

Abstract
Endothelial‐to‐mesenchymal transition (EndoMT) plays an important role in pulmonary hypertension (PH) but the molecular mechanisms regulating EndoMT remain to be defined. We demonstrate that the axis of the transcription factors PPARγ (Peroxisome Proliferator‐Activated Receptor gamma) and ETV2 (ETS variant 2) play important roles in the pathogenesis of PH. Decreased levels of the expression of PPARγ and ETV2 along with reduced endothelial and increased EndoMT markers are consistently observed in lungs and pulmonary artery endothelial cells (PAECs) of idiopathic pulmonary arterial hypertension patients, in hypoxia‐exposed mouse lungs, human PAECs, and in induced‐EndoMT cells. Etv2 +/− mice spontaneously developed PH and right ventricular hypertrophy (RVH), associated with increased EndoMT markers and decreased EC markers. Interestingly, chronic hypoxia exacerbated right ventricular systolic pressure and RVH in Etv2 +/− mice. PPARγ transcriptionally activates the ETV2 promoter. Consistently, while mice overexpressing endothelial PPARγ increases the expression of ETV2 and endothelial markers with reduced EndoMT markers, endothelial PPARγ KO mice show decreased ETV2 expression and enhanced EndoMT markers. Inducible overexpression of ETV2 under induced‐EndoMT cell model reduces number of cells with mesenchymal morphology and decreases expression of mesenchymal markers with increased EC makers, compared to control. Therefore, our study suggests that PPARγ‐ETV2 signaling regulates PH pathogenesis through EndoMT.
Keywords: endothelial‐to‐mesenchymal transition, ETV2, hypoxia, PPARγ, pulmonary hypertension
INTRODUCTION
Pulmonary hypertension (PH), defined as an elevation of the mean pulmonary artery pressure >20 mmHg including a pulmonary vascular resistance ≥ 3 wU (Woods units), causes high morbidity and mortality. 1 , 2 , 3 , 4 PH is characterized by pulmonary endothelial dysfunction and abnormal proliferation of pulmonary vascular wall cells, vascular remodeling, and muscularization of small pulmonary vessels. 2 , 5 , 6 These structural and functional alterations in the pulmonary vasculature increase pulmonary vascular resistance resulting in progressive right‐sided heart failure and death. 7 , 8 Chronic hypoxia is a critical factor leading to vascular remodeling and stimulates steady state inflammation via subclinical vascular endothelial dysfunction. 9 Understanding the mechanisms behind the structural and functional alterations in the pulmonary vasculature opens avenues for targeted therapies. However, there are no specific therapies to reverse pathological vascular remodeling, the major cause of PH, in part because the pathogenesis of PH is poorly understood. Therefore, strategies with novel insights into PH to enhance the current understanding are urgently needed.
Recent studies show that endothelial to mesenchymal transition (EndoMT) contributes to the pathogenesis of PH. EndoMT is a process where endothelial cells (ECs) lose endothelial and gain mesenchymal phenotypic and functional features. The phenotypic and functional switch can ultimately lead to pulmonary vascular remodeling, which is characterized by extensive accumulation of cells expressing smooth muscle actin (αSMA) within the microvessels of the hypertensive lung. 10 , 11 Lineage tracking analysis in mice demonstrated that EndoMT contributes to a spectrum of structural changes including thickening of the adventitial, medial and intimal layers of the pulmonary artery wall, medial hypertrophy of muscular arteries, and muscularization of small pulmonary arterioles. 12 Furthermore, studies in mouse models of PH 13 and histological analysis of human patient samples 14 suggest EndoMT as a potential mechanism of distal pulmonary arteriole muscularization. 14 , 15 However, the molecular mechanisms behind this process have yet to be elucidated and constitute the focus of the current study.
ETS Variant Transcription Factor 2 (ETV2), also known as ER71, an ETS transcription factor functions as a critical regulator of the cardiovascular system. 15 We have demonstrated that Etv2 deficient mouse embryos exhibited complete absence of the embryonic vasculature and died in utero as early as embryonic day 10.5. 16 Similar results were reported from other studies using alternative strategies such as gene trap and knockin/knockout approaches. 17 , 18 Given the conserved functions from zebrafish 19 and Xenopus er71, 20 it is clear that ETV2 is indispensable for cardiovascular development. We have also shown that endothelial Etv2 is required for new vessel formation in response to injury and that delivery of Etv2 into ischemic hindlimbs promotes perfusion recovery with concomitant neovascularization in adults. 21 Furthermore, studies including ours have revealed that ETV2 alone is sufficient to convert human dermal fibroblasts into functional ECs. 22 , 23 Taken together, these findings strongly suggest that ETV2 is a master regulator of EC fate.
Peroxisome Proliferator‐Activated Receptor gamma (PPARγ) is a ligand‐activated transcription factor of the nuclear hormone receptor superfamily. 24 Its expression and activity is reduced in the pulmonary vasculature of patients with severe PH. 25 Furthermore, EC‐targeted depletion of PPARγ cause spontaneous PH in mice. 26 In contrast, activation of PPARγ with thiazolidinedione attenuates PH and vascular remodeling in essentially every experimental model of PH in which they have been tested. 27 These results suggest a pivotal role for PPARγ in the pathogenesis of PH. In this study, we investigated novel functions of the of PPARγ‐ETV2 axis in regulating EndoMT in PH.
MATERIALS AND METHODS
Control and idiopathic pulmonary arterial hypertension (IPAH) lung tissues and pulmonary artery endothelial cells (PAECs)
We obtained peripheral lung tissues from failed donor (control) or IPAH patient specimens collected by the Pulmonary Hypertension Breakthrough Initiative (PHBI). IPAH samples were derived from 2 male and 3 female patients, 24–56 years old whereas control specimens were derived from 2 male and 3 female subjects, 29–55 years old who were control. Human PAECs (HPAECs) isolated from the lungs of IPAH subjects as described 28 were generously provided by Dr. Harry Karmouty‐Quintana (University of Texas Health Science Center at Houston, Houston, TX). We purchased independent primary HPACEs isolated from healthy donors (ScienCell, Research Laboratories, Carlsbad, CA, USA).
Mouse models
All mice are fully backcrossed into a C57BL/6 background with a minimum of 10 generations. The Etv2+/− mice, ePPARγKO or littermate control mice and ePPARγOX mice or littermate control (FulCon). 18 , 29 , 30 The ePPARγKO and ePPARγOX mice are normal in appearance and behavior. All animal studies were approved by the Institutional Animal Care and Use Committee of Emory University or the Atlanta Veterans Affairs Healthcare System.
In vivo mouse model of PH
Male C57BL/6J mice, aged 8–12 weeks, were treated three times with weekly injections of the VEGF receptor antagonist, Sugen 5416 (SU, 20 mg/kg, subcutaneous injection) and exposed to hypoxia (HYP/SU, 10% O2) or normoxia (NOR/SU, 21% O2) for 3 weeks as reported. 30 To assess PH, right ventricular systolic pressure (RVSP) and right ventricular hypertrophy (RVH) were measured in NOR/SU and HYP/SU‐treated mice and in male and female Etv2 +/− mice as reported. 30 All animals were given unrestricted access to water and standard mouse chow.
In vitro cell models
For the hypoxia experiment with ECs, HPAECs were exposed to NOR or HYP (1% O2) conditions for 72 h as reported. 31 To induce EndoMT, HPAECs (passage 2–6, ScienCell, Carlsbad, CA, USA) were incubated with 0.1 ng/mL interleukin‐1 beta (IL‐1β), 10 ng/mL tumor necrosis factor alpha (TNF‐α), and 10 ng/mL transforming growth factor beta (TGFβ) (i‐EndoMT) or DMSO (CON) for 72 h. Levels of HPAEC mRNAs associated with EndoMT were determined using qRT‐PCR.
Luciferase‐based promoter assay
HEK/293 T cells (1.3 × 105/well of a 24‐well plate) were transfected with 2 µg PPARγ expression plasmid (pcDNA3.1‐FLAG‐PPARγ), 200 ng pGL3‐basic‐ETV2 promoter, 32 and 30 ng pRL‐null using lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA). Forty‐eight hours later, cells were harvested, and luciferase activity was measured using the Dual‐Luciferase reporter assay system (Promega, Madison, WI, USA) according to the manufacturer's instructions. Firefly luciferase values were divided by Renilla luciferase values to normalize transfection efficiency. Rosiglitazone (10 µM) was added into the culture 12 h before cell harvest.
Scratch wound assay
HPAEC (3 × 105/well) derived from three independent individuals were cultured in six well plates with ECM media (ScienCell, Carlsbad, CA) then treated with TGF‐β (10 ng/ml), TNF‐α (10 ng/ml) and IL‐1β (0.1 ng/ml) for 24 h or 72 h. Then, resulting cells (2.8 × 104/well) were harvested and cultured overnight to reach a confluent layer in a well separated by an insert (Culture–Insert 2 Well, ibidi GmbH, Germany). Subsequently, the insert was removed to generate the cell‐free gap and 1 ml of ECM media added (ScienCell, Carlsbad, CA, USA). Images were taken at 0 and 5 h after incubation using a phase‐contrast microscope. The area of the cell‐free gap was calculated by Image J.
Immunohistochemistry and immunocytochemistry
For each lung, sections from six mice per group were stained with hematoxylin and eosin for immunobiological analysis of pulmonary tissue. For immunocytochemistry, cells in a slide glass were fixed with 4% paraformaldehyde and washed with 1x PBS for 5 times. Subsequently, cells were incubated with mouse anti‐SLUG, mouse anti‐TWIST, rabbit anti‐PECAM1, or mouse anti‐VE‐Cad antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), followed by anti‐mouse Alexa 488 or anti‐rabbit 567 antibody (Li‐Cor, Lincoln, NE, USA). The images were taken using Olympus IX51 (Evident, Tokyo, Japan).
Collagen gel cell contraction assays
Suspension of i‐EndoMT cells (5 × 105) was mixed with collagen solution in a 1:4 ratio of cell suspension and collagen mixture provided in the cell contraction assay (Cell Biolabs, San Diego, CA, USA) according to the manufacturer's instructions. The gels were imaged at 0 h and 120 h and analyzed by ImageJ software.
ETV2 or PPARγ gain and loss of function
For in vitro ETV2 or PPARγ loss of function, HPAECs were transfected with scrambled or ETV2 or PPARγ RNAi duplexes (20 nM, Integrated DNA Technologies, Coralville, IA) using Lipofectamine 3000 transfection reagent (Invitrogen) according to the manufacturer's instructions. 6 h after transfection, the transfection media were replaced with EGM containing 5% FBS and the cells were then subjected to normoxia (NOR, 21% O2) or hypoxia (HYP, 1% O2) for 72 h. HPAEC lysates were then prepared for PPARγ, ETV2, SLUG, TWIST1, PECAM1/CD31, and VE‐Cad/CDH5 levels using qRT‐PCR analysis. To overexpress ETV2, HPAECs were transfected with ETV2 plasmid constructs (1 µg, oxETV2) or empty vector. For overexpression of PPARγ, HPAECs were transfected with adenovirus containing a PPARγ plasmid (AdPPARγ, 25 multiplicity of infection, MOI) or control GFP plasmid as we previously reported. 30 Six hours later, media were replaced with fresh 5% FBS EGM, and HPAEC were then subjected to normoxia (NOR, 21% O2) or hypoxia (HYP, 1% O2) for 72 h. Subsequently, HPAEC lysates were then prepared for PPARγ, ETV2, SLUG, TWIST1, PECAM1/CD31, and VE‐Cad/CDH5 levels using qRT‐PCR analysis.
mRNA quantitative real‐time polymerase chain reaction (qRT‐PCR) analysis
To measure PPARγ, ETV2, SLUG, TWIST1, PECAM1/CD31, and VE‐Cad/CDH5 levels, total RNAs in IPAH lungs, IPAH ECs, HPAECs, mouse lungs or i‐EndoMT cells were isolated using the mirVana kit (Invitrogen). PPARγ, ETV2, SLUG, TWIST1, PECAM1/CD31, and VE‐Cad/CDH5 mRNA levels in the same sample were determined and quantified using specific mRNA primers as previously described. 33 GAPDH mRNA levels were used as a control.
Western blot analysis
Protein homogenates from mouse lungs were subjected to Western blot analysis as reported. 33 Primary antibodies purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA) included: SLUG mouse monoclonal antibody (1:500 dilution, Cat # SC‐166476, 30 kDa), TWIST1 mouse monoclonal antibody (1:500 dilution, Cat # SC‐81417, 28 kDa), PECAM1 rabbit polyclonal antibody (1:500 dilution, Cat # SC‐8306, 130 kDa) and VE‐Cad rabbit polyclonal antibody (1:500 dilution, Cat # SC‐28644, 130 kDa). GAPDH rabbit polyclonal antibody (1:10,000 dilution, Cat # G9545, 37 kDa) was purchased from Sigma‐Aldrich (St. Louis, MO, USA). Relative protein levels were visualized using Li‐Cor proprietary software, quantified Image J software, and normalized to GAPDH levels within the same lane since GAPDH levels are stable in normoxic or hypoxic conditions.
Statistical analysis
All data are presented as mean ± standard error of the mean (SE). Data were analyzed using analysis of variance. Post hoc analysis used the Student Neuman‐Keuls test to detect differences between specific groups. To test for normality, we employed the Shapiro‐Wilk test. Since the p‐values were greater than the chosen alpha level (α = 0.05), this indicated that the data did not significantly deviate from normality. If the data is not normally distributed, we performed a Mann‐Whitney U test. The level of statistical significance was taken as p < 0.05. Statistical analyses were carried out using GraphPad Prism, Version 8.0 software (LaJolla, CA, USA).
RESULTS
ETV2 expression is downregulated in PH in vivo and in vitro
We first determined whether the levels of markers of EndoMT and ECs are changed in human and experimental PH. As shown, the expression of EndoMT markers including SLUG and TWIST1 was increased in lungs (Figure 1a) and PAECs (Figure 1b) isolated from patients with IPAH, whereas the expression of endothelial markers, PECAM1/CD31 and VE‐Cad/CDH5 was decreased. A similar finding was observed in mice following the induction of PH with hypoxia and the VEGF receptor antagonist, Sugen 5416 (HYP/SU) treatment (Figure 1c). Hypoxia alone was also sufficient to increase EndoMT markers and decrease endothelial markers in human PAECs (HPAECs) in vitro (Figure 1d). These results suggest that EndoMT indeed occurs in the pathogenesis of PH and could have potential role in process. Previous studies showed that lack of Etv2 leads to a complete absence of ECs 16 , 17 , 18 and that forced expression of ETV2 converted non‐ECs into ECs 22 , 23 suggesting that ETV2 has a determinant role in regulating EC functions and fates. Thus, we hypothesized that the expression of ETV2 is downregulated in PH lungs leading to the transition of EC to mesenchymal cells. Interestingly, we found that ETV2 levels were significantly reduced in both lungs (Figure 1e) and PAECs (Figure 1f) isolated from IPAH patients, in lung tissues from HYP/Su mice (Figure 1g), and in hypoxia‐exposed HPAECs (Figure 1h) supporting the nothion that ETV2 plays a role in PH.
Figure 1.
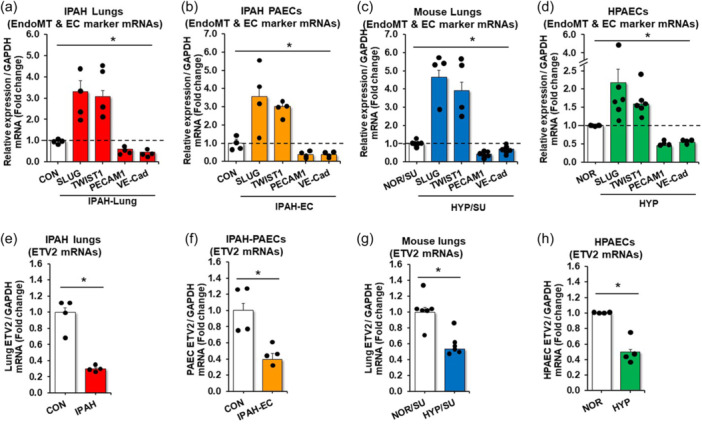
Expression of ETV2 decreases in PH patients and in mice upon hypoxia. RNAs isolated from IPAH lungs (a, e) or IPAH PAECs (b, f) were subjected to qRT‐PCR. The results were expressed relative to GAPDH mRNA ± SE as fold‐change versus CON. *p < 0.05 vs NOR, n = 4/group. (c, g) Whole lungs were collected from mice under normoxic (21%) with sugen (NOR/SU, 20 mg/kg) or hypoxic (10% O2) with sugen (HYP/SU) for 3‐weeks. Levels of lung EndoMT, EC marker, and ETV2 were measured with qRT‐PCR and expressed relative to lung GAPDH mRNA *p < 0.05 vs NOR, n = 4–6/group. (d, h) HPAECs were exposed to normoxia (NOR, 21% O2) or hypoxia (HYP, 1% O2) for 72 h. Levels of HPAEC EndoMT, EC marker, and ETV2 were measured with qRT‐PCR. All bars represent the mean EndoMT or EC marker mRNA levels relative to GAPDH ± SE expressed as fold‐change versus NOR. *p < 0.05 versus NOR, n = 3–6/group.
Reductions in ETV2 increase EndoMT markers and PH in vivo
To determine the functional significance of ETV2 in the pathogenesis of PH, we examined whether loss of ETV2 can lead to the development of PH. Since Etv2 ‐/‐ mice are embryonic lethal, 16 we used male and female Etv2 +/− mice which are fertile, normal in appearance and behavior, and display no overt overall or vascular phenotype. 21 However, we found that Etv2 +/‐ mice (12–16 weeks old) spontaneously developed mild PH (Figure 2a) as demonstrated by increased RVSP and RVH (Figure 2b), whereas no differences in heart rate were observed between littermate control and Etv2 +/− mice. Interestingly, chronic hypoxia exacerbated RVSP and RVH in Etv2 +/− mice. Importantly, enhanced positivity of αSMA, an indicator of vascular remodeling (Figure 2c) was observed in Etv2 +/− mice, compared to control. The expression of EndoMT markers was increased and the expression of endothelial markers were reduced in the lungs of Etv2 +/− mice, compared to contral (Figure 2d and E). Furthermore, we performed siRNA‐mediated ETV2 depletion in HPAEC and found augmented expression of EndoMT markers and reduction of endothelial markers in the cells (Figure 2f). Lentiviral‐mediated‐ETV2 overexpression in HPAECs led to downregulation of the EndoMT markers and was able to increase the expression of PECAM1 and VE‐Cad/CDH5 in HPAECs under hypoxic conditions (Figure 2g). Collectively, these results suggest that endothelial ETV2 play an important role in the progression of PH.
Figure 2.
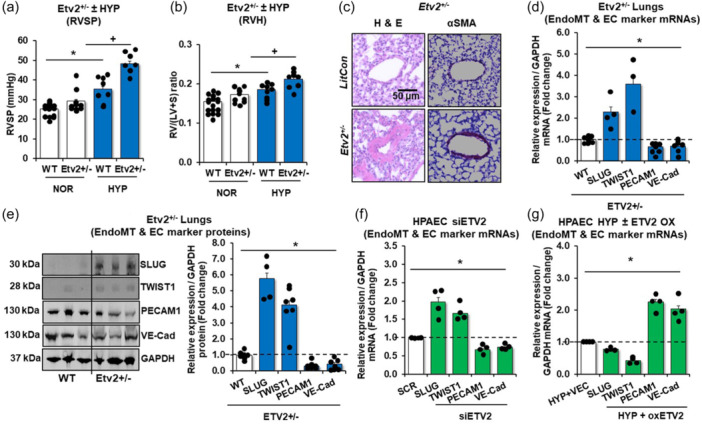
Reduction of ETV2 leads to the development of PH with increased expression of mesenchymal markers. Etv2 +/− and littermate control (WT) mice were exposed to normoxic (21%) with sugen (NOR/SU, 20 mg/kg) or hypoxic (10% O2)/sugen (HYP/SU) mice for 3‐weeks. (a) Right ventricular systolic pressure (RVSP) was recorded in anesthetized mice with a pressure transducer. Each bar represents the mean RVSP in mmHg ± SE. n = 7–17/group. (b) The ratio of the weight of the right ventricle to the left ventricle + septum [RV: (LV + S)] is presented as an index of right ventricular hypertrophy (RVH). n = 8–9/group. (c) Representative images of small mouse arterioles following H&E or αSMA staining. (d, e) Levels of lung EndoMT or EC markers were measured with qRT‐PCR (d) or with Western blot analysis (e). The qRT‐PCR results were expressed relative to GAPDH mRNA and Western blot analysis results to GAPDH protein ± SE as fold‐change versus CON. *p < 0.05 vs NOR, n = 3‐7/group. (f) For ETV2 knockdown experiments, HPAECs were treated with scrambled (SCR) or ETV2 (20 nM) siRNAs for 6 h then incubated for an additional 72 h. Each bar represents mean ± SE. EndoMT or EC markers level relative to GAPDH were expressed as fold‐change vs cells treated with SCR. n = 3–6/group. P value *p < 0.05 vs SCR. (g) For ETV2 gain of function, HPAECs were treated with ETV2 plasmid constructs (oxETV2, 1 µg) or vector (VEC) constructs for 6 h, then incubated for an additional 72 h of normoxia (NOR) or hypoxia (HYP) exposure. qRT‐PCR was performed for EndoMT, or EC markers. Each bar represents mean ± SE EndoMT or EC markers level relative to GAPDH expressed as fold‐change vs cells treated with VEC. n = 3–6/group, *p < 0.05 vs HYP/VEC.
PPARγ functions as a direct upstream regulator of ETV2 expression
Studies have demonstrated that activation of the nuclear hormone receptor PPARγ attenuates PH whereas loss of PPARγ promotes PH. 27 As illustrated in Figure E1 in the online data supplement, PPARγ levels were reduced in lungs and PAECs isolated from IPAH patients, in HYP/SU mouse lungs, and in hypoxia‐exposed HPAECs. As shown in Figures 1 and 2, the expression of ETV2 is well correlated with pathogeneic features of PH, thus suggesting a potential link between PPARγ and ETV2 in PH. Consistent with this postulate, our in silico analysis identified two putative PPREs (PPARγ Responsive Elements) in the ETV2 promoter region (NM_014209, data not shown). Overexpression of PPARγ in HPAECs using adenovirus‐mediated PPARγ transduction (AdPPARγ, Figure 3a) led to a significant increase in ETV2 expression (Figure 3b). Next, to determine whether PPARγ can transcriptionally activate the expression of ETV2, we performed a luciferase‐based ETV2 promoter assay and found that AdPPARγ enhanced the activity of the ETV2 promoter (Figure 3c). The observed promoter activity was further increased upon treatment with the PPARγ ligand, rosiglitazone (RSG, 10 µM), compared with non‐treated, control plasmid (Mock)‐transfected HPAECs (Figure 3c). Consistantly, AdPPARγ overexpression with RSG treatment increased HPAEC ETV2 expression in normoxic conditions and restored ETV2 levels in hypoxic conditions (Figure 3d). Adenovirus‐mediated‐PPARγ overexpression with RSG treatment in HPAECs also mitigated the hypoxia‐induced increase in EndoMT markers and the reduced expression of EC markers (Figure 3e). Importantly, lung tissue isolated from endothelial‐targeted PPARγ overexpressing (ePPARγOX) mice (Figure 3f) showed enhanced expression of both Etv2 (Figure 3g) and EC markers with reduced expression of EndoMT markers (Figure 3h). To further determine the relationship between PPARγ and ETV2, we performed a series of loss of function studies. First, HPAECs were treated with PPARγ siRNA resulting in ~60% knockdown of PPARγ (Figure 4a). Reduction of PPARγ effectively attenuated the level of ETV2 mRNAs (Figure 4b), whereas PPARγ expression did not change in HPAECs transfected with ETV2 siRNA (Figure 4c). Similarly, loss of PPARγ in ECs of endothelial‐targeted PPARγ knockout (ePPARγKO) mice (Figure 4d) led to reduced ETV2 expression (Figure 4e) as well as EC markers and increased mesenchymal markers (Figure 4f) in lung homogenates in vivo. Collectively, these findings indicate that PPARγ functions as a direct upstream regulator of ETV2 expression in ECs, regulating PH.
Figure 3.
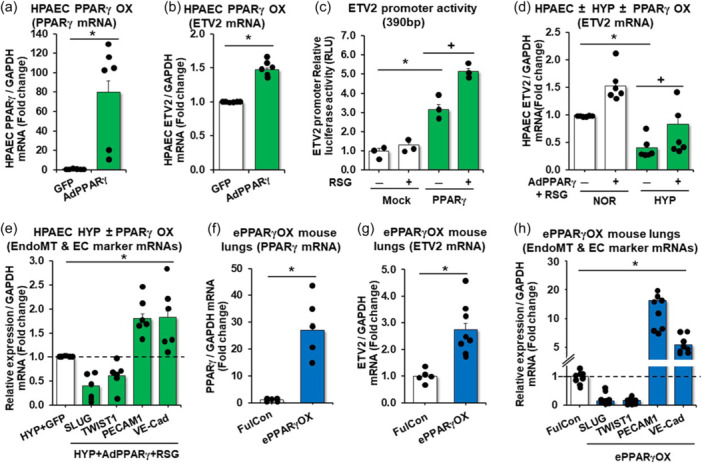
PPARγ increases the expression of ETV2 in HPAECs and PPARγ gain‐of‐function increases ETV2 and endothelial makers and attenuates the expression of EndoMT markers. (a, b) HPAECs treated with AdPPARγ (25 MOI) or green fluorescent protein (GFP) constructs were subjected to qRT‐PCR analysis. n = 3–5/group, *p < 0.05 vs GFP. (c) PPARγ expressing plasmid (PPARγ) or control plasmid (Mock) was co‐transfected with pGL3‐basic‐ETV2 promoter and treated with dimethyl sulfoxide (RSG/−) or rosiglitazone (RSG/+) (10 µM), then incubated for 72 h. Firefly luciferase activity was normalized by Renilla luciferase activity. n = 3–5/group, *p < 0.05 vs Mock/RSG(−). + p < 0.05 vs PPARγ/RSG(−). (d) HPAECs were treated with AdPPARγ + RSG for 6 h, then incubated with fresh medium for an additional 72 h under normoxic (NOR) or hypoxic (HYP) condition. RSG was treated for last 24 h. (e) HPAECs treated with AdPPARγ or green fluorescent protein (GFP) constructs with RSG (10 µM) were cultured under hypoxic condition and subjected to qRT‐PCR analysis. Each bar represents mean ± SE PPARγ, ETV2, EndoMT or EC markers level relative to GAPDH expressed as fold‐change vs cells treated with GFP. n = 5–6/group, *p < 0.05 vs HYP/GFP. (f–h) whole lungs were collected from littermate control (FulCon) or endothelial‐targeted PPARγ overexpression (ePPARγOX) mice. Levels of lung PPARγ (f) or ETV2 (g) or EndoMT and EC markers (h) were measured with qRT‐PCR and expressed relative to GAPDH mRNA *p < 0.05 vs FulCon, n = 5–8/group.
Figure 4.
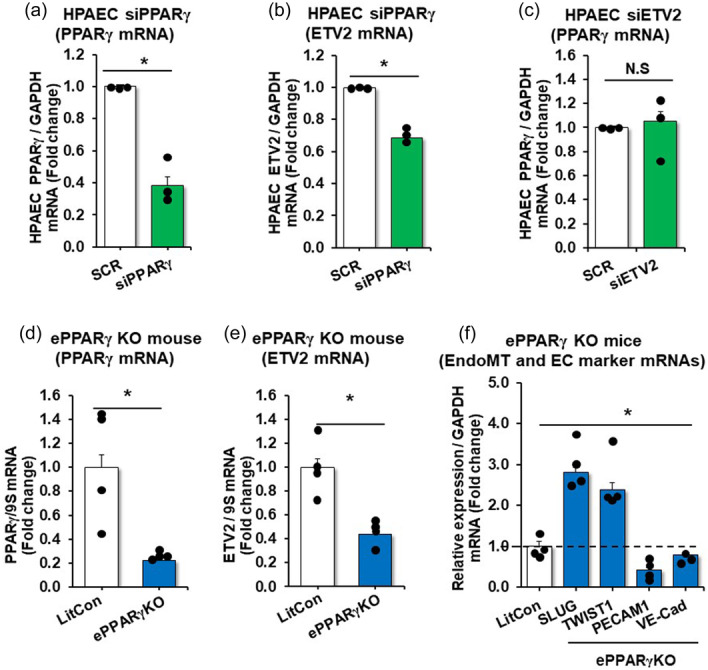
Endothelial depletion of PPARγ reduces ETV2, PECAM1, and VE‐Cad expression and increases levels of EndoMT markers in mouse lungs. (a, b) HPAECs were treated with scrambled (SCR) or PPARγ (20 nM) siRNAs (a, b) or ETV2 (20 nM) siRNAs (c) for 6 h, washed with fresh medium, then incubated for an additional 72 h. Each bar represents mean ± SE PPARγ or ETV2 level relative to GAPDH expressed as fold‐change vs cells treated with SCR. n = 3/group, *p < 0.05 vs SCR. (d–f) Whole lungs were collected from littermate control (LitCon) or endothelial‐targeted PPARγ knockout (ePPARγKO) mice. Lung levels of PPARγ (d), ETV2 (e), or EndoMT and EC markers (f) mRNA levels were measured using qRT‐PCR in littermate control (LitCon) or endothelial‐targeted PPARγ knockout (ePPARγKO) mice and expressed relative to GAPDH mRNA *p < 0.05 vs LitCon, n = 4/group.
In vitro model of EndoMT
To further investigate the role of ETV2 in EndoMT, we refined a model to induce EndoMT (termed induced‐EndoMT, i‐EndoMT) in HPAEC in vitro. 34 HPAECs were treated with inflammatory mediators (IL‐1β, TNF‐α, and TGFβ) in dose‐ranging regimens (Figure E2 in the online data supplement). Figure 5a illustrates that these inflammatory mediators, which are increased in PH, 35 , 36 , 37 caused HPAEC to lose cobblestone morphology and become elongated and spindle‐shaped, consistent with EndoMT. Under these conditions, the expression of PPARγ (Figure 5b), ETV2 (Figure 5c), and EC markers was decreased (Figure 5d), but EndoMT markers were increased (Figure 5d). In collagen gel contraction assays, i‐EndoMT cells displayed enhanced gel contraction (dashed line in photomicrograph) to a fraction of the control gel contour (line graph) consistent with mesenchymal functional characteristics (Figure 5e ). Similarly, the scratch wound healing assays demonstrated that i‐EndoMT promoted cell migration into a monolayer wound, which is also consistent with mesenchymal cell function (Figure 5f). These findings suggest that previously described upregulated pro‐inflammatory pathways in PH are sufficient to reduce endothelial PPARγ and ETV2 expression and induce EndoMT.
Figure 5.
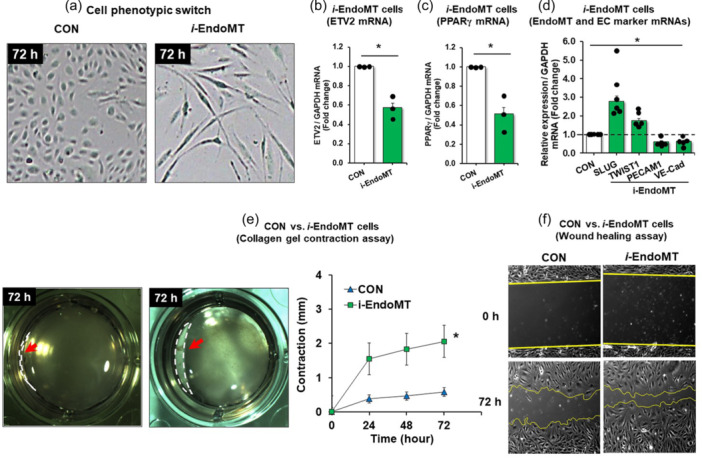
Establishment of EndoMT (i‐EndoMT) model. (a–f) i‐EndoMT cells were induced by addition of 0.1 ng/mL interleukin‐1 beta (IL‐1β), 10 ng/mL tumor necrosis factor alpha (TNF‐α), and 10 ng/mL transforming growth factor beta (TGFβ) to HPAECs up to 72 h. Morphology change (a), mean HPAEC PPARγ (b), ETV2 (c), EndoMT or EC marker (d) levels were measured with qRT‐PCR. All bars represent the mean PPARγ, ETV2, EndoMT or EC marker mRNA levels relative to GAPDH ± SE expressed as fold‐change versus CON. *p < 0.05 versus CON, n = 3/group. (e) Cell contraction assay. CON or i‐EndoMT cells were harvested at 72 h after the treatment and resuspended with a total of 5 × 105 cells in a 1:4 ratio of cell suspension and collagen mixture provided in the cell contraction assay. The gels were imaged at 0 h and 72 h post‐incubation and analyzed by ImageJ software. n = 3/group. Red arrow (dashed line in photomicrograph) indicates the cell contraction. (f) Wound healing assay. CON or i‐EndoMT cells harvested at 72 h posttreatment were harvested and replated in a wound healing chamber. Images were taken 5 h later.
ETV2 overexpression partly converts i‐EndoMT and IPAH lung fibroblasts
To further explore the ability of ETV2 to change or convert EndoMT, HPAECs were cultured with the i‐EndoMT factors together with Doxycycline (Dox) inducible lentiviral particles of ETV2 for 6 days. As shown in Figure 6a, inducible overexpression of ETV2 leads to a significantly reduced number of cells with mesenchymal morphology, compared to control. Cells overexpressing ETV2 under i‐EndoMT had decreased expression of mesenchymal markers with increased EC makers, compared to control (Figure 6b and c). Similar results were found when PPARγ was overexpressed (Figure 6d). Importantly, overexpression of ETV2 increased the expression of EC makers with reduced mesenchymal makers in lung fibroblasts derived from IPAH patients (Figure 6e). These results suggest that the ETV2 as a direct downstream target of PPARγ plays important functions in EndoMT, and thus potentially PH pathogenesis.
Figure 6.

Overexpression of ETV2 induces endothelial cell phenotype. (a, b) HPAECs infected lentiviral particles of doxycycline‐inducible ETV2 (lenti‐ETV2) were incubated with 0.1 ng/mL IL‐1β, 10 ng/mL TNF‐α, and 10 ng/mL TGFβ for 72 h. The resulting cells were then treated ± Dox up to 6 days. A representative image of the resulting cells was shown in (a) (left panels). The cells were randomly selected and counted, the ratio of cobblestone morphology to elongated‐spindle‐shaped phenotype (a, right panel) was determined, and immunocytochemistry was performed (b). (c, d) HPAECs transfected with ETV2 plasmid constructs (oxETV2, 1 µg) or vector (VEC) constructs (c) or infected with AdPPARγ (25 MOI) (d) or green fluorescent protein (GFP) constructs were incubated with 0.1 ng/mL IL‐1β, 10 ng/mL TNF‐α, and 10 ng/mL TGFβ to HPAECs for 72 h. RNAs from the resulting cells were subjected to qRT‐PCR analysis. Twenty‐4 h before cell harvest (d), the cells were treated with rosiglitazone (RSG, 10 μM). Each bar represents the mean ± SE EndoMT or EC marker level relative to GAPDH as indicated. *p < 0.05 versus CON/VEC or i‐EndoMT/GFP. n = 3–4/group. (e) Lung fibroblasts of IPAH patients were infected with AdETV2 and 3 days later, the resulting cells were subjected to qRT‐PCR analysis. The results are presented as fold‐change versus CON. *p < 0.05 vs NOR, n = 5/group. (f) A hypothetical schema defining the role of PPARγ/ETV2 on EndoMT in PH pathogenesis.
DISCUSSION
The current study provides several novel findings that could advance the current understanding of PH pathogenesis; 1) ETV2 is downregulated in lungs and PAECs from patients with IPAH, in lungs of mice with PH caused by HYP/Su, in hypoxia‐exposed HPAECs, and in i‐EndoMT cells. 2) Reduction of ETV2 (i.e., Etv2 +/− mice) leads to a spontaneous development of PH, augmented RVH, and increased expression of EndoMT markers. 3) PPARγ acts as an upstream regulator of ETV2 expression. 4) Sustained expression of PPARγ or ETV2 in ECs inhibits the progression of EndoMT. Collectively, these findings suggest the novel function of the PPARγ‐ETV2 axis in regulating EndoMT (Figure 6f).
PH is a chronic cardiopulmonary disorder that causes significant morbidity and mortality. 1 , 3 , 38 , 39 Current targeted therapies in PH fail to reverse pulmonary vascular remodeling and are frequently not recommended for patients with more common forms of PH. 40 , 41 These observations indicate that detailed and novel insights into PH pathogenesis may permit more effective therapeutic strategies. Recent studies using cell lineage tracking analysis have shown that EndoMT contributes to the initiation and progression of PH. 10 , 14 , 34 This role of EndoMT was further supported by the colocalization of von Willebrand factor and αSMA in pulmonary endothelium from HYP/Su rodent models and PAH patients. 34 However, the molecular mechanisms underlying EndoMT in the pathogenesis of PH remain largely unknown. In this study, we propose the PPARγ‐ETV2 axis as a key mechanism in regulating EndoMT in PH.
ETV2 functions as a master regulator for EC generation and function. 15 , 42 While deficiency in Etv2 leads to a complete lack of vascular ECs, 16 , 17 forced expression of ETV2 is sufficient to reprogram non‐ECs including mesenchymal cells such as human dermal fibroblasts into functional ECs. 22 , 23 These findings suggested that substantial loss of EC in the progression of PH could be due to reduced expression of ETV2. In agreement with this hypothesis, we found a significant reduction in ETV2 expression in clinical and experimental PH lung samples. Reduction of ETV2 (Etv2 +/− mice) was sufficient to induce spontaneous PH and RVH in mice as well as upregulated expression of EndoMT makers and concomitant reductions of EC markers. These results strongly suggest the functional significance of ETV2 in PH. Although the precise molecular mechanisms by which ETV2 regulates PH is unknown, it is clear that ETV2 acts as a direct transcriptional regulator of diverse EC genes including CDH5 and CD31. 43 In addition, a recent report showed that ETV2 can regulate the expression of Robo4, one of the critical EC genes via DNA methylation. 44 Furthermore, we showed that ETV2 functions with valproic acid, a histone acetylation modifier in direct cell reprogramming. 23 These findings suggest that decreased expression of ETV2 in PH induces inactivation of EC gene expression and loss of normal EC function. Currently, it is not known whether increased mesenchymal cells in PH are the direct outcome of ETV2 downregulation or secondary to the loss of ECs. Therefore, determining the molecular mechanisms of ETV2‐EndoMT would be an important next step for PH.
The current study extends the field by providing novel insights into the regulation of ETV2 expression in PH. Given the established role of PPARγ in PH pathobiology, we performed in silico analysis of the upstream promoter region of ETV2 and identified several putative PPARγ binding sites. Our results showed that PPARγ can transcriptionally activate ETV2 expression. This finding was further supported by a series of experiments in vivo and in vitro. Mice lacking endothelial Pparγ exhibited a reduced level of expression of Etv2 in lungs, while mice overexpressing endothelial Pparγ showed augmented level of Etv2. Overexpression or knockdown experiments with PPARγ in vitro further substantiated these results. Collectively, these data suggest that PPARγ regulates PH in part through direct transcriptional activation of ETV2. Therefore, further studies to determine the functional consequences of the PPARγ‐ETV2 axis in PH in vivo will be warranted.
One of the key findings of this study was to refine an in vitro model, which can mimic EndoMT in vivo. A recent study demonstrated that a combination of inflammatory mediators (IL‐1β, TNF‐α, and TGFβ) induced EndoMT. 34 The current study revisited the report and demonstrated that these three inflammatory mediators phenotypically switched not only endothelial cells (cobblestone morphology) to mesenchymal cells (elongated, spindle‐shaped myofibroblastic cell morphology) in vitro, but also induced functional alterations such as enhanced contractile and migratory phenotypes in i‐EndoMT cells. Consistent with these phenotypic and functional features, i‐EndoMT cells expressed higher levels of EndoMT markers with downregulated levels of ETV2 and PPARγ. Importantly, we found that overexpression of PPARγ or ETV2 was able to reduce the expression of mesenchymal markers in cells undergoing i‐EndoMT, suggesting the potential role of PPARγ and ETV2 in EndoMT.
Recent studies have shown that ETV2 can directly reprogram non‐ECs into ECs. 22 , 23 In addition, delivery of lentiviral ETV2 into ischemic hindlimbs or infarcted heart promotes recovery of ischemic damage as evidenced by enhanced perfusion and cardiac functions, respectively. 21 , 45 Interestingly, massive neovascularization was also accompanied in the damaged tissues upon the overexpression of ETV2, suggesting its potent function in endothelial reprogramming and functional recovery of vascular defects. In this study, we showed that PAH‐derived lung myofibroblasts acquire EC characteristics while losing a mesenchymal phenotype when ETV2 was overexpressed. These results suggest that ETV2 could be a therapeutic target for PH in potentially converting mesenchymal cells to functional ECs.
In summary, to our knowledge, the current study provides for the first‐time evidence revealing novel functions of the PPARγ/ETV2 axis in regulating EndoMT during the pathogenesis of PH. Thus, the outcome of the proposed studies will advance current knowledge of EndoMT in PH and provide a new therapeutic paradigm for treating PH patients.
GUARANTOR STATEMENT
Not applicable.
AUTHOR CONTRIBUTIONS
Conception, hypothesis delineation, and design, Changwon Park and Bum‐Yong Kang; experiments, acquisition of data, analysis, and interpretation, Dong Hun Lee, Minseong Kim, Sarah S. Chang, Raham Lee, Andrew J. Jang, Juyoung Kim, Jing Ma, Michael J. Passineau, Raymond L. Benza, Harry Karmouty‐Quintana, Wilbur A. Lam, Benjamin T. Kopp, Roy L. Sutliff, C. Michael Hart, Changwon Park, and Bum‐Yong Kang; writing the manuscript, Dong Hun Lee, Minseong Kim, Sarah S. Chang, Wilbur A. Lam, C. Michael Hart, Changwon Park, and Bum‐Yong Kang.
ETHICS STATEMENT
All animal studies were approved by the Institutional Animal Care and Use Committee of Emory University or the Atlanta Veterans Affairs Healthcare System (IACUC No. V022‐17).
AUTHOR DISCLOSURES
Changwon Park has a United States Patent (No. 10,023,842 B2. Title: Endothelial and endothelial‐like cells produced from fibroblasts and uses related thereto), but the patent has nothing to do with the current manuscript. The other authors have no competing interests. The contents of this report represent the views of the authors and do not represent the views of the Department of Veterans Affairs or the United States Government.
Supporting information
Supporting information.
Supporting information.
Supporting information.
ACKNOWLEDGMENTS
This study was supported by funding from VA BLR&D Merit Review Award (I01 BX004263 to CMH), NIH National Heart, Lung, and Blood Institute R01 grants (HL102167 to CMH and RLS, HL119291 to CP, and HL133053 to BYK). Grants in Aid (Louisiana State University, HSC, Shreveport) to CP. Data/Tissue samples provided by PHBI under the Pulmonary Hypertension Breakthrough Initiative (PHBI). Funding for the PHBI is provided under an NHLBI R24 grant, #R24HL123767, and by the Cardiovascular Medical Research and Education Fund (CMREF).
Lee DH, Kim M, Chang SS, Lee R, Jang AJ, Kim J, Ma J, Passineau MJ, Benza RL, Karmouty‐Quintana H, Lam WA, Kopp BT, Sutliff RL, Hart CM, Park C, Kang B‐Y. PPARγ/ETV2 axis regulates endothelial‐to‐mesenchymal transition in pulmonary hypertension. Pulm Circ. 2024;14:e12448. 10.1002/pul2.12448
Dong Hun Lee, Minseong Kim and Sarah S. Chang authors contributed equally to this work.
Contributor Information
Changwon Park, Email: changwon.park@lsuhs.edu.
Bum‐Yong Kang, Email: Bum-Yong.Kang@emory.edu.
REFERENCES
- 1. Galie N, Manes A, Negro L, Palazzini M, Bacchi‐Reggiani ML, Branzi A. A meta‐analysis of randomized controlled trials in pulmonary arterial hypertension. Eur Heart J. 2008;30(4):394–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, Williams PG, Souza R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53(1):1801913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lilienfeld DE, Rubin LJ. Mortality from primary pulmonary hypertension in the United States, 1979‐1996. Chest. 2000;117(3):796–800. [DOI] [PubMed] [Google Scholar]
- 4. Ruopp NF, Cockrill BA. Diagnosis and treatment of pulmonary arterial hypertension: a review. JAMA. 2022;327(14):1379–1391. [DOI] [PubMed] [Google Scholar]
- 5. Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. 2008;118(7):2372–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thompson AAR, Lawrie A. Targeting vascular remodeling to treat pulmonary arterial hypertension. Trends Mol Med. 2017;23(1):31–45. [DOI] [PubMed] [Google Scholar]
- 7. Vonk Noordegraaf A, Westerhof BE, Westerhof N. The relationship between the right ventricle and its load in pulmonary hypertension. J Am Coll Cardiol. 2017;69(2):236–243. [DOI] [PubMed] [Google Scholar]
- 8. Yerabolu D, Weiss A, Kojonazarov B, Boehm M, Schlueter BC, Ruppert C, Günther A, Jonigk D, Grimminger F, Ghofrani HA, Seeger W, Weissmann N, Schermuly RT. Targeting Jak‐Stat signaling in experimental pulmonary hypertension. Am J Respir Cell Mol Biol. 2021;64(1):100–114. [DOI] [PubMed] [Google Scholar]
- 9. Napoli C, Loscalzo J. Nitric oxide and other novel therapies for pulmonary hypertension. J Cardiovasc Pharmacol Ther. 2004;9(1):1–8. [DOI] [PubMed] [Google Scholar]
- 10. Arciniegas E, Frid MG, Douglas IS, Stenmark KR. Perspectives on endothelial‐to‐mesenchymal transition: potential contribution to vascular remodeling in chronic pulmonary hypertension. Am J Physiol‐Lung Cell Mol Physiol. 2007;293(1):L1–L8. [DOI] [PubMed] [Google Scholar]
- 11. Sohal SS. Epithelial and endothelial cell plasticity in chronic obstructive pulmonary disease (COPD). Respir Investig. 2017;55(2):104–113. [DOI] [PubMed] [Google Scholar]
- 12. Suzuki T, Carrier EJ, Talati MH, Rathinasabapathy A, Chen X, Nishimura R, Tada Y, Tatsumi K, West J. Isolation and characterization of endothelial‐to‐mesenchymal transition cells in pulmonary arterial hypertension. Am J Physiol‐Lung Cell Mol Physiol. 2018;314(1):L118–L126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Qiao L, Nishimura T, Shi L, Sessions D, Thrasher A, Trudell JR, Berry GJ, Pearl RG, Kao PN. Endothelial fate mapping in mice with pulmonary hypertension. Circulation. 2014;129(6):692–703. [DOI] [PubMed] [Google Scholar]
- 14. Ranchoux B, Antigny F, Rucker‐Martin C, Hautefort A, Péchoux C, Bogaard HJ, Dorfmüller P, Remy S, Lecerf F, Planté S, Chat S, Fadel E, Houssaini A, Anegon I, Adnot S, Simonneau G, Humbert M, Cohen‐Kaminsky S, Perros F. Endothelial‐to‐mesenchymal transition in pulmonary hypertension. Circulation. 2015;131(11):1006–1018. [DOI] [PubMed] [Google Scholar]
- 15. Oh SY, Kim JY, Park C. The ETS factor, ETV2: a master regulator for vascular endothelial cell development. Mol Cells. 2015;38(12):1029–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee D, Park C, Lee H, Lugus JJ, Kim SH, Arentson E, Chung YS, Gomez G, Kyba M, Lin S, Janknecht R, Lim DS, Choi K. ER71 acts downstream of BMP, Notch, and Wnt signaling in blood and vessel progenitor specification. Cell Stem Cell. 2008;2(5):497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ferdous A, Caprioli A, Iacovino M, Martin CM, Morris J, Richardson JA, Latif S, Hammer RE, Harvey RP, Olson EN, Kyba M, Garry DJ. Nkx2‐5 transactivates the ets‐related protein 71 gene and specifies an endothelial/endocardial fate in the developing embryo. Proc Natl Acad Sci. 2009;106(3):814–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kataoka H, Hayashi M, Nakagawa R, Tanaka Y, Izumi N, Nishikawa S, Jakt ML, Tarui H, Nishikawa SI. Etv2/ER71 induces vascular mesoderm from Flk1+PDGFRα+ primitive mesoderm. Blood. 2011;118(26):6975–6986. [DOI] [PubMed] [Google Scholar]
- 19. Sumanas S, Lin S. Ets1‐related protein is a key regulator of vasculogenesis in zebrafish. PLoS Biol. 2005;4(1):e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Neuhaus H, Müller F, Hollemann T. Xenopus er71 is involved in vascular development. Dev Dyn. 2010;239(12):3436–3445. [DOI] [PubMed] [Google Scholar]
- 21. Park C, Lee TJ, Bhang SH, Liu F, Nakamura R, Oladipupo SS, Pitha‐Rowe I, Capoccia B, Choi HS, Kim TM, Urao N, Ushio‐Fukai M, Lee D, Miyoshi H, Kim BS, Lim DS, Apte RS, Ornitz DM, Choi K. Injury‐Mediated vascular regeneration requires endothelial ER71/ETV2. Arterioscler Thromb Vasc Biol. 2016;36(1):86–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morita R, Suzuki M, Kasahara H, Shimizu N, Shichita T, Sekiya T, Kimura A, Sasaki K, Yasukawa H, Yoshimura A. ETS transcription factor ETV2 directly converts human fibroblasts into functional endothelial cells. Proc Natl Acad Sci. 2015;112(1):160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lefever TW, Lee YOK, Kovach AL, Silinski MAR, Marusich JA, Thomas BF, Wiley JL. Delivery of nicotine aerosol to mice via a modified electronic cigarette device. Drug Alcohol Depend. 2017;172:80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Duan SZ, Usher MG, Mortensen RM. Peroxisome proliferator‐activated receptor‐γ–mediated effects in the vasculature. Circ Res. 2008;102(3):283–294. [DOI] [PubMed] [Google Scholar]
- 25. Ameshima S, Golpon H, Cool CD, Chan D, Vandivier RW, Gardai SJ, Wick M, Nemenoff RA, Geraci MW, Voelkel NF. Peroxisome proliferator‐activated receptor gamma (PPARγ) expression is decreased in pulmonary hypertension and affects endothelial cell growth. Circ Res. 2003;92(10):1162–1169. [DOI] [PubMed] [Google Scholar]
- 26. Guignabert C, Alvira CM, Alastalo TP, Sawada H, Hansmann G, Zhao M, Wang L, El‐Bizri N, Rabinovitch M. Tie2‐mediated loss of peroxisome proliferator‐activated receptor‐γ in mice causes PDGF receptor‐β‐dependent pulmonary arterial muscularization. Am J Physiol‐Lung Cell Mol Physiol. 2009;297(6):L1082–L1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tseng V, Sutliff RL, Hart CM. Redox biology of peroxisome Proliferator‐Activated receptor‐gamma in pulmonary hypertension. Antioxid Redox Signal. 2019;31(12):874–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Masri FA, Xu W, Comhair SAA, Asosingh K, Koo M, Vasanji A, Drazba J, Anand‐Apte B, Erzurum SC. Hyperproliferative apoptosis‐resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am J Physiol‐Lung Cell Mol Physiol. 2007;293(3):L548–L554. [DOI] [PubMed] [Google Scholar]
- 29. Kleinhenz JM, Kleinhenz DJ, You S, Ritzenthaler JD, Hansen JM, Archer DR, Sutliff RL, Hart CM. Disruption of endothelial peroxisome proliferator‐activated receptor‐γ reduces vascular nitric oxide production. Am J Physiol‐Heart Circ Physiol. 2009;297(5):H1647–H1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kang BY, Park KK, Kleinhenz JM, Murphy TC, Green DE, Bijli KM, Yeligar SM, Carthan KA, Searles CD, Sutliff RL, Hart CM. Peroxisome proliferator‐activated receptor γ and microRNA 98 in Hypoxia‐Induced Endothelin‐1 signaling. Am J Respir Cell Mol Biol. 2016;54(1):136–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kang BY, Park KK, Green DE, Bijli KM, Searles CD, Sutliff RL, Hart CM. Hypoxia mediates mutual repression between microRNA‐27a and PPARγ in the pulmonary vasculature. PLoS One. 2013;8(11):e79503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Koyano‐Nakagawa N, Shi X, Rasmussen TL, Das S, Walter CA, Garry DJ. Feedback mechanisms regulate ets variant 2 (Etv2) gene expression and hematoendothelial lineages. J Biol Chem. 2015;290(47):28107–28119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kang BY, Kleinhenz JM, Murphy TC, Hart CM. The PPARγ ligand rosiglitazone attenuates hypoxia‐induced endothelin signaling in vitro and in vivo. Am J Physiol‐Lung Cell Mol Physiol. 2011;301(6):L881–L891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Good RB, Gilbane AJ, Trinder SL, Denton CP, Coghlan G, Abraham DJ, Holmes AM. Endothelial to mesenchymal transition contributes to endothelial dysfunction in pulmonary arterial hypertension. Am J Pathol. 2015;185(7):1850–1858. [DOI] [PubMed] [Google Scholar]
- 35. Ahmed M, Zaghloul N, Zimmerman P, Casanova NG, Sun X, Song JH, Hernon VR, Sammani S, Rischard F, Rafikova O, Rafikov R, Makino A, Kempf CL, Camp SM, Wang J, Desai AA, Lussier Y, Yuan JXJ, Garcia JGN. Endothelial eNAMPT drives EndMT and preclinical PH: rescue by an eNAMPT‐neutralizing mAb. Pulm Circ. 2021;11(4):20458940211059712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Girgis RE, Ma SF, Ye S, Grigoryev DN, Li D, Hassoun PM, Tuder RM, Johns RA, Garcia JGN. Differential gene expression in chronic hypoxic pulmonary hypertension. Chest. 2005;128(6 Suppl):579S. [DOI] [PubMed] [Google Scholar]
- 37. Moreno‐Vinasco L, Gomberg‐Maitland M, Maitland ML, Desai AA, Singleton PA, Sammani S, Sam L, Liu Y, Husain AN, Lang RM, Ratain MJ, Lussier YA, Garcia JGN. Genomic assessment of a multikinase inhibitor, sorafenib, in a rodent model of pulmonary hypertension. Physiol Genom. 2008;33(2):278–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Adams AB, Durham MM, Kean L, Shirasugi N, Ha J, Williams MA, Rees PA, Cheung MC, Mittelstaedt S, Bingaman AW, Archer DR, Pearson TC, Waller EK, Larsen CP. Costimulation blockade, busulfan, and bone marrow promote titratable macrochimerism, induce transplantation tolerance, and correct genetic hemoglobinopathies with minimal myelosuppression. J Immunol. 2001;167(2):1103–1111. [DOI] [PubMed] [Google Scholar]
- 39. Salvaterra CG, Rubin LJ. Investigation and management of pulmonary hypertension in chronic obstructive pulmonary disease. Am Rev Respir Dis. 1993;148(5):1414–1417. [DOI] [PubMed] [Google Scholar]
- 40. George MG, Schieb LJ, Ayala C, Talwalkar A, Levant S. Pulmonary hypertension surveillance. Chest. 2014;146(2):476–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Poor HD, Girgis R, Studer SM. World health organization group III pulmonary hypertension. Prog Cardiovasc Dis. 2012;55(2):119–127. [DOI] [PubMed] [Google Scholar]
- 42. Lee DH, Kim TM, Kim JK, Park C. ETV2/ER71 transcription factor as a therapeutic vehicle for cardiovascular disease. Theranostics. 2019;9(19):5694–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu F, Li D, Yu YYL, Kang I, Cha MJ, Kim JY, Park C, Watson DK, Wang T, Choi K. Induction of hematopoietic and endothelial cell program orchestrated by ETS transcription factor ER71/ETV2. EMBO Rep. 2015;16(5):654–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tanaka T, Izawa K, Maniwa Y, Okamura M, Okada A, Yamaguchi T, Shirakura K, Maekawa N, Matsui H, Ishimoto K, Hino N, Nakagawa O, Aird WC, Mizuguchi H, Kawabata K, Doi T, Okada Y. ETV2‐TET1/TET2 complexes induce endothelial cell‐specific Robo4 expression via promoter demethylation. Sci Rep. 2018;8(1):5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee S, Lee DH, Park BW, Kim R, Hoang AD, Woo SK, Xiong W, Lee YJ, Ban K, Park HJ. In vivo transduction of ETV2 improves cardiac function and induces vascular regeneration following myocardial infarction. Exp Mol Med. 2019;51(2):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Supporting information.