

Abstract
Glioblastoma (GBM), characterized by high infiltrative capacity, is the most common and deadly type of primary brain tumor in adults. GBM cells, including therapy‐resistant glioblastoma stem‐like cells (GSCs), invade the healthy brain parenchyma to form secondary tumors even after patients undergo surgical resection and chemoradiotherapy. New techniques are therefore urgently needed to eradicate these residual tumor cells. A thiol‐Michael addition injectable hydrogel for compatibility with GBM therapy is previously characterized and optimized. This study aims to develop the hydrogel further to capture GBM/GSCs through CXCL12‐mediated chemotaxis. The release kinetics of hydrogel payloads are investigated, migration and invasion assays in response to chemoattractants are performed, and the GBM‐hydrogel interactions in vitro are studied. With a novel dual‐layer hydrogel platform, it is demonstrated that CXCL12 released from the synthetic hydrogel can induce the migration of U251 GBM cells and GSCs from the extracellular matrix microenvironment and promote invasion into the synthetic hydrogel via amoeboid migration. The survival of GBM cells entrapped deep into the synthetic hydrogel is limited, while live cells near the surface reinforce the hydrogel through fibronectin deposition. This synthetic hydrogel, therefore, demonstrates a promising method to attract and capture migratory GBM cells and GSCs responsive to CXCL12 chemotaxis.
Keywords: cell migration, collagen‐hyaluronic acid hydrogel, controlled delivery, CXCL12 chemotaxis, glioblastoma cells, glioblastoma stem‐like cells, thiol‐Michael addition hydrogel
This study focuses on developing a hydrogel to attract and entrap glioblastoma (GBM) and GBM stem‐like cells. Results from in vitro studies indicate that the sustained release of CXCL12 chemokines from the synthetic hydrogel induces the migration and invasion of the malignant cells into the hydrogel through an amoeboid blebbing migration mode.
1. Introduction
Glioblastoma (GBM) is a grade IV astrocytoma and the most malignant type of primary brain tumor, possessing an incident rate of 3.22 cases per 100 000 people.[ 1 ] Even after conventional therapies, such as maximum surgical resection, followed by adjuvant radiotherapy and chemotherapy are applied to target proliferating cells,[ 2 ] secondary tumors still recur and lead to 5‐year survival rates of only 5% post‐diagnosis.[ 3 ] This recurrence can be attributed to the infiltrative capacity and intratumoral heterogeneity of GBM.
The diffuse nature of GBM limits complete tumor removal, and the residual cancer cells can migrate from the resection cavity and invade 20–25 mm deep into healthy brain tissue[ 4 ] without being detected by radiographic imaging.[ 5 ] Nearly half of all GBM patients have tumors that resist treatment inherently, while this resistance is acquired in other instances.[ 6 ] According to Pisco and colleagues, some GBM treatments are reminiscent of a Lamarckian process and induce GBM cells to adapt and acquire resistance,[ 7 ] while Prager and colleagues argue that treatments can serve as a Darwinian process to be selective toward and expand GBM therapy‐resistant subclones.[ 8 ] Further compounding this issue of heterogeneity is the presence of GBM cells with stem‐like properties, known as glioblastoma stem cells (GSCs). GSCs not only overexpress DNA damage repair enzymes and possess the metabolic characteristics necessary to resist chemoradiotherapy but can self‐renew with tumorigenicity to form secondary tumors, while the differential expression of various GSC biomarkers renders it difficult to selectively target them.[ 9 , 10 , 11 , 12 ] Moreover, GSCs are more migratory with an elevated invasion potential, enabling these cells to drive GBM recurrence post‐resection,[ 13 ] but we believe this characteristic may also provide a potential avenue for GSC therapeutic targeting. It is currently difficult to eradicate both GBM and GSC populations, as several signaling pathways present in these malignant cells are also conserved within healthy neural stem cells.[ 14 ]
Preclinical models indicate GBM cells can migrate with unidirectional movements at velocities ranging from 2 to 6 µm h−1.[ 15 ] Perivascular migration, also known as vessel co‐option, is the most prevalent method of invasion for certain GBM cell lines.[ 16 ] In this method, pre‐existing vascular structures serve as scaffolds for GBM migration, as the chemoattractant gradients from CXCL12 chemokine production by blood vessels attract the GBM cells.[ 17 ] GBM migration is a complex process impacted by the signaling paths and interactions between the tumor and its extracellular matrix (ECM),[ 18 , 19 ] where invading cells first detach from the primary tumor mass, adhere to the ECM, and then subsequently degrade the ECM to migrate using motility and cell contractility.[ 20 ]
Clinical and lab research during the last three decades has not yielded significant increases in GBM patient survival times.[ 21 ] Several therapies have attempted to mitigate GBM invasion by targeting their migratory potential. For example, the upregulation of chemokine receptors (such as CXCR4) in invasive GBM cells makes them more responsive to chemotactic cues compared to healthy brain cells or non‐invasive GBM cells.[ 22 , 23 ] Although inhibition of the CXCR4 receptors in vitro can reduce GBM migration,[ 24 ] similar results were not obtained in vivo.[ 25 ] These findings are likely due to GBM cells' ability to bind CXCL12 to an additional receptor like CXCR7, which enables the cells to utilize compensation mechanisms to activate one receptor when the other receptor is inhibited.[ 26 , 27 ] However, these limitations present a unique opportunity to exploit the invasive potential of GBM cells for therapeutic applications.
In their work, Kim and colleagues determined that cytokines released by breast carcinoma and melanoma can promote the colonization of circulating cancer cells to self‐seed and reinfiltrate the tumors of origin, thereby contributing to local recurrences in solid tumors after resection.[ 28 ] The concept of ecological trap developed by Van Der Sanden and colleagues builds on this tumor self‐seeding phenomenon for GBM treatment.[ 29 ] Specifically, the researchers purport that gradients and migratory cues can be utilized to guide GBM cells to a particular region to concentrate the cells and eradicate them with localized therapy.[ 29 ] Recent research efforts have attempted to implement the ecological cell trap concept by developing a biomaterial scaffold that can be implanted into the resection cavity post‐surgery to attract and entrap residual GBM cells. Biomaterials, such as biocompatible hydrogels, are promising in GBM therapies due to their potential stimuli‐responsive behavior, capacity to be modified for flexibility in tuning toward specific applications, and ability to be loaded with therapeutic agents for controlled delivery. Autier and colleagues developed a bacterial cellulose‐based scaffold loaded with conditioned media from glioma‐associated stromal cells for implantation in the resection cavity to capture GBM cells with chemotaxis for ablation by stereotactic radiosurgery.[ 30 ] However, the release kinetics demonstrated fast release, as 98.2% of the hydrogel payload comprising the model protein human serum albumin was released within 24 h, which contributed to a limited ability to maintain the chemoattractant gradient for a sustained period of time. Although the Transwell assays demonstrated F98 glioma cells migrated in response to this chemoattractant released by the hydrogel, the scaffold was only able to attract nearby GBM cells due to diffusion limitations up to 5 mm away as reported by organotypic brain slice assays, while cell entrapment was limited to adherence on the gelatinous hydrogel surface only. The non‐degradable nature of the scaffold would require surgical removal in patients after treatment, and ablation with stereotactic radiosurgery may be unable to completely eradicate GSCs, which are resistant to radiotherapy compared to GBM cells.[ 31 ]
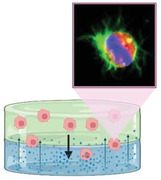
In order to address these challenges, we are developing a synthetic and biodegradable, injectable hydrogel platform to attract and entrap GBM/GSCs for their subsequent ablation utilizing both chemical and physical stimuli. In contrast to implantable hydrogels, injectable hydrogels are advantageous, as they increase patient comfort by undergoing sol–gel transition in situ and can conform to the patient‐specific anatomy. After resection of the primary GBM tumor mass, an injectable hydrogel loaded with chemokines can be dispensed into the resection cavity, where the sustained release of the chemokines can generate a chemoattractant gradient to induce the migration of residual GBM and GSCs into the hydrogel, as indicated by the schematic overview for clinical application of this concept in Figure 1 . Our current study thereby focuses on developing this hydrogel platform to release chemokines to attract and capture invasive GSCs and GBM cells. We had previously systematically tuned and characterized nine formulations of a poly(ethylene glycol)‐based injectable hydrogel, which was crosslinked via a thiol‐Michael addition click reaction.[ 32 ] We had determined that the hydrogel formulation comprising 0.175 m NaHCO3(aq) and 50 wt% water content was the most optimal for biological, physical, and chemical compatibility with the GBM microenvironment and our proposed GBM therapy by systematically characterizing the hydrogel swelling ratios, gel fractions, degradations, viscoelastic properties, as well as the hydrogel interactions with normal human astrocytes, on the basis of cytotoxicity and immunogenicity.[ 32 ] Furthermore, the optimized formulation of the hydrogel possessed mesh sizes with well‐cross‐linked networks in the nanometer range, which may help sustain the release of chemokine payloads over time. In this study, we guide GBM/GSC migration with CXCL12‐mediated chemoattraction and develop our synthetic hydrogel further to capture these malignant cells in vitro. We utilize a novel 3D‐engineered model of the GBM tumor microenvironment with a dual‐layered platform comprising a synthetic hydrogel interfaced with an ECM for therapy development and preclinical testing prior to in vivo models. We also investigate the interactions between the malignant cells and this ECM‐synthetic hydrogel platform to elucidate the invasion pathway and identify further research avenues to improve GBM/GSC cell entrapment in the synthetic hydrogel for their subsequent ablation. Specifically, future research will focus on developing our hydrogel further by integrating electrotaxis[ 33 ] to enhance and increase malignant cell invasion into the hydrogels and investigate eradication of the entrapped cells using minimally or non‐invasive ablation technologies such as focused ultrasound[ 34 ] or irreversible electroporation.[ 35 ]
Figure 1.
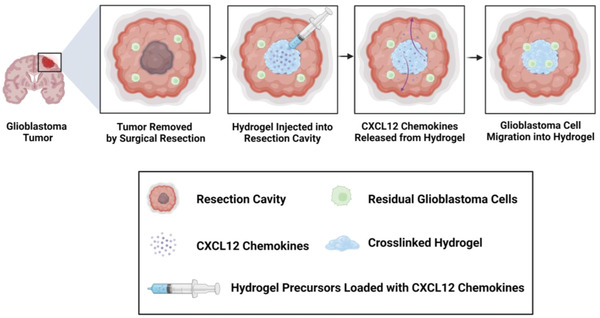
Schematic overview and clinical application of proposed CXCL12‐mediated chemotaxis of residual GBM cells. After resection of the primary tumor mass, an injectable hydrogel loaded with CXCL12 chemokines can be dispensed for crosslinking in the resection cavity in situ. Sustained release of the chemokines can generate a chemotactic gradient and induce the migration of residual GBM cells near the resection cavity for their subsequent invasion into the injectable hydrogel. Upon localization, the malignant cells can be eradicated with a non‐invasive ablation technique such as focused ultrasound. Created with BioRender.com.
2. Results
2.1. FITC‐Dextran Release Kinetics and Predicted Model
In order to assess the kinetics of payloads released from the synthetic hydrogels, fluorescein isothiocyanate (FITC)‐dextran at 10 kDa was loaded at concentrations of either 0.5, 5, or 10 µg mL−1. The FITC‐dextran released from the hydrogels was monitored until complete hydrogel degradation. As indicated by the cumulative release profile in Figure 2a, varying the loading concentration impacted the total cumulative release. Lower loading concentrations led to the more rapid and higher release, as the 0.5 µg mL−1 loading concentration resulted in a final 98.5% cumulative release, 5 µg mL−1 led to 94.1% release, and 10 µg mL−1 led to 80.2% release. In all cases, a burst release was observed during the first 96 h, in which the cumulative releases were 84.4%, 76.5%, and 68.2% with respect to 0.5, 5, and 10 µg mL−1 loading concentrations. Afterward, the release reached a plateau, until the final FITC‐dextran remnants were released and corresponded to complete hydrogel degradation. A higher loading concentration led to a slower degradation of the hydrogels, and the highest loading concentration of 10 µg mL−1 led to the most prolonged and sustained release of FITC‐dextran by releasing any retained FITC‐dextran in its network at the 384 h time point, ≈24–48 h beyond the complete degradation time points observed for the lower payload concentration hydrogels. While the experimental data indicated the release profiles followed a sigmoidal curve shape, the computational model based on the diffusivity data from the 5 µg mL−1 loading concentration predicted a hyperbolic release curve with 90.9% cumulative release by 336 h (Figure 2a). This predicted cumulative release profile was the same regardless of the initial loading concentration. Figure 2b illustrates the total amount of FITC‐dextran released for every time point. After the initial exponential release during the first 24 h, the highest loading concentration of 10 µg mL−1 resulted in the highest amount of FITC‐dextran released per day, while 0.5 µg mL−1 loading concentration resulted in considerably lower amounts of release per day. 5 µg mL−1 loading concentrations led to a release amount in between these two extremes per day and was determined to be the most optimal loading concentration for subsequent CXCL12‐based studies.
Figure 2.
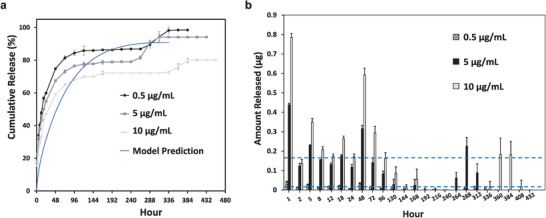
Release kinetics of 10 kDa FITC‐dextran payload from synthetic hydrogels submerged in 1× phosphate buffered saline solution (PBS) at 37 °C for initial loading concentrations of 0.5, 5, and 10 µg mL−1. a) Comparison of cumulative release over time for different hydrogel payload concentrations and predicted cumulative release profile from the computational model based on Fickian diffusion and hydrogel degradation. b) Total amount of FITC‐dextran released over time from hydrogels for each loading concentration. All data are expressed as mean ± SD (n = 4). Blue dashed lines correspond to thresholds of 0.01 and 0.16 µg.
2.2. CXCL12 Release from Synthetic Hydrogels
Synthetic hydrogels were loaded with 5 µg mL−1 of CXCL12 and maintained in 1× phosphate‐buffered saline (PBS) at 37 °C to monitor the chemokine release over 72 h. An enzyme‐linked immunosorbent assay (ELISA) was used to quantify the concentration of CXCL12 released from the hydrogel and into the PBS over time. The cumulative release profile from Figure 3a demonstrated that the CXCL12 release from the hydrogels was slow and sustained, with ≈9.8 ng of CXCL12 released by 24 h. As indicated by Figure 3b, the variation in the amount of CXCL12 released from the hydrogels increased over time.
Figure 3.
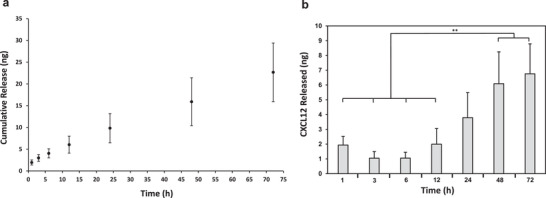
CXCL12 release from synthetic hydrogels submerged in 1× PBS at 37 °C for loading concentration of 5 µg mL−1. a) Cumulative release (ng) of CXCL12 over 72 h. b) Amount of CXCL12 (ng) released for each time point from hydrogels. All data are expressed as mean ± SD (n = 4) by one way analysis of variance (ANOVA) and Tukey's post‐hoc analysis. **p < 0.01.
2.3. CXCL12‐Mediated Migration of GBM Cells and GSCs
The feasibility of using the synthetic hydrogel to release chemoattractants and induce the migration of the GBM/GSCs was next assessed. A Transwell migration assay was set up according to the schematic diagram in Figure 4a. Either GBM or GSC U251 cells were loaded onto the upper chamber of the insert and the number of cells that migrated through the membrane and onto the underside in response to the chemoattractant on the bottom chamber was quantified. All cell migration data were normalized to the corresponding control group comprising only the migration media as the chemoattractant. According to Figure 4b, the highest migration index of the GBM cells across both time points was in response to the positive control comprising CXCL12 in solution at a concentration of 0.2 µg mL−1. The GBM cells had a significantly higher migratory response to U251 GBM hydrogel‐encapsulated cells compared to the control group. After 48 h, the GBM cells also migrated significantly more in response to CXCL12 released from hydrogels compared to the control group. Similar to the GBM cells, the U251 GSCs were most responsive across both time points for the positive control comprising CXCL12 in solution at high concentrations of 0.2 µg mL−1. However, the GSCs were only significantly more responsive to GBM‐loaded hydrogels compared to the control during the first 24 h only (Figure 4c). The GSCs maintained migratory responses to CXCL12 released from hydrogels during both time points, although this significant difference against the control group was lower during the 48 h time point compared to the 24 h time point. The GSC migration index during 48 h of incubation with CXCL12 in solution was 10.2 units higher than the 24 h time point. In contrast, the corresponding increase in the GBM migration index for CXCL12 in solution was more modest at 4.5 units. For GBM and GSCs across both time points, the migration index in response to the control migration media was not significantly different from the empty hydrogel group.
Figure 4.
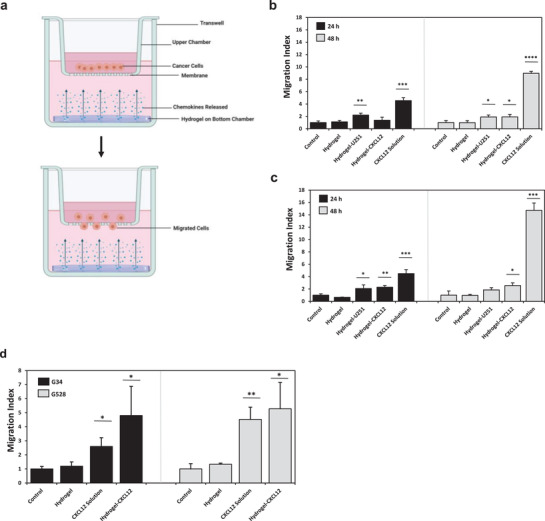
Glioblastoma and glioblastoma stem‐like cell migration in response to chemoattraction in vitro. a) Schematic overview of Transwell setup for migration assay. Created with BioRender.com. 20 000 U251 GBM, U251 GSC, G34, or G528 cells were loaded on the top chamber. The number of cells that migrated through the Transwell insert (8 µm pore) to the underside in response to the chemoattractant on the bottom chamber was quantified. Chemoattractant groups on the bottom chamber were either the migration media (control) comprising Dulbecco's Modified Eagle Medium (DMEM) supplemented with 0.1% bovine serum albumin for U251 cells or complete Neurobasal media without growth factors for G34/G528 cells, empty synthetic hydrogels in migration media (hydrogel), synthetic hydrogels loaded with 5 µg mL−1 of CXCL12 in migration media (hydrogel‐CXCL12), or 0.2 µg mL−1 of CXCL12 in migration media (CXCL12 solution). Transwell assays with the U251 cells had an additional chemoattraction condition comprising synthetic hydrogels loaded with 3 × 106 U251 GBM cells in migration media (hydrogel‐U251). The number of cells quantified was averaged from ten random fields of view per sample at 10× with a confocal microscope. Migration index reported as ratio of number of migrated cells from the sample to the number of migrated cells from the control group. Migration index of b) U251 GBM cells at 24 or 48 h time point, c) U251 GSCs at 24 or 48 h time point, and d) G34 and G528 GSCs at 24 h time point. Data shown are mean ± SD (n = 3), *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by Student's t‐test against the corresponding control group.
The Transwell migration assay was repeated for a 24 h time point with primary, patient‐derived G34 and G528 GSCs with conditions comprising the control migration media, empty hydrogel, CXCL12 in solution at 0.2 µg mL−1, and CXCL12 loaded into hydrogels at 5 µg mL−1. The results, as indicated by Figure 4d, revealed that both patient‐derived GSCs had significantly higher migratory responses from chemokines released from the hydrogels compared to the control group. For each cell type, the hydrogel loaded with CXCL12 induced a migration response that was similar to the positive control comprising CXCL12 in the solution. G528 cells had a higher migration index in response to both CXCL12 in solution and CXCL12 released from the hydrogels compared to the G34 cells. In particular, the migration index of G528 cells in response to hydrogel‐CXCL12 was 5.3, which was ≈0.5 migration index units higher than the G34 cells' response to hydrogel‐CXCL12.
2.4. CXCL12‐Mediated Invasion of U251 Cells
The bioactivity of CXCL12 loaded into the synthetic hydrogels and the feasibility of using this chemokine to induce U251 GBM and GSC invasion in vitro were assessed. A novel dual‐layered hydrogel comprising the collagen‐hyaluronic acid (HA) hydrogel on top and the synthetic hydrogel on the bottom was synthesized in polydimethylsiloxane (PDMS) stamps as indicated by the schematic diagram in Figure 5a to mimic the interface of the ECM and synthetic hydrogel. The ECM hydrogel layer on top encapsulated either U251 GBM or GSCs, while the synthetic hydrogel on the bottom was left empty or loaded with 5 µg mL−1 of CXCL12 chemokines. After 24 h of cell culture, the dual‐layer hydrogel was imaged with confocal microscope reflectance at 640 nm to demarcate the two hydrogel layers and identify the interface. Once this z position was identified, the hydrogels were switched to imaging with the 488 nm and DAPI filters to visualize the cells that invaded into the synthetic hydrogel and assess the cell morphologies. The total number of cells that had invaded at least 20 µm deep into the synthetic hydrogel was quantified for each cell type. No cells had invaded beyond 20 µm deep in the empty synthetic hydrogels (Figure 5c,e), while both GBM and GSC cell invasion into the hydrogels loaded with CXCL12 was observed, as quantified in Figure 5b and depicted by Figure 5d,f. The number of U251 GSCs and GBM cells that had invaded into the synthetic hydrogel from the ECM hydrogel layer in response to CXCL12 was not significantly different from each other. Both GBM and GSCs remained round, as indicated by the green actin staining during the invasion assay endpoint (Figure 5c–f). The maximum depth of invasion into the synthetic hydrogel ranged from 22 to 79 µm for the GBM cells and ranged from 32 to 120 µm for the GSCs.
Figure 5.
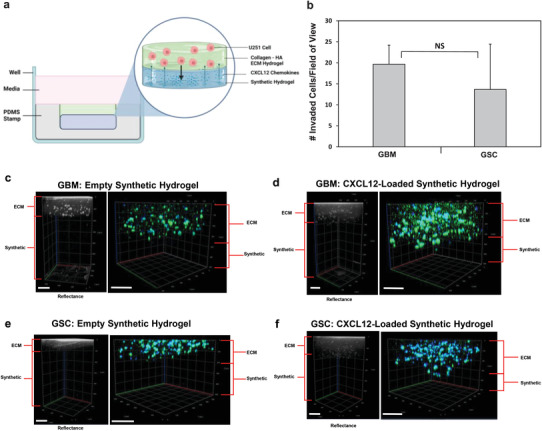
Glioblastoma and glioblastoma stem‐like cell invasion in response to CXCL12‐mediated chemotaxis in vitro. a) Schematic overview of dual layer hydrogel invasion assay setup. Created with BioRender.com. Either empty or 5 µg mL−1 CXCL12‐loaded synthetic hydrogels were synthesized in PDMS molds as the bottom layer. On the top layer, collagen‐HA hydrogels were synthesized encapsulating either U251 GBM or GSCs at 1 × 106 cells/mL. After 24 h of cell culture, reflectance confocal z‐stack imaging at 640 nm was used to demarcate the two hydrogel layers. Blue is DAPI and green is actin filament staining. b) Quantification of GSCs or GBM cells that invaded at least 20 µm (1 cell diameter) deep in synthetic hydrogel due to CXCL12. Data shown are mean ± SD (n = 3) by Student's t‐test. No background invasion for empty synthetic hydrogels was observed. Reflectance and confocal z‐stack images of U251 cell invasion after 24 h in dual‐layer hydrogel for c) GBM cells and empty synthetic hydrogel, d) GBM cells and CXCL12‐loaded synthetic hydrogel, e) GSCs and empty synthetic hydrogel, and f) GSCs and CXCL12‐loaded synthetic hydrogel on the bottom layer. Scale bars represent 200 µm.
2.5. Blebbistatin Treatment and Myosin IIA Immunofluorescence
The U251 GBM invasion assay was repeated with the addition of a blebbistatin treatment group and immunostaining for myosin IIA in order to determine the mechanism of invasion into the synthetic hydrogels. As demonstrated by Figure 6a, no GBM invasion from the ECM hydrogel and into the synthetic hydrogel layer was observed for empty synthetic hydrogels without CXCL12 chemokines. Similarly, no GBM cell migration into the synthetic hydrogels was observed when samples were treated with blebbistatin (Figure 6c). In contrast, cell invasion into the synthetic hydrogel was observed for samples loaded with CXCL12 chemokines without being subjected to blebbistatin treatment (Figure 6b). Cells had also remained round for all three groups during the invasion assay (Figure 6d). A semi‐quantitative analysis was performed to quantify the normalized myosin IIA fluorescence of the U251 GBM cells across the three sample groups during migration through the ECM hydrogel layer according to our previous protocol.[ 32 ] As illustrated by Figure 6e, the normalized myosin IIA fluorescence in samples where invasion occurred (CXCL12‐loaded hydrogels) was significantly higher than in samples where no invasion of the GBM cells was observed (empty and blebbistatin‐treated groups). Images of the ECM hydrogel layer acquired through second harmonic generation with a multiphoton confocal microscope (Figure 6f) indicated that the collagen fibers formed a network in the ECM hydrogel layer. The pores and defects present on the surface of the synthetic hydrogels ranged in size from 0.799 to 16.61 µm, as determined by reflectance imaging with a multiphoton confocal microscope at 543 nm.
Figure 6.
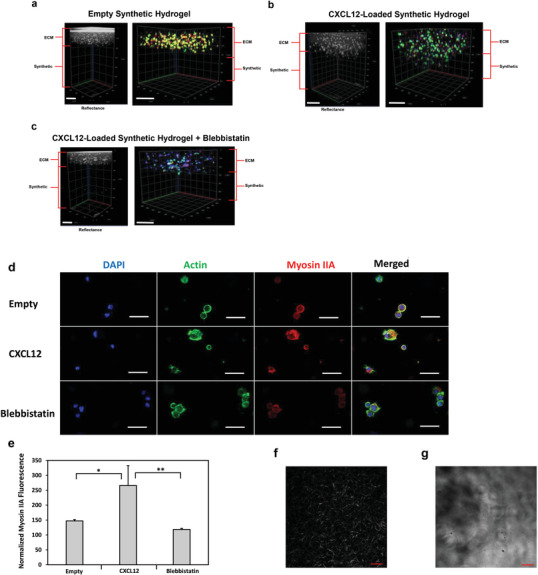
Mechanism of U251 GBM invasion into synthetic hydrogel from collagen‐HA hydrogel layer. Either empty or 5 µg mL−1 CXCL12‐loaded synthetic hydrogels were synthesized in PDMS molds as the bottom layer. On the top layer, collagen‐HA hydrogels were synthesized encapsulating U251 GBM cells at 1 × 106 cells/mL. After 14 h, reflectance confocal z‐stack imaging at 640 nm was used to demarcate the two hydrogel layers. Blue is DAPI, green is actin filament staining, and red is myosin IIA. Cell invasion in response to a) empty synthetic hydrogels, b) CXCL12‐loaded synthetic hydrogels, and c) CXCL12‐loaded synthetic hydrogels subjected to 30 µm of (‐)‐blebbistatin incubation. Scale bars represent 200 µm. d) Representative 1.61 µm optical slice images of the cells in the collagen‐HA layer for each corresponding sample group. Scale bars represent 50 µm. e) Quantification of normalized myosin IIA fluorescence intensities based on three cells from five representative images in the collagen‐HA layer with a confocal microscope at 20×. Data shown are mean ± SD (n = 3) by one way ANOVA and Tukey's post‐hoc analysis. No background invasion for empty synthetic hydrogels or blebbistatin‐treated samples was observed. *p‐value < 0.05, **p‐value < 0.01. f) Representative second harmonic generation image of U251 GBM‐encapsulated collagen‐HA hydrogel after 24 h of culture. Scale bar is 50 µm. g) Representative reflectance (543 nm) image of synthetic hydrogel swelled for 24 h in PBS at 37 °C. Scale bar is 50 µm.
2.6. Immunofluorescence of Stem Cell and Glial Markers during U251 GBM and GSC Interaction with Hydrogel Surface
The GBM and GSC interactions with the ECM and synthetic hydrogel surfaces were examined by assessing nestin and glial fibrillary acidic protein (GFAP) immunofluorescence when the cells were cultured on the surface of each hydrogel type over the course of 24 (Figure 7a) and 48 h (Figure 7b). While both cell types remained round on the synthetic hydrogel surface, the cells formed networks on the control ECM hydrogel surface. A semi‐quantitative analysis revealed that the normalized GFAP fluorescence of the GBM cells cultured on the synthetic hydrogel at the 24 h time point was significantly the highest, more so than the GSCs cultured on the synthetic hydrogel at the 24 h time point, GBM cells cultured on the synthetic hydrogel at the 48 h time point, and GBM cells cultured on the ECM hydrogels at both time points (Figure 7c). In contrast, there was no significant difference in normalized GFAP fluorescence between the two cell types when cultured on the ECM hydrogels, while the GBM cells cultured on synthetic hydrogels at the 48 h time point continued to be significantly higher than the corresponding GSCs on the synthetic hydrogel as well as the GBM cells in the ECM hydrogel at both time points. The normalized nestin fluorescence was significantly lower in the GBM cells compared to the GSCs on the synthetic hydrogel surface during the first 24 h only (Figure 7d). The two cell types also maintained similar levels of normalized nestin fluorescence when cultured on the surface of the ECM hydrogel during both time points and for the synthetic hydrogel at the 48 h time point. The GSC normalized nestin fluorescence was significantly higher in the synthetic hydrogel during 24 h compared to GSCs cultured on the synthetic hydrogel at 48 h and GSCs on ECM hydrogel at both time points.
Figure 7.
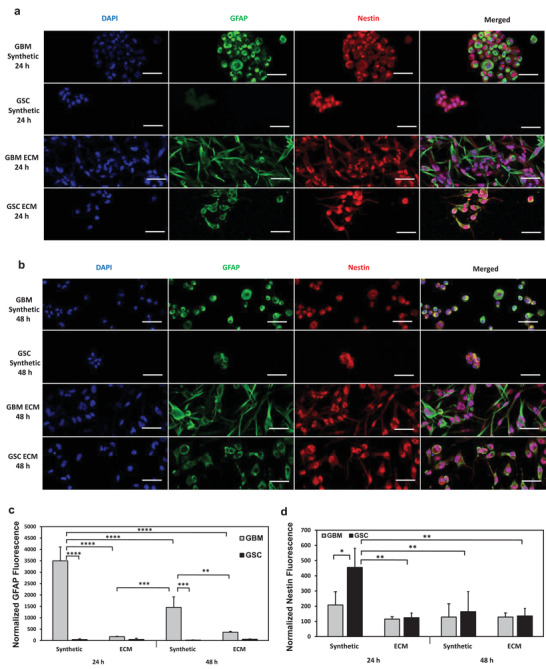
Immunofluorescence analysis of stem cell and glial markers during U251 GBM and GSC interaction with synthetic hydrogel and collagen‐HA hydrogel surfaces. 100 000 U251 GBM cells or GSCs were seeded on the surface of either synthetic hydrogels or collagen‐HA ECM hydrogels. 1.61 µm thick representative optical slice images of the cells focused on the hydrogel surface for a) 24 h time point and b) 48 h time point. Quantification of normalized c) GFAP fluorescence and d) nestin fluorescence. Ten fields of view were randomly selected and three cells were analyzed per field of view. Data shown are mean ± SD (n = 3) by one way ANOVA and Tukey's post‐hoc analysis. *p < 0.05, **p < 0.01, ***p < 0.001,****p < 0.0001. Scale bar is 50 µm.
2.7. Hydrogel Encapsulated U251 GBM Cell Viability
The viability of U251 GBM cells entrapped in the synthetic or ECM hydrogels was compared and assessed through a live/dead assay over the course of 120 h. As indicated by Figure 8a, the GBM cells entrapped in synthetic hydrogels maintained a rounded morphology and tended to cluster together, while cells in the control ECM hydrogels elongated to form networks. The calcein AM green stained live cells were more abundant near the surface of the synthetic hydrogels, while cells deeper beyond 400 µm in the synthetic hydrogels were not viable, as indicated by the propidium iodide staining through z‐stack images (Figure 8b). In contrast, both live and dead cells were spread throughout the entirety of the ECM hydrogels. Therefore, the viabilities of cells within 300 µm of the hydrogel surfaces were quantified for each hydrogel. The viability of GBM cells entrapped within 300 µm in the synthetic hydrogels ranged from 19.5% to 23.3%, which was significantly lower than the viability in ECM hydrogels ranging from 97.1% to 98.3% within the same corresponding depth (Figure 8c). Throughout the duration of the experiment up to 120 h, the cell viabilities in the ECM hydrogel or the synthetic hydrogel within this depth did not significantly differ with time.
Figure 8.
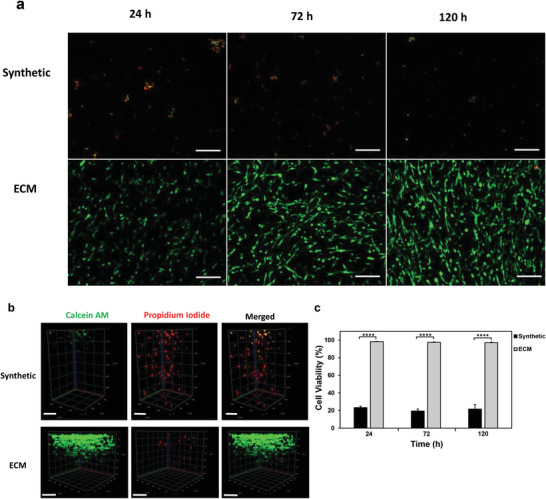
Viability of GBM cells encapsulated in synthetic or collagen‐HA hydrogels. U251 GBM cells were encapsulated in synthetic or collagen‐HA ECM hydrogels at a density of 1 × 106 cells/mL and cultured for up to 120 h in complete DMEM media. a) Representative images of cells encapsulated in both hydrogels subjected to live/dead staining at 24, 72, and 120 h time points. Red stain is propidium iodide (dead cells) and green stain is calcein AM (live cells). Scale bar is 200 µm. Images acquired up to 300 µm deep in each hydrogel. b) Representative z‐stack images of live/dead stained U251 GBM cells encapsulated either in the synthetic hydrogel or collagen‐HA hydrogel during the 120 h time point. Scale bar is 200 µm. c) Viability of U251 GBM cells encapsulated either in collagen‐HA hydrogel or synthetic hydrogel over time. Cell viability is based on the percentage of live cells over total number of cells. Ten fields of view were randomly selected per hydrogel sample to quantify the number of live and dead cells per hydrogel. Acquired images were limited to 300 µm deep in each hydrogel for analysis. All data are averages ± SD (n = 3), ****p‐value < 0.0001 by one way ANOVA and Tukey's post‐hoc analysis. No significant difference in cell viability within ECM hydrogels or synthetic hydrogels over time.
2.8. Degradation of U251 GBM‐Encapsulated Synthetic Hydrogels
The impact of entrapped GBM cells on the hydrogel stability was next assessed. Synthetic hydrogels were either loaded with U251 GBM cells or left empty, and the degradation of the hydrogels in terms of percent mass loss was monitored until complete degradation. As demonstrated by Figure 9a, the empty acellular hydrogels began to degrade significantly faster than cellular hydrogels starting from the first 24 h of cell culture. While acellular hydrogels degraded within 120 h, cellular hydrogels continued to remain intact and stable at the 120 h time point (Figure 9b) until degradation was observed at 168 h. While the percent mass loss in cellular hydrogels steadily increased with each designated time point, the increase in percent mass loss rose from 49.4% to 81.7% between the 72 and 120 h time points for acellular hydrogels, which was a rapid 32.3% increase in the degradation of the hydrogel.
Figure 9.
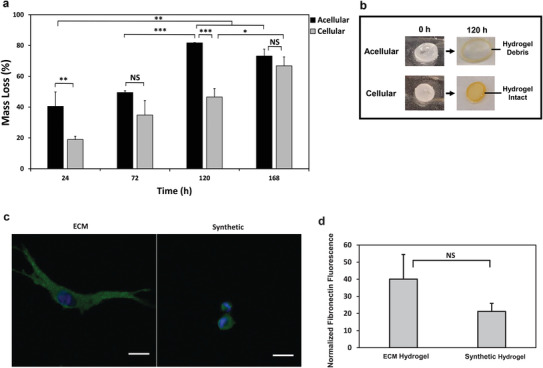
Hydrogel degradation and fibronectin deposition by encapsulated U251 GBM cells. a) Total degradation of either empty synthetic hydrogels (acellular) or synthetic hydrogels encapsulated with 3 × 106 cells/mL of U251 GBM cells (cellular) over time. All data shown are mean ± SD (n = 3) by one way ANOVA and Tukey's post‐hoc analysis. b) Representative images comparing the complete degradation of acellular hydrogels from 0 to 120 h against intact cellular hydrogels from 0 to 120 h during the degradation study. c) Representative 1.61 µm optical slice images of U251 GBM cells encapsulated in synthetic or collagen‐HA ECM hydrogels depositing fibronectin. Cells were encapsulated at a density of 1 × 106 cells/mL and cultured in complete DMEM media for 72 h. Blue is DAPI and green is fibronectin. Scale bar is 20 µm. d) Quantification of normalized fibronectin deposition for U251 GBM cells encapsulated in collagen‐HA ECM hydrogels or synthetic hydrogels. All data shown are mean ± SD (n = 3) by one way ANOVA and Tukey's post‐hoc analysis. Five fields of view were randomly selected per hydrogel and three cells were analyzed per field of view at 20×. Acquired images were limited to 300 µm deep in each hydrogel.
2.9. Fibronectin Deposition
The deposition of fibronectin by GBM cells upon entrapment in the synthetic or control ECM hydrogels was next determined. U251 GBM cells were encapsulated into both synthetic and ECM hydrogels. After 72 h of cell culture in the two hydrogel groups, immunofluorescence staining was used to image the fibronectin deposition by the entrapped cells, and the normalized fluorescence was determined semi‐quantitatively with confocal microscope imaging. As indicated by Figure 9c, U251 GBM cells secreted fibronectin in both the ECM hydrogels and the synthetic hydrogels. A comparison between these two hydrogels indicated that although cells encapsulated in the ECM hydrogels deposited higher normalized fluorescent levels of fibronectin than the cells encapsulated in the synthetic hydrogels, this increase was not statistically significant (Figure 9d). As indicated by confocal z‐stack images in Figure S1, Supporting Information, the fibronectin deposition by cells in the synthetic hydrogel was only up to 400 µm deep in the hydrogel, as beyond this depth, no fibronectin deposition was observed. Therefore, all images were acquired up to 300 µm deep in each type of hydrogel for analysis.
2.10. Hydrogel Electroconductivity
The electroconductivity of both freshly prepared synthetic hydrogels (cured) and synthetic hydrogels maintained in 1× PBS for 24 h (equilibrated) was determined by applying direct current electric fields with ≈50 V. The resulting electroconductivity data are provided in Table 1 and indicated that equilibrated hydrogels possessed an electroconductivity of 0.329 S m−1, which was about 53% higher than freshly cured hydrogels at 0.255 S m−1. The hydrogel precursor solutions, including the Thiocure, poly(ethylene glycol) diacrylate (PEGDA), 0.175 m NaHCO3(aq ), and its corresponding deionized water were also assessed for their electroconductive properties. The results indicated that while Thiocure and PEGDA were not conductive, the base solution was highly conductive (1.23 S m−1), indicating the hydrogel conductivities were derived from the base solution.
Table 1.
Electroconductivity of synthetic hydrogels and synthetic hydrogel precursors
| Sample | Electroconductivity [S m−1] |
|---|---|
| Equilibrated hydrogels | 0.329 ± 0.008 |
| Cured hydrogels | 0.255 ± 0.006 |
| PEGDA | 0.000007 ± 0.000005 |
| Thiocure | 0.0001 |
| 0.175 m NaHCO3(aq) | 1.23 ± 0.05 |
| Deionized water | 0.00029 ± 0.00003 |
Equilibrated hydrogels were maintained in 1× PBS for 24 h at 37 °C and allowed to reach room temperature prior to testing electroconductivity. All other samples tested were maintained at room temperature. Equilibrated and cured hydrogel electroconductivities were determined through direct current electric fields of 50 V. Electroconductivity of precursor solutions was determined with a portable conductivity probe. All electroconductivities were determined with samples prepared in triplicate and data reported are averages with the corresponding standard deviations.
3. Discussion
Despite patients undergoing surgical resection and chemoradiotherapy, residual GBM cells lead to tumor recurrence. This challenge can be addressed by exploiting GBM cells' infiltrative capacity to redirect their migration for subsequent eradication. CXCL12 is the most extensively studied chemokine for inducing GBM migration.[ 36 ] This study further developed our previously optimized thiol‐Michael addition injectable hydrogel[ 32 ] comprising 50 wt% hydration level and 0.175 m NaHCO3(aq) to sustain the release of CXCL12 payloads and induce GBM/GSC migration. We investigated GBM/GSC invasion into the hydrogel, the mechanism of invasion for cells that migrated and became entrapped, and GBM‐hydrogel interactions.
10 kDa FITC‐dextran was used to model the release kinetics of proteins like CXCL12, which is also positively charged with a molecular weight of 8 kDa.[ 37 ] All three payload concentrations demonstrated biphasic release profiles. The hydrogel mesh sizes are 5.6 ± 0.6 nm,[ 32 ] while dextran possesses hydrodynamic radii around 2.3 nm.[ 38 ] The small hydrogel mesh sizes retained the FITC‐dextran payload and generated a barrier for a sustained, lower burst release by diffusion during the first 96 h compared to hydrogels reported in the literature, which released 90.7% of proteins within 4 h.[ 30 ] The rapid diffusion during the first 24 h (Figure 2a) was likely due to micrometer‐sized pores and defects releasing molecules near the surface. 5 µg mL−1 was the most optimal loading concentration, as it sustained release for 336 h with a high final cumulative release of 94.1% while yielding release amounts ranging from 0.01 to 0.16 µg/day (as indicated by the blue dashed line thresholds in Figure 2b), which is within the therapeutic range of CXCL12 elution necessary to induce migration.[ 39 ]
FITC‐dextran release was not exclusively by diffusion, as the bumps in the cumulative release profiles after the plateau (Figure 2a) corresponded to complete hydrogel degradation, indicating some FITC‐dextran payload was retained and not released until hydrogel disintegration. The payload concentration tuned the release profile, with a higher concentration leading to longer sustained release, albeit with a less total cumulative release, and a slower hydrogel degradation rate. Introducing payloads into hydrogel polymer networks can decrease degradation rates, as these molecules may serve as additional crosslinks requiring a longer degradation time to hydrolyze.[ 40 ] Since our profiles alluded to potential solute‐hydrogel interactions, we developed a simplified computational model based on second‐order Fickian diffusion and hydrogel degradation to enhance our understanding of the release kinetics and compare the predicted and experimental data.
The time‐varying diffusivities determined from the cumulative release data for 5 µg mL−1 payloads were used as the model input, as described by Sheth and colleagues previously.[ 41 ] The computational model aligned with the experimental cumulative release, but did not predict biphasic release, while a sensitivity analysis revealed the initial loading concentrations did not impact the cumulative release profile. The experimental release kinetics data deviated from the predicted model, therefore, indicating dextran‐hydrogel interactions were present due to non‐specific binding in the hydrogel. Particle diffusion through polymer networks is a complex process entailing hydrodynamic interactions, obstruction effects, thermodynamic agitation, electrostatic interactions, hydrophobic effects, hydrogen bonding, and van der Waals interactions.[ 42 , 43 ] Dextran is not a neutral molecule during diffusion through hydrogels,[ 42 ] and non‐specific binding within the matrix may have contributed to the tunability in release kinetics.
We therefore next assessed the release of CXCL12 loaded into the synthetic hydrogels at 5 µg mL−1. As indicated by Figure 3, the hydrogel sustained slow release of the chemokine payload with ≈7–10 ng release per day. The chemokine payload release was an order of magnitude less and with greater variation across replicates than FITC‐dextran, indicating potential CXCL12‐hydrogel interactions were more prevalent than dextran‐hydrogel interactions. These interactions may be strongly electrostatic since CXCL12 possesses a much higher surface positive charge density compared to other chemokines[ 44 ] and may bind tightly to the acidic, negatively charged thiol groups in the hydrogel. While CXCL12 hydrodynamic radius is also less than 3 nm,[ 45 ] dextran is a linear polysaccharide, and proteins like chemokines may form tertiary or quaternary structures for more complex interactions with the hydrogel matrices.[ 46 ] CXCL12‐hydrogel interactions may be advantageous, as gradual CXCL12 release can sustain the chemotactic gradient for a longer duration compared to the burst release observed in the literature for porous hydrogels.
The Transwell migration assay revealed CXCL12 released from the synthetic hydrogels induced GBM/GSC migration. Since previous research demonstrated CXCL12 concentrations ranging from 0.02 to 0.2 µg mL−1 induce GBM migration, the Transwell assay comprising CXCL12 in solution was set to the maximum concentration of 0.2 µg mL−1 for high chemoattractive potency as a positive control for inducing GBM/GSC migration.[ 47 , 48 ] Both U251 GSCs and GBM cells migrated in response to factors secreted from the hydrogel‐entrapped U251 GBM cells, illustrating GBM cells localizing into the hydrogel may induce more GBM migration as a positive feedback loop. Interestingly, the GBM‐loaded hydrogels induced significant GBM migration within the first 24 h, while CXCL12‐hydrogels did not induce significant migration until 48 h. Multiple factors secreted by the entrapped GBM cells may synergistically be more potent as chemoattractants. For example, conditioned media from glioma‐associated stromal cells containing fibronectin, CXCL12, and hepatocyte growth factors possess more potent chemoattractive properties compared to purified CXCL12.[ 30 ] Chemokine cellular secretion rates range from 10−8 to 10−6 ng h−1/cell,[ 49 ] which can maintain a steady chemoattractant gradient from the hydrogel‐entrapped GBM cells. GSCs migrated significantly more in response to GBM‐entrapped hydrogels compared to the control during the 24 h time point only. Future research will explore loading other chemokines such as CX3CL1, which can mediate GSC migration,[ 50 ] to induce the migration of heterogeneous malignant cells.
The Transwell results were validated with primary, patient‐derived G34 (mesenchymal subtype) and G528 (classical subtype) GSCs (Figure 4d) known to be chemotactic to CXCL12 via the CXCR4 receptor.[ 51 ] These cells were characterized previously and bear similarity to neural stem cells while recapitulating the gene expression, genotype, and in vivo biology of their corresponding human, GBM parental primary tumors.[ 52 ] Both cell types migrated significantly more in response to CXCL12‐hydrogels compared to the control, indicating the chemokines released can attract other GSC subtypes besides the U251 cells. G528 cells were more migratory in response to CXCL12 compared to G34 cells, with this difference being statistically more significant for the positive control CXCL12 in solution. This difference may be attributed to the higher percentage of G528 cells expressing CXCR4 receptors compared to G34 cells.[ 53 ] While the G34 and G528 cells had similar migratory responses to CXCL12 in solution and CXCL12‐hydrogels, the U251 GBM/GSCs were more responsive to the positive control comprising CXCL12 in solution at both time points. This finding may be attributed to the hydrogel‐cell interactions impacting U251 GSC stemness and U251 GBM malignancy, which is investigated with immunofluorescent staining of glial and stem cell markers and discussed in more detail in a subsequent section. These interactions are not reflected in the Transwell assay, as there was no significant difference in the migration index between the control and empty hydrogel groups. 2D migration across Transwell membranes is less physiologically relevant for non‐adherent or loosely adherent cells such as GSCs.[ 54 ] 3D ECM‐based hydrogels can better mimic in vivo migration due to the presence of chemical cues, while chemokines binding to anionic glycosaminoglycans in the ECM is another important consideration.[ 44 ]
A novel dual‐layered ECM and synthetic hydrogel platform was therefore developed (Figure 5a) to recapitulate the in vivo interface between the ECM and synthetic hydrogel during the invasion. The top ECM layer comprising collagen I and hyaluronic acid matrix was loaded with either U251 GBM or GSCs, while the bottom synthetic hydrogel layer was left empty or loaded with CXCL12 at 5 µg mL−1. We aimed to determine whether GBM/GSCs possessed the propensity to migrate from the ECM and into the synthetic hydrogel with CXCL12‐mediated chemotaxis. HA is the most abundant (30–50%) molecule in the glioma tumor microenvironment,[ 55 ] while collagen I is present in the brain‐vascular migration microenvironment.[ 56 ] Although collagen I is not abundant in normal brain tissue, its expression is upregulated in GBM,[ 57 ] where it is more filamentous and increases the invasion potential of GBM cells.[ 58 ] Both molecules contribute to a pro‐invasive tumor microenvironment,[ 56 , 59 ] and the collagen‐HA hydrogel recapitulates the flow velocity and Young's modulus observed in vivo in the brain.[ 60 ] For all subsequent studies (such as nestin and GFAP immunofluorescence, GBM viability, and fibronectin deposition) this ECM hydrogel was used as a control to compare GBM/GSC behavior in the synthetic hydrogel against behavior in the GBM microenvironment. Collagen‐HA hydrogels have been extensively characterized as in vitro models to investigate GBM behavior and invasion.[ 61 , 62 , 63 ] Moreover, the incorporation of collagen I into the HA matrix does not significantly alter GBM invasion or morphologies.[ 60 ]
Reflectance confocal imaging at 640 nm distinguished synthetic and ECM hydrogel layers (Figures 5 and 6) based on differences in opacity. CXCL12 released from the synthetic hydrogels generated a chemokine gradient and directed U251 GBM/GSC invasion into the synthetic hydrogels, while empty hydrogels did not attract invasion. Only cells that had invaded at least 20 µm (one cell diameter) into the synthetic hydrogel were considered to avoid quantifying cells localizing on the synthetic hydrogel surface only. The number of invaded GBM and GSCs was not significantly different due to the high variation in the GSC group (Figure 5b). We hypothesized U251 GSCs may be differentiating upon interaction with the synthetic hydrogel, as there was a loss in migration index at 48 h compared to 24 h when chemoattractants were released from the hydrogels (Figure 4c), which is discussed later.
Mesenchymal GBM motility resembles fibroblasts with elongation, crawling, actin filament formation, ECM remodeling through matrix metalloproteinase secretion, and strong cell‐ECM interactions.[ 64 ] However, the GBM cells demonstrated amoeboid migration during invasion (Figure 5c–f), which entailed a rounded morphology with membrane blebbing, weak ECM‐cell interactions due to low integrin expression, cortical actin formation, and a lack of matrix metalloproteinase‐based ECM degradation.[ 65 , 66 ] Cells utilized this primitive and efficient migration mode to squeeze through narrow spaces at high velocity,[ 67 , 68 ] even as they responded to chemoattractant cues.[ 65 ] The hydrogels with HA content led to GBM amoeboid migration phenotypes in the ECM layer,[ 56 ] while the lack of integrin and ECM ligand interactions in the synthetic hydrogel also contributed to amoeboid migration upon entrapment, as free thiols in the synthetic hydrogel reduced cell spreading and entailed a rounded morphology.[ 69 ]
We hypothesized cells used amoeboid blebbing and myosin IIA activation to invade through pores present on the synthetic hydrogel surface and sought to determine the mechanism of invasion through blebbistatin treatment. The brain parenchyma comprises submicrometer‐sized extracellular spaces with tightly packed glial and neuronal processes.[ 70 ] Glioma cells utilize amoeboid blebbing to extend their leading cytoplasmic process, exhibit a burst of forward movement, and then deform their cell body and nucleus in an hourglass shape to squeeze through these tight pores.[ 71 ] Myosin II is required for nuclear translocation and cytoplasmic contractile forces, with the myosin IIA isoform uniformly expressed and upregulated in tumors.[ 71 ] Furthermore, myosin II in specific physical environments and under the presence of chemoattractants impacts GBM migration.[ 72 ] We, therefore, investigated the immunofluorescence of GBM cells expressing myosin IIA during the invasion. Blebbistatin is a cell‐permeable, small allosteric inhibitor of myosin II[ 73 ] that blocks cell membrane blebbing.[ 74 ] Blebbistatin dose concentration and treatment time were limited to the range appropriate for U251 cells as determined previously to mitigate any potential cytotoxic effects.[ 71 , 75 ]
Blebbistatin treatment inhibited U251 GBM invasion into the synthetic hydrogels, even under the presence of CXCL12 (Figure 6c), which conformed with a study from Ivkovic et al. where 30 µm blebbistatin inhibited 95% of glioma migration.[ 75 ] CXCL12‐hydrogels without blebbistatin demonstrated GBM cells invaded the synthetic hydrogel (Figure 6b) and resulted in significantly higher normalized myosin IIA fluorescence compared to the blebbistatin and control groups without invasion (Figure 6e). Myosin II is hence blocked with blebbistatin and is necessary for GBM cells to invade into the synthetic hydrogel. Indeed, blebbistatin only inhibits U251 migration when myosin II activity is required for motility, such as when cells need to squeeze through spatial constraints imposed by Transwell assays or aligned nanofibers, as opposed to instances without any geometric barriers to migration like a wound healing assay in 2D.[ 76 ]
All cells maintained a rounded morphology, while only invading cells in the hydrogel‐CXCL12 without blebbistatin condition demonstrated membrane blebbing protrusions (Figure 6d). These bleb‐like protrusions driven by actomyosin contractility enable cells to sense their microenvironment through mechanotransduction and penetrate into tight spaces.[ 77 ] The low collagen fiber density in the ECM hydrogels (Figure 6f) can be attributed to HA, which further induced amoeboid migration. The lack of collagen fiber remodeling further confirmed cells used amoeboid migration to squeeze through the collagen fibers with minimal interactions or ECM degradation.[ 78 ] Multiphoton confocal reflectance imaging at 543 nm revealed the presence of micrometer‐sized pores and defects on the hydrogel surface (Figure 6g). Although the hydrogel mesh size was in the nanometer range,[ 32 ] these larger pores allowed a limited number of GBM cells to squeeze inside by activating myosin IIA during CXCL12‐mediated migration and invasion. We had previously determined the synthetic hydrogel possessed a tan δ (ratio of viscous to elastic response) value of 0.34 in the elastic range.[ 32 ] Cells can invade into pores smaller than the cells if the hydrogel possesses a low viscous component that enables polymer chains to disentangle, deform, and enlarge the pores upon cell interactions.[ 79 ] However, the limited number of these micrometer‐sized pores on the hydrogel surface may have contributed to the limited number of GBM/GSC invasions observed. Future research will study if physical stimuli, like focused ultrasound or electric fields,[ 80 ] can be applied to spatiotemporally control the delivery of CXCL12 retained in the hydrogel as well as the hydrogel pore sizes and porosity to improve cell entrapment.
The nestin and GFAP immunofluorescences revealed GSCs maintained their stemness while interacting with the synthetic hydrogel for the first 24 h, after which point stemness decreased. We had previously characterized the expression of three stem cell markers (CD133, CD44, nestin) and the glial marker GFAP when U251 cells are cultured either under spheroid forming (GSCs) or normal culture conditions (GBM cells), which had revealed U251 GSCs express lower levels of GFAP and higher levels of nestin compared to U251 GBM cells.[ 81 ] Our current study shows GSCs maintained significantly lower levels of GFAP and higher levels of nestin during the first 24 h compared to the GBM cells when cultured on the synthetic hydrogel. During the 48 h time point for synthetic hydrogels, this difference in normalized GFAP fluorescence was lower, while nestin levels were not significantly different altogether. U251 GSCs may be differentiating over time upon interaction with the synthetic hydrogel, which may have contributed to their lower migratory response to the CXCL12 released from hydrogels at 48 h compared to 24 h (Figure 4c).
The normalized GFAP fluorescence of both cells was significantly lower when cultured on ECM hydrogels compared to the synthetic hydrogels. GFAP expression is negatively correlated with astrocytoma grade, which is indicative of tumor malignancy.[ 82 ] Hence, collagen‐HA hydrogels maintained GBM malignancy, while the cells may have become more senescent upon interaction with the synthetic hydrogel. Such a finding will need to be explored in follow‐up studies by assessing metabolic changes in GBM/GSCs upon interaction with the synthetic hydrogels using in vivo, rodent GBM resection models. ECM hydrogels led to no significant difference in the GFAP or nestin levels between the two cell types. Collagen I supports neural cell differentiation in vitro,[ 83 ] while HA supports neural stem cell differentiation.[ 84 ] Collagen‐HA hydrogels may have provided the microenvironment for potential GSC differentiation over time and contributed to GSC heterogeneity (Figure 5b). The range of maximum distance GSCs had invaded (spanning 88 µm) into the synthetic hydrogel was larger compared to the GBM cells (spanning 55 µm). GSCs in the ECM layer may have possessed different differentiation statuses during the invasion, which likely impacted invasive potential. Indeed, GBM invasion under interstitial flow in a 3D hydrogel is dependent on their differentiation status, as GSCs can invade 300 µm deeper into collagen‐Matrigel hydrogels compared to GBM cells.[ 85 ] The dual‐layer hydrogel platform, therefore, recapitulated an in vivo tumor microenvironment for studying heterogeneous malignant cell response to CXCL12‐mediated chemotaxis.
U251 GBM/GSCs did not invade beyond 120 µm deep into the hydrogel and were therefore limited to near the surface, which may be attributed to the low cell viability upon invasion. Synthetic hydrogel‐encapsulated GBM cells only near the surface maintained viability, while cells deeper inside the hydrogel were exclusively dead (Figure 8b). Even 300 µm deep into the hydrogels, U251 GBM cells remained viable and formed networks for up to 120 h when entrapped in the ECM hydrogels (Figure 8a,c), while cells were less viable at around 20% throughout the 120 h when encapsulated in the synthetic hydrogels.
Solano and coworkers discovered grafting arginine‐glycine‐aspartate (RGD) peptides into alginate macroporous hydrogels promote F98 glioma cell adherence with passive migration.[ 86 ] However, these peptides do not enhance invasion, as cells adhere only to the hydrogel surface to proliferate. We observed GBM cells can go beyond only localizing onto our synthetic hydrogel surface by invading inside through CXCL12‐mediated chemotaxis. The entrapment of cells deep into the hydrogel also led to passive eradication. However, future research will need to investigate if ablation can eradicate live cells near the hydrogel surface. One limitation is that our cell viability assay was performed for GBM cells entrapped in the hydrogel during the synthesis process. We previously determined the cured synthetic hydrogel is not cytotoxic to normal human astrocytes cultured on its surface.[ 32 ] It is therefore possible the low viabilities were due to the sol–gel transition promoting programmed cell death during synthesis for hydrogel‐encapsulated GBM cells.[ 87 ] However, it is more likely that mass transfer, oxygen, and nutrient diffusion limitations imposed by the polymer network induced GBM apoptosis and resulted in the cell viability gradient, resulting in unviable cells deep in the hydrogel matrix. Cell death due to sol–gel transition would have resulted in uniformly low viabilities throughout the hydrogel. CXCL12 retained in the hydrogel matrix and its impact on GBM viability should also be considered, as its release was slow and sustained due to the potential non‐specific binding of the chemokines in the hydrogel, while CXCL12 affects GBM proliferation and survival.[ 47 , 88 ] Future research with an in vivo, rodent GBM resection preclinical model will need to assess GBM cell viabilities upon invasion into the CXCL12‐loaded hydrogels while simultaneously decoupling the sol–gel transition process from cytotoxicity assays.
The synthetic hydrogels lose 74% of total mass when incubated in 1× PBS at 37 °C for 15 days.[ 32 ] While these in vitro conditions mimicked cerebrospinal fluid, the impact of cell entrapment and hydrogel degradation by enzymolysis should also be considered.[ 89 ] The dual hydrogel layer invasion studies demonstrated GBM/GSCs can invade into the synthetic hydrogel with CXCL12‐mediated chemotaxis over 24 h. Hence, the synthetic hydrogels were encapsulated with 3 × 106 U251 GBM cells to mimic the typical in vivo density of GBM cells in the brain,[ 90 ] which served as the maximum possible cell invasion into the hydrogel post‐resection. Hydrogels without cells (acellular) were a control group to decouple the degradation due to serum/DMEM from the degradation due to encapsulated cells. Cell culture conditions led to faster degradation within 168 h compared to degradation in PBS only,[ 32 ] as the higher DMEM pH enhanced ester hydrolysis.[ 91 ] Interestingly, cellular hydrogels degraded slowly compared to acellular hydrogels. While hydrogel‐entrapped GBM cells may have promoted an acidic environment to slow material degradation, encapsulated U251 GBM cells may have also deposited their own ECM components. Glioma cells secrete ECM molecules, such as HA and fibronectin, during migration to increase their invasiveness and mobility.[ 92 ]
We, therefore, assessed fibronectin secretion by hydrogel‐encapsulated U251 GBM cells in the ECM and synthetic hydrogels. Fibronectin was selected since HA deposition would be difficult to distinguish from native HA present in the ECM matrix. As indicated by Figure S1, Supporting Information, fibronectin deposition by GBM cells in the synthetic hydrogels was limited to within 400 µm of the hydrogel surface, corresponding to the depth at which live cells were observed (Figure 8b). The fibronectin deposition, as quantified by normalized fluorescent levels, was similar in both synthetic and ECM hydrogels (Figure 9c,d) within the 300 µm depth limitation set by the study. Live GBM cells entrapped near the surface secreted ECM molecules, which helped to reinforce the synthetic hydrogel to counteract degradation effects induced by the local environment. Bryant and Anseth determined that PEG‐based hydrogels enable ECM deposition even without integrin‐binding ligands.[ 93 ] GBM cell invasion can hence potentially slow the hydrogel degradation process to allow more malignant cells to localize before ablation is applied.
The thiol‐Michael addition injectable hydrogel demonstrated the potential to attract and entrap invasive GBM/GSCs through CXCL12‐mediated chemotaxis. Our novel 3D dual‐layer hydrogel tumor model is an in vitro platform that can isolate CXCL12‐responsive GBM/GSCs from a heterogeneous population to study cancer cell migration. Although beyond the scope of the current study, future research will need to investigate GBM hydrogel entrapment under more clinically relevant settings with an in vivo, rodent, GBM resection model. Such studies will elucidate the impact of interstitial diffusion and convection,[ 94 ] which may increase release rates of hydrogel payloads as well as regulate GBM invasion and chemokine distributions in the brain.[ 60 ] Preclinical models can also investigate hydrogel degradation in the tumor microenvironment and the potential selectivity toward entrapping malignant cells over healthy brain cells. In our current study, we performed invasion assays with U251 GSC and GBM cells to directly compare CXCL12‐mediated hydrogel entrapment potential for GBM cells and GBM cells at a stem‐like cell state. GSC differentiation over time likely contributed to the similarity in the number of invaded U251 GSCs and GBM cells in the synthetic hydrogel (Figure 5b). However, this similarity may have also stemmed from the fact that both cell types were derived from the same U251 cell line. To address this limitation and gain further insights into the hydrogel GBM cell entrapment strategy, future research should utilize in vivo preclinical models to deduce the invasion responses of a heterogeneous population of cells present near the resection cavity including healthy brain cells and CXCR4‐ GBM/GSCs.
While invasion beyond 24 h will need to be investigated in the future, the limited ability of the synthetic hydrogel to entrap many GBM/GSCs from the ECM hydrogel (Figure 5d,f) may be attributed to two reasons. First, although U251 cells express significantly higher CXCR4 than normal glial cells,[ 95 ] CXCL12‐mediated chemotaxis is limited to CXCR4+ cells. Second, the slow, sustained release of CXCL12 from the synthetic hydrogels (Figure 3) due to chemokine‐hydrogel interactions may be a limiting factor. Additionally, other CXCR4+ migratory cells in the brain aside from GBM/GSCs may become responsive to CXCL12 released from the hydrogel.[ 29 ] These limitations may be addressed by implementing electrotaxis with chemotaxis to synergistically and selectively induce GBM/GSC migration post‐resection for optimal entrapment of malignant cells in the synthetic hydrogel.
Sublethal, direct current electric fields enable cell‐specific migratory responses with a voltage‐dependent directional bias through electrotaxis[ 96 ] including for GBM tumor aggregates,[ 97 ] cell lines,[ 97 ] and GSCs.[ 98 ] Cured and equilibrated hydrogels possessed electroconductivities ranging from 0.255 to 0.329 S m−1, which is within the range necessary for controlled delivery of therapeutic hydrogel payloads upon electric stimulation.[ 80 ] The hydrogels can potentially be used for electrotaxis, as conductivities around 1.5 S m−1 can model electrolyte solutions during computational simulations of GBM electrotaxis.[ 98 , 99 ] Our results indicated the GSC population is heterogeneous during the invasion. Since electric fields can tune cell migration direction, speed, and biases,[ 100 ] electrotaxis can direct GBM migration to confer patient‐specificity and may guide a greater number of malignant cells deeper into the synthetic hydrogel. Therefore, we aim to conduct further research to determine if electrotaxis and chemotaxis can synergistically improve the hydrogel entrapment of GBM/GSCs for their subsequent ablation with non‐invasive techniques like focused ultrasound.
4. Conclusion
This study demonstrated that a thiol‐Michael addition injectable hydrogel can attract and promote the invasion of GBM and GSCs into the hydrogel through CXCL12‐mediated chemotaxis. The initial loading concentration of hydrogel payloads tuned the release kinetics, with experimental data and computational modeling indicating that the model molecule FITC‐dextran interacted with the hydrogel matrix. We demonstrated CXCL12 loaded into the hydrogel at the optimized 5 µg mL−1 concentration is released slowly in a sustained manner with about 7–10 ng of release per day. Transwell migration assays illustrated U251 GBM/GSCs and primary, patient‐derived G34 and G528 GSCs migrated in response to CXCL12 released from the synthetic hydrogel. We showed with a novel, in vitro, dual‐layer hydrogel platform that invasive U251 GBM/GSCs have the propensity to migrate from the collagen‐HA ECM layer and invade into the synthetic hydrogel through CXCL12‐mediated chemotaxis. Myosin IIA activation enabled cells to squeeze through subnuclear‐sized pores on the synthetic hydrogel surface to invade inside by amoeboid migration. Fibronectin deposition by viable malignant cells entrapped near the synthetic hydrogel surface helped reinforce the hydrogel to slow down its rate of degradation, while cells entrapped deep inside the hydrogel displayed limited viabilities. A decrease in U251 GSC stemness and GBM malignancy upon interaction with the synthetic hydrogel was observed, thereby indicating potential direct therapeutic effects of the hydrogel. Overall, this injectable hydrogel demonstrates promise to promote invasion and entrapment of GBM/GSCs with CXCL12‐mediated chemotaxis. Future research will need to investigate the feasibility of implementing electrotaxis to further improve this hydrogel‐based GBM/GSC entrapment strategy for subsequent ablation of captured cells to help mitigate tumor recurrence in GBM patients post‐resection.
5. Experimental Section
Synthetic Hydrogel Synthesis
The synthetic injectable hydrogels were prepared as described previously.[ 32 ] Briefly, Thiocure ETTMP 333L, a trithiol crosslinker ethoxylated trimethylolpropane tri‐3‐mercaptopropionate (generously donated by Bruno Bock Thiochemicals), as well as PEGDA, at 575 g mol−1 numbered average weight from Sigma‐Aldrich were both brought to room temperature. Aqueous 0.175 m NaHCO3 (Fisher Chemical) was prepared by dissolving the appropriate mass in deionized water. 1.38 mL hydrogel volumes were prepared in a 1:1 thiol to acrylate stochiometric ratio by using 0.389 g of Thiocure, 0.300 g of PEGDA, and 0.690 mL of the 0.175 m NaHCO3(aq). These ratios were scaled accordingly to adjust the final desired volumes of hydrogel solutions. PEGDA was first dissolved in the NaHCO3(aq), and vortexed for 10 s, after which point the Thiocure was injected into the precursor solution and mixed with a stir rod for 20 s. All hydrogels were crosslinked at 37 °C.
FITC‐Dextran Release from Hydrogels
The synthetic hydrogels were loaded with various concentrations of a model molecule and the release kinetics profiles were assessed to determine the impact on the payload concentration. FITC dextran (Sigma‐Aldrich) at 10 kDa was dissolved in 1× PBS solution containing calcium and magnesium and prepared with serial dilutions at concentrations ranging from 100 to 0.0001 µg mL−1 for a standard curve. The fluorescence of these solutions was measured with a spectrophotometer (SpectraMax M2e Molecular Devices) at 490 nm excitation and 520 nm emission. The resulting standard curve relating the FITC‐dextran fluorescence to its corresponding concentration was used to determine the FITC‐dextran concentrations present in PBS solution upon release from the hydrogels. Hydrogels were prepared as described in the previous section and crosslinked at 500 µL volumes in 24 well plate wells. FITC‐dextran solutions in 1× PBS were prepared and loaded into the hydrogels to obtain final payload concentrations of 0.5, 5, or 10 µg mL−1 in the hydrogels. The concentrations were varied from 0.5 to 10 µg mL−1 to identify an optimal FITC‐dextran loading concentration, as previous research had demonstrated that chemokines loaded into hydrogels at concentrations of 0.3[ 79 ] and 10 µg mL−1[ 101 ] induced cancer cell migration. The volume of FITC‐dextran solutions loaded into the hydrogels was limited to 10 µL to avoid disrupting the native gelation and hydration levels of the synthetic hydrogels. FITC‐dextran was mixed into the hydrogel precursor solution containing the PEGDA‐NaHCO3(aq) precursor solution, prior to the addition of Thiocure, to entrap the solute before gelation. Four replicate hydrogels were prepared for each FITC‐dextran loading concentration. Additionally, PBS and 10 µg mL−1 FITC‐dextran in PBS solutions were also prepared as a negative and positive control, respectively. Sample hydrogels were submerged in 1.5 mL of 1× PBS containing magnesium and calcium and maintained in the dark at 37 °C for the duration of the experiment. At designated time points (1, 2, 5, 8, 12, 18, and 24 h, and then every 24 h until complete hydrogel degradation), all of the PBS solution containing the released solute was collected and refreshed. In triplicate subsamples, the fluorescence of the PBS solution containing the released solute for each sample was determined at 490 nm excitation and 520 nm emission. The standard curve was used to determine the final concentration of FITC‐dextran present in the solution at each time point. These concentrations were then used to determine the amount of FITC‐dextran released at each time point. In addition, the percentage of FITC‐dextran released at each time point in relation to the total amount of FITC‐dextran initially loaded into each hydrogel was also determined to report the cumulative release (%) of FITC‐dextran at each designated time point.
Computational Model of FITC‐Dextran Release Kinetics
The experimental release studies were next used to determine the diffusivity of the FITC‐dextran payload and consequently develop a computational model to predict the theoretical FITC‐dextran release profiles from the hydrogels. The swelling properties and ratios of the hydrogel formulations comprising 0.175 m NaHCO3(aq) and 50 wt% water content were previously monitored every 24 h for 9 days, at which point the hydrogels reached equilibrium swelling.[ 32 ] The characteristic swelling ratio (Q) at each time point from the swelling study was therefore used to estimate the hydrogel swelled thickness (δ max in mm) based on the initial hydrogel thickness (δ in mm) from the release study according to Equation (1)[ 102 ]
| (1) |
The swelled hydrogel thicknesses from these time points were then averaged and determined to be 3.54 mm. This average thickness was implemented into the diffusion equations to model the predicted cumulative release, which is discussed in greater detail below. Sheth and colleagues[ 41 ] also determined from their study that implementing the average hydrogel thickness helps to accurately model release kinetics from a swelling and degrading hydrogel with time‐varying thicknesses.
The hydrogel was approximated as a thin slab with diffusion in only one direction since the bottom surface and sides adhered to the well plate during the experiments. The solute release equation developed by Ritger and Peppas[ 103 ] was applied to determine the FITC‐dextran diffusivity at each time point. These diffusivities were based on the cumulative release fraction from the experimental release profiles of FITC‐dextran loaded into the hydrogel at a concentration of 5 µg mL−1. Hence, for each time point, the corresponding diffusivity was determined according to Equation (2), where M t is the total mass (g) of solute released at time t (h) from the hydrogel, M ∞ is the total mass of solute (g) loaded into the hydrogel, D is the diffusion coefficient in mm2 h−1, and h is the average thickness of the hydrogel in mm
| (2) |
According to Ritger and Peppas,[ 103 ] if the hydrogel aspect ratio is equal to or larger than 1, then Equation (2) is valid for the first 65–75% cumulative release. The hydrogel aspect ratio (diameter/height) was determined to be 3.96, and therefore Equation (2) was applied to determine the diffusion coefficients for the first 96 h when ≈75% of the solute was released from the hydrogel, as indicated by the cumulative release profile from Figure 2a. To calculate the diffusion coefficient for time points beyond 96 h and therefore late‐time cumulative release, the equation developed by Park and colleagues[ 104 ] and utilized by Fu and Kao[ 105 ] was applied according to Equation (3) for 120 to 216 h as follows
| (3) |
Controlled diffusion‐based models were implemented across all time points to determine the diffusion coefficients, instead of incorporating anomalous transport or controlled swelling. According to Ritger and Peppas,[ 106 ] for hydrogels with swelling ratios less than 1.33, the swelling can be considered moderate‐low and solute diffusion is primarily Fickian. Since the swelling studies had previously indicated the highest swelling ratio was less than 1.33,[ 32 ] the Fickian diffusion transport equations were applicable.
The diffusion data obtained from the experimental release profiles were next curve fitted to an exponential family. Although Sheth and coworkers[ 41 ] found that curve fitting experimental diffusivity data to an exponential family or quadratic splines yielded similar results, quadratic splines may not work well for limited timepoints and can also yield oscillatory and unrealistic curves. The exponential 3P tool in JMP software was used to curve fit the diffusion coefficients against time according to Equation (4), where a is the asymptote, b is the scale, and c is the decay/growth rate.
| (4) |
The resulting curve fitted equation was D (t) = 0.013 + 0.0572 × e −0.06t and fit the data with an R 2 value of 0.952. The experimental diffusivity data and curve fitted data are both shown in Figure S2, Supporting Information. MATLAB was used to then model the corresponding predicted cumulative release profile based on this curve‐fitted diffusivity exponential equation.
FITC‐dextran released from the hydrogel in the model was assumed to be based on Fick's Second Law of Diffusion, and the method primarily developed by Sheth and colleagues[ 41 ] was followed. In the model, the diffusivity was time‐varying due to hydrolytic bulk degradation. The partial differential equation which represented the solute release profile was provided by Equation (5), where c is the concentration of the solute in mol L−1, t is time in minutes, D(t) is the time‐varying diffusivity of the solute in m2 min−1, and x is the time‐varying thickness of the hydrogel with degradation in m.
| (5) |
In the model, time was varied from 0 to 336 h, at which point the cumulative release for the hydrogels loaded at 5 µg mL−1 concentration reached a maximum. The spatial domain of x in Equation (5) was − h ≤ x ≤ h, where h is the half‐thickness of the gel. Since the constant average thickness of the hydrogel throughout degradation was determined to be 3.54 mm, the value of h was set to 1.77 mm. In their model of drug release from porous and biodegradable polymer matrices idealized as cylinders with length of L, Lemaire and colleagues used the symmetry at the midpoint z = L/2 to simplify their problem further by only considering half of the total length from 0 < z < L/2.[ 107 ] As such, due to the symmetry in the hydrogel slab model, the computational model in this study will consider the spatial domain of x from x = 0 to x = h due to the symmetry at the center of the slab.
In order to solve the system, a set of Dirichlet boundary conditions was applied. The solute concentration was assumed to be zero at the gel boundaries according to Sheth and colleagues,[ 41 ] since the hydrogel volume was smaller than the PBSsolution volume to generate an infinite sink. As such, c(h,t) = 0. Under symmetric geometry, one common boundary condition applied to drug‐releasing hydrogels is setting the derivative to 0 at the symmetric point. For example, Raman et al. investigated diffusion from a hydrogel with spherical geometry comprising radius R, with r as the radial position.[ 108 ] At the symmetric point r = 0 in the center, . Hence, the second boundary condition in this model was set to at x = 0. An initial condition was also applied, where at t = 0, c(x,0) = c o for 0 ≤ x ≤ h(0), where c o is the initial concentration of FITC‐dextran loaded into the hydrogel, which was 5 µg mL−1 in the case of our model. This initial condition assumed that the solute was distributed uniformly throughout the hydrogel upon loading, before beginning the release experiments, and also corresponded to the solute concentration loaded into the hydrogel.
A numerical method and computational approach were applied to solve this problem and Equation (5). Specifically, spatial discretization in x with centered finite difference approximation was performed in MATLAB to obtain an ordinary differential equation, which was solved with MATLAB solver ode45. A total of 10 nodes was used with the method of lines approach to conserve accuracy while also minimizing the computation time. An array of D(t) was created based on the exponential equation. This array was the same size as the array created to compute and store the dc/dt values in the function that used the method of lines to solve the ODE, to enable the values of D that step through time to be placed as inputs into the function that computes c through time according to Equation (5).
The model considered the concentration in the hydrogel over time from the center of the hydrogel to the right boundary. As such, the concentration matrix when Equation (5) was solved was integrated from 0 to h to obtain a single profile of total concentration, c, in the hydrogel from 0 to 336 h. In order to obtain the final cumulative concentration released, the following Equation (6) was implemented to obtain the normalized concentration released (c r) from 0 to 1 in accordance with the data reported. Since c was already normalized in the computational model, the released concentration can be obtained by simply subtracting c from 1. The cumulative release in terms of percentage can therefore be obtained by simply multiplying c r by 100
| (6) |
Since the hydrogel was previously demonstrated to degrade with time in PBS,[ 32 ] a simple degradation term was also incorporated into Equation (6) to account for the FITC‐dextran release due to bulk hydrolytic degradation. This degradation component was based on the nonlinear Kopcha model, which considered the relative relaxation of the hydrogel and diffusion of the solute component according to Equation (7), where Q is the percent of the solute released due to the degradation at time t in h, A is the diffusion exponent (1 for a slab), and B is the erosion constant.[ 109 ] Here, B was taken to be 0.172% mass loss per hour, which was approximated from the 4.13% mass loss per day in 0.175 m NaHCO3(aq) hydrogels with 50 wt% water content as determined in our previous degradation study conducted in PBS.[ 32 ]
| (7) |
Quantifying CXCL12 Release from Synthetic Hydrogels
Four replicates of synthetic hydrogels were synthesized at 0.69 mL final volumes in 6‐dram vials. The hydrogels were loaded with CXCL12 at 5 µg mL−1 by adding the appropriate volume of recombinant human SDF‐1α (PeproTech) reconstituted in 1× PBS into the PEGDA‐NaHCO3(aq) precursor solution and mixing gently by stirring prior to the addition of Thiocure. 3 mL of 1× PBS prewarmed to 37 °C was added to each dram vial and the hydrogels were incubated at 37 °C. At designated time points (1, 3, 6, 12, 24, 48, and 72 h), all of the PBS was collected and stored at −20 °C until analysis. The dram vials were refreshed with 3 mL of 1× PBS at 37 °C at each time point. To quantify the concentration of CXCL12 released, ELISA was performed. The frozen samples were brought to room temperature and the CXCL12 concentration was quantified with Human CXCL12/SDF‐1 DuoSet ELISA kit (R&D Systems) according to the manufacturer's protocol. Absorbance values of the resulting samples from ELISA were quantified with a spectrophotometer (SpectraMax M2e Molecular Devices) with readings taken at 450 nm and reference readings taken at 540 nm to consider wavelength corrections. The 4P logistic sigmoid curve fit function from JMP was used to generate a calibration curve from the standards and determine the corresponding concentrations of CXCL12 in the samples based on the absorbance values.
Cell Culture
The human GBM cell line U251‐MG (Sigma‐Aldrich) was cultured in complete Dulbecco's Modified Eagle Medium (DMEM) (ATCC) containing 4 mm l‐glutamine, 4500 mg L−1 glucose, 1 mm sodium pyruvate, and 1500 mg L−1 sodium bicarbonate supplemented with 10% fetal bovine serum (Tissue Culture Biologicals) and 1% penicillin‐streptomycin (Corning). Cells were maintained in 5% CO2 and 37 °C until 80% confluence was reached, at which point the cells were passaged. For all migration and invasion assays, adherent cells were lifted from flasks by incubating with non‐enzymatic cell dissociation solution (ATCC) for 10 min at 37 °C during passaging. Otherwise, 0.25% trypsin‐EDTA (Lonza) was used to collect adherent cells. In order to culture spheroid‐forming GBM cells with stem‐like cell properties, a previously established protocol was applied to isolate cancer stem cells from the U251 cell line under serum‐free culture conditions.[ 110 ] Briefly, U251 GBM cells subcultured to new flasks were cultured under normal conditions for 24 h to allow adherence before being switched to serum‐free culture conditions by maintaining the cells in neural stem cell media containing DMEM/F‐12 (ATCC), 1× B‐27 (Gibco), 20 ng mL−1 recombinant human epidermal growth factor (Life Technologies), 20 ng mL−1 human basic fibroblast growth factor (Acro Biosystems), 10 ng mL−1 human leukemia inhibitory factor (Acro Biosystems), and 4 U l−1 insulin (Sigma‐Aldrich). Thereafter, the GSC population was isolated by mechanical agitation to dislodge the spheroids, which were subsequently filtered through a 37 µm pore size reversible strainer (StemCell Technologies). Mechanical agitation was used to further breakdown spheroids to single cells, which were cultured in suspension on non‐tissue culture‐treated flasks. The GSCs were continuously cultured under serum‐free neural stem cell media conditions at 37 °C with 5% CO2 and subcultured every 9–10 days to avoid a necrotic core.[ 81 ] The patient‐derived primary G34 and G528 glioma stem cells were provided through a collaboration with Dr. Benjamin Purow (University of Virginia) and were originally a kind gift from Dr. Jakub Godlewski. These cells were cultured according to a previously established protocol that had also characterized them.[ 52 ] Briefly, these GSCs were cultured in 1× Neurobasal Medium (Life Technologies) supplemented with Glutamax (Gibco) at 0.5 mm, B‐27 without Vitamin A (Gibco) at 0.5×, N2 (Gibco) at 0.5×, human basic fibroblast growth factor (Life Technologies) at 25 ng mL−1, and human epidermal growth factor (Life Technologies) at 25 ng mL−1. Spheroids were allowed to reach between 200 and 500 µm before passaging or being used for the Transwell assay. Mechanical agitation was used to dislodge spheroids and subsequently breakdown spheroids into single cells, which were then isolated from media with centrifugation and then cultured in suspension on non‐tissue culture‐treated flasks and maintained at 37 °C with 5% CO2.
Transwell Migration Assay
Tissue culture treated, polycarbonate Transwell inserts (Corning) with 8 µm pore size were placed over 24 well plate wells and used in migration assays for both the U251 GSC and GBM cells. Migration media comprised DMEM supplemented with 0.1% bovine serum albumin (BSA) (Fisher BioReagents) and 1% penicillin‐streptomycin. Sterile synthetic hydrogels were prepared under sterile conditions according to our previous protocol.[ 32 ] Briefly, the 0.175 m NaHCO3(aq) solution was prepared using sterile deionized water. This base solution, the Thiocure precursor, and the PEGDA precursor solutions were all sterile filtered with 0.22 µm sterile filter units (Millex) prior to proceeding with the rest of the synthesis process. Hydrogels were loaded with CXCL12 chemokines during synthesis as described in the previous section. Five different conditions were prepared as the chemoattractant on the bottom chamber of the Transwell assay setup (Figure 4a), including just migration media, empty synthetic hydrogels, synthetic hydrogels encapsulated with 5 µg mL−1 of CXCL12, synthetic hydrogel encapsulating U251 GBM cells at a density of 3 × 106 cells/mL, and 0.2 µg mL−1 of CXCL12 solution in migration media. All of the hydrogel samples were submerged in migration media as well. For the top chamber, 20 000 U251 cells suspended in migration media were seeded on the upper side of the Transwell membrane insert. These cells were allowed to settle for 10 min in the incubator at 37 °C prior to introducing them to the wells containing the chemoattractant in the bottom chamber. For each of these five conditions, three replicates of samples were prepared. Samples were prepared independently for each of the two time points (24 and 48 h) for each cell type (U251 GBM and GSCs). Samples were maintained at 37 °C with 5% CO2 until the designated time point, at which point a cotton swab was used to gently remove cells and the media from the upper side of the membrane insert. Samples were then washed twice with 1× PBS, fixed with 10% formalin (Sigma‐Aldrich) for 20 min, permeabilized with 0.5% TritonX‐100 (Polysciences Inc.) for 20 min, blocked with 1% BSA for 1 h, incubated with 0.1 µg mL−1 of DAPI (Sigma‐Aldrich) for 15 min, and then dried for 30 min at room temperature. Samples were washed twice in 1× PBS in between each of these steps. Afterward, a scalpel was used to remove the membrane insert from the Transwell, which was then placed over a 25 µL droplet of PBS on a No. 1 glass coverslip (Corning) for imaging. Samples were imaged with a Zeiss LSM 800, axio observer Z1/7 inverted confocal microscope with a Plan‐Apochromat 10× objective lens at 0.45 numerical aperture. Ten fields of view were randomly selected to count the number of DAPI‐positive cells that had migrated to the underside of the membrane insert for each sample. The number of cells that migrated per field of view was quantified and averaged for each sample group. The final average number of migrated cells per sample group was normalized to the average number of migrated cells in the control group comprising just the migration media as the chemoattractant for each designated time point and cell type to determine the migration index. Statistical analyses were performed for each condition against the control migration media with a Student's t‐test to determine the chemoattractive potential of each sample compared to the background migration of the cells.[ 79 ]
The U251 GBM and GSC Transwell assay results were validated by repeating the Transwell migration assay with the patient‐derived, primary G34, and G528 glioma stem cells. For each cell type, four different conditions were prepared as the chemoattractant, including just migration media, empty synthetic hydrogels submerged in migration media, CXCL12 loaded into synthetic hydrogels at 5 µg mL−1 concentration and submerged in migration media, and CXCL12 in migration media at 0.2 µg mL−1 concentration. Sterile synthetic hydrogels were prepared as described before and each chemoattractant condition was prepared as described before in the same Transwell inserts. The migration media comprised the complete Neurobasal medium without the epidermal and basic fibroblast growth factors to mitigate any impact on migration. Three replicate samples of the Transwell assay were prepared for each of these conditions and each cell type. The top chamber comprised 20 000 cells (either G34 or G528) suspended in the migration media, which was allowed to settle for 10 min at 37 °C prior to adding the insert onto the bottom chamber with the chemoattractant. Samples were maintained at 37 °C and 5% CO2 for 24 h, at which point the samples were prepared for imaging by removing cells from the upper side of the insert with a cotton swab, fixing, permeabilizing, blocking, and DAPI staining as described previously. Samples were imaged with the confocal microscope and the migration index was quantified in the same manner as described for the U251 cells.
Extracellular Matrix Hydrogel Synthesis
Sterile collagen–HA ECM hydrogels were prepared according to a previous protocol.[ 111 ] Briefly, U251 GBM or GSCs at a concentration of 1 × 106 cells/mL were seeded into the ECM hydrogels comprising 0.12% rat tail type I collagen (Corning) and 0.2% thiolated hyaluronic acid (Advanced Biomatrix), with 0.2% PEGDA (Advanced Biomatrix). Hydrogels were crosslinked for 30 min at 37 °C under 5% CO2 before 1 mL of the appropriate media was added for cell culture.
U251 GBM and GSC Invasion Assay
PDMS mold cutouts were prepared at a ratio of 10:1 (Dow SYLGARD 184) with a 10 mm diameter and 1 mm depth according to a previous protocol.[ 112 ] These PDMS stamps were then autoclaved, plasma treated for 5 min (Harrick Plasma), and each side was sterilized under UV exposure for 1 h and then placed into 24 well plate wells. Subsequently, the stamps were treated for 10 min with 1% polyethylenimine, treated with 0.1% glutaraldehyde for 20 min, washed with sterile deionized water, and then dried for 30 min under sterile conditions. Sterile synthetic hydrogels were synthesized and either left empty or loaded with CXCL12 at 5 µg mL−1 as described previously and dispensed at 50 µL volumes to crosslink directly in the PDMS molds at 37 °C. ECM hydrogels encapsulating either U251 GBM or GSCs at 1 × 106 cells/mL were dispensed at 50 µL volumes directly on the surface of the synthetic hydrogels in the PDMS molds and crosslinked in the incubator as described previously, after which point 1 mL of migration media was added to each sample. For each condition (empty control hydrogels or chemokine‐loaded hydrogels) and cell type (GBM or GSCs), three replicates were prepared. This dual‐layer hydrogel platform setup for the invasion assay is illustrated by the schematic diagram in Figure 5a. Samples were maintained at 37 °C in 5% CO2 for 24 h, after which point the media was removed and samples were fixed, permeabilized, and blocked as described previously in the Transwell migration assay setup. Afterward, samples were washed twice again in PBS and incubated for 1 h with the staining buffer comprising Alexa Fluor 488 phalloidin (1:200, Life Technologies) and 0.1 µg mL−1 DAPI in 1% BSA solution. Samples were washed twice in 1× PBS and imaged with confocal microscopy. The hydrogel‐PDMS composites were carefully taken out of the well plate and flipped onto a 25 µL droplet of PBS on a No. 1 glass coverslip. Reflectance imaging at 640 nm with 41 µm pinhole at 10× was first conducted in order to identify the demarcation between the ECM and synthetic hydrogel layers based on a difference in opacity. Reflectance was used to set the top and bottom z positions for the entire range through the dual‐layer hydrogels, including the surface of the ECM hydrogel on the top layer and the bottom of the synthetic hydrogel layer, when the PDMS mold was visible. Z‐stacks with slices of 4.37 µm thickness were used to image the entire interval from the ECM hydrogel surface, through the synthetic hydrogel layer, and to the start of the PDMS molds. Once the demarcation of the two hydrogel layers was identified through this 3D rendered z‐stack image, the filter was changed to Alexa Fluor 488 and DAPI to image the cells in the hydrogel layers. Z‐stack images were taken at 10× and 37 µm pinhole with optical slices 3.71 µm thick. These z‐stack intervals began on the same z position as that which was determined by the reflectance imaging to be the top of the ECM hydrogel surface, while last z positions were set to 200–400 µm deep into the synthetic hydrogel layer in order to image invading cells. The number of invaded cells at least 20 µm (one cell diameter) deep into the synthetic hydrogels was quantified with ImageJ software for each sample.
Blebbistatin Treated Invasion Assay and Myosin IIA Immunofluorescence
Treated and sterile PDMS molds were prepared as described previously. Sterile dual‐layer hydrogels comprising the synthetic hydrogel on the bottom and ECM hydrogel on top were synthesized in PDMS molds as described previously in the invasion assay setup. A total of three different conditions were prepared, each with hydrogel samples in triplicate, including a negative control comprising empty synthetic hydrogels in migration media, a positive control with synthetic hydrogels loaded with 5 µg mL−1 CXCL12 in migration media, and synthetic hydrogels loaded with 5 µg mL−1 CXCL12 in migration media containing 30 µm (‐)‐blebbistatin (Sigma‐Aldrich). In all of these dual‐layer hydrogels, U251 GBM cells at a density of 1 × 106 cells/mL were embedded in the ECM hydrogel layer. After 14 h of incubation, the media from all samples was removed and samples were washed, fixed, permeabilized, and blocked as described in the Transwell migration assay setup. Samples were then incubated with recombinant Alexa Fluor 647 anti‐non‐muscle myosin IIA conjugated antibody (1:100, Abcam Inc) for 1 h at room temperature. Afterward, samples were washed twice and incubated for 1 h at room temperature with a staining buffer comprising 0.1 µg mL−1 DAPI, Alexa Fluor 488 phalloidin (1:200), and 1% BSA. Samples were washed twice and imaged with confocal microscopy in the same manner as the U251 GBM and GSC invasion assay reported in the previous section. Similarly, the number of invaded cells at least 20 µm deep into the synthetic hydrogels was quantified with ImageJ software for each sample. Five fields of view from the ECM hydrogel layer were randomly selected per sample z‐stack for further analysis. For each field of view, three cells were selected and the normalized myosin IIA fluorescence was determined semi‐quantitatively with analysis from Zeiss Zen Blue 3.3 software. The normalized fluorescence intensities were quantified by determining the ratio of the average fluorescence intensity per pixel for each cell to the average background fluorescence intensity in the same field of view according to the previous protocol.[ 32 ] This method of semi‐quantitative analysis was implemented to take into consideration any changes in the microscope setting with time according to previous protocols.[ 113 , 114 ] Representative 1.61 µm optical slice images were acquired with a Plan‐Apochromat 20× objective lens comprising a numerical aperture of 0.8 and a 35 µm pinhole.
ECM Hydrogel Collagen Fiber and Synthetic Hydrogel Surface Pore Imaging
An LSM 880 multiphoton confocal microscope was used to image the collagen fibers in the ECM hydrogels. In triplicate, ECM hydrogels loaded with 1 × 106 U251 GBM cells/mL were synthesized and crosslinked at 100 µL volumes in sterile, treated PDMS molds. The samples were cultured at 37 °C and 5% CO2 for 24 h, after which point the samples were washed, fixed, permeabilized, blocked, and stained with DAPI as reported in previous sections. The chameleon laser at 780 nm under the non‐descanned mode was used, and samples were imaged with second‐harmonic generation. Images were acquired with a Plan‐Apochromat 20× objective lens with a numerical aperture of 0.8 from five fields of view selected at random. The multiphoton confocal microscope with laser HeNe543 and Channel 2 was also utilized for reflectance imaging of pores on the synthetic hydrogel surface at 543 nm. In triplicate, 100 µL of the empty synthetic hydrogels were also synthesized and crosslinked directly in PDMS molds at 37 °C. Hydrogels were swelled for 24 h in 1× PBS at physiologic temperature prior to imaging. Five random fields of view were selected for imaging the hydrogel surface at 20×. The diameters of pores and defects on the hydrogel surface were obtained through image analysis in Zen Blue, where ten pores or defects were selected per field of view for quantitative analysis of the sizes.
Immunofluorescence of Stem Cell and Glial Markers upon GBM/GSC Interaction with Hydrogels
Sterile and treated PDMS molds were prepared as described previously. 100 µL of either sterile synthetic hydrogels or ECM hydrogels were dispensed and crosslinked separately in the PDMS molds at 37 °C as described previously. All hydrogels were then hydrated with 1 mL of either complete DMEM media for GBM cell samples or 1 mL of GSC serum‐free media for samples intended for GSC culture. Afterward, the media was removed and 100 000 U251 GSC or GBM cells were seeded on top of the hydrogel surface. Samples were incubated for 10 min at 37 °C and 5% CO2 to allow the cells to settle on the surface. Subsequently, appropriate volumes of either complete DMEM or GSC media were added to the samples and the cells were cultured on the hydrogel surface until the designated time point (24 or 48 h). For both time points, a total of four different conditions each with three replicates of hydrogels for both cell types were prepared: GBM cells cultured on the ECM hydrogel, GSCs cultured on the ECM hydrogel, GBM cells cultured on the synthetic hydrogel, and GSCs cultured on the synthetic hydrogel. At the designated time point, the media from the samples was removed and samples were washed, fixed, permeabilized, and blocked as reported in the Transwell migration assay previously. The samples were then incubated for 1 h at room temperature with the primary antibody buffer comprising rabbit nestin polyclonal antibody (1:200, Proteintech) and GFAP mouse monoclonal antibody GA5 (1:200, Life Technologies). After washing with PBS, samples were then incubated for 1 h at room temperature with the secondary antibody staining buffer comprising goat anti‐rabbit secondary antibody Alexa Fluor 647 (1:500, Invitrogen) and IgG1 cross‐adsorbed goat anti‐mouse secondary antibody Alexa Fluor 488 (1:500, Life Technologies). Samples were then washed again with PBS, incubated for 30 min with 0.1 µg mL−1 DAPI, and washed again prior to imaging with the confocal microscope. Optical slice images 1.61 µm thick and focused on the hydrogel surface were acquired with a 20× objective lens comprising a numerical aperture of 0.8 and a 35 µm pinhole. Ten random fields of view were selected per sample. For each field of view, three cells were selected and the normalized nestin or GFAP fluorescence intensities were determined semi‐quantitatively as reported in the previous section. Samples of both ECM and synthetic hydrogels with no primary antibody incubation were also prepared as negative controls to confirm the lack of non‐specific binding and staining with the secondary antibodies.
U251 Cell Viability in Hydrogels
Sterile and treated PDMS molds were prepared as described previously. Both ECM and synthetic hydrogels at 100 µL volumes and encapsulated with 1 × 106 U251 GBM cells/mL were dispensed separately and directly crosslinked in the PDMS stamps. Three replicates of each hydrogel type were prepared for each time point including 24, 72, and 120 h. 1 mL of complete DMEM media was added to each well after hydrogels were crosslinked, and samples were maintained at 37 °C under 5% CO2 until the designated time point, after which point the media was removed, samples were washed with sterile 1× PBS, and incubated in the dark at 37 °C for 1 h with the live/dead staining buffer comprising calcein green AM (1:500, Invitrogen) and propidium iodide (1:65, Life Technologies) in 1× PBS. Samples were subsequently washed with sterile 1× PBS and the hydrogel‐PDMS stamps were taken out of the well and flipped onto a No. 1 glass coverslip for imaging with the confocal microscope. For each hydrogel, samples were imaged under the 514 and 617 nm wavelengths with z‐stacks for depths up to 600 µm (ECM hydrogels) and 1 mm (synthetic hydrogels). These images were obtained with a 10× objective lens with a numerical aperture of 0.45 and optical slices with a thickness of 3.45 µm. Samples were also imaged with an N‐Achroplan 5× objective lens at 0.15 numerical aperture with 32 µm pinhole to acquire representative images up to 300 µm deep in each hydrogel. Ten fields of view were randomly selected per sample to image and quantify live/dead cells. Images of negative controls of both ECM and synthetic hydrogels without any encapsulated cells confirmed the lack of non‐specific background staining. The number of calcein stained cells (C c) and propidium iodide (C p) stained cells per field of view was quantified with an automated algorithm in ImageJ using green and red channels, respectively, and used to determine the percent cell viability of the U251 GBM cells encapsulated in the hydrogels according to Equation (8) as follows
| (8) |
Hydrogel Degradation
Sterile hydrogels of 500 µL volumes were synthesized in 24 well plate molds and either were left empty or encapsulated with U251 GBM cells at a density of 3 × 106 cells/mL. Three replicates of both cellular and acellular hydrogels were prepared for each time point including 24, 72, 120, and 168 h. These hydrogels were then transferred to sterile, vented 6‐dram vials maintained in 3 mL of complete DMEM media at 37 °C in 5% CO2. During the designated time point, media from the corresponding hydrogels was removed and the hydrogels were washed twice with deionized water to remove any salts and proteins before being placed in a watch glass to air dry for 24 h at room temperature and then dried in vacuo at room temperature for 48 h or more until completely dried. The final dried masses were recorded (m f). The media was completely refreshed every other day for all samples. Another set of both cellular and acellular hydrogels with three replicates for each was also prepared. These hydrogels were not subjected to any media and were air dried and then dried in vacuo as described to obtain the final dried masses of cured hydrogels (m o). The percent degradation of the hydrogels at each time point was then calculated according to Equation (9) as follows
| (9) |
Fibronectin Deposition
Sterile and treated PDMS molds were prepared as described previously. In triplicate, collagen‐HA hydrogels and synthetic hydrogels were prepared separately and dispensed at 100 µL volumes to crosslink directly in the PDMS molds. Each hydrogel was encapsulated with 1 × 106 GBM cells/mL and cultured in 1 mL of complete DMEM media at 37 °C and 5% CO2. Media for all samples was refreshed after 2 days of cell culture. After 72 h of cell culture, the media from all samples was removed and samples were washed, fixed, permeabilized, and blocked as described previously in the Transwell migration assay. Subsequently, samples were incubated overnight at 4 °C with fibronectin primary mouse antibody 2F4 (1:100, Novus Biologicals Inc), after which point samples were washed twice with PBS and incubated for 1 h at room temperature with goat anti‐mouse IgG1 cross‐adsorbed Alexa Fluor 488 secondary antibody (1:1000, Life Technologies). Samples were again washed twice with PBS and incubated for 30 min at room temperature with 0.1 µg mL−1 DAPI and washed twice again before imaging with a confocal microscope. Z‐stacks were obtained with 3.73 µm slices at 10× to image up to 400 µm depths into both hydrogels. Five fields of view were randomly selected per hydrogel sample for imaging at 20×, and 1.61 µm thick optical slice images were acquired with a 37 µm pinhole. These representative images were limited to within 300 µm depths into each hydrogel. Three cells were selected for analysis per field of view, and the normalized fibronectin fluorescence was determined semi‐quantitatively for each cell as described in the previous section on myosin IIA staining. Samples without any primary antibody incubation were also prepared and imaged to confirm minimal to lack of non‐specific binding of primary antibody to both ECM and synthetic hydrogels.
Electroconductivity
In triplicate, synthetic hydrogels were synthesized and dispensed in 900 µL volumes to crosslink directly in electroporation cuvettes (Sigma‐Aldrich) with 0.4 cm gap width. Hydrogels were incubated in 900 µL of 1× PBS containing calcium and magnesium ions (to mimic cerebrospinal fluid) for 24 h. Subsequently, direct current electric field pulses were applied to the samples with an ECM 830 electro square porator (BTX) at 50 V with pulse lengths of 100 µs and 3 pulses at 1 s intervals with an attenuation of 1/100. An oscilloscope (Wavesurfer 3024z) was used to determine the final voltage (V in volts) and resulting current (I in amperes) values to calculate the resistance (R in ohms) according to Equation (10) as follows
| (10) |
The pulsing scheme was applied to each sample thrice. The resistances and geometry of the cuvettes were then used to determine the conductivity of the hydrogels (C in S m−1) according to Equation (11), where t is the gap between plates (0.004 m) and A is the area of the plates (0.02 × 0.01 m)
| (11) |
Freshly prepared synthetic hydrogels not subjected to 24 h of incubation in PBS were also pulsed using the same procedure to determine the conductivity of cured hydrogels. For the hydrogel precursor liquid solutions, including 0.175 m NaHCO3(aq), PEGDA575, Thiocure, and deionized water, an Oakton PCTS 50 conductivity probe was used to determine the electroconductivities. The precursor solutions were freshly prepared in triplicate to report the final average conductivity values. All samples (cured hydrogels, equilibrated hydrogels, and precursor solutions) were brought to room temperature prior to subjecting them to pulsing or the conductivity probe.
Statistics
All results are reported as averages ± standard deviations based on replicates in experiments. All experiments were performed in triplicate or more. Statistical analyses were performed through JMP software using one way analysis of variance (ANOVA) with Tukey's post‐hoc analysis or Student's t‐test. Differences in data were deemed statistically significant based on p‐values < 0.05.
Conflict of Interest
Z.M.K., S.S.V., T.E.L., and E.V. are inventors on a pending patent related to this work. The authors declare no other financial or commercial conflicts of interest.
Supporting information
Supporting Information
Acknowledgements
This research was funded by the NSF CAREER Award [Grant CBET‐1652112] (SSV), NSERC Postgraduate Doctoral Scholar Award (ZMK), P.E.O. Scholar Award (ZMK), and Virginia Tech Graduate Research Development Program grant (ZMK). The authors would like to acknowledge Yanping Liang for culturing the G34 and G528 cells, Dr. Kinsley Tate for technical training on synthesizing the collagen‐HA hydrogels, Dr. Caleb Stine for guidance on setting the parameters for reflectance imaging on the multiphoton microscope, and Dr. Rosalyn Hatlen for guidance on setting the parameters for second harmonic generation on the multiphoton microscope. The authors thank Dr. Rafael Davalos for access to the BTX electro square porator and acknowledge Zaid Salameh and Raffae Ahmad for technical training on using the BTX electro square porator. Graphical abstract was designed with BioRender.com.
Khan Z. M., Munson J. M., Long T. E., Vlaisavljevich E., Verbridge S. S., Development of a Synthetic, Injectable Hydrogel to Capture Residual Glioblastoma and Glioblastoma Stem‐Like Cells with CXCL12‐Mediated Chemotaxis. Adv. Healthcare Mater. 2023, 12, 2300671. 10.1002/adhm.202300671
Data Availability Statement
The data that supports the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Ostrom Q. T., Cioffi G., Waite K., Kruchko C., Barnholtz‐Sloan J. S., Neuro. Oncol. 2021, 23, iii1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stupp R., Brada M., van den Bent M. J., Tonn J. C., Pentheroudakis G., Ann. Oncol. 2014, 25, iii93.24782454 [Google Scholar]
- 3. Ostrom Q. T., Cote D. J., Ascha M., Kruchko C., Barnholtz‐Sloan J. S., JAMA Oncol. 2018, 4, 1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hatoum A., Mohammed R., Zakieh O., Cancer Manag. Res. 2019, 11, 1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lara‐Velazquez M., Al‐Kharboosh R., Jeanneret S., Vazquez‐Ramos C., Mahato D., Tavanaiepour D., Rahmathulla G., Quinones‐Hinojosa A., Brain Sci. 2017, 7, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Oliva C. R., Nozell S. E., Diers A., S. G. McClugage, 3rd , Sarkaria J. N., Markert J. M., Darley‐Usmar V. M., Bailey S. M., Gillespie G. Y., Landar A., Griguer C. E., J. Biol. Chem. 2010, 285, 39759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pisco A. O., Brock A., Zhou J., Moor A., Mojtahedi M., Jackson D., Huang S., Nat. Commun. 2013, 4, 2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Prager B. C., Bhargava S., Mahadev V., Hubert C. G., Rich J. N., Trends Cancer 2020, 6, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lathia J. D., Mack S. C., Mulkearns‐Hubert E. E., Valentim C. L. L., Rich J. N., Genes Dev. 2015, 29, 1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pistollato F., Abbadi S., Rampazzo E., Persano L., Puppa A. D., Frasson C., Sarto E., Scienza R., D'Avella D., Basso G., Stem Cells 2010, 28, 851. [DOI] [PubMed] [Google Scholar]
- 11. Li Z., Bao S., Wu Q., Wang H., Eyler C., Sathornsumetee S., Shi Q., Cao Y., Lathia J., McLendon R. E., Hjelmeland A. B., Rich J. N., Cancer Cell 2009, 15, 501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bao S., Wu Q., McLendon R. E., Hao Y., Shi Q., Hjelmeland A. B., Dewhirst M. W., Bigner D. D., Rich J. N., Nature 2006, 444, 756. [DOI] [PubMed] [Google Scholar]
- 13. Cheng L., Wu Q., Guryanova O. A., Huang Z., Huang Q., Rich J. N., Bao S., Biochem. Biophys. Res. Commun. 2011, 406, 643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jackson M., Hassiotou F., Nowak A., Carcinogenesis 2015, 36, 177. [DOI] [PubMed] [Google Scholar]
- 15. Alieva M., Leidgens V., Riemenschneider M. J., Klein C. A., Hau P., van Rheenen J., Sci. Rep. 2019, 9, 2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baker G. J., Yadav V. N., Motsch S., Koschmann C., Calinescu A. A., Mineharu Y., Camelo‐Piragua S. I., Orringer D., Bannykh S., Nichols W. S., deCarvalho A. C., Mikkelsen T., Castro M. G., Lowenstein P. R., Neoplasia 2014, 16, 543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Seano G., Jain R. K., Angiogenesis 2020, 23, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Conte M., Casas‐Tintò S., Soler J., PLoS Comput. Biol. 2021, 17, e1008632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ridley A. J., Schwartz M. A., Burridge K., Firtel R. A., Ginsberg M. H., Borisy G., Parsons J. T., Horwitz A. R., Science 2003, 302, 1704. [DOI] [PubMed] [Google Scholar]
- 20. Onishi M., Ichikawa T., Kurozumi K., Date I., Brain Tumor Pathol. 2011, 28, 13. [DOI] [PubMed] [Google Scholar]
- 21. Tamimi A. F., Juweid M., in Epidemiology and Outcome of Glioblastoma, (Ed: De Vleeschouwer S.), Codon Publications, Brisbane, Australia: 2017, pp. 143–153. [PubMed] [Google Scholar]
- 22. Mair D. B., Ames H. M., Li R., Mol. Biol. Cell 2018, 29, 2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ehtesham M., Winston J. A., Kabos P., Thompson R. C., Oncogene 2006, 25, 2801. [DOI] [PubMed] [Google Scholar]
- 24. Zagzag D., Esencay M., Mendez O., Yee H., Smirnova I., Huang Y., Chiriboga L., Lukyanov E., Liu M., Newcomb E. W., Am. J. Pathol. 2008, 173, 545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brennecke P., Arlt M. J., Campanile C., Husmann K., Gvozdenovic A., Apuzzo T., Thelen M., Born W., Fuchs B., Clin. Exp. Metastasis 2014, 31, 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Najberg M., Haji Mansor M., Boury F., Alvarez‐Lorenzo C., Garcion E., Front. Pharmacol. 2019, 10, 887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen Q., Zhang M., Li Y., Xu D., Wang Y., Song A., Zhu B., Huang Y., Zheng J. C., Stem Cells 2015, 33, 2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim M. Y., Oskarsson T., Acharyya S., Nguyen D. X., Zhang X. H., Norton L., Massagué J., Cell 2009, 139, 1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. van der Sanden B., Appaix F., Berger F., Selek L., Issartel J.‐P., Wion D., Future Oncol. 2013, 9, 817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Autier L., Clavreul A., Cacicedo M. L., Franconi F., Sindji L., Rousseau A., Perrot R., Montero‐Menei C. N., Castro G. R., Menei P., Acta Biomater. 2019, 84, 268. [DOI] [PubMed] [Google Scholar]
- 31. De Bacco F., D'Ambrosio A., Casanova E., Orzan F., Neggia R., Albano R., Verginelli F., Cominelli M., Poliani P. L., Luraghi P., Reato G., Pellegatta S., Finocchiaro G., Perera T., Garibaldi E., Gabriele P., Comoglio P. M., Boccaccio C., EMBO Mol. Med. 2016, 8, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Khan Z. M., Wilts E., Vlaisavljevich E., Long T. E., Verbridge S. S., Acta Biomater. 2022, 144, 266. [DOI] [PubMed] [Google Scholar]
- 33. Lange F., Venus J., Abady D. S. E., Porath K., Einsle A., Sellmann T., Neubert V., Reichart G., Linnebacher M., Köhling R., Kirschstein T., Life 2022, 12, 580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Edsall C., Khan Z. M., Mancia L., Hall S., Mustafa W., Johnsen E., Klibanov A. L., Durmaz Y. Y., Vlaisavljevich E., Ultrasound Med. Biol. 2021, 47, 620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Latouche E. L., Arena C. B., Ivey J. W., Garcia P. A., Pancotto T. E., Pavlisko N., Verbridge S. S., Davalos R. V., Rossmeisl J. H., Technol. Cancer Res. Treat. 2018, 17, 153303381878528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yao X. H., Liu Y., Chen K., Gong W., Liu M. Y., Bian X. W., Wang J. M., Int. Immunopharmacol. 2011, 11, 1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dalonneau F., Liu X. Q., Sadir R., Almodovar J., Mertani H. C., Bruckert F., Albiges‐Rizo C., Weidenhaupt M., Lortat‐Jacob H., Picart C., Biomaterials 2014, 35, 4525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wei M.‐T., Elbaum‐Garfinkle S., Holehouse A. S., Chen C. C.‐H., Feric M., Arnold C. B., Priestley R. D., Pappu R. V., Brangwynne C. P., Nat. Chem. 2017, 9, 1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Atterberry P. N., Roark T. J., Severt S. Y., Schiller M. L., Antos J. M., Murphy A. R., Biomacromolecules 2015, 16, 1582. [DOI] [PubMed] [Google Scholar]
- 40. Van Tomme S. R., van Nostrum C. F., de Smedt S. C., Hennink W. E., Biomaterials 2006, 27, 4141. [DOI] [PubMed] [Google Scholar]
- 41. Sheth S., Barnard E., Hyatt B., Rathinam M., Zustiak S. P., Front. Bioeng. Biotechnol. 2019, 7, 410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hagel V., Haraszti T., Boehm H., Biointerphases 2013, 8, 36. [DOI] [PubMed] [Google Scholar]
- 43. Masaro L., Zhu X. X., Prog. Polym. Sci. 1999, 24, 731. [Google Scholar]
- 44. Wang Y., Irvine D. J., Biomaterials 2011, 32, 4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Venturoli D., Rippe B., Am. J. Physiol. Renal. Physiol. 2005, 288, F605. [DOI] [PubMed] [Google Scholar]
- 46. O'Shea T. M., Aimetti A. A., Kim E., Yesilyurt V., Langer R., Adv. Mater. 2015, 27, 65. [DOI] [PubMed] [Google Scholar]
- 47. Bajetto A., Barbieri F., Dorcaratto A., Barbero S., Daga A., Porcile C., Ravetti J. L., Zona G., Spaziante R., Corte G., Schettini G., Florio T., Neurochem. Int. 2006, 49, 423. [DOI] [PubMed] [Google Scholar]
- 48. Rubin J. B., Kung A. L., Klein R. S., Chan J. A., Sun Y., Schmidt K., Kieran M. W., Luster A. D., Segal R. A., Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 13513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.a) Chieppa M., Bianchi G., Doni A., Del Prete A., Sironi M., Laskarin G., Monti P., Piemonti L., Biondi A., Mantovani A., Introna M., Allavena P., J. Immunol. 2003, 171, 4552; [DOI] [PubMed] [Google Scholar]; b) Lebre M. C., Burwell T., Vieira P. L., Lora J., Coyle A. J., Kapsenberg M. L., Clausen B. E., De Jong E. C., Immunol. Cell Biol. 2005, 83, 525. [DOI] [PubMed] [Google Scholar]
- 50. Locatelli M., Boiocchi L., Ferrero S., Martinelli Boneschi F., Zavanone M., Pesce S., Allavena P., Maria Gaini S., Bello L., Mantovani A., Eur. Cytokine Network 2010, 21, 27. [DOI] [PubMed] [Google Scholar]
- 51. Kingsmore K. M., Logsdon D. K., Floyd D. H., Peirce S. M., Purow B. W., Munson J. M., Integr. Biol. 2016, 8, 1246. [DOI] [PubMed] [Google Scholar]
- 52. Lee J., Kotliarova S., Kotliarov Y., Li A., Su Q., Donin N. M., Pastorino S., Purow B. W., Christopher N., Zhang W., Park J. K., Fine H. A., Cancer Cell 2006, 9, 391. [DOI] [PubMed] [Google Scholar]
- 53. Allison K., Logsdon D., Floyd D., Peirce‐Cottler S., Purow B., Munson J., Integr. Biol. 2016, 8, 1246. [DOI] [PubMed] [Google Scholar]
- 54. Visweshwaran S. P., Maritzen T., MethodsX 2019, 6, 2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gladson C. L., J. Neuropathol. Exp. Neurol. 1999, 58, 1029. [DOI] [PubMed] [Google Scholar]
- 56. Cui Y., Cole S., Pepper J., Otero J. J., Winter J. O., Biomater. Sci. 2020, 8, 4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chernov A. V., Baranovskaya S., Golubkov V. S., Wakeman D. R., Snyder E. Y., Williams R., Strongin A. Y., J. Biol. Chem. 2010, 285, 19647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Huijbers I. J., Iravani M., Popov S., Robertson D., Al‐Sarraj S., Jones C., Isacke C. M., PLoS One 2010, 5, e9808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cha J., Kang S.‐G., Kim P., Sci. Rep. 2016, 6, 24912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Munson J. M., Bellamkonda R. V., Swartz M. A., Cancer Res. 2013, 73, 1536. [DOI] [PubMed] [Google Scholar]
- 61. Yang Y.‐l., Sun C., Wilhelm M. E., Fox L. J., Zhu J., Kaufman L. J., Biomaterials 2011, 32, 7932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rao S. S., Dejesus J., Short A. R., Otero J. J., Sarkar A., Winter J. O., ACS Appl. Mater. Interfaces 2013, 5, 9276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cha J., Kang S. G., Kim P., Sci. Rep. 2016, 6, 24912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kaufman L. J., Brangwynne C. P., Kasza K. E., Filippidi E., Gordon V. D., Deisboeck T. S., Weitz D. A., Biophys. J. 2005, 89, 635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sahai E., Marshall C. J., Nat. Cell Biol. 2003, 5, 711. [DOI] [PubMed] [Google Scholar]
- 66. Wyckoff J. B., Pinner S. E., Gschmeissner S., Condeelis J. S., Sahai E., Curr. Biol. 2006, 16, 1515. [DOI] [PubMed] [Google Scholar]
- 67. Te Boekhorst V., Friedl P., Adv. Cancer Res. 2016, 132, 209. [DOI] [PubMed] [Google Scholar]
- 68. Eichinger L., Pachebat J. A., Glöckner G., Rajandream M. A., Sucgang R., Berriman M., Song J., Olsen R., Szafranski K., Xu Q., Tunggal B., Kummerfeld S., Madera M., Konfortov B. A., Rivero F., Bankier A. T., Lehmann R., Hamlin N., Davies R., Gaudet P., Fey P., Pilcher K., Chen G., Saunders D., Sodergren E., Davis P., Kerhornou A., Nie X., Hall N., Anjard C., et al, Nature 2005, 435, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cha J., Kim P., ACS Appl. Mater. Interfaces 2021, 13, 31371. [DOI] [PubMed] [Google Scholar]
- 70. Thorne R. G., Nicholson C., Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Beadle C., Assanah M. C., Monzo P., Vallee R., Rosenfeld S. S., Canoll P., Mol. Biol. Cell 2008, 19, 3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lee W., Lim S., Kim Y., PLoS One 2017, 12, e0171312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Limouze J., Straight A. F., Mitchison T., Sellers J. R., J. Muscle Res. Cell Motil. 2004, 25, 337. [DOI] [PubMed] [Google Scholar]
- 74. Straight A. F., Cheung A., Limouze J., Chen I., Westwood N. J., Sellers J. R., Mitchison T. J., Science 2003, 299, 1743. [DOI] [PubMed] [Google Scholar]
- 75. Ivkovic S., Beadle C., Noticewala S., Massey S. C., Swanson K. R., Toro L. N., Bresnick A. R., Canoll P., Rosenfeld S. S., Mol. Biol. Cell 2012, 23, 533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Agudelo‐Garcia P. A., De Jesus J. K., Williams S. P., Nowicki M. O., Chiocca E. A., Liyanarachchi S., Li P. K., Lannutti J. J., Johnson J. K., Lawler S. E., Viapiano M. S., Neoplasia 2011, 13, 831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Paluch E., Piel M., Prost J., Bornens M., Sykes C., Biophys. J. 2005, 89, 724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wolf K., Mazo I., Leung H., Engelke K., von Andrian U. H., Deryugina E. I., Strongin A. Y., Bröcker E. B., Friedl P., J. Cell Biol. 2003, 160, 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Giarra S., Ierano C., Biondi M., Napolitano M., Campani V., Pacelli R., Scala S., De Rosa G., Mayol L., Carbohydr. Polym. 2018, 191, 112. [DOI] [PubMed] [Google Scholar]
- 80. Khan Z. M., Wilts E., Vlaisavljevich E., Long T. E., Verbridge S. S., Macromol. Biosci. 2022, 22, 2100355. [DOI] [PubMed] [Google Scholar]
- 81. Alinezhadbalalami N., Douglas T. A., Balani N., Verbridge S. S., Davalos R. V., Electrophoresis 2019, 40, 2592. [DOI] [PubMed] [Google Scholar]
- 82. Sereika M., Urbanaviciute R., Tamasauskas A., Skiriute D., Vaitkiene P., J. Cancer 2018, 9, 4496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Tsou Y.‐H., Khoneisser J., Huang P.‐C., Xu X., Bioact. Mater 2016, 1, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wang T.‐W., Spector M., Acta Biomater. 2009, 5, 2371. [DOI] [PubMed] [Google Scholar]
- 85. Namba N., Chonan Y., Nunokawa T., Sampetrean O., Saya H., Sudo R., Tissue Eng., Part A. 2021, 27, 467. [DOI] [PubMed] [Google Scholar]
- 86. Solano A. G., Dupuy J., Therriault H., Liberelle B., Faucheux N., Lauzon M. A., Virgilio N., Paquette B., Carbohydr. Polym. 2021, 266, 118115. [DOI] [PubMed] [Google Scholar]
- 87. Ribeiro V. P., Silva‐Correia J., Gonçalves C., Pina S., Radhouani H., Montonen T., Hyttinen J., Roy A., Oliveira A. L., Reis R. L., Oliveira J. M., PLoS One 2018, 13, e0194441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Würth R., Bajetto A., Harrison J. K., Barbieri F., Florio T., Front. Cell Neurosci. 2014, 8, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Xu Q., He C., Zhang Z., Ren K., Chen X., ACS Appl. Mater. Interfaces 2016, 8, 30692. [DOI] [PubMed] [Google Scholar]
- 90. Dragoj M., Stojkovska J., Stanković T., Dinić J., Podolski‐Renić A., Obradović B., Pešić M., Brain Sci. 2021, 11, 1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Rodriguez E. J., Marcos B., Huneault M. A., J. Appl. Polym. Sci. 2016, 133, 45. [Google Scholar]
- 92. Bilozur M. E., Hay E. D., Dev. Biol. 1988, 125, 19. [DOI] [PubMed] [Google Scholar]
- 93. Bryant S. J., Anseth K. S., J. Biomed. Mater Res. 2002, 59, 63. [DOI] [PubMed] [Google Scholar]
- 94. Pritchard C. D., O'Shea T. M., Siegwart D. J., Calo E., Anderson D. G., Reynolds F. M., Thomas J. A., Slotkin J. R., Woodard E. J., Langer R., Biomaterials 2011, 32, 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Liu F., Yang B., Bio. Med. Res. Int. 2020, 2020, 7930160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Barnes F. S., Greeenebaum B., Handbook of Biological Effects of Electromagnetic Fields – Two volume Set, CRC Press, Boca Raton, FL: 2006. [Google Scholar]
- 97. Lyon J. G., Carroll S. L., Mokarram N., Bellamkonda R. V., Sci. Rep. 2019, 9, 5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Huang Y.‐J., Hoffmann G., Wheeler B., Schiapparelli P., Quinones‐Hinojosa A., Searson P., Sci. Rep. 2016, 6, 21583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Tsai H. F., C I. J., Shen A. Q., APL Bioeng. 2020, 4, 036102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Tsai H.‐F., IJspeert C., Shen A. Q., APL Bioeng. 2020, 4, 036102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Tang L., United States Patent PCT/US2013/065803, 2015.
- 102. Varshosaz J., Hajian M., Drug Delivery 2004, 11, 53. [DOI] [PubMed] [Google Scholar]
- 103. Ritger P. L., Peppas N. A., J. Controlled Release 1987, 5, 23. [Google Scholar]
- 104. Park W. S. W. S. K., Park H., Biodegradable Hydrogeis for Drug Delivery, CRC Press, Boca Raton, FL: 1993. [Google Scholar]
- 105. Fu Y., Kao W. J., Expert Opin. Drug Delivery 2010, 7, 429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ritger P. L., Peppas N. A., J. Controlled Release 1987, 5, 37. [Google Scholar]
- 107. Lemaire V., Bélair J., Hildgen P., Int. J. Pharm. 2003, 258, 95. [DOI] [PubMed] [Google Scholar]
- 108. Raman C., Berkland C., Kim K., Pack D. W., J. Controlled Release. 2005, 103, 149. [DOI] [PubMed] [Google Scholar]
- 109. Vigata M., Meinert C., Hutmacher D. W., Bock N., Pharmaceutics 2020, 12, 1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Yu S. C., Ping Y. F., Yi L., Zhou Z. H., Chen J. H., Yao X. H., Gao L., Wang J. M., Bian X. W., Cancer Lett. 2008, 265, 124. [DOI] [PubMed] [Google Scholar]
- 111. Harris A. R., Yuan J. X., Munson J. M., Methods 2018, 134–135, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Ivey J. W., Latouche E. L., Sano M. B., Rossmeisl J. H., Davalos R. V., Verbridge S. S., Sci. Rep. 2015, 5, 17157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Galarza S., Crosby A. J., Pak C., Peyton S. R., Adv. Healthcare Mater. 2020, 9, 1901419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Placone A. L., McGuiggan P. M., Bergles D. E., Guerrero‐Cazares H., Quiñones‐Hinojosa A., Searson P. C., Biomaterials 2015, 42, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that supports the findings of this study are available from the corresponding author upon reasonable request.