

Summary
MicroRNAs, crucial in regulating protein-coding gene expression, are implicated in various diseases. We performed a genome-wide association study of plasma miRNAs (ex-miRNAs) in 3,743 Framingham Heart Study (FHS) participants and identified 1,027 cis-ex-miRNA-eQTLs (cis-exQTLs) for 37 ex-miRNAs, with 55% replication in an independent study. Colocalization analyses suggested potential genetic coregulation of ex-miRNAs with whole blood mRNAs. Mendelian randomization indicated 29 ex-miRNAs potentially influencing 35 traits. Notably, the chromosome 14q23 and 14q32 miRNA clusters emerged as the top signal, contributing over 50% of the significant cis-exQTL results, and were associated with a diverse range of traits including platelet count. Correlations of 10 ex-miRNAs (such as miR-376c-3p) in 14q32 with platelet count and volume were confirmed in FHS participants. These findings shed light on the genetic basis of ex-miRNA expression and their involvement in complex traits.
Subject areas: Body substance sample, Quantitative genetics, Association analysis, Genomics, Transcriptomics
Graphical abstract
Highlights
-
•
1,027 cis-exQTLs for 37 ex-miRNAs were identified, with 55% replication by independent study
-
•
Chr. 14q23 and 14q32 miRNA clusters show enriched genetic regulation and functional roles
-
•
Ten ex-miRNAs associate with platelet traits by MR and cross-sectional analyses
-
•
Computational analyses suggest potential co-regulation of ex-miRNAs with blood mRNAs
Body substance sample; Quantitative genetics; Association analysis; Genomics; Transcriptomics
Introduction
MicroRNAs (miRNA) are a class of small non-coding RNA molecules of approximately 22 nucleotides that play a crucial role in the regulation of gene expression.1 miRNAs act by binding to complementary sequences on messenger RNAs (mRNAs), which can cause the degradation of these mRNAs or prevent their translation into proteins.2 Circulating miRNAs, also known as extracellular miRNAs (ex-miRNAs), are miRNAs that are secreted from cells and can be detected in plasma or various biological fluids. These ex-miRNAs can act as signaling molecules and play important roles in intercellular communication and the regulation of gene expression in distant cells.3,4 Recently, ex-miRNAs have emerged as potential diagnostic and prognostic biomarkers of disease due to their stability and the ease of their detection and quantification.5 Aberrant expression of ex-miRNAs has been observed in various diseases, including cancer,6 cardiovascular disease,7,8,9 and neurodegenerative disorders.10 Despite the potential diagnostic and therapeutic applications of ex-miRNAs, the underlying mechanisms governing their contributions to disease pathogenesis are not fully understood.
Expression quantitative trait loci (eQTLs) are genetic variants that regulate gene expression. These variants can be located near the gene (cis) or at a distance (trans) and affect gene expression levels. Identifying genetic variants associated with ex-miRNA expression levels (known as ex-miRNAs eQTLs, or exQTLs for short) may provide insights into the genetic regulatory mechanisms of ex-miRNAs and improve our understanding of the mechanisms underlying miRNA-mediated intercellular communication and gene regulation. Moreover, linking exQTLs with complex traits and diseases is crucial for understanding the underlying mechanisms of disease pathogenesis in which ex-miRNAs play a role. This knowledge will facilitate the development of biomarkers for disease diagnosis and prognosis and highlight promising therapeutic targets.
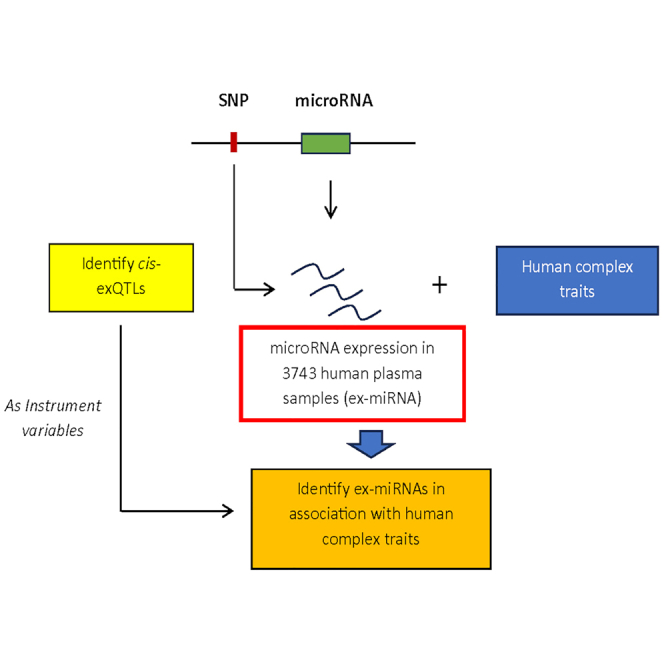
In a previous study, we focused on identifying eQTLs for whole blood miRNAs and reported 5,269 cis-miRNA-eQTLs in 5,239 whole blood samples from Framingham Heart Study (FHS) participants.11 While other studies have explored exQTLs and their associations with complex traits such as cardiovascular diseases,8,12,13 these studies were fraught with certain limitations and challenges including the need for larger sample sizes, replication of findings in diverse populations, and further exploration of downstream functions and pathways influenced by ex-miRNAs. To address these knowledge gaps, our current project utilized genotypes from whole-genome sequencing (WGS) and plasma expression levels of 282 mature miRNAs (in this study, unless otherwise specified, the terms “miRNA” or “ex-miRNA” refer to mature miRNAs) in 3,743 participants from the FHS. We carried out a comprehensive genome-wide association study (GWAS) of ex-miRNAs to identify cis-exQTLs, and followed this up with independent external validation in the Rotterdam Study (RS). Leveraging the cis-exQTLs as instrumental variables, we conducted two-sample Mendelian randomization (MR) analyses to identify ex-miRNAs with putatively causal effects on a variety of traits. Furthermore, we explored the correlation of ex-miRNAs with mRNAs expressed in whole blood from the same participants, and identified colocalized cis-eQTLs that influenced the expression of nearby mRNAs in blood and plasma. One of the traits for which putatively causal relations and colocalization were identified was platelet count. We further explored these results by testing associations of implicated ex-miRNAs with corresponding platelet traits in FHS participants. An overview of the study workflow is presented in Figure 1.
Figure 1.

Flowchart of study design
Employing GWAS, we identified genome-wide significant exQTL variants, followed by independent replication in RS cohort. Leveraging cis-exQTLs as instrumental variables, we identified potential causal ex-miRNAs for human complex diseases and traits. We also evaluated ex-miRNAs’ associations with platelet traits to support the MR findings.
Results
Genome-wide mapping exQTLs
miRNA-seq performed on plasma-derived RNA from 40 FHS participants identified 331 expressed miRNAs (ex-miRNAs).14 We employed qRT-PCR to quantify the expression levels of 282 ex-miRNAs in plasma samples collected from 4400 FHS participants. Of the 4400 participants, 3793 have both ex-miRNA data and genotyping data available for exQTL analysis (mean age 40 years, 54% female). Figure S1 illustrates the abundance and distribution of the 282 miRNAs in the study population, including186 miRNAs expressed in ≥3108 samples (70% of studied samples). Clinical characteristics of the participants are presented in Table S1.
cis-exQTLs were defined as SNPs located within 1 Mb upstream or downstream of the start site of the mature miRNA being targeted. Our analysis identified 1,027 cis-exQTLs that were significantly associated with 37 mature miRNAs at a threshold of P < 5x10−8, including 2,475 miRNA-SNP pairs (Figures 2A and S2; Table S2). We detected 40 independent cis-exQTL SNPs using conditional analysis (these genetic variants denote independent associations with ex-miRNA expression and are not solely due to linkage disequilibrium [LD] with other SNPs in the region). Table 1 provides a summary of the 22 sentinel cis-exQTLs, each representing all cis-exQTLs for an miRNA or multiple miRNAs in a given locus. This sentinel variant is the variant with the strongest association signal in the region. The most significant results include 506 cis-exQTL SNPs (1 sentinel SNP, 15 independent SNPs) for miR-625-3p, which is located on chromosome 14q23 (Figure 2B), and 282 cis-exQTL SNPs (3 sentinel SNP, 8 independent SNPs) for 17 miRNAs located in the chromosome 14q32 cluster (Figure 2C). These miRNAs shared cis-exQTLs (Figure S3). Our analysis also identified 7,154 trans-exQTLs that were significantly associated with 125 miRNAs, including 7,518 miRNA-SNP pairs at a threshold of p < 1.78x10−10 (Table S3).
Figure 2.

Manhattan plot illustrating cis-exQTLs
(A) Manhattan plot displaying all cis-exQTLs identified across the genome.
(B) Regional plot highlighting the cis-exQTLs for miR-625-3p in 14q25.
(C) Regional plot highlighting the 14q32 miRNA cluster locus, providing detailed insight into the genetic associations of interest within this region. In Figure 2A, the green dots represent SNPs located within 2 MB regions associated with ex-miRNAs exhibiting significant cis-exRNA eQTLs. The remaining dots are displayed in alternating black or gray colors to differentiate chromosomal regions. In Figures 2B and 2C, miRNA genes with cis-exRNA eQTLs are highlighted in red. The 15 miRNA genes highlighted in red in Figure 2C represents 17 mature miRNAs with cis-exQTLs reported in this study, including miR-127-3p, miR-134-5p, miR-136-3p/5p, miR-154-5p, miR-369-3p/-5p, miR-376a-3p, miR-376c-3p, miR-382-5p, miR-431-5p, miR-433-3p, miR-485-3p, miR-494-3p, miR-495-3p, miR-543, and miR-656-3p. The dots above the red line represent associations with a significance level of P < 5e-8, while those above the blue line represent associations with a significance level of p < 0.05.
Table 1.
Sentinel cis-exQTL variants and their associated ex-miRNAs
| miRNA | Sentinel SNP | Chr | SNP Position (hg38) | Effect Allele | Other Allele | EAF | FHS |
RS |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Betaa | SE | p value | Betaa | SE | p value | |||||||
| miR-1290 | rs7524223 | 1 | 18906196 | G | A | 0.009 | −1.44 | 0.26 | 4.3E-08 | – | – | – |
| miR-9-3p | rs139213408 | 1 | 156878571 | C | T | 0.005 | −3.63 | 0.62 | 6.0E-09 | – | – | – |
| miR-181a-5p | rs10919655 | 1 | 199279533 | G | A | 0.339 | −0.33 | 0.05 | 6.5E-11 | 0.11 | 0.013 | 5.2E-16 |
| miR-181b-5p | rs138900185 | 1 | 198702776 | A | G | 0.006 | −1.78 | 0.29 | 6.7E-10 | – | – | – |
| miR-562 | rs766631473 | 2 | 232828371 | C | CT | 0.005 | −15.01 | 2.11 | 2.3E-12 | – | – | – |
| miR-582-5p | rs5024245 | 5 | 60250066 | A | G | 0.481 | −0.42 | 0.06 | 1.0E-12 | – | – | – |
| miR-584-5p | rs36047 | 5 | 149061758 | T | C | 0.355 | −0.32 | 0.05 | 1.7E-10 | 0.23 | 0.026 | 6.9E-19 |
| miR-605-5p | rs60202580 | 10 | 51908729 | C | A | 0.009 | −13.82 | 1.98 | 1.5E-11 | – | – | – |
| miR-130a-3p | rs7117896 | 11 | 57621078 | A | G | 0.277 | −0.32 | 0.06 | 2.8E-08 | 0.30 | 0.023 | 7.1E-39 |
| miR-16-1-3p | rs116939667 | 13 | 49344847 | T | A | 0.006 | −2.58 | 0.47 | 3.6E-08 | – | – | – |
| miR-4306 | rs140415911 | 13 | 99168139 | C | T | 0.013 | 1.22 | 0.22 | 4.7E-08 | – | – | – |
| miR-625-3p (14q23)b | rs2127870 | 14 | 65330128 | C | G | 0.748 | 1.01 | 0.06 | 2.2E-59 | 0.36 | 0.022 | 3.9E-60 |
| miR-369-5p (14q32) | rs7144855 | 14 | 100400292 | C | G | 0.004 | −6.80 | 0.86 | 3.0E-15 | – | – | – |
| miR-136-5p (14q32) | rs11622192 | 14 | 100549853 | A | G | 0.221 | 0.58 | 0.10 | 6.4E-9 | – | – | – |
| miR-127-3p, miR-134-5p, miR-136-3p, miR-154-5p, miR-369-3p, miR-376a-3p, miR-376c-3p, miR-382-5p, miR-431-5p, miR-433-3p, miR-485-3p, miR-494-3p, miR-495-3p, miR-543, miR-656-3p (14q32) | rs12881545 (rs12881760 or rs72700576, r2 = 1) | 14 | 100709875 | C | G | 0.643 | −0.56 | 0.05 | 3.5E-24 | 0.25 | 0.028 | 1.8E-19 |
| miR-629-5p | rs78212770 | 15 | 70079438 | G | C | 0.019 | 1.06 | 0.18 | 7.1E-09 | – | – | – |
| miR-125a-3p | rs1181776908 | 19 | 51311263 | G | A | 0.003 | −5.38 | 0.96 | 2.4E-08 | – | – | – |
| miR-373-3p | rs145507432 | 19 | 53293768 | C | T | 0.013 | −7.73 | 1.17 | 1.4E-10 | – | – | – |
| miR-1323 | rs562999849 | 19 | 54390570 | A | AC | 0.006 | −11.05 | 1.98 | 3.9E-08 | – | – | – |
| miR-499a-3p | rs1433162886 | 20 | 35110006 | C | A | 0.004 | −15.17 | 2.48 | 1.9E-09 | – | – | – |
| miR-499a-5p | rs1343050945 | 20 | 34837329 | A | C | 0.004 | −2.24 | 0.38 | 4.5E-09 | – | – | – |
| miR-133a-3p, miR-1-3p | rs12624608 | 20 | 62558091 | A | C | 0.242 | −0.67 | 0.07 | 1.41E-22 | 0.17 | 0.025 | 4.3E-12 |
EAF: effect allele frequency; Beta: effect beta value; SE: standard error.
Please note that in FHS, expression levels of ex-miRNAs were quantified using Ct values. It’s essential to grasp that in this context, a higher Ct value signifies a lower expression level of the miRNA. Thus, when interpreting the results presented in this table, a positive beta value indicates that the effect allele increased miRNA Ct values (resulting in decreased expression), and vice versa. Conversely, in RS, expression levels of ex-miRNAs were reported using conventional values derived from miRNA-seq, where higher values indicate higher expression levels. Consequently, positive beta values in RS signify that the effect allele increased miRNA expression.
In RS, expression levels of miR-625-5p were measured instead of miR-625-3p. It’s noteworthy that the effect direction of cis-exQTLs with miR-625-5p in RS is opposite to that observed with miR-625-3p in FHS.
A sensitivity analysis of exQTLs was conducted specifically in FHS Third Generation cohort participants of European ancestry (N = 3416). Comparing the T values of exQTLs identified in all participants versus those in the Third Generation participants, we observed nearly identical T values for cis-exQTLs (Figure S4).
External replication of exQTLs
We sought to replicate the FHS exQTLs in the RS. The RS utilized a miRNA-seq platform to measure ex-miRNA expression levels in plasma samples in 2,178 RS participants (mean age 72, 57% female). Genotypes in the RS were obtained from array-based genotyping and imputed genotypes for minor allele frequency (MAF) > 0.01, which differed from the sequencing variants used in our study.13 Among the total of 994 cis-exQTLs at an MAF>0.01 detected in our study, 548 (55%) were also cis-exQTLs in RS, another 46 were proxy SNPs for cis-exQTLs in RS with high LD (r2 > 0.80). Notably, the miRNA clusters located on chromosome 14q23 and 14q32 emerged as the top signals in RS, collectively contributing to 54% of the significant cis-exQTL results in RS. Within the 14q23 region, RS assessed the expression levels of miR-625-5p rather than miR-625-3p. Interestingly, 371 cis-exQTLs showed significance in both studies (i.e., for miR-625-3p in FHS and for miR-625-5p in RS), albeit with opposite effect sizes. However, for the remaining cis-exQTLs, the direction of effect sizes remained consistent between FHS and RS. We were unable to replicate trans-ex-miRNA-SNP pairs in RS. For this reason, the subsequent analyses focus only on cis-exQTLs.
Functional and genomic regulatory features of cis-exQTLs
Of the 1,027 cis-exQTLs in the FHS, only 1 was found within miRNA seed regions (rs78212770 for miR-629-5p). The majority of cis-exQTLs (N = 438, 43%) were intronic or intergenic variants (N = 268, 26%), while some were within exons (N = 35), UTRs (N = 8), and up/downstream of protein-coding genes (N = 73). None of the cis-variants were considered deleterious, as none had a CADD score greater than 15.
Our study identified 33 cis-exQTLs with a MAF<0.01, which are considered uncommon or rare. Previous GWAS and eQTL analyses have shown that rare variants have larger effect sizes than common variants. Consistent with these findings, we observed that low frequency cis-exQTLs also had larger effect sizes (as shown in Figure S5), with a median effect size of 8.22 for variants with MAF<0.01, compared to 0.48 and 0.76 and for cis-variants with MAF within the ranges of 0.01–0.1 and 0.1–0.5, respectively.
Correlation of ex-miRNAs with whole-blood mRNAs
We further investigated the correlation between ex-miRNAs and whole-blood mRNA expression in 2,721 individuals from the FHS. The levels of whole blood mRNA were measured using RNAseq.15 Our analysis revealed a total of 44,720 significant associations (FDR <0.01) between ex-miRNAs and whole-blood mRNAs, involving 255 ex-miRNAs and 8,886 mRNAs (see Table S4). The r2 values for these associations are low (median value is 0.015); only 2166 pairs (5%) displayed r2 > 0.1. Among the 44,720 significant associations, 91% were positively associated (higher miRNA levels were associated with higher mRNA levels), and 9% were negatively associated. Among these mRNAs, 5,279 were protein-coding, 989 were long non-coding RNAs (lncRNAs), and 5,621 belong to other categories. On average, each miRNA was associated with 175 mRNAs. Notably, over 83% of the miRNAs had fewer than 200 coexpressed mRNAs, and a few were coexpressed with more than 1,000 mRNAs (Figure S6). To gain further insights into the functional implications of these associations, we performed Gene Ontology (GO) enrichment analysis on coexpressed protein-coding mRNAs for each of miRNAs. Many miRNAs shared multiple biological processes and pathways, including epidermis development, cilium movement/assembly, regulation of cell motility, extracellular matrix organization, and multiple metabolic processes (Figure 3).
Figure 3.

Significantly enriched gene ontology biological process terms for coexpressed protein-coding genes for ex-miRNAs
We identified a total of 1,114 miRNA-protein-coding mRNA coexpressed pairs, where the mRNA was confirmed as a target gene of the corresponding miRNAs. These targeted pairs accounted for only 4% of the total coexpression results. The large number of coexpressed miRNA-mRNAs pairs exhibiting relatively few miRNA targets led us to speculate that there might exist specific co-regulation relationships between ex-miRNAs and whole-blood mRNAs.
Among the 1,027 cis-exQTLs, 823 (80%) overlapped with cis-eQTLs for nearby whole-blood mRNAs including 6430 SNP-mRNA pairs (Table S5). The direction of beta coefficients for SNP-ex-miRNA pairs in plasma and SNP-mRNA pairs in blood was consistent for 5094 pairs (79%), indicating that nearby ex-miRNAs and mRNAs may be concordantly regulated by the same genetic variants. The colocalization analysis further revealed 19 ex-miRNAs that exhibited genetic variants in close proximity to their associated whole-blood mRNAs, including miR-584-5p for SH3TC2, miR-625-3p for LINC02324, miR-130a-3p for TIMM10, CTNND1, SERPING1, LPXN, CLP1 and SLC43A1, and the 14q32 miRNA cluster for MEG3, MEG8, MEG9, RF01872, DLK1, SLC25A47, AL118558.3 and AL118558.4 (Table S6).
Correlation of ex-miRNAs with clinical traits
Of the 1,027 cis-exQTLs, 114 SNPs were associated with 62 clinical traits at P < 5x10−8 in the GWAS Catalog16 (Table S7). To further investigate potential causal associations between ex-miRNAs and clinical traits, we conducted two-sample MR tests for more than 1,000 traits whose full GWAS results were available (this analysis included 2261 full sets of GWAS results, see Methods). At FDRMR<0.05, our analysis revealed that 29 ex-miRNAs were putatively causal for 35 traits (Table S8). One of the most noteworthy findings in our study is the MR causal relations for miRNAs in the 14q32 miRNA cluster (i.e., miR-127-3p) with a variety of traits, including asthma, breast cancer, platelet count, and testosterone levels. miR-130a-3p (chr11) was putatively causally linked with a variety of traits including asthma, breast cancer, platelet count, pappalysin-1 levels, and neuroticism.
In the MR analysis, 12 ex-miRNAs were assessed for causal links to platelet count.17,18 Among these, 10 resided in 14q32, while the remaining two were miR-130a-3p and miR-625-3p. We expanded our analysis to investigate the cross-sectional correlation between ex-miRNAs and platelet traits, including PLT and mean platelet volume (MPV), utilizing participants from the FHS with ex-miRNA and platelet trait data. Employing an FDR threshold of less than 0.05, we identified 50 ex-miRNAs associated with PLT and 97 ex-miRNAs associated with MPV (Figures 4A and 4B; Table S9). Remarkably, 10 out of 12 ex-miRNAs with causal links to platelet count by MR (except for miR-130a-3p and miR-625-3p) were significant in cross-sectional associations with PLT, and all 12 ex-miRNAs were significantly associated with MPV (Tables 2 and S10). The effect sizes of ex-miRNAs on PLT estimated through MR analysis consistently aligned with the observed associations for all 10 ex-miRNAs (i.e., increased expression levels of ex-miRNAs were linked to increased PLT and decreased MPV). Figure 4C illustrated an example of associations of exQTLs, miR-494-3p, and platelet traits.
Figure 4.

Associations of ex-miRNA with platelet traits
(A and B) Volcano plots depicting ex-miRNA differential expression for (A) platelet count (PLT) and (B) mean platelet volume (MPV).
(C) Shows the associations of exQTLs, ex-miRNAs, and platelet traits, with beta and p values presented for miR-494-3p as a representative miRNA in 14q32. Given that ex-miRNA expression levels were assessed through Ct values (higher values indicating lower expression), positive associations correspond to negative beta estimates (red dots), and vice versa (blue dots). The term “non-significant” (green dots) refers to ex-miRNAs with an FDR >0.05. MR: Mendelian Randomization.
Table 2.
Associations of sentinel SNPs, ex-miRNAs and platelet counts, and Mendelian randomization
| Exposure | Chr Region | Sentinel exQTL | Effect/Other allele | Association of SNP – ex-miRNA |
bAssociation of SNP - PLT |
MR: ex-miRNA--> PLT |
Association of ex-miRNA-PLT |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Betaa | SE | P-value | Betaa | SE | p-value | Betaa | SE | P-value | Betaa | SE | P-value | FDR | ||||
| miR-130a-3p | 11q12 | rs7117896 | A/G | −0.32 | 0.06 | 2.8E-08 | 0.02 | 2.1E-03 | 3.33E-23 | −0.06 | 1.3E-02 | 1.2E-06 | −1.6E-03 | 8.9E-04 | 0.08 | 0.24 |
| miR-625-3p | 14q23 | rs2127870 | C/G | 1.01 | 0.06 | 2.2E-59 | 0.03 | 2.3E-03 | 1.34E-31 | 0.03 | 2.7E-03 | <1.0E-16 | −1.7E-03 | 8.9E-04 | 0.06 | 0.19 |
| miR-431-5p | 14q32 | rs2183632 | T/C | −0.49 | 0.07 | 2.0E-12 | 0.02 | 2.0E-03 | 1.15E-17 | −0.04 | 6.5E-03 | 5.1E-08 | −4.4E-03 | 1.0E-03 | 1.3E-05 | 4.4E-04 |
| miR-543 | 14q32 | rs12881545 | C/G | −0.51 | 0.06 | 1.1E-17 | 0.05 | 2.0E-03 | 4.3E-117 | −0.09 | 1.1E-02 | 6.7E-16 | −4.6E-03 | 9.1E-04 | 4.5E-07 | 6.0E-05 |
| miR-127-3p | 14q32 | rs12881545 | C/G | −0.66 | 0.08 | 1.6E-16 | 0.05 | 2.0E-03 | 4.3E-117 | −0.07 | 8.8E-03 | 6.0E-15 | −3.5E-03 | 1.2E-03 | 3.1E-03 | 0.02 |
| miR-376c-3p | 14q32 | rs12881545 | C/G | −0.58 | 0.06 | 4.5E-23 | 0.05 | 2.0E-03 | 4.3E-117 | −0.08 | 8.5E-03 | <1.0E-16 | −3.2E-03 | 8.4E-04 | 1.5E-04 | 2.4E-03 |
| miR-376a-3p | 14q32 | rs12881545 | C/G | −0.54 | 0.05 | 1.3E-22 | 0.05 | 2.0E-03 | 4.3E-117 | −0.08 | 9.2E-03 | <1.0E-16 | −3.4E-03 | 8.8E-04 | 1.3E-04 | 2.4E-03 |
| miR-494-3p | 14q32 | rs12881545 | C/G | −0.57 | 0.06 | 1.6E-19 | 0.05 | 2.0E-03 | 4.3E-117 | −0.08 | 9.4E-03 | <1.0E-16 | −3.0E-03 | 9.7E-04 | 2.1E-03 | 1.6E-02 |
| miR-495-3p | 14q32 | rs12881545 | C/G | −0.56 | 0.05 | 3.5E-24 | 0.05 | 2.0E-03 | 4.3E-117 | −0.08 | 8.7E-03 | <1.0E-16 | −3.8E-03 | 8.5E-04 | 7.3E-06 | 3.9E-04 |
| miR-136-3p | 14q32 | rs12881760 | C/G | −0.53 | 0.05 | 4.2E-22 | 0.05 | 2.0E-03 | 3.1E-117 | −0.09 | 9.5E-03 | <1.0E-16 | −2.7E-03 | 7.9E-04 | 7.4E-04 | 9.0E-03 |
| miR-369-3p | 14q32 | rs12881760 | C/G | −0.61 | 0.05 | 2.6E-29 | 0.05 | 2.0E-03 | 3.1E-117 | −0.07 | 7.3E-03 | <1.0E-16 | −2.8E-03 | 8.3E-04 | 8.2E-04 | 9.6E-03 |
| miR-433-3p | 14q32 | rs12881760 | C/G | −0.77 | 0.10 | 2.5E-15 | 0.05 | 2.0E-03 | 3.1E-117 | −0.06 | 7.8E-03 | 5.3E-14 | −5.4E-03 | 1.4E-03 | 1.4E-04 | 2.4E-03 |
Please note that the expression levels of ex-miRNAs were reported using Ct values. It’s important to understand that in this context, a higher Ct value indicates a lower expression level of the miRNA. Therefore, when interpreting the results in this table, a positive beta value indicates a negative association between the expression levels of the miRNA and the effect allele, or a negative association between the expression levels of the miRNA and PLT, or vice versa.
The GWAS of platelet count was from.17
Discussion
We investigated the associations between genetic variants and plasma miRNA expression (or ex-miRNAs), along with the relationships between ex-miRNA and whole blood mRNA coexpression in a large population. Additionally, we explored the links between ex-miRNA and various clinical traits. This research uncovered crucial insights into ex-miRNA regulation, potential biomarkers, and the molecular basis of clinical phenotypes.
A major finding of our study was the identification of numerous cis-exQTLs, which revealed intricate genetic control of ex-miRNAs. Notably, the 14q32 miRNA cluster was enriched for cis-exQTLs, suggesting shared genetic regulation. This cluster also emerged as the top signal in RS, reinforcing its genetic control and likely functional significance. This cluster also demonstrated enriched cis-miR-eQTLs in whole blood in our prior study.
Coexpression analysis between ex-miRNAs and whole-blood mRNA pointed to potential regulatory links. Nevertheless, despite the abundance of coexpressed miRNA-mRNA pairs, our validation endeavors underscored a narrow subset of coexpressed pairs involving targeted interactions between miRNAs and protein-coding mRNAs (comprising less than 4% of the total). This stark disparity between the extensive pool of coexpressed miRNA-mRNA pairs and the restricted count of confirmed target interactions leads us to speculate on the possibility that these relationships could represent a broader regulatory phenomenon. The observed expression patterns may indeed signify miRNAs targeting specific pathways. This perspective is supported by similar findings in other studies.19,20,21,22,23
Acknowledging the six-year time gap between whole blood sample collection for miRNA and mRNA quantification and plasma collection for ex-miRNA quantification as a major limitation in our study is crucial. This gap could introduce variability due to changes in sample storage over time, potentially influencing the accuracy and reliability of our results. Additionally, the biological milieu might have changed during this time frame, affecting the results. Future studies with more concurrent data collection methods or efforts to minimize time gaps between different analyses could provide a more robust understanding of the biological phenomena under investigation. Furthermore, the Paxgene system preserves total RNA from whole blood, encompassing RNA from both plasma and cells. While plasmatic vesicles are primarily enriched with small RNA moieties rather than mRNAs, we acknowledge that our experimental strategy may have limitations, and future research in need to address these issues more effectively.
The 14q32 miRNA cluster stands out as one of the largest polycistronic clusters, encompassing a total of 54 miRNAs in humans. This particular miRNA cluster has garnered attention due to its substantial involvement as a regulator of neovascularization.24 Beyond its role in neovascularization, this region serves additional pivotal functions in various pathological processes including oncogenesis, malignant growth, proliferation, cell survival, cellular interactions with the extracellular matrix, and potentially even processes within the central nervous system.24 Employing MR analysis, our investigation unveiled a significant insight: numerous ex-miRNAs residing within this genomic region appear to exert causal influence on a diverse array of clinical traits. Notably, these traits encompass asthma, breast cancer, platelet traits, and testosterone levels. In follow up of these findings, we proceeded to validate the relationship between these ex-miRNAs and platelet traits through a cross-sectional analysis among FHS participants. All 10 miRNAs identified in this region through MR analysis were validated by the cross-sectional analysis. The effect sizes of ex-miRNAs on platelet traits estimated through MR analysis consistently aligned with the observed associations.
Platelets play a pivotal role in both the physiological process of hemostasis and the pathological progression of thrombosis. Platelets serve as reservoirs for a diverse array of molecules, including proteins and RNAs, with a notable presence of miRNAs.25 Upon activation, platelets release microparticles containing a wealth of proteins, inflammatory factors, and noncoding RNAs. A significant portion of ex-miRNAs in plasma originates from platelets, primarily secreted within vesicles upon platelet activation. miRNAs such as miR-495 have been shown to modify the expression of platelet proteins influencing platelet reactivity.19 miR-376c has been reported in relation with platelet activation by affecting PAR4-mediated pathway which is a membrane receptor for thrombin.26 In this study, we identified 10 ex-miRNAs (including miR-495 and miR-376c) located in the 14q32 region that exhibited significant positive associations with PLT and negative associations with MPV. Many other miRNAs have been implicated in various aspects of cardiovascular pathology, including thrombosis and inflammation, which are closely associated with platelet function. For example, research has shown that miR-494 expression is upregulated in atherosclerotic lesions and may contribute to plaque development and instability by modulating endothelial cell function, vascular smooth muscle cell proliferation, and inflammatory responses. miR-494 expressed in platelet and its dysregulation has been linked to platelet activation and thrombosis.27 Those miRNAs could offer valuable insights for the development of novel antiplatelet therapeutic strategies for cardiovascular disease.
Prior studies have suggested that the presence of residual platelets in plasma may potentially influence the variability in ex-miRNA expression and the resulting outcomes.28 To mitigate this concern, we implemented measures to minimize the potential impact of platelet contamination, as described in our previous work.14 Furthermore, none of the miRNAs with cis-exQTLs or associated with platelet counts and volume in this study were among the most studies intraplatelet miRNAs (the top five are miR-126, miR-223, miR-21, miR-96, and miR-204).29 Additionally, associations between ex-miRNAs and platelet traits were identified through both two-sample MR and cross-sectional association analyses. The MR analysis provided evidence of the genetic effects of ex-miRNAs on platelet traits, supporting the validity of the associations reported in our study and minimizing the likelihood of technical artifacts. Based on the above, we do not believe that our results will be affected by potential platelet contamination.
Our MR analysis also revealed miR-130a-3p in association with multiple diseases or traits. Published functional studies support the association of miR-130a-3p with lung function and asthma30 and breast cancer.31 The overlap of cis-exQTLs of miR-130a-3p with pQTLs of Pappalysin-1 was validated in the RS study.13 miR-130a-3p has been found to be related with neural stem cell,32 Alzheimer’s disease,33 and cerebral syndrome34,35 which may provide some supports for their association with neuroticism, intelligence, feeling hurt/worry, and cognitive performance in our MR analysis. The "potential" causality results as we claimed in this study were based on two-sample MR analysis. Further validation, including longitudinal studies and functional studies, is needed to validate the causal roles of ex-miRNAs in diseases.
Parent-of-origin effects (POE) have been observed in the 14q32 region, notably impacting gene expression of key genes in a parent-of-origin specific manner, including DLK1, RTL1, MEG3, MEG8 and MEG9. These effects have been associated with diverse physiological processes, such as age at menarche and platelet phenotypes.36 Coexpression and colocalization analysis suggested that miRNAs and mRNAs in these cluster are coregulated. It is conceivable that miRNAs within this region may also be subject to parent-of-origin regulation. A study in mice demonstrated that miRNAs within this cluster are influenced by a germline-derived differentially methylated region (IG-DMR), located about 200 kb upstream from the miRNA cluster.37 The expression of these miRNAs is contingent upon the inheritance of this regulatory element from either the maternal or paternal chromosome. When the IG-DMR is deleted from the maternally inherited chromosome, it results in the suppression of maternally expressed imprinted genes. We hypothesized that this region might harbor cis-genetic variants responsible for regulating miRNA expression in a parent-of-origin-specific manner. Regrettably, our study lacks sufficient samples within families to comprehensively explore this hypothesis.
The cis-eQTLs identified in our study are replicated well in an independent study (i.e., RS). We encountered challenges when attempting to replicate trans-exQTLs, which are genetic variants linked to the expression levels of distant ex-miRNAs. The regulation of ex-miRNA expression is intricate, involving numerous factors such as transcription factors, epigenetic modifications, chromatin structure, and intercellular communication. It remains uncertain whether trans-exQTLs play a causal role in ex-miRNA expression changes or if they are merely correlated markers. This complexity hinders the replication of trans-exQTLs across diverse experimental contexts and studies, primarily due to technical variations and genetic heterogeneity. To enhance the replicability of trans-exQTLs, we believe that studies of diverse populations and investment in larger sample sizes are essential steps. Additionally, the methods of measuring miRNA and DNA sequences differ significantly in our study and in RS, potentially introducing inherent biases or errors that could affect the validity of replication. Therefore, further replication may be necessary.
In conclusion, our study comprehensively analyzed exQTLs in miRNAs and their links to clinical traits and mRNA expression. These findings warrant functional validation studies and can help guide future miRNA research in disease and therapy. Despite the limitations of our study, including the necessity for replication and functional validation, our work offers valuable insights into the intricate genetic regulation of miRNA expression and its relevance to human phenotypes.
Limitations of the study
While our study has yielded significant insights into the genetic regulation of ex-miRNAs and their associations with clinical traits and mRNA expression, several limitations should be noted. First, the six-year time gap between the collection of whole blood samples for mRNA quantification and plasma collection for ex-miRNA quantification could underestimate the extent of correlation between miRNAs and mRNAs. Additionally, biological changes over this period might have influenced the outcomes. Second, our analysis was constrained by the use of the PAXgene system, which preserves total RNA from whole blood, including RNA from both plasma and cells. While plasma vesicles are primarily enriched with small RNA moieties rather than mRNAs, our experimental strategy may not have fully captured this distinction, necessitating more refined approaches in future research. Third, our study’s findings related to cis-exQTLs were well-replicated, but replicating trans-exQTLs proved challenging. The intricate regulation of ex-miRNA expression, involving numerous factors like transcription factors, epigenetic modifications, chromatin structure, and intercellular communication, complicates replication across different experimental contexts and populations. Larger sample sizes and studies involving diverse populations are essential to enhance the replicability of trans-exQTLs. Despite these limitations, our study provides valuable insights into the genetic regulation of ex-miRNA expression and its implications for human phenotypes, warranting further functional validation and replication efforts.
Resource availability
Lead contact
Requests for further information and resources should be directed to the lead contact, D.L. (levyd@nih.gov).
Materials availability
This study did not generate new materials.
Data and code availability
-
•
The datasets used for analysis in the present study can be accessed at the dbGAP repository under the accession number phs000007.v33.p14.
-
•
All algorithm and software used are published, established methods and R packages as described in the key resources table and following Methods section.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Acknowledgments
The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health; or the U.S. Department of Health and Human Services.
The Framingham Heart Study is funded by National Institutes of Health contract N01-HC-25195 and HHSN268201500001I. The laboratory work for this investigation was funded by the Division of Intramural Research, National Heart, Lung, and Blood Institute, National Institutes of Health. The analytical component of this project was funded by the Division of Intramural Research, National Heart, Lung, and Blood Institute, and the Center for Information Technology, National Institutes of Health, Bethesda, MD.
Author contributions
D.L. designed, directed, and supervised the project. J.F. directed and supervised the ex-miRNA experiments. T.H. and D.L. drafted the manuscript. T.H., R.J., J.R., and M.C, conducted the analyses in the FHS. R.M., A.D., and M.G. performed replication analysis. All authors participated in revising and editing the manuscripts. All authors have read and approved the final version of the manuscript.
Declaration of interests
The authors declare no conflict of interest.
STAR★Methods
Key resources table
Experimental model and study participant details
The FHS is a long-term observational study with a focus on cardiovascular disease that started in 1948. The study began by recruiting an original cohort of residents of Framingham, MA, with the aim of identifying risk factors for CVD. Later on, in 1971, the FHS Offspring cohort was established, consisting of a subset of children of the original cohort and their spouses. To further expand the study, the Third Generation cohort was established in 2002 and recruited children of the FHS Offspring Cohort. In order to increase the diversity of the FHS study population and accurately reflect the evolving demographics of Framingham, the first Omni cohort was established in 1995. This cohort was comprised of participants from various ethnic backgrounds, including African American, Hispanic, Asian, Indian, Pacific Islander, and Native American communities. All FHS participants underwent examinations at approximately 2 to 4-year intervals since the study’s inception. These examinations entailed the comprehensive collection of various biosamples including whole blood and plasma.
In this investigation, plasma samples for ex-miRNA measurement were obtained from 4,036 participants in the FHS Third Generation cohort who attended examination cycle 1 (2002–2005), and from 404 participants of the OMNI II cohort who attended examination cycle 1 (2003–2005). There were 3,416 participants from the Third Generation and 377 from the OMNI II cohort having ex-miRNA data and genotyping data available for exQTL analysis.
Whole blood samples for both whole blood miRNA and mRNA (RNA-seq) measurements were collected from participants in the FHS Offspring cohort attending examination cycle 8 (2005–2008), and from the Third Generation cohort during examination cycle 2 (2008–2010). For correlation analysis between ex-miRNA and whole blood miRNA, 3,196 samples from the FHS Third Generation cohort were utilized, including those with available data for both ex-miRNA and whole blood miRNA. Furthermore, for correlation analysis between ex-miRNA and whole blood mRNA, 2,721 samples from the FHS Third Generation cohort were utilized, including those with available data for both ex-miRNA and whole blood miRNA. It is noteworthy that there was a 6-year gap between the two time points of plasma and whole blood sample collection, which may potentially overlook dynamic molecular changes that could have occurred during the time frame in the correlation analysis. All participants provided written consent for genetic research.
Method details
Plasma ex-miRNA measurement
We previously implemented an extracellular RNAseq pipeline using 40 plasma samples from FHS Offspring cohort participants as described previously.14 This strategy yielded 331 expressed human miRNAs that were selected for quantitative reverse transcription-polymerase chain reaction (qRT-PCR) measurements in 4,440 FHS Third Generation and Omni cohort participants. The 282 miRNAs in the present investigation are miRNAs that were expressed in at least one plasma sample.
qRT-PCR is a sensitive and specific method for measuring RNA expression levels. The ThermoFisher MagMAX mirVana Total RNA Isolation Kit (Applied Biosystems; catalog #A27828) was utilized for the isolation of plasma miRNAs. EDTA was added to the proteinase K (PK) digestion and elution buffer at a final concentration of 1 mM. Modifications were made to the protocol by increasing the volume of magnetic beads to 30 μL, the volume of the lysis buffer to 195 μL, and the volume of isopropanol added to the beads/lysis buffer mix to 375 μL. C. elegans miRNA 39.3p was added to the lysis buffer as an internal control after being phosphorylated. Subsequently, samples were processed using the ThermoFisher Flex Magnetic Particle Processor, 96DW. Following elution, 25 μL of miRNAs were dried using Biotage SPE instruments and stored at −80°C until further use. ThermoFisher TaqMan Advanced miRNA cDNA synthesis kits (catalog #A28007) were used for conversion of isolated miRNAs to cDNAs. Preamplification involved the addition of 5 μLPoly(A) Reaction Mix to the dried miRNA sample followed by 16 cycles of amplification as per vendor instructions. Applied Biosystems ProFlex (96-well PCR System Catalog #4484075) or Quant Studio 3 PCR system (Product A28137) was used for both syntheses and preamplification. The Fluidigm BioMark HD instrument (#BMKHD-BMKHD) and ThermoFisher TaqMan Advanced miRNA assays (#A25576) were employed for multiplex quantification of miRNAs in 96 samples x 96 samples. Samples and assays were loaded onto the integrated fluidic circuit (IFC) chips using the Fluidigm HX (#68000112) instrument. The miRNA expression levels were measured using cycle threshold (Ct) values. Single copy can be detected at threshold Ct < 34.
Whole genome sequencing (WGS)
Using genomic DNA extracted from whole blood buffy coat, FHS participants underwent WGS as part of the NHLBI’s TOPMed program.38 DNA fragmentation and library construction were carried out using standard procedures, and the sequencing was performed by the Broad Institute of MIT and Harvard, which is a TOPMed reference laboratory. HiSeq X, together with sequencing software HiSeq Control Software (HCS) version 3.3.76, was used for the sequencing process, followed by analysis using RTA2 (Real Time Analysis). The DNA sequence reads were aligned to human genome build GRCh38, using a common pipeline that was utilized by all TOPMed WGS centers. A sample’s sequence was considered complete when the mean coverage of nDNA was more than 30x. For this analysis, genetic variants generated from TOPMed Freeze 10a were used, and we focused on 19 million SNPs that had a minor allele count of at least 10 and at Hardy-Weinberg equilibrium (HWE) p-value >1e-10.
Whole blood mRNA expression
Peripheral whole-blood samples were collected in PAXgene tubes (Asuragen, Inc., Austin, TX, USA). Total RNA was extracted using a standard protocol with the PAXgene Blood RNA Kit. After allowing tubes to thaw for 16 h at room temperature, white blood cell pellets were collected following centrifugation and washing. Subsequently, the cell pellets were lysed in a guanidinium-containing buffer. The extracted RNA was tested for its quality by determining absorbance readings at 260 and 280 nm using a NanoDrop ND-1000 UV spectrophotometer. The Agilent Bioanalyzer 2100 microfluidic electrophoresis (Nano Assay and the Caliper LabChip system) was used to determine the integrity of total RNA. All RNA samples were sequenced by an NHLBI TOPMed program reference laboratory (Northwest Genomics Center) following the TOPMed RNA-seq protocol. All RNA-seq data were processed by University of Washington Northwest Genome Center. The raw reads (in FASTQ file) were aligned using the GRCh38 reference build to generate BAM files using STAR v2.6.1day. RNA-SeQC v2.3.3 was used for processing of RNA-seq data by the TOPMed RNA-seq pipeline to derive standard quality control metrics from aligned reads. Gene-level expression quantification was provided as read counts and transcripts per million (TPM). GENCODE 30 annotation was used for annotating gene-level expression. For more comprehensive information on the RNA-seq and the subsequent data processing steps, please refer to the provided ref.15
Platelet traits
We collected standard blood platelet (PLT) and mean platelet volume (MPV) measurements from the Third Generation cohort participants attending exam 2. These measurements were obtained using a complete blood count and analyzed with the Coulter HmX hematology analyzer (Beckman Coulter, Inc.). Our analysis focused exclusively on individuals who had measurements of ex-miRNAs. We also excluded individuals who had been previously diagnosed with leukemia or lymphoma before the examination, as these conditions have been associated with low blood cell counts, and also excluded individuals with PLT levels exceeding 1000x109/L. Following these exclusions, our study included 2398 individuals for further analysis of PLT and 2,389 for MPV analysis.
Quantification and statistical analysis
Identification of exQTLs
A linear mixed model was employed to assess the association of each ex-miRNA with each polymorphic SNP with a minor allele count of at least 10. The model was adjusted for covariates including age, sex, high-density lipoprotein (HDL), total cholesterol (TC), triglyceride (TG), and 5 genetic principle components as fixed effects, with cohort included as a random effect. As ex-miRNAs can be bound to and guided by lipoproteins, the regression models included lipid covariates including HDL, TC and TG. cis- and trans-exQTLs were identified based on the location of SNPs relative to ex-miRNAs. cis-was defined as an SNP within 1 megabase (Mb) of the miRNA, while trans-was defined as an SNP outside of this range. The statistical significance thresholds for cis- and trans-were set at P < 5x10−8 and P < (5x10−8/282) = 1.8x10−10, respectively. The exQTL analysis was performed using a custom Java script developed in-house. The NIH-supported STRIDES cloud infrastructure (cloud.nih.gov/) was utilized for all analyses, with the aid of a program based on a graphical Processing Unit (GPU) to facilitate computation.
We utilized GCTA-COJO (Genome-wide Complex Trait Analysis)41 to perform a multi-SNP-based conditional and joint association analysis, aiming to identify association signals that are conditionally independent within 1 MB of the peak SNPs. This analysis employed stepwise selection to choose SNPs based on their conditional P-values and provided the joint effects of the selected SNPs after optimizing the model. To compute the LD reference panel, we used genetic data from FHS WGS data from TOPMed Freeze 10a (N = ∼7,000). Our analysis incorporated several filters, including a MAF threshold of >0.001, a conditional p-value threshold of 5 × 10−8, a collinearity threshold of 0.9, and an assessment window of 10,000 base pairs.
Functional annotation of exQTLs
We annotated SNPs by adding functional information to variants using Ensembl Variant Effect Predictor (VEP, version 109) and SnpEff 5.1.d (Build 2022-04-19).45 VEP and SnpEff take information from a multiple annotation database and determine the effect of genetic variants on genes, transcripts, and protein sequence, as well as regulatory regions. The reference genome is GRCh38. We used Combined Annotation Dependent Depletion (CADD) to predict functional consequences of identified allelic variants. CADD is an integrative annotation built from more than 60 genomic features and can score whole human genome ∼29.4 million variants.39 A scaled CADD score of 10 or 20 means that a variant is among the top 10% or 1% of deleterious variants in the human genome. SNPs scored CADD >15 were considered putatively deleterious variants.
We used the NHGRI-EBI GWAS catalog to annotate clinical diseases and traits associated exQTLs.16 The NHGRI-EBI GWAS catalog is a curated collection of all human genome-wide association studies, produced by a collaboration between EMBL-EBI and NHGRI.16 Significant associations of 196,868 unique SNPs with 4,417 unique clinical phenotypes (P < 5x10−8) were downloaded from the GWAS catalog.
Mendelian randomization (MR) analysis
To investigate the putatively causal relation of ex-miRNAs to clinical phenotypes, we performed two-sample MR analyses by utilizing cis-exQTLs as instrument variables (IVs), ex-miRNAs as exposures, and clinical traits as outcomes. MR is a statistical method used to investigate the causal relationship between an exposure and an outcome. In two-sample MR, genetic variants associated with the exposure are identified in one sample, while genetic variants associated with the outcome are identified in another sample. The effect of the exposure on the outcome is then estimated by combining the effect sizes of the genetic variants on the exposure and the outcome using statistical methods.47
To facilitate this analysis, we utilized our in-house developed analytical pipeline called MR-Seek (https://github.com/OpenOmics/mr-seek.git). To estimate associations and effect sizes between SNPs and traits, we relied on publicly available GWAS databases. Specifically, we included 2,261 published sets of GWAS results for more than 1000 clinical traits from the NHGRI-EBI GWAS catalog.16 As most ex-miRNAs have only one independent cis-exQTL, we employed the Wald MR method using the peak cis-exQTL (a single genetic variant that exhibits the strongest association signal with ex-miRNA expression, as determined by the variant with the lowest SNP-ex-miRNA p-value) as an IV for each test.48 Significance levels of MR results were determined based on the Benjamini-Hochberg corrected FDR with a threshold of <0.05. The MR analysis of platelet count were further replicated using GWAS of platelet count.17
Colocalization analysis
Colocalization tests were conducted on each pair of ex-miRNA and whole blood mRNA, which shared at least one cis-eQTL (i.e., cis-miR-eQTL-p is the same as the cis-mRNA-eQTL-w). A Bayesian colocalization method implemented in the coloc.abf() function in an R package was implemented to assess the likelihood of a specific variant acting as a causal factor for the expression levels of an ex-miRNA and whole blood mRNA pair.42 Each cis-eQTL was assigned to one of the five hypotheses: H0: there exist no causal variants for either the plasma or the whole blood expression; H1: there exists a causal variant for the plasma expression only; H2: there exists a causal variant for the whole blood expression only; H3: there exist two distinct causal variants, one for the plasma expression and one for the whole blood expression; or H4: there exists a single causal variant for both the plasma and the whole blood expression. The result of this procedure is five posterior probabilities (PP0, PP1, PP2, PP3 and PP4) for each hypothesis. In this study, the association results between SNPs and ex-miRNA, and between SNPs and whole blood miRNA or mRNA within a 2 Mb region were used as input. Prior probabilities were set as default values, that is p1 and p2 are both set to 1E−4, and p12 is set to 1E−5. An SNP was considered to be colocalized for an ex-miRNA with a whole blood miRNA (or mRNA) if the posterior probability (PP4) was greater than or equal to 80%.
cis-mRNA-eQTL-w was calculated in 2622 samples whose WGS and whole blood RNA-seq data were both available as described previously.15
Coexpression analysis
Coexpression analysis of ex-miRNA with whole blood mRNA was performed on all profiled protein-coding or non-coding mRNAs (N = 37,773) and 282 ex-miRNAs, utilizing data from 2,732 FHS participants where both ex-miRNA and whole blood mRNA data were available. Linear mixed models were utilized to perform pairwise association analyses of ex-miRNAs and whole blood mRNAs. Covariates considered in the models included age, sex, imputed blood cell types for whole blood samples, technique covariates (e.g., batch) for miRNA and mRNA measurements, surrogate variables (SVs) of RNAseq data, and family ID. These SVs were computed using the 'SVA' R package.43 To control for multiple testing, the Benjamini–Hochberg method was applied to calculate FDR. Subsequently, coexpression pairs with an FDR <0.01 were considered statistically significant and selected for further analysis.
miRNA targets
For the coexpressed miRNA–mRNA pairs, we used miRDB version 6.0 to evaluate whether the mRNAs were the corresponding targets for the miRNAs.46 miRDB is a comprehensive database, offers an extensive collection of predicted and experimentally validated miRNA-target interactions, making it a valuable tool for understanding the regulatory roles of miRNAs in gene expression. Leveraging the latest version 6.0 of miRDB ensures that we incorporate the most up-to-date and accurate information in our analysis, providing a robust foundation for our research.
Gene ontology (GO) enrichment analysis
GO biological process (GO-BP) enrichment analysis was performed for mRNA which coexpressed with miRNAs using the R package enrichR.44 Fisher’s exact tests were utilized to assess whether the set of mRNAs showed enrichment for genes involved in certain GO-BP terms. The significant threshold used a corrected FDR<0.05 for multiple terms.
Replication of exQTLs
We replicated the significant cis- and trans-exQTLs identified in FHS by investigating the SNP-ex-miRNA associations in a separate cohort of 2,178 participants from the RS. The methods used in this replication study are described in detail in the ref.13
RS is a prospective population-based cohort study that focuses on middle-aged and elderly individuals residing in the suburb of Ommoord, Rotterdam, the Netherlands. Genotyping in the RS cohort was carried out using either the HumanHap550 Duo 446 BeadChip (Illumina, San Diego, California) or the Global Screening Array 447 (GSAMD-v3) Illumina array. Imputation of genotypes was performed using the MaCH/minimac software with reference to the 1000 Genomes dataset. Genetic variants with a minor allele frequency <0.05 and imputation quality <0.7 were excluded from the analysis after genotype imputation. To measure plasma miRNA levels, the HTG EdgeSeq miRNA Whole Transcriptome Assay, capable of quantitatively detecting the expression of 2,083 human miRNA transcripts, was used (Molecular Diagnostics, Tuscon, AZ, USA). Additionally, the Illumina NextSeq 500 sequencer (Illumina, San Diego, CA, USA) was employed to facilitate this analysis. To assess the associations between each SNP and each of the 2,083 miRNAs, multiple linear regression analyses were conducted on the subset of 2,178 participants randomly selected from three sub-cohorts within the Rotterdam Study. These analyses were adjusted for age, sex, sub-cohort membership, and the first five principal components (to account for population stratification).
Associations of ex-miRNA with platelet traits
For association analyses of each ex-miRNA with platelet traits including PLT and MPV, covariates included age, age2, sex, HDL, TC, and TG as fixed effects, with family ID as a random effect. The significance threshold used a corrected FDR<0.05.
Published: September 18, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.110988.
Contributor Information
Tianxiao Huan, Email: huant@nih.gov.
Daniel Levy, Email: levyd@nih.gov.
Supplemental information
References
- 1.Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 2.Bartel D.P. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Brien K., Breyne K., Ughetto S., Laurent L.C., Breakefield X.O. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat. Rev. Mol. Cell Biol. 2020;21:585–606. doi: 10.1038/s41580-020-0251-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garcia-Martin R., Wang G., Brandão B.B., Zanotto T.M., Shah S., Kumar Patel S., Schilling B., Kahn C.R. MicroRNA sequence codes for small extracellular vesicle release and cellular retention. Nature. 2022;601:446–451. doi: 10.1038/s41586-021-04234-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mori M.A., Ludwig R.G., Garcia-Martin R., Brandão B.B., Kahn C.R. Extracellular miRNAs: from biomarkers to mediators of physiology and disease. Cell Metab. 2019;30:656–673. doi: 10.1016/j.cmet.2019.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim S., Jeon O.H., Jeon Y.-J. Extracellular RNA: Emerging roles in cancer cell communication and biomarkers. Cancer Lett. 2020;495:33–40. doi: 10.1016/j.canlet.2020.09.002. [DOI] [PubMed] [Google Scholar]
- 7.Jones Buie J.N., Goodwin A.J., Cook J.A., Halushka P.V., Fan H. The role of miRNAs in cardiovascular disease risk factors. Atherosclerosis. 2016;254:271–281. doi: 10.1016/j.atherosclerosis.2016.09.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nikpay M., Beehler K., Valsesia A., Hager J., Harper M.-E., Dent R., McPherson R. Genome-wide identification of circulating-miRNA expression quantitative trait loci reveals the role of several miRNAs in the regulation of cardiometabolic phenotypes. Cardiovasc. Res. 2019;115:1629–1645. doi: 10.1093/cvr/cvz030. [DOI] [PubMed] [Google Scholar]
- 9.Creemers E.E., Tijsen A.J., Pinto Y.M. Circulating microRNAs: novel biomarkers and extracellular communicators in cardiovascular disease? Circ. Res. 2012;110:483–495. doi: 10.1161/CIRCRESAHA.111.247452. [DOI] [PubMed] [Google Scholar]
- 10.Sheinerman K.S., Toledo J.B., Tsivinsky V.G., Irwin D., Grossman M., Weintraub D., Hurtig H.I., Chen-Plotkin A., Wolk D.A., McCluskey L.F., et al. Circulating brain-enriched microRNAs as novel biomarkers for detection and differentiation of neurodegenerative diseases. Alzheimer's Res. Ther. 2017;9:89. doi: 10.1186/s13195-017-0316-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huan T., Rong J., Liu C., Zhang X., Tanriverdi K., Joehanes R., Chen B.H., Murabito J.M., Yao C., Courchesne P., et al. Genome-wide identification of microRNA expression quantitative trait loci. Nat. Commun. 2015;6:6601. doi: 10.1038/ncomms7601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akiyama S., Higaki S., Ochiya T., Ozaki K., Niida S., Shigemizu D. JAMIR-eQTL: Japanese genome-wide identification of microRNA expression quantitative trait loci across dementia types. Database. 2021;2021 doi: 10.1093/database/baab072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghanbari M., Mustafa R., Mens M., van Hilten A., Huang J., Roshchupkin G., Huan T., Broer L., Elliott P., Levy D. An Atlas of Genetic Regulation and Disease Associations of microRNAs. medRxiv. 2022 doi: 10.1101/2022.11.10.22282180. Preprint at. [DOI] [Google Scholar]
- 14.Freedman J.E., Gerstein M., Mick E., Rozowsky J., Levy D., Kitchen R., Das S., Shah R., Danielson K., Beaulieu L., et al. Diverse human extracellular RNAs are widely detected in human plasma. Nat. Commun. 2016;7 doi: 10.1038/ncomms11106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu C., Joehanes R., Ma J., Wang Y., Sun X., Keshawarz A., Sooda M., Huan T., Hwang S.-J., Bui H., et al. Whole genome DNA and RNA sequencing of whole blood elucidates the genetic architecture of gene expression underlying a wide range of diseases. Sci. Rep. 2022;12 doi: 10.1038/s41598-022-24611-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buniello A., MacArthur J.A.L., Cerezo M., Harris L.W., Hayhurst J., Malangone C., McMahon A., Morales J., Mountjoy E., Sollis E., et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47:D1005–D1012. doi: 10.1093/nar/gky1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen M.-H., Raffield L.M., Mousas A., Sakaue S., Huffman J.E., Moscati A., Trivedi B., Jiang T., Akbari P., Vuckovic D., et al. Trans-ethnic and ancestry-specific blood-cell genetics in 746,667 individuals from 5 global populations. Cell. 2020;182:1198–1213.e14. doi: 10.1016/j.cell.2020.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vuckovic D., Bao E.L., Akbari P., Lareau C.A., Mousas A., Jiang T., Chen M.-H., Raffield L.M., Tardaguila M., Huffman J.E., et al. The polygenic and monogenic basis of blood traits and diseases. Cell. 2020;182:1214–1231.e11. doi: 10.1016/j.cell.2020.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagalla S., Shaw C., Kong X., Kondkar A.A., Edelstein L.C., Ma L., Chen J., McKnight G.S., López J.A., Yang L., et al. Platelet microRNA-mRNA coexpression profiles correlate with platelet reactivity. Blood. 2011;117:5189–5197. doi: 10.1182/blood-2010-09-299719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu F., Dong H., Mei Z., Huang T. Investigation of miRNA and mRNA Co-expression Network in Ependymoma. Front. Bioeng. Biotechnol. 2020;8:177. doi: 10.3389/fbioe.2020.00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Do D.N., Dudemaine P.-L., Fomenky B.E., Ibeagha-Awemu E.M. Integration of miRNA weighted gene co-expression network and miRNA-mRNA co-expression analyses reveals potential regulatory functions of miRNAs in calf rumen development. Genomics. 2019;111:849–859. doi: 10.1016/j.ygeno.2018.05.009. [DOI] [PubMed] [Google Scholar]
- 22.Liu H., Kohane I.S. Tissue and process specific microRNA–mRNA co-expression in mammalian development and malignancy. PLoS One. 2009;4 doi: 10.1371/journal.pone.0005436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McManus D.D., Rong J., Huan T., Lacey S., Tanriverdi K., Munson P.J., Larson M.G., Joehanes R., Murthy V., Shah R., et al. Messenger RNA and MicroRNA transcriptomic signatures of cardiometabolic risk factors. BMC Genom. 2017;18:139. doi: 10.1186/s12864-017-3533-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dimmeler S., Ylä-Herttuala S. 14q32 miRNA cluster takes center stage in neovascularization. Am. Heart Assoc. 2014;115:680. doi: 10.1161/CIRCRESAHA.114.304973. [DOI] [PubMed] [Google Scholar]
- 25.Landry P., Plante I., Ouellet D.L., Perron M.P., Rousseau G., Provost P. Existence of a microRNA pathway in anucleate platelets. Nat. Struct. Mol. Biol. 2009;16:961–966. doi: 10.1038/nsmb.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edelstein L.C., Simon L.M., Montoya R.T., Holinstat M., Chen E.S., Bergeron A., Kong X., Nagalla S., Mohandas N., Cohen D.E., et al. Racial differences in human platelet PAR4 reactivity reflect expression of PCTP and miR-376c. Nat. Med. 2013;19:1609–1616. doi: 10.1038/nm.3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wezel A., Welten S.M.J., Razawy W., Lagraauw H.M., de Vries M.R., Goossens E.A.C., Boonstra M.C., Hamming J.F., Kandimalla E.R., Kuiper J., et al. Inhibition of microRNA-494 reduces carotid artery atherosclerotic lesion development and increases plaque stability. Ann. Surg. 2015;262:841–848. doi: 10.1097/SLA.0000000000001466. [DOI] [PubMed] [Google Scholar]
- 28.Sunderland N., Skroblin P., Barwari T., Huntley R.P., Lu R., Joshi A., Lovering R.C., Mayr M. MicroRNA biomarkers and platelet reactivity: the clot thickens. Circ. Res. 2017;120:418–435. doi: 10.1161/CIRCRESAHA.116.309303. [DOI] [PubMed] [Google Scholar]
- 29.Gutmann C., Mayr M. Circulating microRNAs as biomarkers and mediators of platelet activation. Platelets. 2022;33:512–519. doi: 10.1080/09537104.2022.2042236. [DOI] [PubMed] [Google Scholar]
- 30.Shi J., Chen M., Ouyang L., Wang Q., Guo Y., Huang L., Jiang S. miR-142-5p and miR-130a-3p regulate pulmonary macrophage polarization and asthma airway remodeling. Immunol. Cell Biol. 2020;98:715–725. doi: 10.1111/imcb.12369. [DOI] [PubMed] [Google Scholar]
- 31.Poodineh J., Sirati-Sabet M., Rajabibazl M., Mohammadi-Yeganeh S. MiR-130a-3p blocks Wnt signaling cascade in the triple-negative breast cancer by targeting the key players at multiple points. Heliyon. 2020;6 doi: 10.1016/j.heliyon.2020.e05434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li W., Shan B.Q., Zhao H.Y., He H., Tian M.L., Cheng X., Qin J.B., Jin G.H. MiR-130a-3p regulates neural stem cell differentiation in vitro by targeting Acsl4. J. Cell Mol. Med. 2022;26:2717–2727. doi: 10.1111/jcmm.17285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y., Shi M., Hong Z., Kang J., Pan H., Yan C. MiR-130a-3p has protective effects in alzheimer’s disease via targeting DAPK1. Am. J. Alzheimers Dis. Other Demen. 2021;36 doi: 10.1177/15333175211020572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu J., Gao F. Circulating miR-130a-3p is elevated in patients with cerebral atherosclerosis and predicts 2-year risk of cerebrovascular events. BMC Neurol. 2022;22:308. doi: 10.1186/s12883-022-02829-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen S.P., Chang Y.A., Chou C.H., Juan C.C., Lee H.C., Chen L.K., Wu P.C., Wang Y.F., Fuh J.L., Lirng J.F., et al. Circulating microRNAs associated with reversible cerebral vasoconstriction syndrome. Ann. Neurol. 2021;89:459–473. doi: 10.1002/ana.25965. [DOI] [PubMed] [Google Scholar]
- 36.Hofmeister R.J., Rubinacci S., Ribeiro D.M., Buil A., Kutalik Z., Delaneau O. Parent-of-Origin inference for biobanks. Nat. Commun. 2022;13:6668. doi: 10.1038/s41467-022-34383-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seitz H., Royo H., Bortolin M.-L., Lin S.-P., Ferguson-Smith A.C., Cavaillé J. A large imprinted microRNA gene cluster at the mouse Dlk1-Gtl2 domain. Genome Res. 2004;14:1741–1748. doi: 10.1101/gr.2743304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taliun D., Harris D.N., Kessler M.D., Carlson J., Szpiech Z.A., Torres R., Taliun S.A.G., Corvelo A., Gogarten S.M., Kang H.M., et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature. 2021;590:290–299. doi: 10.1038/s41586-021-03205-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rentzsch P., Witten D., Cooper G.M., Shendure J., Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47:D886–D894. doi: 10.1093/nar/gky1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burgess S., Thompson S.G. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur. J. Epidemiol. 2017;32:377–389. doi: 10.1007/s10654-017-0255-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang J., Lee S.H., Goddard M.E., Visscher P.M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Giambartolomei C., Vukcevic D., Schadt E.E., Franke L., Hingorani A.D., Wallace C., Plagnol V. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10 doi: 10.1371/journal.pgen.1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leek J.T., Johnson W.E., Parker H.S., Jaffe A.E., Storey J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–883. doi: 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A., et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cingolani P., Platts A., Wang L.L., Coon M., Nguyen T., Wang L., Land S.J., Lu X., Ruden D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen Y., Wang X. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020;48:D127–D131. doi: 10.1093/nar/gkz757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pierce B.L., Burgess S. Efficient design for Mendelian randomization studies: subsample and 2-sample instrumental variable estimators. Am. J. Epidemiol. 2013;178:1177–1184. doi: 10.1093/aje/kwt084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wald A. The fitting of straight lines if both variables are subject to error. Ann. Math. Statist. 1940;11:284–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The datasets used for analysis in the present study can be accessed at the dbGAP repository under the accession number phs000007.v33.p14.
-
•
All algorithm and software used are published, established methods and R packages as described in the key resources table and following Methods section.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.