the 253 by applying an 80% sequence identity cutoff. Numbering and secondary structure elements derive from the structure of B. pertussis Bug27 (PDB: 2qpq). Conserved residues putatively involved in the quinolinate binding site are marked by triangles with the same color coding as panel A.