

Abstract
Prion diseases result from the misfolding of the physiological prion protein (PrPC) to a pathogenic conformation (PrPSc). Compelling evidence indicates that prevention and/or reduction of PrPSc replication are promising therapeutic strategies against prion diseases. However, the existence of different PrPSc conformations (or strains) associated with disease represents a major problem when identifying anti-prion compounds. Efforts to identify strain-specific anti-prion molecules are limited by the lack of biologically relevant high-throughput screening platforms to interrogate compound libraries. Here, we describe adaptations to the protein misfolding cyclic amplification (PMCA) technology (able to faithfully replicate PrPSc strains) that increase its throughput to facilitate the screening of anti-prion molecules. The optimized PMCA platform includes a reduction in sample and reagents, as well as incubation/sonication cycles required to efficiently replicate and detect rodent-adapted and cervid PrPSc strains. The visualization of PMCA products was performed via dot blots, a method that contributed to reduced processing times. These technical changes allowed us to evaluate small molecules with previously reported anti-prion activity. This proof-of-principle screening was evaluated for six rodent-adapted prion strains. Our data show that these compounds targeted either none, all or some PrPSc strains at variable concentrations, demonstrating that this PMCA system is suitable to test compound libraries for putative anti-prion molecules targeting specific PrPSc strains. Further analyses of a small compound library against deer prions demonstrate the potential of this new PMCA format to identify strain-specific anti-prion molecules. The data presented here demonstrate the use of the PMCA technique in the selection of prion strain-specific anti-prion compounds.
Keywords: anti-prion molecules, in vitro screening, prion strains, prions, protein misfolding cyclic amplification (PMCA)
1 |. INTRODUCTION
The cellular prion protein (PrPC) is highly conserved in mammals.1 PrPC is present in various tissues but features most prominently in the brain, spinal cord, and lymphoid organs.2 Remarkably, PrPC can misfold into an aggregation-prone conformation (PrPSc) that is able to self-propagate, leading to invariably fatal neurodegenerative disorders known as transmissible spongiform encephalopathies (TSEs), or prion diseases.3 Prion diseases are associated with genetic, acquired, and sporadic origins.4–6 Although rare in humans, these diseases may exhibit notable incidence in economically relevant livestock such as cattle (bovine spongiform encephalopathy, BSE)7,8 sheep and goats (scrapie),9 and cervids (chronic wasting disease, CWD).10–12 At present, only CWD has been observed beyond captive environments13; and in turn, has attracted considerable attention from the scientific and non-scientific communities alike due to its unclear potential for transmission to humans.14–19 Considering the growing geographical distribution of CWD, as well as the fatal and economic impact of the BSE epidemic that occurred at the end of the last century, major investigative and regulatory efforts have sought to expand knowledge of prion disease mechanisms and improve surveillance procedures.20 Unfortunately, despite several efforts, all prion diseases are without a known cure.
Numerous research laboratories have explored different strategies to prevent or delay the clinical onset of prion diseases. Many of these approaches have been tested using in vitro systems or animal models. Several reports describe extension of the expected incubation periods but not total remission of the disease when animals are exposed to high doses of the infectious agent.21–28 Therapeutic strategies investigated for TSEs can be broadly classified into five categories, as follows: (i) inhibition of prion replication,29–31 (ii) modulation of cell signaling pathways,32,33 (iii) PrPC competition/depletion approaches,31,34 (iv) immunizations,35 and (v) molecular chaperones.36,37 Some of these experimental approaches have demonstrated a delay in disease progression,30,38 and some prophylactic treatments offer partial protection against the infectious agent.39–41 Other approaches have suggested suppression of prion replication as a relevant therapeutic avenue in prion diseases.42 Nonetheless, it is important to note that most of these treatments have been tested on laboratory-adapted rodent prions; and thus, their relevance to naturally occurring animal or human prion diseases is uncertain.
Efforts to develop therapeutic strategies against prion diseases must consider the mechanisms underlying the array of clinicopathological manifestations observed within and between species. Compelling evidence indicates that the wide phenotypical variability of prion diseases arises from distinct conformational variants or “strains” of PrPSc.43,44 Indeed, prion diseases are shown to be best understood as a function of the associated PrPSc strain(s), where inter-species or inter-polymorphic transmission, or spontaneous emergence, can result in the adaptation of unique or more than one variety of prions displaying a diverse array of clinical manifestation and pathological features.45 Given the general presumption that different prion strains arise from distinct conformations acquired by PrPSc,43,44,46,47 a therapeutic approach that shows promise against one prion strain may not prove effective against another.48
Animal models of prion diseases have been crucial to advancing therapeutic efforts; however, the associated maintenance costs and the long incubation periods make bioassays largely impractical to test a sufficient range of compounds against the expanding list of known PrPSc strains. To address this issue, we propose that the protein misfolding cyclic amplification (PMCA) technique can be adapted to screen small molecule libraries to identify anti-prion molecules. PMCA is a prion replication technique that replicates infectious prions in an accelerated manner.49 Considering its ultra-sensitive nature, short assay times, and technical simplicity, PMCA is an ideal technique to test small molecule libraries. Importantly, PMCA has been demonstrated to maintain the strain-specific properties of the PrPSc input.50 Therefore, this technique may additionally help to identify strain-specific anti-prion compounds.
The aim of this study was to adapt the PMCA technology for higher throughput, facilitating the screening of small-molecule libraries for strain-specific anti-prion compounds. Specifically, PMCA was adapted to a 96-well plate format for six well characterized rodent prion strains, and one cervid prion isolate. This modified PMCA setup, hereafter referred to as 96wp-PMCA, includes substantial reduction in reagent volumes and processing times. The 96wp-PMCA was first tested with known anti-prion and anti-amyloid molecules. As expected, some of these compounds displayed strain-specific anti-prion activity. Next, we demonstrated the suitability of the 96wp-PMCA to screen a commercially available compound library against chronic wasting disease (CWD) prions, which are responsible for one of the most worrisome animal prion disease at present. Overall, the data presented in this study provides technical innovations on prion replication methods that could be used and adapted for therapeutic and diagnostic efforts.
2 |. METHODS
2.1 |. Preparation of prion infected brain extracts
Brain extracts containing infectious prions were collected from terminally ill rodents inoculated with the following prion strains: RML, ME7, and 301C from mice, and HY, 139H, and SSLOW from Syrian hamsters. Seeds were prepared via the homogenization of prion-infected brain tissues at a concentration of 10% weight/volume (w/v) in phosphate-buffered saline (PBS, Fisher Scientific, Hampton, NH, USA) supplemented with an EDTA-free protein inhibitor cocktail (Roche, Little Falls, NJ, USA). The homogenization was performed using ice-cooled Potter-Elvehjem tissue grinders (DWK Life Sciences, Vineland, NJ, USA). Next, samples were centrifuged at 800 g for 45 s at 4°C, and the resulting supernatants were mixed, aliquoted, and stored at −20°C until ready to be used. A similar procedure was performed to obtain CWD prions-bearing brain extracts. This was done by homogenizing the brain of a terminally ill tg1536 mouse (expressing cervid prion protein51) infected with the brain of an experimentally infected, terminally ill white-tailed deer homozygous for glycine at position 96 of the prion protein.
2.2 |. Preparation of PMCA substrate
Brain homogenates from prion-free wild-type mice, Syrian hamsters or a prnp KO mouse were prepared as previously described.49,52 Briefly, the brains were weighted and homogenized as noted above, PMCA conversion buffer containing 1% Triton-X (Milipore Sigma, Burlington, MA, USA), and 150mM NaCl (Fisher Scientific, Hampton, NH, USA) was used. Homogenates were prepared to a final concentration of 10% (w/v). Next, the homogenates were centrifuged at 800g for 1 min at 4°C, and the resulting supernatant was aliquoted and stored at −80°C until use. For assays including CWD prions, we used PMCA substrate derived from homozygous tg1536 mice51 encoding the cervid PrPC. The tg1536 substrate was further supplemented with digitonin (Thermo Fisher Scientific, Waltham, MA, USA) and EDTA (Roche, Little Falls, NJ, USA) at final concentrations of 0.025% (v/v) and 6mM, respectively.
2.3 |. 96-well plate PMCA (96wp-PMCA)
The PMCA protocol was modified based on established procedures,49 and the modifications are provided in detail in the results section of this manuscript. In summary, the final and optimized protocol is as follows: each well of a 96-well plate (Thomas Scientific, Swedesboro, NJ, USA) was loaded with two teflon bead PTFE Grade Balls 3/32″ (Hoover Precision Products, Cumming, GA, USA). Prions were added to each well at varying concentrations (range: 1 × 10−2 to 1 × 10−4 brain homogenate equivalent after dilution in PMCA substrate). A total volume of 50 μL was used for each reaction. Samples were covered with 12-strip lids (Thomas Scientific, Swedesboro, NJ, USA) prior to being placed in a water bath sonicator (Qsonica, Newtown, CT, USA). The PMCA procedure lasted 24 h and each PMCA round consisted of continuous cycles of 29 min and 40 s of incubation, followed by 20 s of sonication within a 37°C water bath incubator. Additionally, the PMCA reactions were supplemented with varying concentrations of either ethanol (EtOH) or dimethyl sulfoxide (DMSO), along with small molecules dissolved in these solvents at concentrations ranging from 0.1 to 100 μM (dissolved in 1% EtOH or DMSO). Negative controls consisted of PMCA reactions without prion supplementation, whereas positive controls included PMCA reactions supplemented with prions of known seeding activity. Each PMCA reaction described in this study was performed in an inoculum/substrate homologous fashion (e.g., mouse substrate for RML, ME7, and 301C prions; Syrian hamster substrate for HY, 139H, and SSLOW prions; and deer substrate for CWD prions).
2.4 |. Screening of a small molecule library for CWD prions
To demonstrate the suitability of the 96wp-PMCA system for CWD research, brain extracts containing CWD prions were added to each well at a concentration of 1 × 10−4 brain homogenate equivalent diluted in PMCA buffer supplemented with 5 mM EDTA (Roche, Little Falls, NJ, USA) and 0.025% digitonin (Thermo Fisher Scientific, Waltham, MA, USA). These were tested against a small library of small molecules. The library is a commercially available anti-neurodegenerative compound collection (MedChem Express) with many of the compounds having different targets. These include ion channels, different relevant receptors, autophagy, apoptosis, and synaptic integrity, among other relevant processes. Some of the compounds included in this library have also been tested against the aggregation of tau and amyloid beta. Additional information can be found at https://www.medchemexpress.com/screening/neurodegenerative-disease-related-compound-library.html. The test compounds were included at a final concentration of 100 μM.
2.5 |. Proteinase K digestion
PMCA products, or the brain extract of a wild-type mouse (later used as PMCA susbtrate), were treated with proteinase K (PK, Sigma-Aldrich, Saint Louis, MO, USA) at a final concentration of 100 μg/mL prior to transfer to an Eppendorf® thermomixer. PK digestion was conducted by incubation at 37°C with a rotation speed of 450 rpm for 1 h. To halt the PK reaction, 5 mM phenylmethane sulfonyl fluoride (PMSF, Roche, Little Falls, NJ, USA) was added to each well and incubated at 70°C for 10 min. Subsequently, the samples were evaluated for the presence of PK-resistant prions using either dot blot or western blot analyses.
2.6 |. Dot blot analysis
Five μL of PK-digested PMCA-products were directly applied onto a nitrocellulose membrane (GE Healthcare Amersham, Chicago, IL, USA) using a Bio-Dot Apparatus (Bio-Rad Laboratories, Hercules, CA, USA). The membranes were dried using a blow dryer and then transferred to an incubation chamber containing 10 mL of 3 M guanidinium thiocyanate (Chem-impex int’l INC, Wood Dale, IL, USA) for 10 min. Next, membranes were rinsed three times with 50 mL of washing buffer (0.05% Tween (Milipore Sigma, Burlington, MA, US) in PBS) and then transferred to a blocking solution consisting of 5% (w/v) dry non-fat milk (Lab Scientific, Danvers, MA, USA) dissolved in washing buffer. The membranes were blocked for 1 h on a rocking platform at room temperature. Following, membranes were incubated with monoclonal 6D11 anti-prion antibody (1:10 000, Biolegend, San Diego, CA, USA) in 5% (w/v) blocking buffer for 1 h on a rocking platform at room temperature. After three washes (5–10 min each) using washing buffer, membranes were incubated with a horseradish peroxidase-linked polyclonal anti-mouse IgG (whole molecule)—peroxidase antibody produced in sheep (1:3000, Sigma-Aldrich, Saint Louis, MO, USA) diluted in washing buffer for 1 h on a rocking platform at room temperature. The membranes were washed as described for the previous step, and protein bands were visualized using an ECL kit according to the manufacturer’s instructions. Signals were considered positive if they provided densitometric values over 20% of the background (measured in the same membrane).
2.7 |. Western blotting
Western blots were conducted using a similar procedure as dot blot with the difference that PK-digested samples were fractioned in NuPAGE 12% Bis–Tris gels (Invitrogen, Carlsbad, CA, USA) and then transferred to nitrocellulose membranes (GE Healthcare Amersham, Chicago, IL, USA). Details on the western blot procedure can be found in our previous publications.53,54 Membranes were blocked using 5% w/v non-fat milk solution and probed with monoclonal purified 6D11 antibody (Biolegend, San Diego, CA, USA) at a 1:10 000 dilution. Besides the step involving incubation with the guanidinium hydrochloride solution, the rest of the process is as described for dot blotting.
3 |. RESULTS
3.1 |. Optimization of the PMCA technique to increase its throughput
Platforms aiming to screen small molecule libraries should include easy manipulation, short readout times, low reaction volumes, and the ability to simultaneously screen multiple compounds, among other features amicable for the testing of numerous variables. Unfortunately, the conventional PMCA procedure49 does not fully comply with these requirements. For example, PrPSc detection in PMCA is typically detected via western blots.49 To address this limitation, we standardized the use of dot blots as they are faster than western blots and suitable for high throughput, allowing the analysis of a larger number of specimens while using smaller sample volumes.55 Our first objective was to determine the minimum volume of PMCA product required for detection via dot blot procedure. To this end, we used either 3, 5, or 7 μL of proteinase K (PK)-digested brain extract from an RML-infected mouse in a dot blot procedure as described in Methods (Figure S1). These volumes are lower than those used for detection in western blotting (~15–30 μL).49 We found that signals were not detected in all samples when only 3 μL were used (Figure S1A). In contrast, when 5 and 7 μL were tested, consistent PrPSc signals across all samples were observed (Figure S1B,C). These findings were consistent in multiple replicates, demonstrating reproducibility. Together, these data indicated that 5 μL is the minimum sample volume required for reproducible detection of PrPSc via dot blots. Further, because the detection method for the 96wp-PMCA required minimal consumption of samples, this platform is optimal for investigative efforts involving a low quantity of analytes and/or additional quantitative/qualitative readouts.
Another limitation of the conventional PMCA protocol involves the reaction times that include several days of incubation/sonication cycles and multiple rounds.49 To improve the assay for the screening of large compound libraries, we aimed to reduce the time of the PMCA procedure for uniform detection across a 96 well plate format. For that purpose, we tested the amplification of RML prions (1 × 10−4 dilution) in a single PMCA round for either 24 or 48 h. Our results showed that both PMCA times were sufficient for consistent detection of PrPSc amplification across the 96 well plates using dot blots (Figure S2). Therefore, 24 h were chosen to further improve the 96wp-PMCA platform. It is important to mention that multiple 96 well plates from different brands were tested in this experiment. Not all of them were useful in the 96wp-PMCA format, and the one providing the best performance was selected for these and further analyses.
Next, we sought to determine the lowest concentration of PrPSc seeds required for uniform amplification across all positions in the 96-well plate. To this end, we tested four concentrations of RML prions (10−2, 10−3, and 10−4 brain homogenate equivalents) (Figure S3). PMCA products from each dilution were subjected to PK treatment prior to detection using dot blot analyses. We found that the 10−4 dilution was optimal for the detection of RML prions across the 96-well plate (Figure S3 and Figure 1A). Considering this as the highest dilution tested, this parameter was used for further experiments amplifying rodent prions. Additionally, we made other crucial modifications to the PMCA procedure for adaptation to the 96wp-PMCA platform (Table 1). These included the reduction of the number of Teflon beads from 3 to 2 units, and minimization of the total reaction volume per well (from 100 to 50 μL). Collectively, these adjustments allowed the adaptation of the conventional PMCA technique to a 96wp-PMCA format that facilitates the rapid screening of small molecules against various PrPSc strains.
FIGURE 1.

Representative panels of optimized 96wp-PMCA assays. In vitro replication of (A) RML or (B) SSLOW prions. Left panels explain how seeded (black circles) and unseeded (white circles) reactions were positioned across the 96 well plate. Right panels demonstrate actual results. All samples shown in this experiment were PK treated before visualized in dot blots, as described in Methods.
TABLE 1.
Parameters of conventional PMCA compared with those of the optimized 96wp-PMCA.
| PMCA (based on 3 rounds) | 96wp-PMCA (based on 1 round) | |
|---|---|---|
| Platform | PCR tubes | 96-well plate |
| Volume | 100 μL | 50 μL |
| # of teflon beads | 3 beads | 2 beads |
| Sonication time | 6 days | 1 day |
| Volume of sample used for analysis | 19 μL | 5 μL |
| Signal analysis | Western Blot | Dot Blot |
| Total time | 8 days | 2 days |
Using the parameters established for RML prions in the 96wp-PMCA, we evaluated the in vitro amplification performance of two additional mouse-adapted (301C and ME7) and three Syrian hamster-adapted (HY, SSLOW, and 263K) PrPSc strains. Negative controls (no PrPSc seeds) were included to detect possible cross-contamination during the PMCA procedure and sample handling. Following the previous standardization with the RML prions, all prion strains were tested at a 10−4 dilution. This dilution was also selected as no PrPSc signals were detected in these samples before PMCA in western blots (data not shown). Similarly, as observed for RML prions, uniform amplification in all wells containing the rodent-adapted PrPSc strains was achieved (Figure 1B and Figures S4 and S5). Importantly, none of the negative controls displayed PrPSc signals after PMCA, demonstrating the specificity of our assay. These findings indicate that the 96wp-PMCA platform allows the amplification of structurally heterogeneous rodent-adapted PrPSc strains.
3.2 |. Screening of known anti-prion and anti-amyloid molecules against rodent-adapted PrPSc strains to query the fidelity of the 96wp-PMCA system
Solubility is a relevant factor to consider when testing the activity of a potential therapeutic molecule. According to their particular properties, drug candidates can be prepared in a variety of solvents to ensure uniform dispersion. Here, we assessed the performance of 96wp-PMCA in the presence of two common drug solvents, namely dimethyl sulfoxide (DMSO) and ethanol (EtOH). These experiments were performed using different concentrations of these solvents (ranging from 8% to 0.0625% v/v) on 96wp-PMCA reactions seeded with either mouse-adapted RML and 301C strains or the hamster-adapted HY and 263K strains. Our results revealed that PMCA is not affected by the presence of DMSO (Figure S6). EtOH demonstrated some inhibition of PMCA reactions, but only at higher concentrations (4%–8% v/v). Considering these results, a small library of compounds of previously reported anti-prion or anti-amyloid activity were tested after dissolving them in either 1% v/v EtOH or DMSO.
The compounds tested in this screening included astemizole, curcumin, quinacrine dihydrochloride, imatinib, resveratrol, azure A, azure B, azure C, thioflavin, tetracycline, tannic acid, tetradrine, quinacrine mustard, rhodamine and thionine acetate.30,56–67 Most anti-prion molecules were selected based on their effectiveness against RML prions, as previously reported in a cell-based assay.21 On the other hand, the six anti-amyloid compounds have been previously tested against misfolded proteins associated with Parkinson’s and Alzheimer’s diseases (α-synuclein and amyloid-β, respectively).68–70 For the mouse-adapted agents, we found that two anti-prion molecules (Congo Red and tannic acid) showed inhibitory effects against all three PrPSc strains (Figure 2); however, we observed a difference in the lowest inhibitory concentration for Congo Red, but not tannic acid. Specifically, 10 μM of Congo Red were sufficient to inhibit the amplification of RML prions, whereas a higher concentration of this molecule (100 μM) was required to inhibit the replication of the 301C and ME7 strains (Figure 2). Among the six anti-amyloid molecules tested, four inhibited replication of all mouse PrPSc strains, namely, azure A, azure B, azure C, and thionine acetate. The concentrations of azure B and azure C needed to inhibit the replication of RML (Figure 2A) and 301C (Figure 2B) prions varied between 1 and 10 μM when the compound was diluted in DMSO. Interestingly, the concentrations needed to reach anti-prion effect for azure A and azure B in ME7 prions substantially decreased when the compounds were dissolved in EtOH (Figure 2C, Figure 3 and Figure S7).
FIGURE 2.
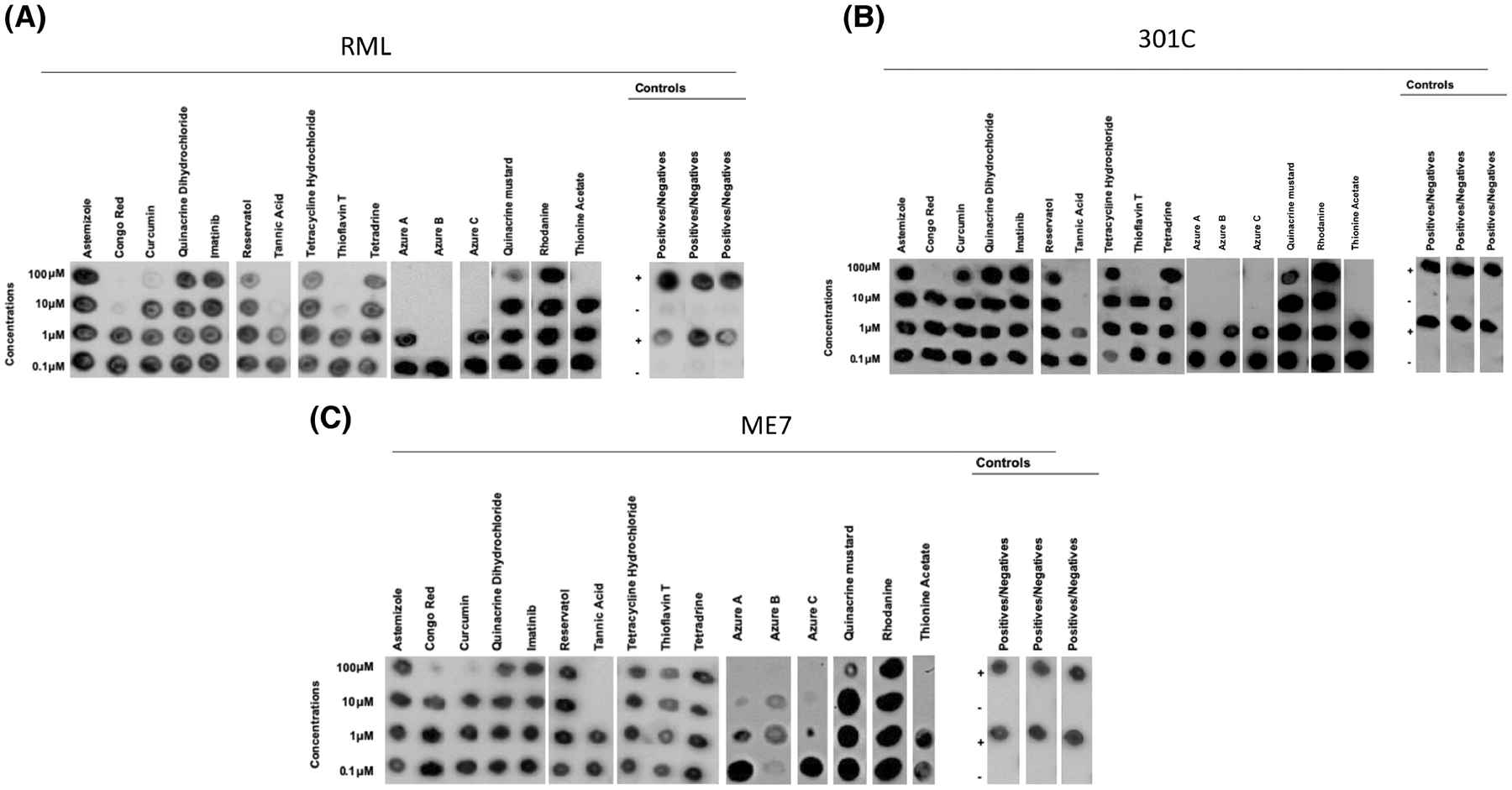
Specific activity of small molecules of known anti-prion and anti-amyloid activities in mouse prion strains. Sixteen small molecules with proven anti-prion or anti-amyloid activities (dissolved in DMSO) were tested at four different concentrations for their anti-prion activities against the RML (A), 301C (B) and ME7 (C) prion strains using the 96wp-PMCA. Each molecule was tested in concentrations ranging from 100 to 0.1 μM. Controls for solvent representative for all strains, positive samples and negative samples without compounds were included to monitor the assay. Dot blots were modified for labeling. The panels shown in this figure are representative from three independent assays.
FIGURE 3.
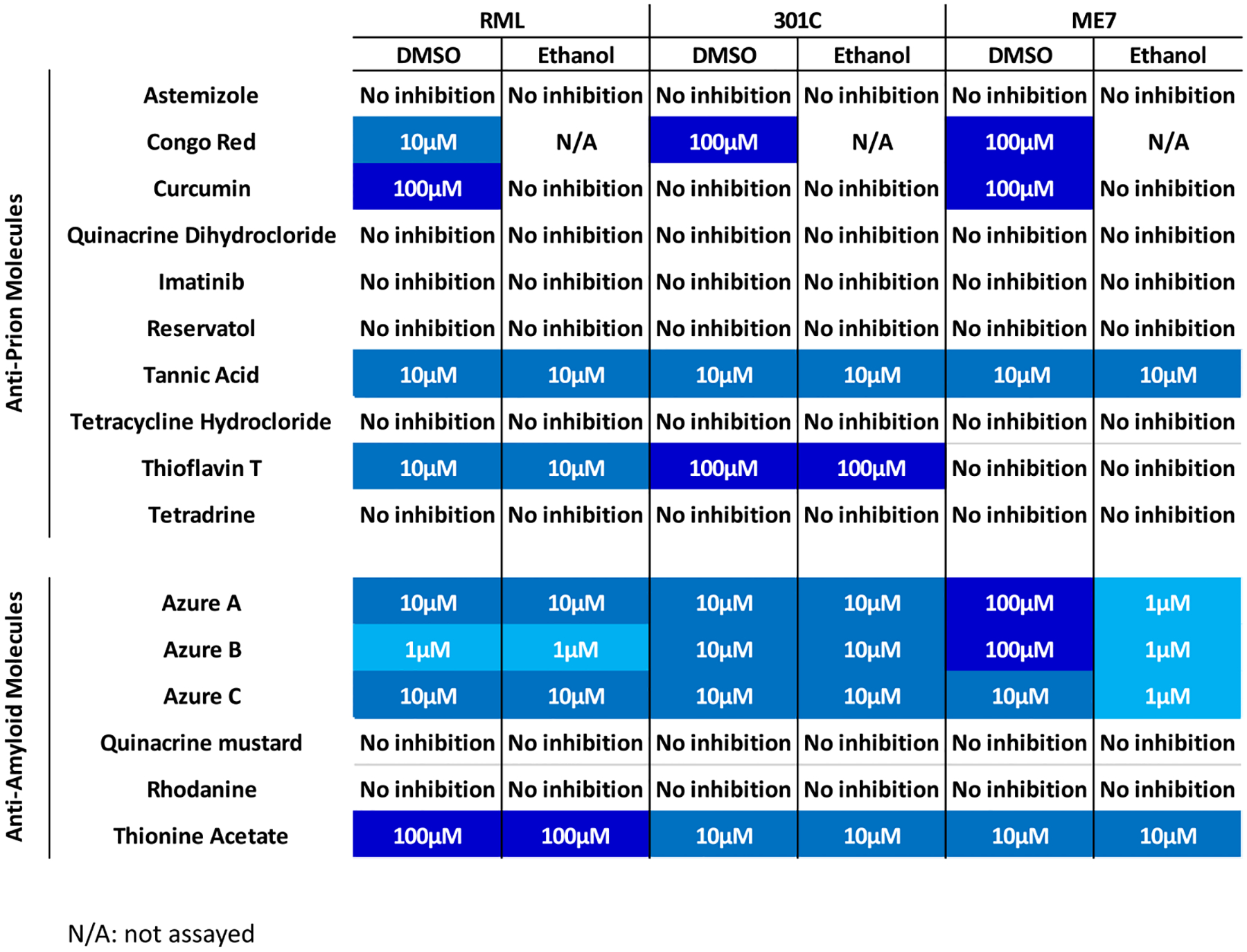
Lowest concentration of molecules able to halt the in vitro replication of three mouse prion strains.
For the hamster-adapted PrPSc strains, amplification was inhibited by the anti-prion molecules Congo Red, curcumin, thioflavin-T, and tannic acid (Figure 4, Figure 5 and Figure S8). Congo red, tannic acid, and thioflavin T exerted an inhibitory effect for the three hamster strains in concentrations ranging between 100 and 10 μM. Tannic acid was apparently most effective against 263K prions as inhibition was observed at 10 μM in both solvents, whereas in HY this molecule shows variable effectiveness depending on the solvent used. The strain-specific response of this compound was observed when tested against SSLOW prions, as this compound was effective for this prion strain only at 100 μM in both solvents (Figure 4, Figure 5 and Figure S8). Additionally, curcumin was effective against 263K prions at 100 μM when dissolved in both solvents (Figure 4, Figure 5 and Figure S8); and interestingly, this compound inhibited HY-induced misfolding at 100 μM when dissolved in EtOH only (Figure 5 and Figure S8). Quinacrine mustard demonstrated anti-prion activity against HY and SSLOW prions at 100 μM in both solvents. This compound was also effective against 263K prions at the same concentration but only when dissolved in EtOH (Figure 5 and Figure S8). Interestingly, this small molecule was not active against any mouse-adapted prion strain. Among the other anti-amyloid compounds, azure C displayed the lowest inhibitory concentration against the hamster-adapted agents, as it inhibited HY-induced misfolding at 1 μM (Figure 5). The inhibitory effect of this molecule was also observed for the other hamster prion strain but at a higher concentration (10 μM for SSLOW and 100 μM for 263K). Azure A showed an inhibitory effect against the three hamster prion strains. A prion strain-specific behavior was observed for thionine acetate. Although this molecule is active against all hamster prion strains was most efficient against SSLOW prions when dissolved in DMSO. Finally, azure B inhibited prion replication for all the hamster-adapted strains at 10 μM, although its efficacy decreased for the 263K strain when dissolved in EtOH (Figure 4, Figure 5 and Figure S8).
FIGURE 4.
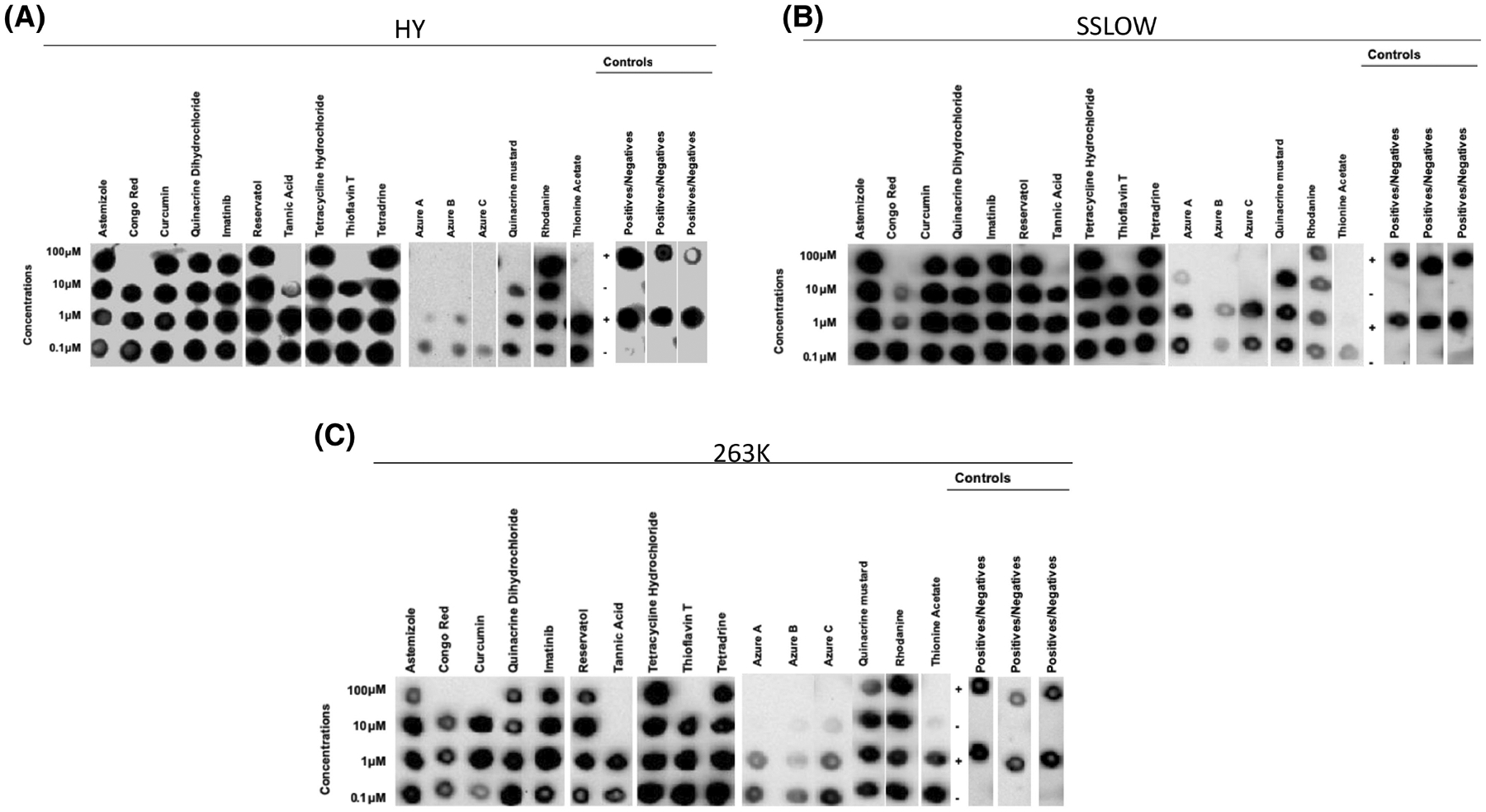
Specific activity of small molecules of known anti-prion and anti-amyloid activity in Syrian hamster prion strains. Sixteen small molecules with proven anti-prion or anti-amyloid activity (dissolved in DMSO) were tested at four different concentrations for their anti-prion activities against the HY (A), SSLOW (B) and 263K (C) prion strains using the 96wp-PMCA. Each molecule was tested in concentrations ranging from 100 to 0.1 μM. Controls for solvent representative for all strains, positive samples and negative samples without compounds were included to monitor the assay. Dot blots were modified for labeling. The panels shown in this figure are representative from three independent assays.
FIGURE 5.
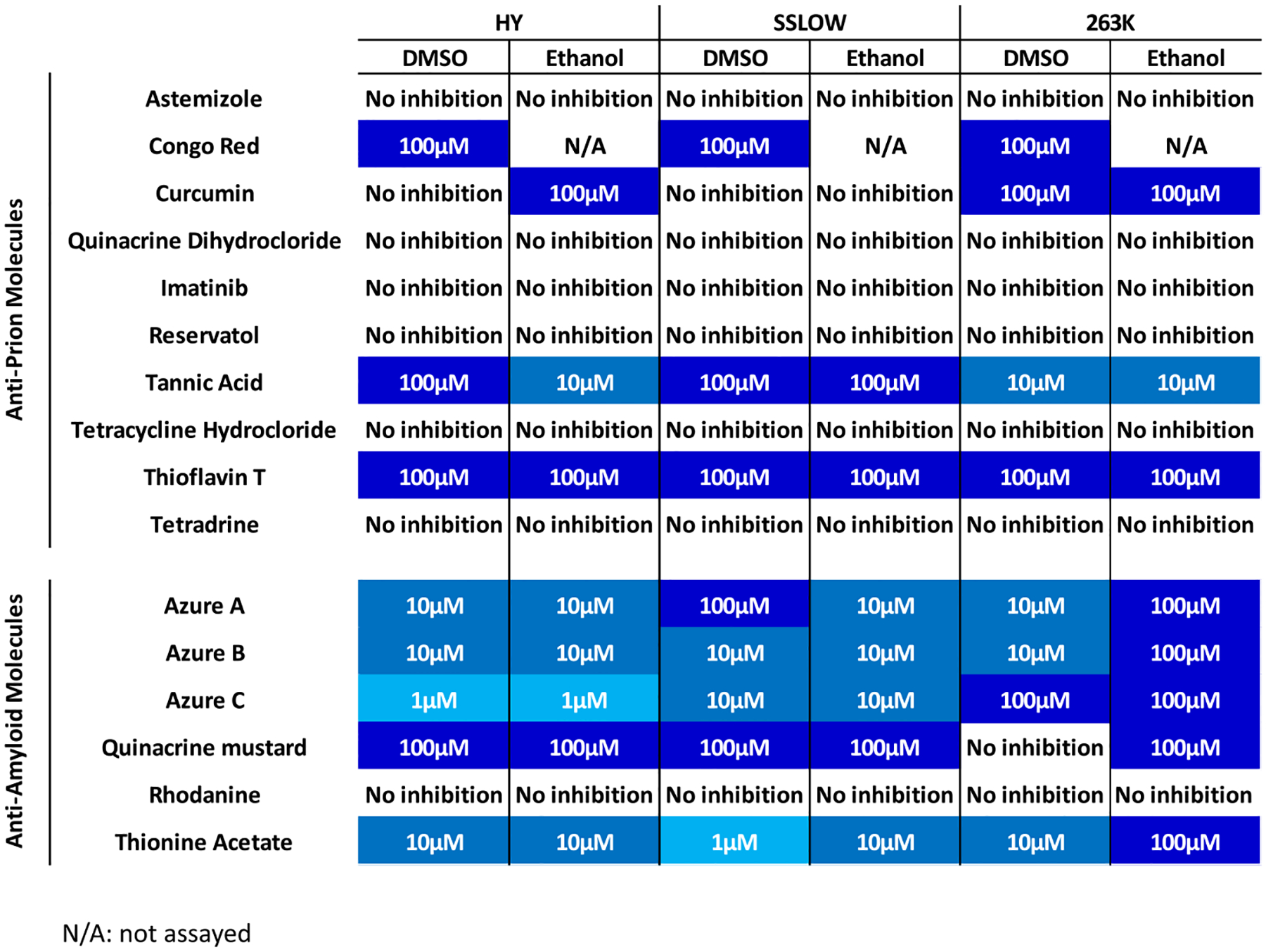
Lowest concentration of molecules able to halt the in vitro replication of three Syrian hamster prion strains.
Together, the data presented above confirms the anti-prion activity of previously reported molecules to support the use of the 96wp-PMCA system in the screening of small-molecule libraries for anti-prion compounds. Moreover, these results demonstrate that our system can identify prion strain-specific anti-prion compounds.
3.3 |. Screening of a small compound library for anti-prion molecules active against CWD prions using the 96wp-PMCA
To demonstrate the applicability of the 96wp-PMCA platform, we screened a small, commercially available compound library for neurodegenerative diseases (n = 204 compounds) for the identification of potential anti-prion molecules targeting CWD prions (responsible for a natural prion disease of growing relevance). We identified seven compounds with anti-prion activity against CWD prions (bemesetron, (−)-Securine, UM-164, Hoechst 34580, clorgyline, ZLN005, and zingerone) (Figure 6). Remarkably, only one of these compounds had been previously reported as an anti-prion molecule (Hoechst 34580).71
FIGURE 6.
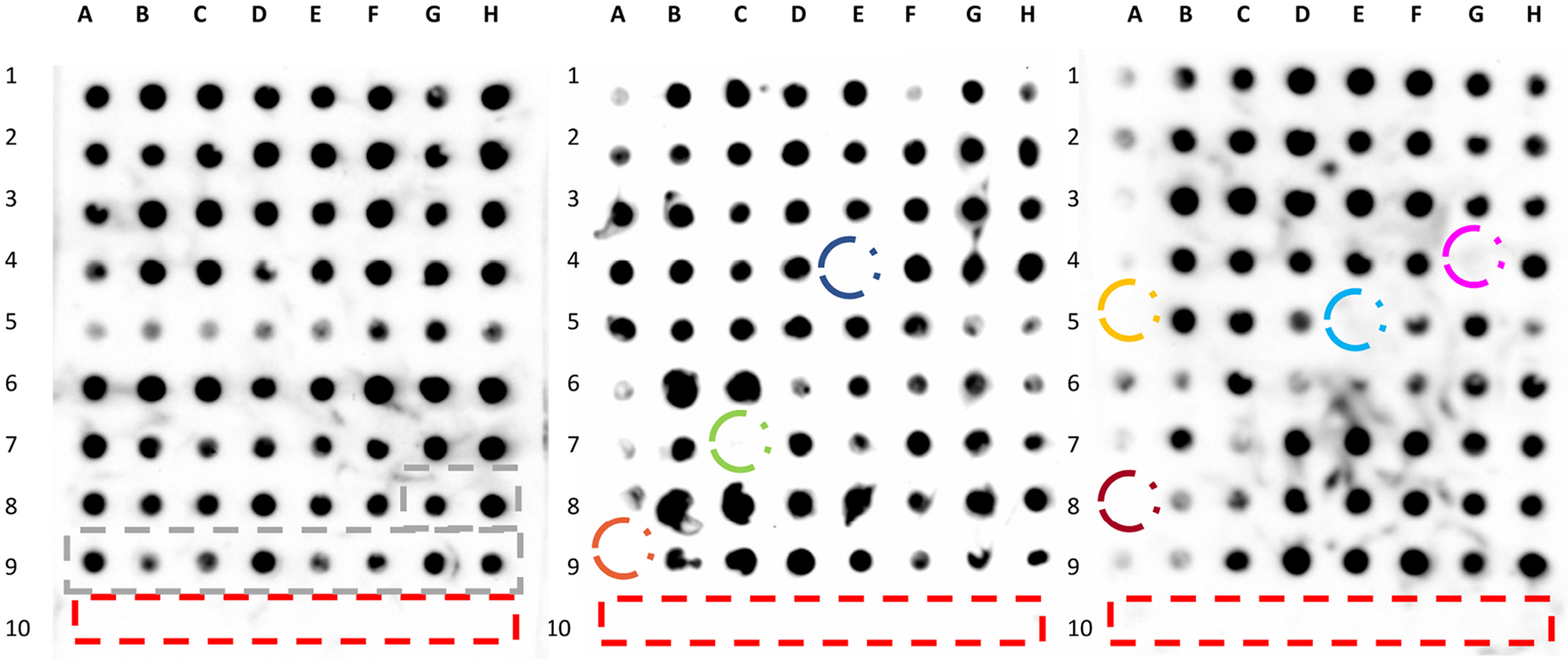
Screening of a small compound library to identify molecules active against CWD prions. A small library of compounds (described in Methods, n = 204 molecules) was tested against CWD prions. All molecules were tested at a concentration of 100 μM. Unseeded PMCA reactions, used as negative controls, are marked noted in the red rectangles. Blacked rectangles depict PMCA reactions were no molecules were added (positive controls). Compounds identified with anti-prion activity in this assay were circled and include clorgyline hydrochloride (blue), ZNL005 (green), zingerone (orange), (−)-securine (pink), bemestron (yellow), UM-164 (light blue) and Hoechst 34580 (terracotta).
Collectively, these findings confirm that the 96wp-PMCA screening platform has the potential to identify small molecules able to inhibit prion replication using an in vitro assay that relies on mimicking faithfully prion replication.
4 |. DISCUSSION
The PMCA technology was deemed optimal for adaptation to increase its throughput. The technical optimizations described in this article substantially reduced the time of sample processing compared with conventional PMCA, as well as decreased the materials needed. This new PMCA platform is obviously more cost-effective than in vivo procedures, as well as more translationally relevant than other in vitro alternatives due to its ability to replicate the strain-specific properties of prions.50,72,73 Specifically, the development of the 96wp-PMCA platform required reduction of the assay time and total volume per reaction, lowering the number of Teflon beads in each well, and changing the technique used to detect PrPSc signals, among other modifications (summarized in Table 1). Ultimately, these changes facilitated a considerable increase in the number of samples tested relative to the conventional procedure, while allowing 96wp-PMCA to retain comparable fidelity.
From a mechanistic point of view, it is possible that anti-prion molecules may exert their effects by multiple pathways, including (i) binding to PrPC and preventing its conversion to PrPSc, (ii) interacting with PrPSc and inhibiting the recruitment of PrPC into the growing aggregates, (iii) targeting the unique glycosylation preferences of each PrPSc strain to mitigate PrPC misfolding, (iv) disaggregate previously formed prion aggregates, and/or (v) binding to yet unknown co-factors present in brain homogenate that could influence the misfolding of PrPC into PrPSc. Considering the nature of the PMCA procedure, this technique is expected to identify molecules acting at all these different levels. Along this line, removing the PrPC from the substrate results, as expected, in PMCA products that devoid of PK-resistant PrP (Figure S9). To validate the potential use of the 96wp-PMCA procedure, we tested a series of previously reported anti-prion and anti-amyloid molecules. We hypothesized that some of these compounds have variable activity against different prion strains due to the conformational differences between them. Consequently, the structural motifs of PrPSc reacting against these molecules may vary depending on the prion strain being studied. Among the tested anti-prion molecules, only three showed effectiveness against one or more mouse-adapted PrPSc variants, these being tannic acid, Congo Red, and thioflavin T. The former compound inhibited all three mouse-adapted strains, whereas the latter molecules affected only some (RML and 301C, but not ME7). Although RML and ME7 are both derived from scrapie,45 they originate from different sources and are thought to be structurally heterogeneous, which is reflected in their distinct glycosylation profiles and unique lesion patterns in brains from experimentally infected rodents.50 We believe that the hypothesized structural heterogeneity of RML and ME7 underlies the strain-specific inhibition of Congo Red and thioflavin T. Interestingly, Congo Red inhibited all hamster-adapted PrPSc strains, suggesting that species-specific common structural motifs present in either hamster PrPC, PrPSc, or both, are able to react with this molecule. Nevertheless, the structural heterogeneity of the hamster-adapted agents likely affected the effectiveness of anti-prion compounds, as for example curcumin inhibited the replication of 263 K and HY, but not SSLOW prions.
In addition to anti-prion compounds, we also tested anti-amyloid molecules known to inhibit the aggregation of proteins linked to other protein misfolding disorders (e.g., Alzheimer’s and Parkinson’s diseases, among others). These compounds include azure A, azure B, azure C, and thionine acetate, which belong to the phenothiazine class of drugs70 and have been shown to inhibit Aβ aggregation and tau filament formation. Additionally, we queried rhodanine because of its ability to mitigate tau aggregation,74 and quinacrine mustard due to its inhibitory effects against amyloid-β oligomerization and fibrillization59,75 (not to be confused for quinacrine, previously shown as active against prions66). We found that for mouse-adapted PrPSc strains, two of the anti-amyloid molecules inhibited prion replication at concentrations as low as 1 μM, which contrasts with our observations for anti-prion compounds wherein the lowest effective concentration was 10 μM. The strain-specific activity of the tested compounds is further observed in the fact that quinacrine mustard was active against hamster prions but did not display inhibitory activities against the mouse-adapted PrPSc strains used in this study.
It is relevant to note that the active concentration of the anti-prion and anti-amyloid compounds used against the rodent prion strain varies between PMCA and the previously reported values. For example, some of the listed compounds showed anti-prion activities between 100 and 0.1 μM in cell culture systems, with tannic acid acting at the lowest concentration (Table S1). The anti-amyloid compounds were reported to act in concentrations ranging from 0.09 to 2000 μM, showing azure C as the most potent inhibitor (Table S1). Nevertheless, it is important to mention that we cannot directly extrapolate the data from our analyses with the previously published information. This is due to the potential synergistic activities of these compounds across the cascade of events leading to prion disease (that can be better appreciated in cell cultures but not in PMCA). Moreover, the high efficiency of PMCA in replicating infectious prions provides barriers for the anti-prion activity of compounds. We believe that this is actually advantageous for the screening of anti-prion molecules considering that this platform favors the identification of the best prion-replication inhibitors.
Remarkably, 96wp-PMCA screening of a small compound library (n = 204) to identify anti-prion molecules against CWD prions revealed that seven compounds demonstrated the target effect (bemesetron, (−)-Securine, UM-164, Hoechst 34580, clorgyline, ZLN005, and zingerone). Among these molecules, four were hydrophobic: (−)-Securine, UM-164, ZNL005, and zingerone, a characteristic proposed to be associated with binding of the hydrophobic domain of PrPC, a site crucial for prion misfolding.76,77 Additionally, UM-164 and ZNL005 possess multiple aromatic rings that are known to impede protein aggregation.78 On the other hand, clorgyline and bemestron are hydrophilic compounds that exhibit inhibitory effects which may be due to its interaction(s) with PrPC, whereas Hoechst 34580 is a dye, akin to thioflavin-T, is able to intercalate within the prion fibrils.71
5 |. LIMITATIONS OF THE STUDY
A technical limitation of PMCA as a drug-screening platform lies in its qualitative (and at most, semi-quantitative) nature. Due to its high efficiency, PMCA includes some intrinsic variability. Considering this, readouts should be interpreted in an all-or-nothing manner. This is particularly relevant for this type of prion-inhibition studies. The latter acquires additional relevance considering the use of the variable readouts provided by dot blots.
The main mechanistic limitation of this study is that PMCA, being a cell-free system, is unable to identify molecules involved in potentially beneficial biological events. These include the clearance of misfolded proteins, inhibition of neuronal death, improved phagocytic activity, decrease of overt inflammatory responses, and reduction of PrPC production, among many others. In addition, most of the prion strains tested in this study corresponded to cloned, experimentally generated isolates. Although these rodent prion strains represented a good model to test our hypotheses, future studies should address whether the 96wp-PMCA system described here is useful to identify anti-prion molecules against relevant prion diseases in humans and non-human animals, such as CJD, CWD, and scrapie. Importantly, assessment of the 96wp-PMCA against larger compound libraries is underway to further validate the relevance of this novel protocol for the identification of potential therapeutic agents against prion diseases.
6 |. CONCLUSION
Collectively, the data presented here support our original hypothesis that modifications to the PMCA technique can be used to develop an assay that can efficiently interrogate compound libraries for anti-prion molecules. We also show that some proven anti-prion compounds display variable activity depending on the prion strain being investigated. Finally, this technique allowed us to identify the prion-strain specific activity of certain anti-amyloid molecules that were not previously tested against PrPSc. Future studies using relevant prion strains and larger compound libraries in the 96wp-PMCA may allow the identification of most needed molecules active against these fatal infectious agents. Moreover, the principles described here may be used for other misfolded proteins (amyloid-β, tau, α-synuclein, and others) in which conformational strain variation is linked with different disease phenotypes.
Supplementary Material
FUNDING INFORMATION
This work was funded by grants from the Creutzfeldt-Jakob Disease Foundation and NIH (R01AI132695) to RM, and grant NIH P01AI077774 to CS.
HHS | NIH | National Institute of Allergy and Infectious Diseases (NIAID), Grant/Award Number: R01AI132695 and P01AI077774; Creutzfeldt-Jakob Disease Foundation (CJDF)
Abbreviations:
- 96wp-PMCA
96 well plate – protein misfolding cyclic amplification
- CWD
chronic wasting disease
- DMSO
dimethyl sulfoxide
- EDTA
ethylenediaminetetraacetic acid
- EtOH
ethanol
- PBS
phosphate buffer saline
- PK
proteinase K
- PMCA
protein misfolding cyclic amplification
- PMSF
phenylmethane sulfonyl fluoride
- RML
Rocky Mountain Laboratories
- SSLOW
synthetic strain leading to overweight
- TSE
transmissible spongiform encephalopathy
Footnotes
DISCLOSURES
Dr. Claudio Soto is the inventor of the PMCA technique and the CSO of Amprion, a biotech company aiming to commercialize PMCA as a diagnostic method. Dr. Rodrigo Morales is listed as an inventor in one patent describing the PMCA technique.
ETHICS APPROVAL
Brains from Syrian hamsters, wild-type mice, and transgenic mice expressing the deer prion protein (tg1536) were used to prepare PMCA substrates. Work with animals was approved by the Animal Welfare Committee (AWC) of the University of Texas Health Science Center at Houston.
DATA AVAILABILITY STATEMENT
Reasonable requests of data and/or materials can be submitted to the corresponding author.
REFERENCES
- 1.Wulf M-A, Senatore A, Aguzzi A. The biological function of the cellular prion protein: an update. BMC Biol. 2017;15:34. doi: 10.1186/s12915-017-0375-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Westergard L, Christensen HM, Harris DA. The cellular prion protein (PrPC): its physiological function and role in disease. Biochim Biophys Acta. 2007;1772:629–644. doi: 10.1016/j.bbadis.2007.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liemann S, Glockshuber R. Transmissible spongiform encephalopathies. Biochem Biophys Res Commun. 1998;250:187–193. doi: 10.1006/bbrc.1998.9169 [DOI] [PubMed] [Google Scholar]
- 4.Behan PO. Creutzfeldt-Jakob disease. Br Med J (Clin Res Ed). 1982;284:1658–1659. doi: 10.1136/bmj.284.6330.1658-a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ironside JW. Variant Creutzfeldt-Jakob disease. Haemophilia. 2010;16:175–180. doi: 10.1111/j.1365-2516.2010.02317.x [DOI] [PubMed] [Google Scholar]
- 6.Brown P, Brandel JP, Sato T, et al. Iatrogenic creutzfeldt-Jakob disease, final assessment. Emerg Infect Dis. 2012;18:901–907. doi: 10.3201/eid1806.120116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collee JG, Bradley R. BSE: a decade on–part I. Lancet. 1997;349:636–641. doi: 10.1016/S0140-6736(96)01310-4 [DOI] [PubMed] [Google Scholar]
- 8.Bradbury J Maternal transmission of BSE demonstrated in cattle. Lancet. 1996;348:393. doi: 10.1016/S0140-6736(05)64995-1 [DOI] [PubMed] [Google Scholar]
- 9.Plummer PJG. Scrapie; a disease of sheep; a review of the literature. Can J Comp Med Vet Sci. 1946;10:49–54. [PubMed] [Google Scholar]
- 10.Gilch S, Chitoor N, Taguchi Y, Stuart M, Jewell JE, Schätzl HM. Chronic wasting disease. Top Curr Chem. 2011;305:51–77. doi: 10.1007/128_2011_159 [DOI] [PubMed] [Google Scholar]
- 11.Williams ES. Chronic wasting disease. Vet Pathol. 2005;42:530–549. doi: 10.1354/vp.42-5-530 [DOI] [PubMed] [Google Scholar]
- 12.Miller MW, Williams ES. Chronic wasting disease of cervids. Curr Top Microbiol Immunol. 2004;284:193–214. doi: 10.1007/978-3-662-08441-0_8 [DOI] [PubMed] [Google Scholar]
- 13.Sigurdson CJ, Aguzzi A. Chronic wasting disease. Biochim Biophys Acta. 2007;1772:610–618. doi: 10.1016/j.bbadis.2006.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hannaoui S, Zemlyankina I, Chang SC, et al. Transmission of cervid prions to humanized mice demonstrates the zoonotic potential of CWD. Acta Neuropathol. 2022;144:767–784. doi: 10.1007/s00401-022-02482-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nemani SK, Myskiw JL, Lamoureux L, Booth SA, Sim VL. Exposure risk of chronic wasting disease in humans. Viruses. 2020;12:1454. doi: 10.3390/v12121454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Belay ED, Maddox RA, Williams ES, Miller MW, Gambetti P, Schonberger LB. Chronic wasting disease and potential transmission to humans. Emerg Infect Dis. 2004;10:977–984. doi: 10.3201/eid1006.031082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wadsworth JDF, Joiner S, Linehan JM, et al. Humanized transgenic mice are resistant to chronic wasting disease prions from Norwegian reindeer and moose. J Infect Dis. 2021;226:933–937. doi: 10.1093/infdis/jiab033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kong Q, Huang S, Zou W, et al. Chronic wasting disease of elk: transmissibility to humans examined by transgenic mouse models. J Neurosci. 2005;25:7944–7949. doi: 10.1523/JNEUROSCI.2467-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Osterholm MT, Anderson CJ, Zabel MD, Scheftel JM, Moore KA, Appleby BS. Chronic wasting disease in cervids: implications for prion transmission to humans and other animal species. MBio. 2019;10:10–128. doi: 10.1128/mBio.01091-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Food Safety and Inspection Service, USDA. Prohibition of the use of specified risk materials for human food and requirements for the disposition of non-ambulatory disabled cattle. 2004.
- 21.Kocisko DA, Baron GS, Rubenstein R, Chen J, Kuizon S, Caughey B. New inhibitors of scrapie-associated prion protein formation in a library of 2,000 drugs and natural products. J Virol. 2003;77:10288–10294. doi: 10.1128/jvi.77.19.10288-10294.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fernández-Borges N, Elezgarai S, Eraña H, Castilla J. Animal models for testing anti-prion drugs. Curr Top Med Chem. 2013;13:2504–2521. doi: 10.2174/15680266113136660177 [DOI] [PubMed] [Google Scholar]
- 23.Krance SH, Luke R, Shenouda M, et al. Cellular models for discovering prion disease therapeutics: progress and challenges. J Neurochem. 2020;153:150–172. doi: 10.1111/jnc.14956 [DOI] [PubMed] [Google Scholar]
- 24.Aguzzi A, Lakkaraju AKK, Frontzek K. Toward therapy of human prion diseases. Annu Rev Pharmacol Toxicol. 2018;58:331–351. doi: 10.1146/annurev-pharmtox-010617-052745 [DOI] [PubMed] [Google Scholar]
- 25.Chung E, Prelli F, Dealler S, Lee WS, Chang Y-T, Wisniewski T. Styryl-based and tricyclic compounds as potential anti-prion agents. PLoS One. 2011;6:e24844. doi: 10.1371/journal.pone.0024844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mayer-Sonnenfeld T, Avrahami D, Friedman-Levi Y, Gabizon R. Chemically induced accumulation of GAGs delays PrPSc clearance but prolongs prion disease incubation time. Cell Mol Neurobiol. 2008;28:1005–1015. doi: 10.1007/s10571-008-9274-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bolognesi ML, Legname G. Approaches for discovering anti-prion compounds: lessons learned and challenges ahead. Expert Opin Drug Discov. 2015;10:389–397. doi: 10.1517/17460441.2015.1016498 [DOI] [PubMed] [Google Scholar]
- 28.Relaño-Ginés A, Lehmann S, Bencsik A, Herva ME, Torres JM, Crozet CA. Stem cell therapy extends incubation and survival time in prion-infected mice in a time window–dependant manner. J Infect Dis. 2011;204:1038–1045. doi: 10.1093/infdis/jir484 [DOI] [PubMed] [Google Scholar]
- 29.Massignan T, Sangiovanni V, Biggi S, et al. A small-molecule inhibitor of prion replication and mutant prion protein toxicity. ChemMedChem. 2017;12:1286–1292. doi: 10.1002/cmdc.201700302 [DOI] [PubMed] [Google Scholar]
- 30.Shim KH, Sharma N, An SSA. Prion therapeutics: lessons from the past. Prion. 2022;16:265–294. doi: 10.1080/19336896.2022.2153551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mustazza C, Sbriccoli M, Minosi P, Raggi C. Small molecules with anti-prion activity. Curr Med Chem. 2019;27:5446–5479. do i: 10.2174/0929867326666190927121744 [DOI] [PubMed] [Google Scholar]
- 32.Heiseke A, Aguib Y, Riemer C, Baier M, Schätzl HM. Lithium induces clearance of protease resistant prion protein in prion-infected cells by induction of autophagy. J Neurochem. 2009;109:25–34. doi: 10.1111/j.1471-4159.2009.05906.x [DOI] [PubMed] [Google Scholar]
- 33.Karpuj MV, Gelibter-Niv S, Tiran A, et al. Conditional modulation of membrane protein expression in cultured cells mediated by prion protein recognition of short phosphorothioate oligodeoxynucleotides. J Biol Chem. 2011;286:6911–6917. doi: 10.1074/jbc.M110.194662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Uliassi E, Nikolic L, Bolognesi ML, Legname G. Therapeutic strategies for identifying small molecules against prion diseases. Cell Tissue Res. 2023;392:337–347. doi: 10.1007/s00441-021-03573-x [DOI] [PubMed] [Google Scholar]
- 35.Mead S, Khalili-Shirazi A, Potter C, et al. Prion protein monoclonal antibody (PRN100) therapy for Creutzfeldt–Jakob disease: evaluation of a first-in-human treatment programme. Lancet Neurol. 2022;21:342–354. doi: 10.1016/S1474-4422(22)00082-5 [DOI] [PubMed] [Google Scholar]
- 36.Kuwata K, Nishida N, Matsumoto T, et al. Hot spots in prion protein for pathogenic conversion. Proc Natl Acad Sci USA. 2007;104(29):11921–11926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chaudhuri TK, Paul S. Protein-misfolding diseases and chaperone-based therapeutic approaches. FEBS J. 2006;273: 1331–1349. doi: 10.1111/j.1742-4658.2006.05181.x [DOI] [PubMed] [Google Scholar]
- 38.White AR, Enever P, Tayebi M, et al. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature. 2003;422:80–83. doi: 10.1038/nature01457 [DOI] [PubMed] [Google Scholar]
- 39.Sethi S, Lipford G, Wagner H, Kretzschmar H. Postexposure prophylaxis against prion disease with a stimulator of innate immunity. Lancet. 2002;360:229–230. doi: 10.1016/S0140-6736(02)09513-2 [DOI] [PubMed] [Google Scholar]
- 40.Bade S, Frey A. Potential of active and passive immunizations for the prevention and therapy of transmissible spongiform encephalopathies. Expert Rev Vaccines. 2007;6:153–168. doi: 10.1586/14760584.6.2.153 [DOI] [PubMed] [Google Scholar]
- 41.Heppner FL, Musahl C, Arrighi I, et al. Prevention of scrapie pathogenesis by transgenic expression of anti-prion protein antibodies. Science. 2001;1979(294):178–182. doi: 10.1126/science.1063093 [DOI] [PubMed] [Google Scholar]
- 42.Aguzzi A, Sigurdson CJ. Antiprion immunotherapy: to suppress or to stimulate? Nat Rev Immunol. 2004;4:725–736. doi: 10.1038/nri1437 [DOI] [PubMed] [Google Scholar]
- 43.Poggiolini I, Saverioni D, Parchi P. Prion protein misfolding, strains, and neurotoxicity: an update from studies on mammalian prions. Int J Cell Biol. 2013;2013:1–24. doi: 10.1155/2013/910314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rossi M, Baiardi S, Parchi P. Understanding prion strains: evidence from studies of the disease forms affecting humans. Viruses. 2019;11:309. doi: 10.3390/v11040309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morales R, Abid K, Soto C. The prion strain phenomenon: molecular basis and unprecedented features. Biochim Biophys Acta. 2007;1772:681–691. doi: 10.1016/j.bbadis.2006.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Otero A, Duque Velasquez C, McKenzie D, Aiken J. Emergence of CWD strains. Cell Tissue Res. 2023;392:135–148. doi: 10.1007/s00441-022-03688-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Colby DW, Giles K, Legname G, et al. Design and construction of diverse mammalian prion strains. Proc Natl Acad Sci USA. 2009;106:20417–20422. doi: 10.1073/pnas.0910350106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berry D, Giles K, Oehler A, Bhardwaj S, DeArmond SJ, Prusiner SB. Use of a 2-aminothiazole to treat chronic wasting disease in transgenic mice. J Infect Dis. 2015;212:S17–S25. doi: 10.1093/infdis/jiu656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morales R, Duran-Aniotz C, Diaz-Espinoza R, Camacho MV, Soto C. Protein misfolding cyclic amplification of infectious prions. Nat Protoc. 2012;7:1397–1409. doi: 10.1038/nprot.2012.067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Castilla J, Morales R, Saá P, Barria M, Gambetti P, Soto C. Cell-free propagation of prion strains. EMBO J. 2008;27:2557–2566. doi: 10.1038/emboj.2008.181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Browning SR, Mason GL, Seward T, et al. Transmission of prions from mule deer and elk with chronic wasting disease to transgenic mice expressing cervid PrP. J Virol. 2004;78:13345–13350. doi: 10.1128/JVI.78.23.13345-13350.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen B, Morales R, Barria MA, Soto C. Estimating prion concentration in fluids and tissues by quantitative PMCA. Nat Methods. 2010;7:519–520. doi: 10.1038/nmeth.1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kramm C, Gomez-Gutierrez R, Soto C, Telling G, Nichols T, Morales R. In vitro detection of chronic wasting disease (CWD) prions in semen and reproductive tissues of white tailed deer bucks (Odocoileus virginianus). PLoS One. 2019;14:e0226560. doi: 10.1371/journal.pone.0226560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bravo-Risi F, Soto P, Eckland T, et al. Detection of CWD prions in naturally infected white-tailed deer fetuses and gestational tissues by PMCA. Sci Rep. 2021;11:18385. doi: 10.1038/s41598-021-97737-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Workman RG, Maddison BC, Gough KC. Ovine recombinant PrP as an inhibitor of ruminant prion propagation in vitro. Prion. 2017;11:265–276. doi: 10.1080/19336896.2017.1342919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Teruya K, Iwabuchi S, Watanabe Y, et al. Activities of curcumin-related compounds in two cell lines persistently infected with different prion strains. Biochim Biophys Acta Gen Subj. 2022;1866:130094. doi: 10.1016/j.bbagen.2022.130094 [DOI] [PubMed] [Google Scholar]
- 57.Reddy PH, Manczak M, Yin X, et al. Protective effects of Indian spice curcumin against amyloid-β in Alzheimer’s disease. J Alzheimers Dis. 2018;61(3):843–866. doi: 10.3233/JAD-170512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Karapetyan YE, Sferrazza GF, Zhou M, et al. Unique drug screening approach for prion diseases identifies tacrolimus and astemizole as antiprion agents. Proc Natl Acad Sci USA. 2013;110:7044–7049. doi: 10.1073/pnas.1303510110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Park S, Kim HY, Oh H-A, et al. Quinacrine directly dissociates amyloid plaques in the brain of 5XFAD transgenic mouse model of Alzheimer’s disease. Sci Rep. 2021;11:12043. doi: 10.1038/s41598-021-91563-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kocisko DA, Morrey JD, Race RE, Chen J, Caughey B. Evaluation of new cell culture inhibitors of protease-resistant prion protein against scrapie infection in mice. J Gen Virol. 2004;85:2479–2483. doi: 10.1099/vir.0.80082-0 [DOI] [PubMed] [Google Scholar]
- 61.Yun S-W, Ertmer A, Flechsig E, et al. The tyrosine kinase inhibitor imatinib mesylate delays prion neuroinvasion by inhibiting prion propagation in the periphery. J Neurovirol. 2007;13:328–337. doi: 10.1080/13550280701361516 [DOI] [PubMed] [Google Scholar]
- 62.Hyeon JW, Kim SY, Lee SM, et al. Anti-prion screening for Acridine, dextran, and tannic acid using real time–quaking induced conversion: a comparison with PrPSc-infected cell screening. PLoS One. 2017;12:e0170266. doi: 10.1371/journal.pone.0170266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jeong J-K, Moon M-H, Bae B-C, et al. Autophagy induced by resveratrol prevents human prion protein-mediated neurotoxicity. Neurosci Res. 2012;73:99–105. doi: 10.1016/j.neures.2012.03.005 [DOI] [PubMed] [Google Scholar]
- 64.Biancalana M, Koide S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim Biophys Acta. 2010;1804:1405–1412. doi: 10.1016/j.bbapap.2010.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ladner-Keay CL, Ross L, Perez-Pineiro R, et al. A simple in vitro assay for assessing the efficacy, mechanisms and kinetics of anti-prion fibril compounds. Prion. 2018;12:280–300. doi: 10.1080/19336896.2018.1525254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Korth C, May BCH, Cohen FE, Prusiner SB. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc Natl Acad Sci USA. 2001;98:9836–9841. doi: 10.1073/pnas.161274798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Secker C, Motzny AY, Kostova S, et al. The polyphenol EGCG directly targets intracellular amyloid-β aggregates and promotes their lysosomal degradation. J Neurochem. 2023;166:294–317. doi: 10.1111/jnc.15842 [DOI] [PubMed] [Google Scholar]
- 68.Shahnawaz M, Tokuda T, Waragai M, et al. Development of a biochemical diagnosis of Parkinson disease by detection of α-Synuclein misfolded aggregates in cerebrospinal fluid. JAMA Neurol. 2017;74:163. doi: 10.1001/jamaneurol.2016.4547 [DOI] [PubMed] [Google Scholar]
- 69.Salvadores N, Shahnawaz M, Scarpini E, Tagliavini F, Soto C. Detection of misfolded Aβ oligomers for sensitive biochemical diagnosis of Alzheimer’s disease. Cell Rep. 2014;7:261–268. doi: 10.1016/j.celrep.2014.02.031 [DOI] [PubMed] [Google Scholar]
- 70.Taniguchi S, Suzuki N, Masuda M, et al. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J Biol Chem. 2005;280:7614–7623. doi: 10.1074/jbc.M408714200 [DOI] [PubMed] [Google Scholar]
- 71.Thai NQ, Tseng N-H, Vu MT, et al. Discovery of DNA dyes Hoechst 34580 and 33342 as good candidates for inhibiting amyloid beta formation: in silico and in vitro study. J Comput Aided Mol Des. 2016;30:639–650. doi: 10.1007/s10822-016-9932-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pritzkow S, Gorski D, Ramirez F, Telling GC, Benestad SL, Soto C. North American and Norwegian chronic wasting disease prions exhibit different potential for interspecies transmission and zoonotic risk. J Infect Dis. 2022;225:542–551. doi: 10.1093/infdis/jiab385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Castilla J, Gonzalez-Romero D, Saá P, Morales R, De Castro J, Soto C. Crossing the species barrier by PrP(Sc) replication in vitro generates unique infectious prions. Cell. 2008;134:757–768. doi: 10.1016/j.cell.2008.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bulic B, Pickhardt M, Khlistunova I, et al. Rhodanine-based tau aggregation inhibitors in cell models of tauopathy. Angew Chem Int Ed. 2007;46:9215–9219. doi: 10.1002/anie.200704051 [DOI] [PubMed] [Google Scholar]
- 75.Necula M, Kayed R, Milton S, Glabe CG. Small molecule inhibitors of aggregation indicate that amyloid β oligomerization and fibrillization pathways are independent and distinct. J Biol Chem. 2007;282:10311–10324. doi: 10.1074/jbc.M608207200 [DOI] [PubMed] [Google Scholar]
- 76.Harrison CF, Lawson VA, Coleman BM, et al. Conservation of a glycine-rich region in the prion protein is required for uptake of prion infectivity. J Biol Chem. 2010;285:20213–20223. doi: 10.1074/jbc.M109.093310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hyeon JW, Choi J, Kim SY, et al. Discovery of novel anti-prion compounds using in Silico and in vitro approaches. Sci Rep. 2015;5:14944. doi: 10.1038/srep14944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Blázquez-Sánchez M, de Matos A, Rauter A. Exploring anti-prion glyco-based and aromatic scaffolds: a chemical strategy for the quality of life. Molecules. 2017;22:864. doi: 10.3390/molecules22060864 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Reasonable requests of data and/or materials can be submitted to the corresponding author.